

Abstract
Aims
A physiologically‐based pharmacokinetic (PBPK) model of the vaginal space was developed with the aim of predicting concentrations in the vaginal and cervical space. These predictions can be used to optimize the probability of success of vaginally administered dapivirine (DPV) for HIV prevention. We focus on vaginal delivery using either a ring or film.
Methods
A PBPK model describing the physiological structure of the vaginal tissue and fluid was defined mathematically and implemented in MATLAB. Literature reviews provided estimates for relevant physiological and physiochemical parameters. Drug concentration–time profiles were simulated in luminal fluids, vaginal tissue and plasma after administration of ring or film. Patient data were extracted from published clinical trials and used to test model predictions.
Results
The DPV ring simulations tested the two dosing regimens and predicted PK profiles and area under the curve of luminal fluids (29 079 and 33 067 mg h l–1 in groups A and B, respectively) and plasma (0.177 and 0.211 mg h l–1) closely matched those reported (within one standard deviation). While the DPV film study reported drug concentration at only one time point per patient, our simulated profiles pass through reported concentration range.
Conclusions
HIV is a major public health issue and vaginal microbicides have the potential to provide a crucial, female‐controlled option for protection. The PBPK model successfully simulated realistic representations of drug PK. It provides a reliable, inexpensive and accessible platform where potential effectiveness of new compounds and the robustness of treatment modalities for pre‐exposure prophylaxis can be evaluated.
Keywords: antiretrovirals, pharmacokinetics, pharmacometrics
What is Already Known About this Subject
HIV prevalence is concentrated in sub‐Saharan Africa where it disproportionately impacts young African women and adolescent girls.
Safe and effective vaginal microbicides have the potential to offer female‐controlled HIV protection.
Validated physiologically‐based pharmacokinetic models can address questions regarding the optimal dosing regimen, the optimal formulation and the effect of patient adherence on product effectiveness.
What this Study Adds
A robust physiological and systems modelling framework focusing specifically on vaginally administered dapivirine in different formulations.
Accurate in silico prediction of dapivirine drug concentrations using a physiologically‐based pharmacokinetic model can potentially reduce both the risk and enormous costs associated with bringing new drug compounds and/or formulations to market.
Introduction
HIV continues to be a major global public health concern affecting approximately 36.7 (34.0–39.8) million people worldwide at the end of 2015 1. Prevalence is clearly concentrated in Sub‐Saharan Africa where it still affects approximately 25.6 (23.1–28.5) million people 1. Infection disproportionately impacts young African women and adolescent girls 2 as they make up a significant vulnerable population who account for >50% of cases in Africa and are most at risk of acquiring new infections 2. While the chance of male to female sexual transmission of HIV is relatively low (0.30% in low income countries 3), almost all methods of HIV protection currently available are dependent on male cooperation (e.g. condoms or voluntary male circumcision). The development of female‐controlled, pre‐exposure prophylaxis (PrEP) has the potential to provide vulnerable women with protection from sexually transmitted HIV infection.
The World Health Organization (WHO) guidelines for PrEP currently recommend the use of daily oral PrEP 4. The effectiveness of daily oral dosing with Truvada (emtricitabine and tenofovir disoproxil fumarate) for PrEP has been studied extensively 5, 6, 7, 8, 9, 10, 11. However, many of these studies suffered from some degree of poor patient adherence (reviewed in 11) and the reported reduction in HIV infection rates varied between 44% 5 and 86% 6, 7. Given that the risk of HIV infection is disproportionately high in young African women and adolescent girls, additional options for PrEP are greatly needed.
The development of safe and effective vaginal microbicides for topical PrEP offers women safe, effective alternatives to oral dosing and according to the WHO such products have “great potential to reduce the number of people newly infected if scaled up strategically among populations at high risk of infection” 2 (see 12, 13, 14, 15 for reviews of vaginal microbicides development). The women at risk are a diverse group including both married and unmarried women. It is therefore logical to assume that they will have a variety of differing personal preferences, particularly with regards to their preferred formulation, the product acceptability, and their general interest or concerns (see for example 16). The need to study and develop topical PrEP dosage form options for women is therefore crucial.
This manuscript focuses on the topical antiretroviral (ARV) drug, dapivirine (DPV). DPV is a non‐nucleoside reverse transcriptase inhibitor that blocks HIV's ability to replicate in healthy cells by irreversibly binding and inhibiting HIV reverse transcriptase and thus preventing conversion of viral RNA to previral DNA 17. The metabolism of orally administered DPV appears to be primarily by cytochrome P450 (CYP) isozymes in the liver 18 and while oral absorption is poor, it has been shown to be a promising drug candidate for topical PrEP formulations. DPV has been studied in the clinic in three vaginally applied dosage forms, an intravaginal ring, film and gel.
The DPV ring incorporates drug into a flexible silicone matrix polymer capable of sustaining long‐term ARV release locally 19, 20. Women are able to insert the ring themselves vaginally, where it slowly delivers DPV directly to the site of potential infection until it is removed 1 month later. Early clinical trials showed the ring's long‐term safety and efficacy 20, 21, 22, 23, 24, 25, while later trials found the ring moderately reduced HIV incidence by 27–31%, compared to the placebo, with efficacy improving with increased patient adherence 26, 27. Two open‐label extension studies are currently gathering real world data on the DPV ring including extended safety data, how, when and why women use the ring, the level of patient adherence and the associated ring efficacy 28, 29.
In comparison to the ring, quick dissolving DPV films are a much newer microbicide dosage form which to date have been evaluated in the clinic for coitally dependent protection from HIV infection. It is hoped that films may provide a more acceptable method of vaginal drug delivery than previously seen with gels, given that films can be discreetly used without the leakage issues associated with gel products. The FAME‐02 first‐in‐human study compared the safety, pharmacokinetics (PK) and pharmacodynamics (PD) of gel and film formulations of DPV 30. They concluded that DPV films could effectively deliver ARVs to genital tissue at concentrations sufficient to prevent HIV infection and similar to those seen in sustained delivery systems 30. The FAME‐02B study investigated the multicompartment PK (blood, cervical tissue and cervicovaginal fluid) and ex vivo PD (HIV tissue explant challenge) over one week in women using a single vaginal DPV film or gel product 31. They found both the film and gel were well tolerated and concluded that the PK findings of FAME‐02B were consistent with those measured after seven daily doses (FAME‐02) 31.
Physiologically‐based (PB) PK modelling and simulations can be used to predict the PK profile of a drug in specific tissues and organs by combining physiology, population and drug characteristics. They are particularly useful during the early stages of drug discovery where information on the compound is limited since these models can be informed by in vitro and preclinical data. A validated PBPK model has the potential to aid in the development of vaginally administered drugs, particularly in terms of suggesting the recommended dosing regimen for first‐in‐human studies and further evaluating and optimizing clinical trial design. Moreover, PBPK modelling has become increasingly accepted by regulatory agencies as a means of informing clinical studies a priori, and as such it has become a useful tool in drug development 32, 33. To date, multiple software packages have been developed to allow users to run PBPK simulations 34, 35, 36, 37, 38. However, these models have primarily focused on drugs administered via more traditional routes (i.e. oral, intramuscular, intravenous administration) with the most advanced model methodologies relating to describing oral gut metabolism and transporters. To our knowledge, there are currently no PBPK models that allow the user to administer drugs vaginally.
Vaginal dosing presents a unique set of challenges for PBPK model development. In this case, vaginal administration aims to achieve high concentrations in the local vaginal tissue (to prevent new infections by HIV) and low systemic concentrations (to reduce the chance of adverse events). This requires a detailed, well‐defined description of drug release from different formulations into the vaginal luminal fluids and movement into and through local tissues. Work dedicated to describing the physiology and blood flow rates of vaginal tissue, especially in nonpregnant women, is limited. Similarly, work to investigate potential changes in the vaginal ecology and physiology associated with different drugs and diseases states is particularly under‐investigated. This is perhaps not surprising when one considers the difficulty in obtaining consent to do trials in this area of the body and the highly inaccessible nature of the tissues. Further confounding the model development process is characterization of drug absorption, distribution and elimination. For example, while the absorption profile of a drug administered orally may be well characterized, changing the route of administration can lead to drastically different absorption profiles. There may be flip‐flop kinetics to consider along with increased/decreased exposure in local tissues. Similarly, drug clearance may be well‐known following oral dosing but vaginally dosing aims to dramatically reduce the concentration of drug in the systemic circulation and so it is more important to determine whether the drug can be metabolized and/or eliminated before reaching the systemic circulation (see discussion for an example in DPV).
This paper describes the development of a PBPK model of the vaginal space and specifically focuses on simulating DPV delivered vaginally by either a ring 21 or film 30. Our aim was to provide a robust modelling framework that can be applied to vaginally administered antiretroviral drugs to quantitatively predict local tissue concentrations in the cervicovaginal tract, plasma concentrations and their potential future effectiveness. While we recognize the importance of clinical trials, particularly during drug/formulation development, they require a huge investment of both time and money. We hope that the addition of this validated PBPK model can provide answers to questions regarding the optimal dosing regimen for drugs, the optimal formulation for the drugs, and the effect of patient adherence on product effectiveness.
Methods
A PBPK model characterizing the physiological structure of the vaginal tissue and luminal fluid in humans was developed using a series of ordinary differential equations and implemented in the MATLAB software (version R2017a; Mathworks, Natick, MA, USA).
Structural model
The structural compartments of the PBPK model are shown in Figure 1. Briefly, the model includes five tissue/organ compartments representing the vaginal epithelium, vaginal stroma, lungs, liver and the rest of body. All model compartments were assumed to be homogeneous and well mixed. In this model, drug enters the system via the dosage form (i.e. ring or film), directly into the vaginal luminal fluid compartment. Drug was assumed to leave the system via the liver only. Movement of drug through vaginal tissue was assumed to happen both passively and actively while movement through the systemic circulation was mediated by blood flow rates and partition coefficients. Mass balance equations were written for each compartment; hence, the model is described by a series of differential equations.
Figure 1.
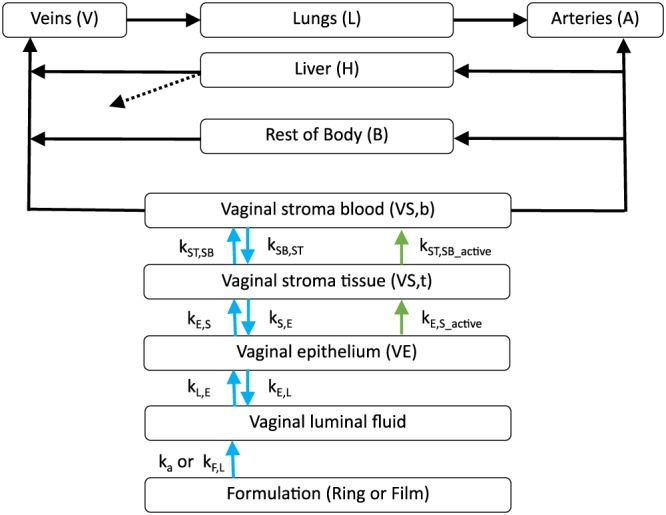
The structural compartments of the physiologically‐based pharmacokinetic model. The compartments represent the ring or film, the vaginal fluid, tissues and organs; connecting arrows represent blood supplies (solid black), elimination of drug (dotted black), passive movement of un‐ionized drug (blue) and active movement of ionized drug (green)
Model equations
A detailed description of all model equations can be found in the supplementary material.
Formulation independent equations
Briefly, drug released from a dosage form (i.e. ring or film) was present in the luminal fluids, epithelium and stroma tissue in both its un‐ionized and ionized form. The change in un‐ionized and ionized drug in the luminal fluid over time was described as:
| (1) |
| (2) |
where D Lumen and D IonLumen represent the amount (mg) of un‐ionized and ionized drug respectively, V Lumen is the volume of the luminal fluid (L), the k parameters are model rate constants (described in Table 2), DR Formulation represents the equation describing drug release from the dosage form (ring/film) into the luminal fluids (described in Equations (6) and (7) respectively), I Lumen is the ionization rate in the lumen and UI is the un‐ionization rate.
Table 2.
Pharmacokinetic parameters
| Description | Abbreviation | Value | CV% | Reference/Assumptions | |
|---|---|---|---|---|---|
| Rate constant (h –1 ) | Film to luminal fluids | kF,L | 4.16 | 30a | 46 |
| Ring to luminal fluids | ka | 7.32 × 10−5 | Variedb | 24 Assuming F = 30%, only 1.2 mg is available over the 28 day period, therefore ka = −ln(23.8/25) / 28 /24 = 7. 32 × 10−5 (h–1) | |
| Decay of ka | ka_exp | 1.2 × 10−3 | 10a | Value identified to replicate trial results | |
| Ring: luminal fluids to epithelium | kL,E_film | 2.773 | 30a | Value identified to replicate trial results. Equivalent to luminal fluid half‐life 0.25 hrs = ln(2)/0.25 = 2.773 | |
| Film: luminal fluids to epithelium | kL,E_ring | 0.058 | 30a | 23 | |
| Epithelium to luminal fluids | kE,L | 0 | 30a | ||
| Epithelium to stroma tissue (passive) | kE,S | 6.79 × 10−3 | 30a | 46 | |
| Epithelium to stroma tissue (active) | kE,S_active | 0.5 | 30a | Value identified to replicate trial results | |
| Stroma tissue to epithelium (passive) | kS,E | 0 | 30a | ||
| Stroma tissue to stroma blood (passive) | kST,SB | 0.122 | 30a | Value identified to replicate trial results | |
| Stroma tissue to stroma blood (active) | kST,SB_active | 0.5 | 30a | Value identified to replicate trial results | |
| Stroma blood to stroma tissue (passive) | kSB,ST | 0.01 | 30a | Value identified to replicate trial results | |
| pKa | pKa | 5.8 | 46 | ||
| Bioavailability (%) | F | 30 | 25–35c | Value identified to replicate trial results | |
| Clearance (l h –1 ) | CL | 4 | 10a | Value identified to replicate trial results | |
| Partition coefficients (tissue/ plasma) | Vaginal stroma | PVS | 6 | 1a | 47 |
| Liver | PH | 4.1 | 1a | 47 | |
| Lungs | PL | 0.18 | 1a | 47 | |
| Body | PB | 8 | 1a | Value identified to replicate trial results | |
| Ionisation Rate | Lumen | ILumen | 0.04 | ‐ | Henderson–Hasselbach equationd |
| Epithelium | IVE | 15.85 | ‐ | Henderson–Hasselbach equationd | |
| Stroma | IS | 15.85 | ‐ | Henderson–Hasselbach equationd | |
| Unionisation rate | UI | 1 | ‐ | Henderson–Hasselbach equationd | |
Coefficient of variation (CV) assumed to be 30% due to lack of data;
Varied according to patient bioavailability;
Random number from a uniform distribution between 25 and 35%;
See main text for further details
Similar equations describing drug movement through the epithelial and stromal tissue in the stromal blood are given in the supplementary material (Equations S3 to S6).
The drug in the systemic circulation was described as:
| (3) |
| (4) |
where the subscripts V, A, VS, CO, B, H and L represent model compartments for the veins, arteries, vaginal stroma, cardiac output, rest of body, liver and lungs, respectively. Q is the blood flow rate (l h–1), D is the amount of drug (mg) and P is the partition coefficient of a given compartment.
The drug concentration in a typical tissue was described as:
| (5) |
where the i subscript refers to the individual tissue being considered, V i is the total volume of the tissue (l), Qi is the tissue blood flow rate or the total cardiac output in lung tissue (l h–1), Di is the amount of drug in tissue (mg) and Pi is the partition coefficient. The liver has an additional clearance term for Equation (5): .
Ring formulation
Drug released from the vaginal ring was described as:
| (6) |
where D Ring is the amount of drug in the ring (mg), k parameters are model rate constants (described in Table 2), and t is the model time. The positive form of this equation describes the drug release, DR Formulation, from the ring formulation to the luminal fluids in Equation (1). The ring simulations (described in section 2.4.1) included use of multiple rings and so the Ring subscript indicates ring specific parameters. The model time was adjusted for rings inserted later in the simulation (i.e. after Day 0) using the model time t minus the time the current ring was inserted t Ring. Further details on how these parameters were derived is included in the section on ring‐dependent parameters below.
Film formulation
Drug release from the film was described as:
| (7) |
where the Film subscript refers to the specific film parameters and V is the volume of the film. The positive form of this equation describes the drug release, DR Formulation, from the film formulation to the luminal fluids in Equation (1). Further description of these parameters is given in below in the section film‐dependent parameters.
Model parameterization
Formulation independent parameters
Extensive literature reviews were performed to determine the PBPK parameters required by the model. The PB parameters included the relative volumes of the model compartments, their associated pH and blood flow rates. The PK parameters included rate constants describing drug movement, the drug's pKa to determine ionization rates, the drug clearance rate and partition coefficients. The final anatomic, physiological and PK parameters and their associated interpatient variability are given in Table 1 and Table 2 respectively.
Table 1.
Physiological parameters
| Description | Abbreviation | Value | CV% | Reference/assumptions | |
|---|---|---|---|---|---|
| Volume (I) | Film | Vfilm | 9.032 × 10−5 | 0 | 46 |
| Luminal fluids | Vlumen | 0.0005 | 71 | 52, 53 | |
| Vaginal epithelium | VVE | 0.077a | Variedb | 39, 40, 42 | |
| Vaginal stroma tissue | VVS,t | 1.078 | ‐ | Estimated based on the relative thickness of the epithelium and the stroma measured in 43 | |
| Vaginal stroma blood | VVS,b | 0.108 | ‐ | Value identified to replicate trial results | |
| Lungs | VL | 1.2 | ‐ | 59 | |
| Liver | VH | 1.7 | ‐ | 59 | |
| Body | VB | 60.5 | ‐ | Inferred from Wt and other volumes | |
| Venous blood | Vv | 3.546 | ‐ | 41 | |
| Arterial blood | VA | 1.746 | ‐ | 41 | |
| Weight (kg) | Wt | 70 | 70 | 10c | |
| Blood flow rate (l h –1 ) | Total cardiac output | QCO | 294.0 | 30d | The cardiac output for the average person at rest = 70 beats min–1 × 70 ml beat–1 = 4900 ml min–1 |
| Vaginal stroma | QVS | 14.3 | ‐ | 45 | |
| Liver | QH | 98.8 | ‐ | 41 | |
| Body | QB | 180.9 | ‐ | QB = QCO – QH – QLu – Qvs | |
| pH | Luminal fluid | ‐ | 4.4 | 1.8 | 60, 61 |
| Rest of body | ‐ | 7.0 | 1.8e | 62 | |
The mean epithelium volume was calculated from the mean cell diameter (63.95 μm), the mean vagina surface area (87.46 cmb), the mean number of cell layers (27) and the mean thickness of a single cell layer (250 μm)
Variability was incorporated into the intermediary parameters assuming 18%, 8.9%, 2.3% and 10% coefficient of variation (CV) for cell diameter, vagina surface area, number of cell layers and thickness of a single cell layer, respectively.
Assumed to be normally distributed
Variability (CV = 30%) was added to the beats per minute and stroke volume for each patient. This was used to update the QCO. The ratio of blood flow rates between QCO and QVS, QH and QB of a typical patient was maintained.
Equivalent to luminal fluid estimate
The parameters describing the vaginal physiology were largely inferred indirectly from other data. For example, the volume of the vaginal epithelium (V VE) was determined from the size of a typical vaginal cell 39, the vaginal surface area 40, the thickness of a single cell layer 41 and the typical number of cell layers 42. Variability in the epithelium volume was incorporated by varying these intermediary parameters according to the coefficient of variation (CV) percentage reported in the literature (Table 1). Gao and Katz 43 measured the thickness of the vaginal epithelium and stroma at 200 μm and 2800 μm respectively. Given their observation that the stroma is approximately 14 times thicker than the epithelium, it was assumed the volume of the stromal tissue (V VS,t) was also 14 times greater, i.e. (0.077 l × 14) = 1.078 l. There are currently no estimates of the volume of blood in the stromal tissue (V VS,b) and so we used estimates of the dermal blood volume measured by Yudovsky and Pilon 44 as a proxy. They developed a method to inversely measure the blood volume percentage in the inner/outer forearm and forehead of patients; measurements varied from 0.78 to 2.06% blood volume. The stromal tissue was expected to have a rich blood supply and so a value of 2% total blood volume was used i.e. total blood volume (5.4 l) × 0.02 = 0.108 l. A sensitivity analysis was performed and the volume had only limited impact on the simulation results. Estimates of the volume for all other physiological compartments were taken directly from the literature (see Table 1). Variability was incorporated into these tissue volumes using the specific patients weight to maintain the ratio of tissue volumes between patients.
The total cardiac output was found by multiplying the heart rate by the stroke volume. For the average patient at rest, the heart rate and stroke volume were determined to be 70 beats min–1 and 70 ml min–1, respectively, which gives a resting heart rate of 4900 ml min–1 or 294 l h–1 (Table 1). The blood flow rates of the lungs and liver were taken from 41. The only available data on blood flow rates in the vaginal stroma came from nine volunteers with normal pregnancies 45. Issa et al. 45 reported the mean blood flow rate in the uterine vein and artery as 274 ml min–1 and 203 ml min–1 respectively. The median value of 238.5 ml min–1 (or 14.3 l h–1; Table 1) was used in our simulations. This is likely to be an overestimate as vaginal blood flow increases during pregnancy but the paper does not report what stage of pregnancy women are in or how much they expect blood flow rates to have increased from the baseline (i.e. nonpregnant flow rates). Variability was incorporated in to the blood flow rates by varying the heart rate and stroke volume (assuming 30% CV and log‐normal distribution) and recalculating the total cardiac output. The ratio of blood flow rates was then maintained by multiplying the mean flow rate percentage by the new total cardiac output.
The ionization rate of drugs in the luminal fluid, epithelium and stroma was derived using the Henderson–Hasselbach equation:
| (8) |
where the pKa is the acid dissociation constant and DPV has a pKa of 5. 8 46. Human tissue, in this case vaginal epithelium and stroma, was assumed to have a pH of 7 (Table 1) which gives a ratio of base: acid of 15.85: 1. Vaginal luminal fluid was assumed to have a pH of 4.4 (Table 1) and an ionization ratio base: acid of 0.04: 1.
The apparent tissue permeability of DPV was determined from a Franz cell model using excised human cervical tissue after first removing excess stroma tissue to isolate the epithelium layer. We used the permeability of epithelium tissue (mean 1.66 × 10−6 cm s–1 46) multiplied by the vaginal surface area (estimated mean of 87.46 cm2 40) to estimate a clearance rate of DPV from the epithelium (i.e. 1.66 × 10−6 × 87.46 = 1.45 × 10−4 cm3 s–1 or 5.23 × 10−4 l h–1) and divided this by the volume of the epithelium to find the rate of DPV movement from the epithelium to the stroma (k E,S). Due to a lack of data describing the passive and active rate of drug movement, we tested different rate constants (see below for specific details) until the simulations were able to replicate the clinical trial results from three different dosing regimens with two different formulations.
The bioavailability of vaginally administered formulations was systematically reduced until the simulated concentrations in the luminal fluid and tissues matched those measured in clinical trials; a value of 30% was chosen. Variability was incorporated by choosing a random number from a uniform distribution between 25–35%. Orally administered DPV appears to be primarily cleared by CYP isozymes in the liver 18 and so we assumed that vaginally administered DPV was also eliminated from the liver only (Figure 1). The rate of metabolic clearance was determined by performing a parameter scan in simbiology, this allows the user to determine the effect of varying the value of a parameter on the species of interest. The final value ensured simulations were able to replicate field data (Table 2).
Partition coefficients for DPV were estimated from data in Neves et al. 47 who administered 25 μl of fluorescent DPV‐loaded nanoparticles intravaginally to 8–12‐week‐old mice. The mice were sacrificed at predetermined time points to determine drug concentration and PK parameters. Figure 4b of 47 shows the DPV levels in different tissues, organs and blood plasma following daily vaginal administration of DPV‐loaded nanoparticles. We digitized these data to determine the mean DPV concentration on days 1 and 14 in each tissue/organ/plasma and used the plasma: tissue ratio to inform the partition coefficients (Table 2). The day 1 and day 14 DPV concentrations were found to be 0.005 μg ml–1 and 0.006 μg ml–1 respectively in plasma, 0.02 μg g–1 and 0.025 μg g–1 in the liver, and 0.028 μg g–1 and 0.17 μg g–1 in the vaginal tissue (Figure 4b of 47). However, the authors were unable to detect DPV in the lungs at either time point and so for the purpose of calculating a partition coefficient we assumed the concentration was below the limit of detection (0.001 μg g–1; reported in the supplementary information of 47). Given that the tissues measured here related to the reproductive system and three of the four organs had undetectable DPV concentrations on either day 1 or 14, the partition coefficient of our rest of body compartment (P B) was assigned by testing different values until simulations were able to replicate observed data.
Figure 4.
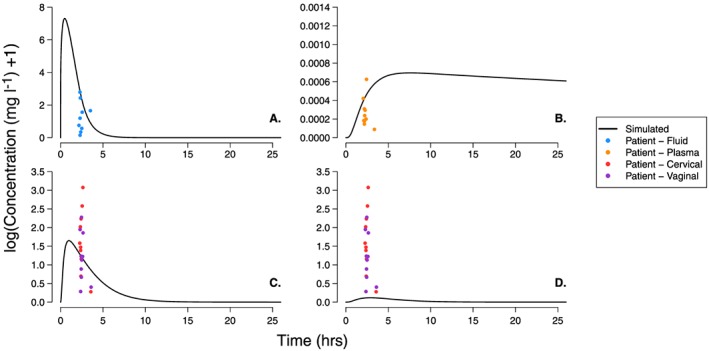
Dapivirine (DPV) concentration–time profiles following administration of a single vaginal film measured in the vaginal fluid, plasma, epithelium tissue and stroma tissue. The black line represents the DPV concentration of a single patient simulated using the physiologically‐based pharmacokinetic parameters from Tables 1 and 2. The coloured dots represent single time point measurements in patients participating in the FAME 02 study; DPV concentration was determined in the vaginal fluid (blue), plasma (orange), cervical tissue (red) and vaginal tissue (purple) for each patient in the hours following administration of dose 7. As the study did not provide information on the proportion of epithelium or stroma tissue present in their tissue samples, we include both vaginal and cervical tissue measurements on the plot. The individual panels show the DPV concentration in: (A) vaginal luminal fluid; (B) plasma; (C) vaginal epithelial tissue; and (D) vaginal stromal tissue
Ring dependent parameters
The DPV ring simulated here is an active vaginal ring (Ring‐004) containing a loading dose of 25 mg of DPV dispersed in a platinum‐catalysed silicone elastomer 21. Nel et al. 24 recently performed a safety, acceptability and adherence study of the DPV ring (Ring‐004) in multiple sub‐Saharan African countries and found that only approximately 4 mg of the 25 mg loaded DPV was released over 28 days. We therefore determined the maximum rate of drug release from the ring to luminal fluids (k a) was –ln (21/25) / 28 / 24 = 2.59 × 10−4 h–1 when assuming 100% bioavailability. To account for the reduced bioavailability, we assume that only 30% (1.2 mg) is released from the ring during the simulation and reduce the drug release rate (k a) accordingly [−ln (23.8/25) / 28 / 24 = 7.32 × 10−5 h–1; Table 2]. Drug was assumed to be released from the surface of the ring first, the rate of release was expected to reduce over time as an area of drug depletion is created at the surface of the ring structure. Subsequent drug must then diffuse through the ring structure before being released and the larger this drug‐depletion zone, the slower the drug is released 19. To mimic this in silico, we allow the initial rate of drug release (k a) to decay exponentially over time according to the rate constant, k a_exp. The value of k a at time t, is therefore:
| (9) |
The value of k a_exp used here was determined by testing multiple values to find which provided the best fit to data (Table 2).
The rate of drug movement from the vaginal fluids to tissue was based on the assumption that all drug released from the ring moves into the tissue. Nel et al. 23 measured the level of DPV in vaginal fluids after ring removal in eight healthy women and determined a mean value of 12–14 h. We used a terminal elimination half‐life of 12 h to describe drug movement from luminal fluids to tissue.
Film‐dependent parameters
Each DPV film had a target loading dose of 1.25 mg and was designed to dissolve rapidly in the vaginal luminal fluids 46. Full details on the development and characterization of the film can be found in Akil et al. 46; a standard class IV United States Pharmacopeia method with a 1% Cremophor aqueous solution was used to test DPV film dissolution. They found DPV was rapidly released from the film, with 50% of the DPV released in <10 min (Figure 5 of 46). We therefore assumed each film had a half‐life of 10 min, which is equivalent to a drug release rate constant (k F,L) of 4.16 h–1.
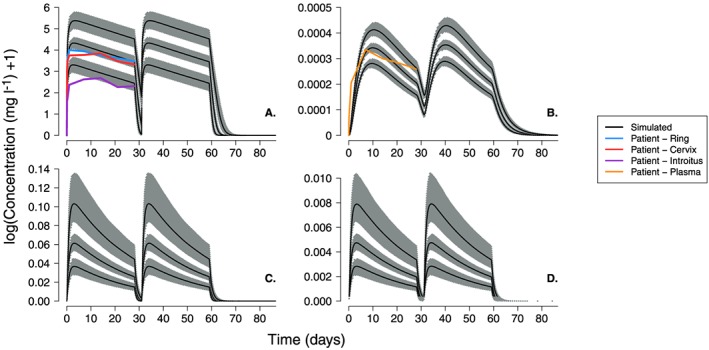
Figure 5.
Dapivirine concentration–time profiles as for Figure 2 but including variability to allow for visual predictive check of simulated profiles. The visual predictive check simulations were run 1000 times for 30 patients with variability incorporated in to all model parameters using the values and associated coefficients of variation given in Tables 1 and 2. In each panel, the grey bands represent the variability in the 5th, 50th and 95th centiles while the black line shows the 50th centile value for each distribution
With the obvious exception of PK parameters that describe the release of drug from the different formulations, the only other parameter that differs between ring and film formulations is the rate of drug movement from the luminal fluids to the epithelium (k L,E). When simulating the ring formulation, the k L,E was informed by the terminal elimination half‐life of DPV in the vaginal fluids (described above). However, using this k L,E rate in the film simulations results in very high, sustained concentrations of DPV in the vaginal fluids and very low tissue/plasma concentrations that were not in line with what was measured during clinical trials. This probably occurs because the film releases the drug very quickly in a short space of time while the ring releases the drug much more slowly over a much longer period. For example, given our assumption of 30% bioavailability (see above), the ring releases 1.2 mg of DPV over 28 days and the film releases 0.375 mg of DPV in approximately 1 h. For simplicity, if we were also to assume a constant release rate of DPV from the ring, we would conclude the ring releases approximately 1.2 (mg) /28 (days) / 24 (h) = 0.0018 mg of DPV h–1 compared to the film's release rate of 0.375 mg h–1. This is equivalent to a 200‐fold increase in the concentration gradient between the luminal fluids to the epithelium following film administration compared to ring administration. As a result, the rate of drug movement from dosage form to lumen in the film simulations was increased to approximately 50‐fold greater than that used in ring simulations to produce simulated drug concentration profiles matched those seen in clinical trials (note, values of approximately 100‐, 150‐ and 200‐fold greater were also tested).
Simulations
The PBPK model was used to simulate DPV concentration–time profiles in the vaginal luminal fluids, tissues and blood plasma following administration of two different DPV formulations. Simulations were implemented in the Simbiology package of Matlab (version R2017a; Mathworks, Natick, MA, USA) for either 100 days (ring simulations) with time steps of 1 h or 100 h (film simulations) with time steps of 0.1 h. The Matlab results were plotted using R (version 3.3.3 48).
Ring simulations
The simulated dosing regimens for the ring study were taken from Nel et al. 21 who followed women using the DPV ring (Ring‐004). The study was a double‐blind randomized, placebo‐controlled trial in which 48 women were assigned to one of two treatment groups. The women in ring group A used two rings over a 56‐day period. The first ring was inserted for 28 consecutive days, upon removal, there was a 3‐day period without a ring and then the second ring was inserted for 28 days. The women in group B used three rings over 57 days. The first ring was inserted for 35 consecutive days, followed by a 3‐day period without a ring, the second ring was inserted for 21 days and on removal of the second ring, a third ring was inserted immediately for 24 h. Each DPV ring contained a loading dose of 25 mg of DPV and was assumed to release approximately 4 mg of drug over 28 days 24. We simulated the results of each regimen in a single patient for 100 days using the mean values of the PBPK parameters described in Table 1 and Table 2. Although patients only had rings inserted for up to 57 days, simulations were run for 100 days to ensure the drug elimination profile in the vaginal fluid, all tissues and plasma was captured. The results were exported to R and the simulated luminal fluid and plasma concentration–time profiles were plotted with the mean patient data (extracted from Figure 2A and 2B of 21 respectively) to visually determine whether simulations were able to capture the mean patient profile. Note that in Nel et al. 21, the original PK profiles plotted in Figure 2A and 2B of 21 include large error bars around each mean value. As the error bars in these plot overlaps substantially, it was unfortunately not possibly to extract and exact estimate of the associated error for each point. Additionally, the luminal concentrations were measured in three locations: the surface of the cervix, introitus and near the site of ring placement. The introitus measurements are sufficiently far away from the luminal fluid and vaginal tissues being simulated here that this study only aims to predict the concentration in the luminal fluids measured in the cervix and near the site of ring placement. We include data measured from luminal fluids on the surface of the introitus only because it was measured in the study and provides an estimate for the amount of drug moving away from the vaginal tissue. Although the DPV tissue concentrations were not reported in patients, the simulated tissue concentrations were plotted for completeness.
Figure 2.
Dapivirine concentration–time profiles following administration of two vaginal rings (Group A dosing regimen in 21) measured in the (A) vaginal fluid, (B) plasma, (C) epithelium tissue and (D) stromal tissue. The first ring was inserted for 28 consecutive days and, upon removal, followed by a 3‐day period without a ring. The second ring was inserted for 28 days before removal. The black line represents the DPV concentration of a single patient simulated using the physiologically‐based pharmacokinetic parameters from Tables 1 and 2. The coloured lines represent the arithmetic mean DPV concentration collected from the vaginal fluid near the site of the ring (blue), from the vaginal fluid on the surface of the cervix (red), the vaginal fluid on the surface of the introitus (purple) and the plasma (orange); patient data represents the mean values extracted was from Figure 2 of 21.
Film simulations
The dosing regimen for the DPV film was taken from the phase I, FAME 02 study; a single‐site, double‐blind, randomised placebo‐controlled trial that compared the safety, PK and PD for DPV dosage forms, a film and gel, with placebo formulations 30. The DPV film had a loading dose of 1.25 mg and with a half‐life of disintegration assumed to be 10 min 46. The study randomized 15 women to the DPV film and each were given seven daily doses of DPV film with doses 1 and 7 administered at the study site and doses 2–6 administered at home 30. Raw data collected directly in the FAME 02 study include single‐time point measurements of the drug concentration in the cervicovaginal lavage, vaginal tissue, cervical tissue and plasma for each patient in the hours following administration of dose 7. The results of a single film application were simulated in a single patient for up to 100 h using the mean PBPK parameters described in Table 1 and Table 2. The simulated results were outputted to R and the cervicovaginal lavage, tissue and plasma concentration–time profiles were plotted with the individual patient data from the FAME 02 study for visual confirmation that the mean parameters were able to capture the general patient profile.
Visual predictive check simulations
After running the individual simulations described above, we ran visual predictive check (VPC) simulations for all ring and film scenarios. All VPC simulations were run 1000 times for 30 patients with variability incorporated using the parameters associated CV values (Table 1 and Table 2) and, assuming a log‐normal distribution (unless otherwise stated), the sampling.
Distribution around the 5th, 50th and 95th percentiles was subsequently plotted. Running a single simulation containing 30 000 patients would allow us to determine only one value for the 5th, 50th and 95th percentiles of the population. This would give an overly optimistic impression of uncertainty – one estimate per percentile – not the uncertainty associated with them. Repeatedly simulating populations of 30 patients provides insight into the simulation predictions at each of the percentiles and whether there was a departure in those percentiles from the observed data. The first VPC simulations were performed to determine whether the model remained predictive when variability was incorporated into parameters that were estimated with some uncertainty and all parameters arbitrarily assigned. This included the volume of the vaginal stroma tissue (V VS,t) and blood (V VS,b), DPV bioavailability (F), which impacts estimates of k F,L, and k a in the film and ring study respectively), DPV (CL), the rest of body partition coefficient (P B) and the following rate constants: k a_exp (ring study only); k L,E_film (film study only); k E,S_active; k ST,SB; k ST,SB_activ;, and k SB,ST (see Tables 1 and 2 for explanation of abbreviations). Sensitivity analyses indicated the concentration in each compartment was sensitive only to parameters defining the rate of drug movement into a compartment, i.e. k L,E_film, k E,S_active and k ST,SB_active for the lumen, epithelium and stroma compartments, respectively (data not shown). The second set of VPC simulations included interpatient variability in all parameters to reflect the true population variability and aimed to show that the model could again capture the patient data from clinical trials.
External model validation
The dosing regimen of a second ring study 23 was used to determine whether the model was capable of predicting the results of a study not included in the original model building process. The chosen study was a double‐blind, randomized, placebo‐controlled trial in which 16 healthy women were randomized 1:1 to use either the active or matching placebo ring for 28 consecutive days 23. The study's active ring was the same as that used in the model building: Ring‐004 containing 25 mg of DPV 21. The ring formulation methods described above and the mean PBPK parameters described in Table 1 and Table 2 were used to simulate the concentration–time profiles of DPV in women for 35 days after the ring was inserted. The resulting profiles were plotted against the average patient profile (extracted from Figure 1 of 23) to visually assess simulation predictions and compared with the select PK parameters given in Table 2 of 23 to allow a quantitative comparison of the model predictions and patient population.
Results
Ring simulation results
A single patient, described using the mean PBPK parameters given in Table 1 and Table 2, was simulated for each of the three dosing regimens and DPV concentrations were estimated in the vaginal fluid, tissue and plasma concentrations over 100 days.
The simulated concentration–time profiles of DPV in the vaginal fluid, plasma, epithelial tissue and stroma tissue, for a single patient, using two or three consecutive DPV rings (regimen A or B) are given in Figures 2 and 3, respectively. Nel et al. 21 determined the DPV concentration in patients’ plasma and vaginal fluid; the vaginal fluid was sampled from the surface of the cervix, the introitus and near the site of ring placement (i.e. vagina). Table 3 shows the results of simulated vaginal fluid and plasma with the reported mean [standard deviation (CV%)] values of: (i) maximum DPV concentration (Cmax); (ii) time to Cmax (Tmax); (iii) DPV concentration prior to ring removal (for all rings); and (iv) DPV concentration immediately following insertion of ring 2. Table 4 shows the area under the concentration–time curve (AUC; mg h l–1) reported in vaginal luminal fluids and plasma after women removed ring one; AUC0–28 for group A and AUC0–35 for group B (reported in Table 2 of 21). This was compared to the corresponding AUC of the extracted (patient data from Figure 2 of 21; see methods) and simulated concentration–time profiles shown in Figure 2 and Figure 3. The simulated AUC of the vaginal fluid was within 1% (group A) and 3–5% (group B) of both the extracted and reported mean AUC (Table 4). Simulations were also able to predict DPV concentrations measured on the surface of the cervix; simulated AUC was approximately 1.1‐fold and 1.3‐fold larger than the extracted and mean reported AUC in groups A and B respectively (Table 4). Notably, DPV concentrations in the luminal fluids sampled from patient's the introitus were approximately 3.5‐fold smaller than measured in the patient's vagina or cervix. The simulated DPV plasma concentration–time profiles are shown on panel B of Figure 2 and Figure 3. While rings one and two were inserted, Table 3 reports concentrations for the simulated profiles and those measured in patients. The simulated AUC differed from the reported and extracted plasma AUCs by 1.01–1.27 fold and 1.16–1.21 fold for groups A and B respectively (Table 4).The simulated plasma concentration following insertion of ring 3, in group B, slightly underestimated the level of DPV accumulation during the 24 h of use (panel B, Figure 3). The DPV concentration in vaginal tissue was simulated and plotted (panels C and D of Figure 2 and Figure 3) but not measured by Nel et al. 21. While the tissue concentration–time profiles follow a similar shape to those measured in the fluids and plasma, the DPV concentration was significantly lower in the stroma tissue compared to the epithelium.
Figure 3.
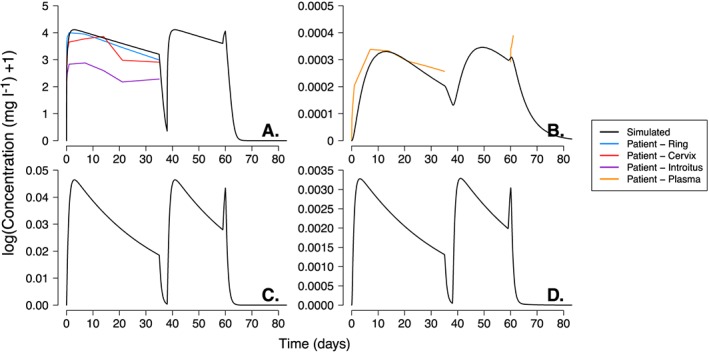
Dapivirine concentration–time profiles following administration of three vaginal rings (Group B dosing regimen in 21) measured in the vaginal fluid, plasma, epithelium tissue and stroma tissue. The first ring was inserted for 35 consecutive days and, upon removal, followed by a 3‐day period without a ring. The second ring was inserted for 21 days and on removal, a third ring was inserted immediately for 24 h. The black line represents the DPV concentration of a single patient simulated using the physiologically‐based pharmacokinetic parameters from Tables 1 and 2. The coloured lines represent the arithmetic mean DPV concentration collected from the vaginal fluid near the site of the ring (blue), from the vaginal fluid on the surface of the cervix (red), the vaginal fluid on the surface of the introitus (purple) and the plasma (orange); patient data represents the mean values extracted was from Figure 2 of 21. The individual panels show the DPV concentration in: (A) vaginal luminal fluid; (B) plasma; (C) vaginal epithelial tissue; and (D) vaginal stromal tissue
Table 3.
Simulated and reported DPV concentration in the luminal fluid and plasma after ring administration for (A) dosing regimen A and (B) dosing regimen B
| A. | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cmax | Tmax | C prior to ring 1 removal | C0 ring 2 | C prior to ring 2 removal | ||||||
| Reporteda | Simulatedb | Reportedc | Simulatedb | Reporteda | Simulatedb | Reporteda | Simulatedb | Reporteda | Simulatedb | |
| Plasma | 3.47e‐4 ± 7.12e‐5 (20.76) | 3.30e‐04 | 170.00 (168–674) | 315.33 | 2.60e‐4 ± 5.83e‐5 (22.40) | 2.48e‐04 | 9.5e‐5 ± 4.99e‐5 (52.46) | 1.65e‐04 | 2.70e‐4 ± 8.46e‐5 (31.28) | 2.49e‐04 |
| Vaginal fluid (cervix) | 61.22 ± 26.09 (42.63) | 60.06 | 24.80 (4.2–506.7) | 71.51 | 27.84 ± 19.28 (69.23) | 28.96 | 0.1856 ± 0.3102 (167.1) | 0.53 | 27.03 ± 12.40 (45.89) | 28.96 |
| Vaginal fluid (introitus) | 21.38 ± 10.54 (49.29) | 336.97 (24.1–675.0) | 10.33 ± 8.235 (79.90) | 0.2810 ± 0.4891 (252.3) | 9.168 ± 6.993 (76.28) | |||||
| Vaginal fluid (near ring) | 65.14 ± 29.83) | 24.32 (4.2–671.8) | 32.41 ± 19.54 (60.30) | 0.8336 ± 2.103 (252.3) | 29.32 ± 16.65 (56.80) | |||||
| B. | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cmax | Tmax | C prior to ring 1 removal | C0 ring 2 | C prior to ring 2 removal | C prior to ring 3 removal | |||||||
| Reporteda | Simulatedb | Reportedc | Simulatedb | Reporteda | Simulatedb | Reporteda | Simulatedb | Reporteda | Simulatedb | Reporteda | Simulatedb | |
| Plasma | 3.59e–4 ± 5.23e–5 (14.60) | 3.30e–04 | 336.00 (168–338) | 317.86 | 2.60e–4 ± 4.56e–5 (15.61) | 2.00e–04 | 1.00e–4 ± 4.32e–5 (43.11) | 1.35e–04 | 2.93e–4 ± 6.85e–5 (23.35) | 2.98e–04 | 3.90e–4 ± 8.64e–5 (22.19) | 3.06e‐04 |
| Vaginal fluid (cervix) | 55.54 ± 24.06 (43.32) | 60.06 | 168.29 (24.2–338.4) | 69.67 | 18.53 ± 12.80 (69.08) | 23.56 | BLQ | 0.43 | 22.42 ± 12.79 (57.05) | 35.65 | 40.06 ± 22.40 (55.90) | 56.88 |
| Vaginal fluid (introitus) | 58.83 ± 145.8 (247.8) | 24.17 (4.1–506.3) | 9.881 ± 4.669 (47.26) | BLQ | 8.327 ± 5.501 (66.07) | 33.46 ± 48.42 (144.7) | ||||||
| Vaginal fluid (near ring) | 67.14 ± 26.84 (39.98) | 168.12 (4.1–506.9) | 20.25 ± 13.17 (65.04) | BLQ | 31.11 ± 19.62 (63.05) | 55.41 ± 42.91 (77.44) | ||||||
Reported values show the mean ± standard deviation (coefficient of variation%) extracted Table 2 of 21; the median or geometric means and standard deviation of the logs were not available in the original publications.
Simulated results come from the single patient simulations using the physiologically‐based pharmacokinetic parameters from Table 1 and Table 2.
Cmax = maximum DPV concentration; Tmax = time to Cmax; C = concentration; C0 = initial concentration; BLQ = below limit of quantification
Table 4.
Simulated and reported DPV area under the concentration–time curve (AUC) in the luminal fluid and plasma after ring administration for dosing regimens A and B
| Group A AUC0–28 (mg h l–1) | Group B AUC0–35 (mg h l–1) | |||||
|---|---|---|---|---|---|---|
| Reporteda | Extractedb | Simulatedc | Reporteda | Extractedb | Simulatedc | |
| Plasma | 0.191 ± 0.038 (20.20) | 0.244 | 0.177 | 0.245 ± 0.0332 (13.52) | 0.256 | 0.211 |
| Vaginal fluid (cervix) | 26 070 ± 12 850 (49.31) | 26 221 | 29 079 | 25 440 ± 11 750 (46.18) | 25 291 | 33 067 |
| Vaginal fluid (introitus) | 7676 ± 3636 (47.37) | 8138 | 21 420 ± 40 180 (187.6) | 9952 | ||
| Vaginal fluid (near ring) | 28 770 ± 14 310 (49.72) | 29 258 | 31 550 ± 12 660 (40.12) | 32 208 | ||
Reported AUC show the mean ± standard deviation (coefficient of variation %) reported in Table 2 of 21; the median or geometric means and standard deviation of the logs were not available in the original publications.
Simulated results come from the single patient simulations using the physiologically‐based pharmacokinetic parameters from Table 1 and Table 2.
AUC = area under the concentration–time curve
Film simulation results
The concentration–time profiles following use of the DPV film was simulated in the vaginal fluid, plasma, epithelium tissue and stroma tissue of a single patient and are shown in Figure 4. The Cmax in the luminal fluid is reached very quickly (approximately 30 min) after film insertion, the concentration then falls to pass through the patient data points measured 2–4 h after film insertion and then below the HIV in vitro IC99 (measure in cervical tissue, 3.3 ng ml–1 21) within 10 h (panel A of Figure 4). The simulated plasma concentration gradually increases to a Cmax of 0.0012 mg l–1 approximately 10 h after film insertion before steadily declining. The plasma concentration remains below the HIV EC99 and above the HIV IC50 (mean IC50 range of 0.09–0.14 ng ml–1 49) for approximately 200 h (panel B of Figure 4). The simulated epithelium and stroma tissue concentrations accumulate very quickly, reaching Cmax approximately 1–3 h after film insertion respectively (panels C and D of Figure 4). DPV concentrations measured in the epithelium pass through the range of values measured in patient vaginal and cervical tissue samples while stroma concentrations are just below. As with the ring simulations, the larger stroma volume means stromal concentrations are approximately 14 times smaller than the those measured in the epithelium.
VPC results
The VPC simulations for the three regimens (Figures 5, 6, 7 and Figure S1 to Figure S3) included 1000 simulations of 30 patients. For each fluid or tissue in a single simulation (i.e. of 30 patients), the 5th, 50th and 95th DPV concentration centile values were determined. The median (50th centile) and range (5th to 95th centile) of these centiles was found across all 1000 simulations and is indicated by the black line and grey bands respectively. So, for example, the top black line and its associated grey band on each plot represent the median and range of the 95th centile DPV concentration.
Figure 6.
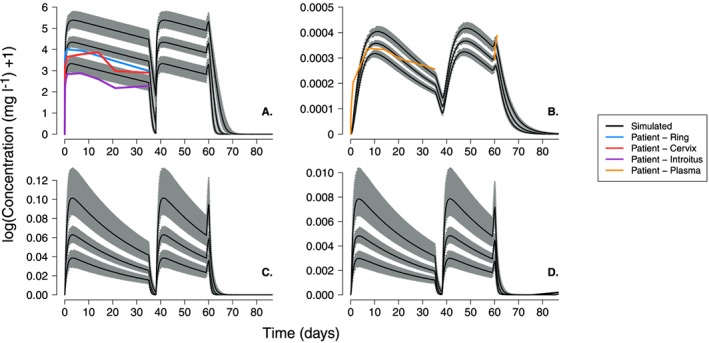
Dapivirine concentration–time profiles as for Figure 3 but including variability to allow for visual predictive check of simulated profiles. The visual predictive check simulations were run 1000 times for 30 patients with variability incorporated in to all model parameters using the values and associated coefficients of variation given in Tables 1 and 2. In each panel, the grey bands represent the variability in the 5th, 50th and 95th centiles while the black line shows the 50th centile value for each distribution
Figure 7.
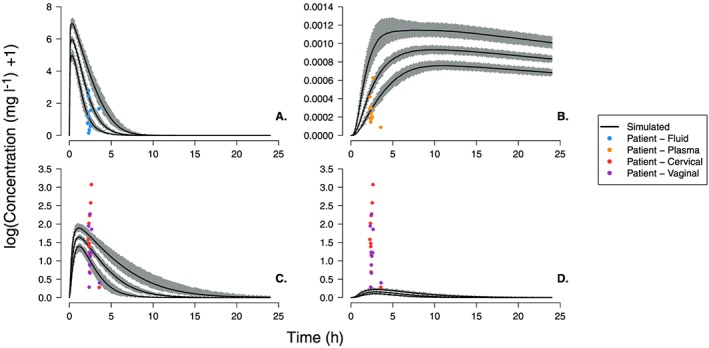
Dapivirine concentration–time profiles as for Figure 4 but including variability to allow for visual predictive check of simulated profiles. The visual predictive check simulations were run 1000 times for 30 patients with variability incorporated in to all model parameters using the values and associated coefficients of variation given in Table 1 and Table 2. In each panel, the grey bands represent the variability in the 5th, 50th and 95th centiles while the black line shows the 50th centile value for each distribution
Ring VPC
The first set of VPC simulations (Figure S1 to Figure S2) included variability in the PBPK parameters that were either estimated with some uncertainty or with uncertainty arbitrarily assigned (see methods). The median 50th centile prediction of DPV concentration in the vaginal fluid almost exactly follows the patient measurements of DPV measured near the ring and cervix (panel A of Figure S1 and Figure S2). As with the single patient simulations, concentrations measured on the surface of the introitus remained below the predicted range. The simulated plasma concentrations were also able to predict patient data when the ring is inserted. However, there are some slight deviations approximately 21 days (~ 500 h) after ring insertion; the fall in the simulated plasma concentrations is slightly quicker than we would expect from the patient measurements. This was particularly evident for the simulations of the group B dosing regimen (Figure S2) where patients wore the first ring for longer (35 days in group B compared to 28 days in group A).
The second set of VPC simulations included variability in all parameters; the results are shown in Figures 5 and 6. Again, the simulated concentration ranges in the vaginal fluids and plasma show close agreement with the patient data, despite the increased level of variability in the patient population. For example, the median 50th centile prediction of DPV concentration in the vaginal fluid almost exactly follows the patient measurements of DPV measured near the ring and cervix. The lower range of the 5th centile values simulated in the vaginal fluids is very close to that measured on the surface of the patients introitus and all other patient data from the ring study are within the simulated range of DPV concentrations.
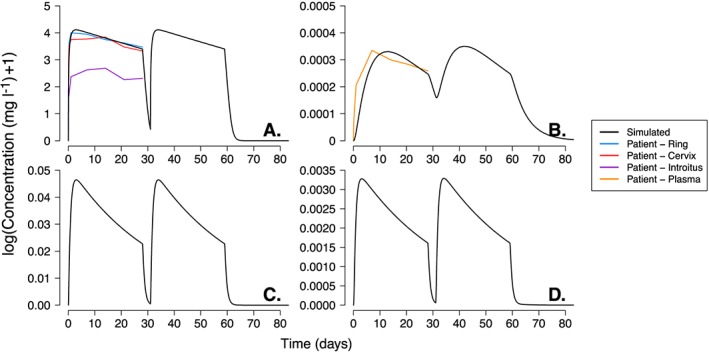
Film VPC
The VPCs of the DPV film (Figures 7 and S3), show the simulated range of DPV in the vaginal fluid and plasma were able to capture the majority of the concentration data points measured in patients. Both sets of VPCs show only a few patient data points (fewer than five) falling below the simulated 5th centile range. The simulated range of epithelium concentrations passes through the middle of the single‐time point patient measurements (panels C and D of Figures 7 and S3) but the range observed in patients vaginal and cervical tissue was much larger (0.3–20.6 mg l–1 and 0.3–8.7 mg l–1 respectively) than simulated range (approximately 1.12–4.75 mg l–1). As with the single patient simulations, the range of stroma concentrations underestimated the DPV concentration measured in patients.
External model validation
The simulated concentration–time profiles of DPV in the vaginal fluid and plasma, epithelial, for a single patient, after using a single DPV ring are given in Figure 8. Nel et al. 24 determined the DPV concentration in patients’ plasma and vaginal fluid; the vaginal fluid was sampled from the surface of the cervix, the introitus and near the site of ring placement (i.e. vagina). These simulations aimed to predict vaginal fluid samples taken from the cervix and near the site of ring placement. Table 5 shows the results of simulated vaginal fluid and plasma with the reported mean [standard deviation (CV%)] or median (range) values of: (i) DPV concentration 1.5 h after ring insertion (C1.5h); (ii) DPV Cmax; (iii) Tmax; (iv) DPV concentration prior to ring removal and; (v) AUC (mg h l–1).
Figure 8.
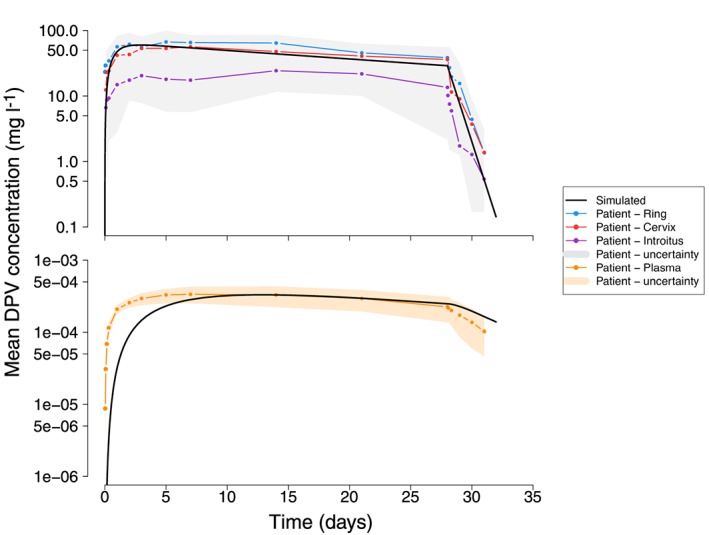
The dapivirine (DPV) concentration–time profiles following insertion of a single vaginal ring for 28 consecutive days measured in the (A) vaginal fluid and (B) plasma. The black line represents the DPV concentration of a single patient simulated using the physiologically‐based pharmacokinetic parameters from Table 1 and Table 2. The coloured lines represent the average DPV concentration collected from patients the vaginal fluid near the site of the ring (blue), from the vaginal fluid on the surface of the cervix (red), the vaginal fluid on the surface of the introitus (purple) and the plasma (orange). The grey and orange bands (panels A and B, respectively) show the range of the patient data error bars extracted from Figure 1 of 23. Note that the original publication 23 does not report whether the data plotted represent the population mean and standard deviation or median and range
Table 5.
Simulated and reported dapivirine concentration and area under the concentration–time curve (AUC) in the luminal fluid and plasma after ring administration
| C 1.5 h after ring insertion | Cmax (mg l–1) | Tmax (h) | C prior to ring removal | vAUC 0‐24 h (mg hr l–1) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Reporteda | Simulatedb | Reporteda | Simulatedb | Reportedc | Simulatedb | Reporteda | Simulatedb | Reporteda | Simulatedb | |
| Plasma | 8.62 e–6 ± 5.38e–6) | 3.23e–08 | 3.55e–4 ± 8.76e–5 (24.78) | 3.31e–04 | 168.54 120.30–336.43) | 311.48 | 2.175e–4 8.238e–5 (37.87) | 2.65E–04 | 3.022e–3 3.891e–4 (12.88) | 2.20E–04 |
| Vaginal fluid (cervix) | 11.980 (6.151) | 4.75 | 66.61 (20.12; 30.21) | 60.03 | 96.60 (4.67–336.47) | 72.87 | 35.78 (18.17; 50.80) | 31.20 | 687.1 (214.1; 21.17) | 625.15 |
| Vaginal fluid (introitus) | 9.416 (3.97) | 31.38 (10.95; 34.90) | 336.39 (48.45–671.40) | 13.26 (11.14; 84.08) | 259.1 (156.9; 60.56) | |||||
| Vaginal fluid (near ring) | 245.50 (130.70) | 79.90 (23.20; 29.03) | 84.68 (24.32–676.70) | 38.59 (13.70; 35.52) | 973.2 (270.2; 27.77) | |||||
Reported values show the mean (± standard deviation; coefficient of variation %) extracted from Table 2 of 21; the median or geometric means and standard deviation of the logs were not available in the original publications.
The simulated concentration–time profiles of DPV in the vaginal fluid closely matched that measured in patients (Figure 8A and Table 5). The simulated AUC of the vaginal fluid was within the reported range of AUC measured on the surface of the cervix and near the ring (Table 5). The simulated DPV plasma concentration–time profiles are shown on panel B of Figure 8 and the Table 5 shows the DPV concentrations and AUC for the simulated profiles and those measured in patients. As with the previous simulations of plasma concentrations, the model predicted a slower rise in plasma levels than that seen in patients (see Tmax and C1.5h, Table 5) but the model was able to almost exactly predict the plasma Cmax [reported Cmax = 3.5 × 10−6 mg l–1 vs. simulated Cmax 3.3 × 10−6 (mg l–1); Table 5].
Discussion
It is clear that achieving safe, effective and affordable female‐driven protection is crucial to reducing HIV infection among women. Accurate models that are able to quantify the effectiveness of vaginal microbicides for PrEP have the potential to aid drug development. This paper details the development, calibration and validation of a PBPK model describing the physiological structure of the vaginal space and the absorption/distribution of vaginally administered drugs using a system of differential equations. The model predictions were tested by simulating the drug concentration–time profiles in the luminal fluids, vaginal tissue and plasma after use of either the DPV ring or film. The results show the final PBPK model, with reduced bioavailability and parameterized using the values in Table 1 and Table 2, was able to reasonably reproduce clinical trial data when simulating DPV delivery via a vaginal ring or film and both qualitatively and quantitatively replicate the results of an independent ring study.
The choice of model PBPK parameters was, where possible, based on the estimates (and their associated variability) reported in the literature. However, data characterizing the vaginal tissue volumes, vaginal blood flow rates and drug movement through the cervicovaginal tract of humans are limited and required some assumptions. For example, the volume of the vaginal epithelium tissue was inferred based on the size of a typical vaginal cell 39, the vaginal surface area 40, the thickness of a single cell layer 41 and the typical number of cell layers 42. The thickness (and hence volume) of the epithelial tissue would also differ throughout a women's menstrual cycle. While this added layer of complexity was not incorporated into this current model, the explicit inclusion of the epithelium tissue allows users to easily incorporate this if they choose to (e.g. through a time‐varying estimate the number of cell layers in the epithelium). This appears to be a reliable approximation for the epithelium but unfortunately, similar details describing the vaginal stroma were much more limited. The volume of the vaginal stroma was therefore based on the observation of Gao and Katz 43 that the stroma is approximately 14 times thicker than the epithelium. The accuracy of this estimate was difficult to test with the data available to us; the ring study did not provide DPV tissue concentrations and the film study provided single time‐point measurements of vaginal and cervical DPV without reference to the proportion of epithelium or stroma tissue present. The results of the film simulations (Figure 7 and Figure S3) suggest that the stromal drug concentration is underestimated by the simulation.
This is likely to be a direct result of the difference in tissue volumes; the stroma tissue was assumed to be 14 times larger than the epithelium and the simulated stroma concentrations are approximately 14 times smaller those simulated in the epithelium (e.g. 0.05 mg l–1/14 = 0.0035 mg l–1). One obvious way to overcome this would be to reduce the volume of the stroma and hence increase the simulated concentrations. However, there are currently no data available to suggest whether this is would be a biologically accurate strategy and we were reluctant to speculate given the equally limited data available describing DPV movement through the stroma. It is also likely that there will be a concentration gradient throughout both vaginal tissues that is not accounted for by the model (which currently assumes compartments are distinct and well mixed); however, the inaccessibility of these tissues would make this difficult to quantify in vivo.
The VPC simulations provided insight into the simulation predictions at each of the percentiles and show there was no great departure in those percentiles from the observed data. To ensure the simulated populations predictions are specific to the population of interest, users can easily incorporate between‐subject differences in drug disposition due to demographic and clinical differences by changing the mean parameter values or variability estimates given in Table 1 and Table 2. For example, the estimate of vaginal surface area used here reflects the mean surface area determined from 62 vinyl polysiloxane casts of the vagina by Pendergrass et al. 40. The original study by Pendergrass et al. 50 obtained the vaginal casts and aimed to compare the vaginal shapes in Afro‐American (n = 23), Caucasian (n = 39) and Hispanic (n = 15) women. They found statistically significant variations in the vaginal shapes and dimensions between the three groups 50. Alongside the mean population estimates of vaginal surface area, the later study 40 also provides data describing the surface area according to the subject's race, parity and vaginal shape. This information can be incorporated into the population simulations through changes in the parameter values to reflect the specific demography of the study population. The only limitation to making the simulations more study‐specific, is the availability of data describing the vaginal space in the target population.
The DPV partition coefficient parameters were estimated from mouse data describing DPV levels in different tissues, organs and blood plasma 47. This relied on two key assumptions: (i) that tissue/organ to plasma ratio is equivalent to the tissue/organ to blood ratio; and (ii) that data generated in a mouse model were representative human partition coefficients. These assumptions were arguably unlikely but without an alternative data source, and with model predictions that were able to replicate field data, they were the best option available. Future applications of this model to other drug compounds would obviously require researchers obtain drug‐specific estimates of these partition coefficient parameters.
Another important assumption made here, was that drug bioavailability was just 30%. This was done to reduce the simulated drug concentrations to within the reported patient range. Without this assumption, the model was able to replicate the shape of the DPV concentration–time profiles in each tissue/fluid but it consistently over‐estimated the amount of drug present. There are a number of possible reasons to explain why the remaining 70% of the drug is unavailable. For example, drug elimination of orally administered DPV occurs primarily in the liver. However, when administered topically, it is possible mucosal tissue or the bacteria present in the luminal fluid may play an important role for local drug metabolism. Zanger et al. 51 determined a small subset of CYP isozymes regulate 70–80% of metabolism for all clinically used drugs, including antiretrovirals. Recently, To et al. 18 demonstrated that CYP enzymes are expressed and active in both vaginal and colorectal tissue. Using tissue biopsies from healthy volunteers, they show both tissues were able to metabolize DPV, producing metabolites that were determined to be CYP‐mediated using human liver microsomes 18. The model currently assumes that any metabolism of DPV occurring in luminal fluid and vaginal tissue is accounted for by reducing the bioavailability parameter. This seems a logical first step as the rate/amount of tissue metabolism is not reported. However, further work to characterize a biotransformation rate of DPV in biologically relevant tissue/fluid would allow for more explicit inclusion of the process in the model.
The reduced bioavailability in this model can also be explained by the movement of DPV into other tissues or fluid leakage. For example, information describing the volume and movement of ambient vaginal fluid is limited, in part, due to the difficultly involved in sampling it. The specific volume of vaginal fluid used here, was based on two studies that determined the median ambient fluid volume was 0.51 ml 52, and reported that patients’ volume ranged from either 0.33–0.69 ml 52 or 0.5–0.75 g (assuming the vaginal fluid density of 1 g cm–3; 53). Masters and Johnson suggested that the presence of vaginal fluid varies along the length of the vaginal canal 54 and, while likely, there is no specific information describing this difference and so this model assumed it was evenly distributed. Owen and Katz 55 previously suggested a maximum fluid volume of approximately 0.75 ml but they reference prior studies suggesting the daily production of vaginal fluid is around 6 g day–1, with approximately 0.5–0.75 g present in the vagina at any one time (see Table 1 of 55). This implies a relatively high rate of fluid turn over and hence a potential for drug loss. The convective effects (or turnover rate) of vaginal fluid also appears to cause rapid distribution of drug across the vaginal surfaces 56 and it seems likely that this carries a significant portion of the drug up into the cervix. Nel et al. 21 show fluid samples taken from the surface of the cervix contained approximately the same concentration of drug as that measured near the ring in the vagina. We chose not to explicitly model the cervical tissue at this time to limit the number of parameters arbitrarily defined. To date, the PB data characterizing cervical tissue volumes, blood flow rates and rates of drug movement (through cervical tissue) is scarce and their inclusion would further increase model uncertainty. It is also possible that DPV moves into colon tissue or the lymphatic system. A preclinical study by Murphy et al. 49, inserted a single 25 mg DPV vaginal ring into four adult female cynomolgus macaques (Macaca fascicularis) for 28 days. After 28 days, animals were necropsied and the DPV concentration characterized in three samples of various tissues (vagina, cervix, uterus, rectum, female genital tract draining lymph nodes and distal lymph nodes). They found DPV was present in the vaginal tissue (1800 ng g–1), the cervix (94 ng g–1) and the rectum (40 ng g–1) but noted no detectable DPV in the uterus, axillary lymph nodes or iliac lymph nodes 49. A study by Nuttall et al. 57, determined drug concentration in the draining lymph nodes of rhesus macaques following once daily administration of [14C]DPV gel for 7 days. They found low levels of DPV and radioactivity were present in the iliac and inguinal lymph nodes at 1 and 24 h after dosing (mean DPV values varied, 3–15 ng g–1, Table 2 of 57). While low, these concentrations are approximately 100‐fold greater than those reported and simulated in the plasma of humans and thus may account for a proportion of unavailable drug.
One current limitation of the model is that the process of in vitro drug release is not differentiated from the formulation and chemical dissolution properties of the formulation in vivo. The current estimate of DPV release rate for the film in vivo was informed by an in vitro DPV film dissolution test which showed 50% of the DPV was released in <10 min 46.
The in vitro experiment was conducted using Cremophor, a nonbiologically relevant media, which is required in the experimental set up to maintain sink conditions due to the hydrophobicity of DPV. The solubility of DPV in this medium is much greater than that anticipated in biological fluids and observations from clinical trials suggest drug release levels, as well as film disintegration rate, are greatly overestimated in experiments with the Cremophor‐containing medium. It was assumed that any effect of the formulation and drug dissolution characteristics were accounted for in the reduced bioavailability.
This paper set out to describe a PBPK model of vaginally administered drugs with specific application to DPV, delivered via a vaginal ring or film. We employed a top‐down approach to model development in which we allowed our understanding of a woman's physiology to dictate the models compartmental structure. However, the limited availability of data describing drug properties lead to difficulties describing drug absorption and movement through vaginal tissues, and thus the model relies on the assumptions outlined above. Describing drug movement using rate constants is not necessarily typical of PBPK models, but in this case, they were often based on some underlying physiological or chemical properties. For example, the rate of drug movement from the luminal fluids to epithelial tissue is based on both vaginal surface area and tissue permeability. The rate constants used herein where chosen to reduce the number of model parameters; however, the user could, of cours,e define these intermediate values and have the model determine a rate of movement if they prefer. This choice has arguably made the resulting model structure more similar to that of a semi‐physiological model rather than a strictly PBPK model. To a certain extent, this limits the degree of generalizability of this model to other drug compounds. However, by highlighting the limitations of the current model structure and describing the types of data required to develop this into a more mechanistic PBPK model, we provide a reliable foundation for future researchers to build on.
Despite these assumptions, the PBPK model described here was able to reproduce patient data and the population variability accurately. Simulations of a single patient using either the ring or film, show DPV concentration–time profiles that closely match the mean profiles extracted from the literature. Following ring insertion, both simulated and patient profiles show DPV concentrations immediately increase rapidly, decrease gradually while the ring is inserted and then fall off rapidly when the ring is removed. The shape of the simulated plasma profiles was similar to that seen in the luminal fluids (i.e. a quick increase following ring insertion, a steady decline over time and then fairly rapid decline after ring removal) but, as seen in patients, systemic concentrations were approximately 4‐fold lower. Following film insertion, DPV was rapidly released into the vaginal fluid and quickly moved into the tissue. Due to the highly hydrophobic nature of DPV 58, movement into the systemic circulation is slow and, as with the ring simulations, the simulated/reported DPV plasma concentration remains much lower than tissue concentrations. The simulated plasma concentration never reached the HIV EC99 remained below the HIV IC50 for approximately 200 h (panel B of Figure 4). The VPC simulations show that, despite the relatively large interpatient variability included in parameters that were either estimated with some degree of uncertainty or arbitrarily assigned, the model was still able to adequately predict the patient concentration–time profiles. The ring VPCs of the epithelium and stroma tissue (panels C and D of Figure S1 and Figure S2), show a relatively large, and sometimes overlapping, range of simulated DPV concentrations. Peak concentrations in simulated stromal tissue varied by as much as 7‐fold compared to approximately 3‐fold variability in the simulated DPV in the vaginal fluid. This is probably due to the higher number of parameters that were estimated with some uncertainty or arbitrarily assigned when determining stromal tissue concentrations. For example, we do not have data directly measuring the volume of stroma or estimating the rate drug moves into and out of the stromal tissue and so it makes sense that when allowing an arbitrary 30% variability in all these parameters it would give the widest range of estimates.
This work provides the first physiological and systems framework that focuses specifically on characterizing the cervicovaginal tract to evaluate vaginally administered DPV in different formulations. Applying this model to other vaginally administered compounds would require users to recalibrate the model in terms of the drug specific parameters. However, the in silico predictions of drug concentration in the vaginal luminal fluid, tissue and plasma of humans presented herein, have the potential to reduce both the risk and enormous costs associated with bringing new drug compounds and/or formulations to market. For example, it allows comparatively quick evaluation of different formulations and dosing regimens, which would lead to more quantitatively informed decisions regarding clinical trial design. This in turn should reduce the risk associated with clinical trials and potentially reduce the number of trials or patients typically required. The model can also be used to evaluate the potential effectiveness of drugs by combining it with PD models of drug effect. Specifically, we have introduced a robust quantitative method that can be used to estimate the efficacy of vaginal PrEP interventions for HIV.
Competing Interests
There are no competing interests to declare.
This research was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health, IPCP Grant U19‐AI120249.
We acknowledge Lindsay Ferguson Kramzer, Pharm.D. Ph.D. (Magee Women's Hospital) for her contribution to the compilation of the FAME‐02 data and Sharon Hillier, Ph.D. (University of Pittsburgh, Magee Women's Hospital) for her intellectual contributions throughout the model development and manuscript preparation.
Contributors
R.B. and L.R. conceived the study. K.K., D.K.S. and R.B. developed the model methodology and interpreted the results. K.K. wrote the computer code, ran the experiments and wrote the first manuscript draft. All authors critically reviewed the manuscript for important intellectual content and approved the final manuscript.
Supporting information
Figure S1 Visual predictive check profiles as for Figure 5 but includes variability in only the parameters that were estimated with some uncertainty and all parameters arbitrarily assigned. The visual predictive check simulations were run 1000 times for 30 patients with variability incorporated in to the estimated model parameters using the values and associated coefficient of variation given in Table 1 and Table 2
Figure S2 Visual predictive check profiles as for Figure 6 but includes variability in only the parameters that were estimated with some uncertainty and all parameters arbitrarily assigned. The visual predictive check simulations were run 1000 times for 30 patients with variability incorporated in to the estimated model parameters using the values and associated coefficient of variation given in Table 1 and Table 2
Figure S3 Visual predictive check profiles as for Figure 7 but includes variability in only the parameters that were estimated with some uncertainty and all parameters arbitrarily assigned. The visual predictive check simulations were run 1000 times for 30 patients with variability incorporated in to the estimated model parameters using the values and associated coefficient of variation given in Table 1 and Table 2
Supporting info item
Kay, K. , Shah, D. K. , Rohan, L. , and Bies, R. (2018) Physiologically‐based pharmacokinetic model of vaginally administered dapivirine ring and film formulations. Br J Clin Pharmacol, 84: 1950–1969. 10.1111/bcp.13625.
References
- 1. UNAIDS . Fact sheet: latest statistics on the status of the AIDS epidemic 2016. Available at http://www.unaids.org/sites/default/files/media_asset/UNAIDS_FactSheet_en.pdf (last accessed 10 April 2017).
- 2. World Health Organization . Global health sector response to HIV, 2000‐2015. 2016: 1–109.
- 3. Boily M‐C, Baggaley RF, Wang L, Masse B, White RG, Hayes RJ, et al Heterosexual risk of HIV‐1 infection per sexual act: systematic review and meta‐analysis of observational studies. Lancet Infect Dis 2009; 9: 118–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. World Health Organization . Guideline on when to start antiretroviral therapy and on pre‐exposure prophylaxis for HIV. 2015: 1–76. [PubMed]
- 5. Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, et al Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med 2010; 363: 2587–2599 10.1056/NEJMoa1011205. PubMed PMID: 21091279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Molina J‐M, Capitant C, Spire B, Pialoux G, Chidiac C, Charreau I, et al On demand PrEP with oral TDF‐FTC in MSM: results of the ANRS Ipergay trial. Conference on Retroviruses and Opportunistic Infections (CROI); Seattle, USA 2015.
- 7. McCormack S, Dunn D. Pragmatic open‐label randomised trial of preexposure prophylaxis: the PROUD study. Conference on Retroviruses and Opportunistic Infections (CROI); Seattle, USA 2015.
- 8. Baeten JM, Donnell D, Ndase P, Mugo NR, Campbell JD, Wangisi J, et al Antiretroviral prophylaxis for HIV prevention in heterosexual men and women. N Engl J Med 2012; 367: 399–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Thigpen MC, Kebaabetswe PM, Paxton LA, Smith DK, Rose CE, Segolodi TM, et al Antiretroviral preexposure prophylaxis for heterosexual HIV transmission in Botswana. N Engl J Med 2012; 367: 423–434. [DOI] [PubMed] [Google Scholar]
- 10. Choopanya K, Martin M, Suntharasamai P, Sangkum U, Mock PA, Leethochawalit M, et al Antiretroviral prophylaxis for HIV infection in injecting drug users in Bangkok, Thailand (the Bangkok Tenofovir study): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet 2013; 381: 2083–2090. [DOI] [PubMed] [Google Scholar]
- 11. Amico KR, Stirratt MJ. Adherence to preexposure prophylaxis: current, emerging, and anticipated bases of evidence. Clin Infect Dis 2014; 59 (Suppl. 1): S55–S60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Friend DR. Recent trends in microbicide formulations. Antiviral Res 2010; 88 (Supplement): S1–S66. [DOI] [PubMed] [Google Scholar]
- 13. Adams JL, Kashuba AD. Formulation, pharmacokinetics and pharmacodynamics of topical microbicides. Best Pract Res Clin Obstet Gynaecol 2012; 26: 451–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morris GC, Lacey CJ. Microbicides and HIV prevention: lessons from the past, looking to the future. Curr Opin Infect Dis 2010; 23: 57–63. [DOI] [PubMed] [Google Scholar]
- 15. Buckheit RW Jr, Watson KM, Morrow KM, Ham AS. Development of topical microbicides to prevent the sexual transmission of HIV. Antiviral Res 2010; 85: 142–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mack N, Evens EM, Tolley EE, Brelsford K, Mackenzie C, Milford C, et al The importance of choice in the rollout of ARV‐based prevention to user groups in Kenya and South Africa: a qualitative study. J Int AIDS Soc 2014; 17 (3 Suppl 2): 19157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. AIDS info drug database. Dapivirine. Available at https://aidsinfo.nih.gov/drugs/523/dapivirine/0/patient (last accessed 21 October 2016).
- 18. Toas EE, Hendrix CW, Bumpus NN. Dissimilarities in the metabolism of antiretroviral drugs used in HIV pre‐exposure prophylaxis in colon and vagina tissues. Biochem Pharmacol 2013; 86: 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Malcolm RK, Edwards KL, Kiser P, Romano J, Smith TJ. Advances in microbicide vaginal rings. Antiviral Res 2010; 88 (Suppl 1): S30–S39. [DOI] [PubMed] [Google Scholar]
- 20. International partnership for microbicides. IPM Product Pipeline Silver Spring, MD 2016. Available at http://www.ipmglobal.org/our-work/product-pipeline (last accessed 19 October 2016).
- 21. Nel A, Haazen W, Nuttall J, Romano J, Mesquita PMM, Herold BC, et al Pharmacokinetics and safety assessment of anti‐HIV dapivirine vaginal microbicide rings with multiple dosing. J AIDS Clin Res 2014; 5: 355–365. [Google Scholar]
- 22. Nel A, Smythe S, Young K, Malcolm K, McCoy C, Rosenberg Z, et al Safety and pharmacokinetics of dapivirine delivery from matrix and reservoir intravaginal rings to HIV‐negative women. Clin Sci 2009; 51: 416–423. [DOI] [PubMed] [Google Scholar]
- 23. Nel A, Haazen W, Nuttall J, Romano J, Rosenberg Z, van Niekerk N. A safety and pharmacokinetic trial assessing delivery of dapivirine from a vaginal ring in healthy women. AIDS 2014; 28: 1479–1487. [DOI] [PubMed] [Google Scholar]
- 24. Nel A, Bekker LG, Bukusi E, Hellstrm E, Kotze P, Louw C, et al Safety, acceptability and adherence of dapivirine vaginal ring in a microbicide clinical trial conducted in multiple countries in Sub‐Saharan Africa. PLoS One 2016; 11: e0147743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen BA, Panther LM, Marzinke MA, Hendrix CW, Hoesley CJ, van der Straten A, et al Phase 1 safety, pharmacokinetics, and pharmacodynamics of dapivirine and maraviroc vaginal rings: a double‐blind randomized trial. J Acquir Immune Defic Syndr 2015; 70: 242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Baeten JM, Palanee‐Phillips T, Brown ER, Schwartz K, Soto‐Torres LE, Govender V, et al Use of a vaginal ring containing dapivirine for HIV‐1 prevention in women. N Engl J Med 2016; 375: 2121–2132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nel A, van Niekerk N, Kapiga S, Bekker LG, Gama C, Gill K, et al Safety and efficacy of a dapivirine vaginal ring for HIV prevention in women. N Engl J Med 2016; 375: 2133–2143. [DOI] [PubMed] [Google Scholar]
- 28. International Partnership For Microbicides . IPM's dapivirine ring may offer significant HIV protection when used consistently. Silver Spring, MD 2016. Available at http://www.ipmglobal.org/content/ipms-dapivirine-ring-may-offer-significant-hiv-protection-when-used-consistently-new-data (last accessed 19 October 2016).
- 29. International Partnership For Microbicides . After The Ring Study: DREAM. An open‐label extension study of IPM's dapivirine ring Silver Spring, MD 2016. Available at http://www.ipmglobal.org/sites/default/files/attachments/publication/dream-fs_july20162.pdf (last accessed 19 October 2016).
- 30. Bunge K, Dezzutti C, Rohan L, Hendrix C, Marzinke M, Richardson‐Harman N, et al A phase 1 trial to assess the safety, acceptability, pharmacokinetics, and pharmacodynamics of a novel dapivirine vaginal film. J Acquir Immune Defic Syndr 2016; 71: 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Robinson JA, Marzinke MA, Bakshi RP, Fuchs EJ, Radebaugh CL, Aung W, et al Comparison of dapivirine vaginal gel and film formulation pharmacokinetics and pharmacodynamics (FAME 02B). AIDS Res Hum Retroviruses 2017; 33: 339–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sager JE, Yu J, Ragueneau‐Majlessi I, Isoherranen N. Physiologically based pharmacokinetic (PBPK) modeling and simulation approaches: a systematic review of published models, applications, and model verification. Drug Metab Dispos 2015; 43: 1823–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhuang X, Lu C. PBPK modeling and simulation in drug research and development. Acta Pharm Sin B 2016; 6: 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. SimulationsPlus . GastroPlus software 2017. Available at http://www.simulations-plus.com/software/gastroplus (last accessed 26 January 2017).
- 35. Institute for Systems Biology . PKSIM software. Available at http://www.systemsbiology.com/ (last accessed 26 January 2017).
- 36. Certara . Simcyp PBPK modeling and simulation. Available at http://www.simcyp.com (last accessed 26 January 2017).
- 37. Fujitsu . ADMEWORKS DDI simulator. Available at http://www.fqs.pl/chemia/produkty/admeworks-ddi-simulator (last accessed 26 January 2017).
- 38. Cyprotex . Cloe® PK ‐ pharmacokinetic prediction using PBPK modeling. Available at http://www.cyprotex.com/insilico (last accessed 26 January 2017).
- 39. Evers H, Birngruber CG, Ramsthaler F, Müller U, Brück S, Verhoff MA. Differentiation of epithelial cell types by cell diameter. Arch Kriminol 2011; 228: 11–19. [PubMed] [Google Scholar]
- 40. Pendergrass PB, Belovicz MW, Reeves CA. Surface area of the human vagina as measured from vinyl polysiloxane casts. Gynecol Obstet Invest 2003; 55: 110–3. [DOI] [PubMed] [Google Scholar]
- 41. Hosseini‐Yeganeh M, McLachlan AJ. Physiologically based pharmacokinetic model for Terbinafine in rats and humans. Antimicrob Agents Chemother 2002; 46: 2219–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Patton DL, Thwin SS, Meier A, Hooton TM, Stapleton AE, Eschenbach DA. Epithelial cell layer thickness and immune cell populations in the normal human vagina at different stages of the menstrual cycle. Am J Obstet Gynecol 2000; 183: 967–973. [DOI] [PubMed] [Google Scholar]
- 43. Gao Y, Katz DF. Multicompartmental pharmacokinetic model of tenofovir delivery by a vaginal gel. PLoS One 2013; 8: e74404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Yudovsky D, Pilon L. Retrieving skin properties from in vivo spectral reflectance measurements. J Biophotonics 2011; 4: 305–314. [DOI] [PubMed] [Google Scholar]
- 45. Issa B, Moore RJ, Bowtell RW, Baker PN, Johnson IR, Worthington BS, et al Quantification of blood velocity and flow rates in the uterine vessels using echo planar imaging at 0.5 tesla. J Magn Reson Imaging: JMRI 2010; 31: 921–927. [DOI] [PubMed] [Google Scholar]
- 46. Akil A, Parniak MA, Dezzuitti CS, Moncla BJ, Cost MR, Li M, et al Development and characterization of a vaginal film containing Dapivirine, a non‐ nucleoside reverse transcriptase inhibitor (NNRTI), for prevention of HIV‐1 sexual transmission. Drug Deliv Transl Res 2011; 1: 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. das Neves J, Araújo F, Andrade F, Amiji M, Bahia MF, Sarmento B. Biodistribution and pharmacokinetics of dapivirine‐loaded nanoparticles after vaginal delivery in mice. Pharm Res 2014; 31: 1834–1815. [DOI] [PubMed] [Google Scholar]
- 48. R Core Team . R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing, 2017. [Google Scholar]
- 49. Murphy DJ, Desjardins D, Dereuddre‐Bosquet N, Brochard P, Perrot L, Pruvost A, et al Pre‐clinical development of a combination microbicide vaginal ring containing dapivirine and darunavir. J Antimicrob Chemother 2014; 69: 2477–2488. [DOI] [PubMed] [Google Scholar]
- 50. Pendergrass PB, Reeves CA, Belovicz MW, Molter DJ, White JH. Comparison of vaginal shapes in Afro‐American, Caucasian and Hispanic women as seen with vinyl polysiloxane casting. Gynecol Obstet Invest 2000; 50: 54–59. [DOI] [PubMed] [Google Scholar]
- 51. Zanger UM, Schwab M. Cytochrome P450 enzymes in drug metabolism: regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol Ther 2013; 138: 103–141. [DOI] [PubMed] [Google Scholar]
- 52. Mitchell C, Paul K, Agnew K, Gaussman R, Coombs RW, Hitti J. Estimating volume of cervicovaginal secretions in cervicovaginal lavage fluid collected for measurement of genital HIV‐1 RNA levels in women. J Clin Microbiol 2011; 49: 735–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tasoglu S, Rohan LC, Katz DF, Szeri AJ. Transient swelling, spreading, and drug delivery by a dissolved anti‐HIV microbicide‐bearing film. Phys Fluids 2013; 25: 31901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Masters WH, Johnson VE. Human Sexual Response. Boston: Little, Brown and Company, 1966. [Google Scholar]
- 55. Owen DH, Katz DF. A vaginal fluid simulant. Contraception 1999; 59: 91–95. [DOI] [PubMed] [Google Scholar]
- 56. Geonnotti AR, Katz DF. Compartmental transport model of microbicide delivery by an intravaginal ring. J Pharm Sci 2010; 99: 3514–3521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Nuttall JP, Thake DC, Lewis MG, Ferkany JW, Romano JW, Mitchnick MA. Concentrations of dapivirine in the rhesus macaque and rabbit following once daily intravaginal administration of a gel formulation of [14C]dapivirine for 7 days. Antimicrob Agents Chemother 2008; 52: 909–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fletcher P, Kiselyeva Y, Wallace G, Romano J, Griffin G, Margolis L, et al The nonnucleoside reverse transcriptase inhibitor UC‐781 inhibits human immunodeficiency virus type 1 infection of human cervical tissue and dissemination by migratory cells. J Virol 2005; 79: 11179–11186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Davies B, Morris T. Physiological parameters in laboratory animals and humans. Pharm Res 1993; 10: 1093–1095. [DOI] [PubMed] [Google Scholar]
- 60. Patton DL, Sweeney YC, Tsai C‐C, Hillier SL. Macaca fascicularis vs. Macaca nemestrina as a model for topical microbicide safety studies. J Med Primatol 2004; 33: 105–108. [DOI] [PubMed] [Google Scholar]
- 61. Wagner G, Levin R. Human vaginal pH and sexual arousal. Fertil Steril 1984; 41: 389–394. [PubMed] [Google Scholar]
- 62. Brandis K. Acid‐base pHysiology 2015. Available at http://www.anaesthesiamcq.com/AcidBaseBook/ABindex.php (last accessed 5 April 2017).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Visual predictive check profiles as for Figure 5 but includes variability in only the parameters that were estimated with some uncertainty and all parameters arbitrarily assigned. The visual predictive check simulations were run 1000 times for 30 patients with variability incorporated in to the estimated model parameters using the values and associated coefficient of variation given in Table 1 and Table 2
Figure S2 Visual predictive check profiles as for Figure 6 but includes variability in only the parameters that were estimated with some uncertainty and all parameters arbitrarily assigned. The visual predictive check simulations were run 1000 times for 30 patients with variability incorporated in to the estimated model parameters using the values and associated coefficient of variation given in Table 1 and Table 2
Figure S3 Visual predictive check profiles as for Figure 7 but includes variability in only the parameters that were estimated with some uncertainty and all parameters arbitrarily assigned. The visual predictive check simulations were run 1000 times for 30 patients with variability incorporated in to the estimated model parameters using the values and associated coefficient of variation given in Table 1 and Table 2
Supporting info item