

Abstract
Ginseng (Panax ginseng C.A. Meyer) is one of the best-selling herbal medicines, with ginsenosides as its main pharmacologically active constituents. Although extensive chemical and pharmaceutical studies of these compounds have been performed, genome-wide studies of the basic helix-loop-helix (bHLH) transcription factors of ginseng are still limited. The bHLH transcription factor family is one of the largest transcription factor families found in eukaryotic organisms, and these proteins are involved in a myriad of regulatory processes. In our study, 169 bHLH transcription factor genes were identified in the genome of P. ginseng, and phylogenetic analysis indicated that these PGbHLHs could be classified into 24 subfamilies. A total of 21 RNA-seq data sets, including two sequencing libraries for jasmonate (JA)-responsive and 19 reported libraries for organ-specific expression analyses were constructed. Through a combination of gene-specific expression patterns and chemical contents, 6 PGbHLH genes from 4 subfamilies were revealed to be potentially involved in the regulation of ginsenoside biosynthesis. These 6 PGbHLHs, which had distinct target genes, were further divided into two groups depending on the absence of MYC-N structure. Our results would provide a foundation for understanding the molecular basis and regulatory mechanisms of bHLH transcription factor action in P. ginseng.
KEY WORDS: Genome-wide characterization, bHLH transcription factors, Panax ginseng, Ginsenosides, Phylogenetic analysis
Graphical abstract
Altogether 169 basic helix-loop-helix type transcription factor genes were identified in the genome of P. ginseng, and phylogenetic analysis indicated that these PGbHLHs could be classified into 24 subfamilies. Six PGbHLH genes from 4 subfamilies were revealed to be potentially involved in the regulation of ginsenoside biosynthesis. These 6 PGbHLHs, which had distinct target genes, were further divided into two groups depending on the absence of MYC-N structure.
1. Introduction
Ginseng (Panax ginseng C.A. Meyer), belonging to the order Apiales and family Araliaceae, has been widely used as a medicinal and functional food for over 5000 years in East Asia. The genus name “Panax” means panacea or cure-all, and the specific epithet “ginseng” comes from the Chinese name “Ren shen” because of its man-like shape1. Now, as one of the best-selling herbal medicines, ginseng is used as a tonic and adaptogenic agent in more than 35 countries around the world2. Because of its multiple clinical and pharmacological effects, numerous chemical and pharmaceutical studies of P. ginseng have been conducted1. Through these investigations, ginsenosides have been shown to be the major pharmacological ingredients and are found almost exclusively in Panax plants3. In recent years, many transcriptome analyses have been performed to profile ginseng gene expression, particularly of genes involved in ginsenoside biosynthesis4. However, because of its large (3.2 Gb) and high-complexity genome, genome-wide investigations of transcription factors in ginseng remain limited. Recent completion of whole-genome sequencing of ginseng has facilitated genome-wide biological gene analysis studies5. Ginsenosides, as triterpene saponins, are mainly biosynthesized utilizing the precursor IPP through themevalonic acid (MVA) pathway in the cytosol and the methylerythritol phosphate (MEP) pathway in the plastid1. After these series of condensation reactions, squalene is generated and subsequently converted into (S)-2,3-oxidosqualene through cyclization5. Various types of ginsenoside precursors, including oleanolic acid and protopanaxadiol (PPD)/ protopanaxatriol (PPT), are formed after multiple oxidations6. As transcription factors processed the mutil-point regulation by binding to the promoter region of target genes, the operating of specific transcription factors becoming an efficient strategy to control the biological process, such as improving the yields of target metabolites7.
The basic helix-loop-helix (bHLH) transcription factor family, which was named after their highly conserved bHLH domain, is one of the largest transcription factor families found in eukaryotic organisms8. These transcription factors are involved in a myriad of regulatory processes, including neurogenesis, myogenesis, and heart development modulation in animals9, 10; phosphate uptake and glycolysis adjustment in yeast11; and secondary metabolism, epidermal differentiation, and environmental factor response regulation in plants12, 13, 14, 15. Typically, a bHLH domain consists of approximately 60 amino acids comprising a stretch of approximately 15 basic amino acids at the N-terminus, followed by two regions of hydrophobic residues predicted to form amphipathic helices separated by an intervening loop16. With the increasing number of completed and draft plant genomes, more than one thousand bHLH transcription factors have been identified not only in crops (soybean, potato, tomato or rice)14, 17 and model plants (Arabidopsis; tobacco)16, 18 but also in pharmaceutical plants (Salvia miltiorrhiza; Panax notoginseng)7, 19. Interestingly, higher plants have more bHLH transcription factors (about 150) and a larger gene family (approximately 26 subfamilies) than most animal species12, 20. With the elucidation of secondary metabolic pathway of plants, the regulatory mechanism and identification of transcription factors regulating specific metabolites have been highly developed16. Several transcription factors specifically modulating plant triterpenoid biosynthesis have recently been identified. For example, Bl (bitter leaf) and Bt (bitter fruit) were found to regulate the accumulation of cucurbitacin triterpenes in the leaves and fruits of Cucurbita pepo, respectively21. TSAR1 and its homolog TSAR2, two jasmonate (JA)-inducible bHLH transcription factors, have been shown to direct the biosynthesis of the triterpenoid saponin in Medicago truncatula22. Very recently, two additional bHLH transcription factors, TSARL1 and TSARL2 were shown to increase the expression of genes in the triterpene biosynthetic pathway, resulting in increased accumulation of triterpene saponins in Chenopodium quinoa23. Also in P. notoginseng, a bHLH transcription factor, PnbHLH1, has been cloned, which improved triterpenoid biosynthesis by interacting with E-box core sequences in promoter region of target genes7. However, little is known about bHLH transcription factors in the regulation of triterpene saponin biosynthesis in P. ginseng.
In this study, bHLH transcription factor genes were identified in the genome of P. ginseng, and phylogenetic analysis was conducted. A total of 21 RNA-seq data sets were selected for expression pattern analyses. Based on gene-specific expression patterns and up-regulated expression patterns in response to methyl jasmonate (MeJA) treatment, 6 bHLH transcription factor identified in P. ginseng (PGbHLH) genes were revealed as potentially involved in the regulation of ginsenoside biosynthesis.
2. Material and methods
2.1. Sequence retrieval and identification
The draft genome of a line IR826 P. ginseng was assembled, with a total of 42,006 protein-coding gene models predicted5. All annotated genes were identified initially by online BLAST against PlantTFDB V4.0 (http://planttfdb.cbi.pku.edu.cn/index.php) resulting in 208 transcription factor candidates24. All amino acid sequences of these candidates were then screened for a bHLH domain with the local PFAM profile hidden Markov model (Pfam: PF00010.24). A total of 169 candidates were considered to be the PGbHLHs (alignment length≥55 aa, E≤0.0001).The bHLH data set of Arabidopsis thaliana was obtained from The Arabidopsis Information Resource (TAIR; http://www.arabidopsis.org/index.jsp). The bHLH data sets of Oropetium thomaeum, Daucus carota, S. miltiorrhiza, Arachis ipaensis and Capsicum annuum were retrieved from the Plant Transcription Factor Data Base (http://planttfdb.cbi.pku.edu.cn/index.php). Subsequently, all these bHLH domains were searched using Pfam (Pfam: PF00010.24) with the same parameters as PGbHLHs. The confirmed bHLHs of each species were counted separately and reorganized for further analysis.
2.2. Multiple alignment and phylogenetic analysis
Multiple protein sequence alignments of bHLH domains were performed using ClustalW in BioEdit Sequence Alignment Editor software with the default parameters. To analyse the conserved amino acid residues, the aligned sequences were visualized with the WEBLOGO program (http://weblogo.berkeley.edu). As reconstruction of evolutionary relationships was performed on the basis of amino acid sequences, only the bHLH domains were used for phylogenetic tree construction. The Jones, Taylor, and Thornton (JTT) model was selected as the best-fitting amino acid substitution model with an estimated proportion of invariable sites (I) and an estimated g-distribution parameter (G). An unrooted phylogenetic tree of 169 PGbHLHs was constructed using MEGA 7.0 using the neighbour-joining method with the following parameters: pairwise deletion option, 1000 bootstrap replicates and Poisson correction distance. The display, annotation and management of the phylogenetic tree were performed by iTOL (Interactive Tree of Life, http://itol.embl.de/).
2.3. Sequence features and gene structure
The exon-intron structure of each PGbHLH gene was determined by aligning the full-length cDNA sequence or predicted coding sequence (CDS) with the genomic sequence in a previously published P. ginseng genome database. The cross-platform program TBtools 0.53 was used to display the gene structure (http://cj-chen.github.io/tbtools). In addition, the theoretical isoelectric point (pI) and molecular weight (MW) of PGbHLH proteins were predicted by the online ExPASy server (https://www.expasy.org/). Conserved motifs of 169 PGbHLHs were identified using MEME (Suite version 4.12.0, http://meme-suite.org/index.html) with the following criteria: expected E-values less than 10−30, any number of repetitions of a motif, and an optimum width of 6–50 amino acids. Subsequently, the MAST program was used to search detected motifs25.
2.4. Digital gene expression analysis
For comparing gene expression patterns among the different organs of P. ginseng, 10 RNA-Seq datasets from NCBI (accession number SRP066368) were analysed26. For analysis among different tissues, 9 RNA-Seq datasets from NCBI (accession numbers PRJNA369187 and PRJNA381509) were used27. The clean reads were separately aligned to assemble the P. ginseng genome in the orientation mode with TopHat software (http://tophat.cbcb.umd.edu/)28. After assembling transcripts by Cufflinks and combining by Cuffmerge (http://cufflinks.cbcb.umd.edu/), the expression level for each transcript was calculated using fragments per kilobase of exon per million mapped reads (FRKM) method to identify differentially expressed genes (DEGs) among the different samples29. Cuffdiff was used for differential expression analysis30. The DEGs were selected using the following criteria: the logarithm of the fold change >2 and the false discovery rate (FDR) <0.05.
For MeJA treatment analysis, callus from IR826 ginseng induced with a previously published method was used4. Newly established callus was moved to solid Murashige and Skoog media (MS media) supplemented with 1.0 mg/L 2,4-dichlorophenoxy acetic acid (2,4-D) and 0.1 mg/L kinetin (KT) then subcultured once every 2 weeks. After 2 weeks of culture, the callus was transferred to solid MS media supplemented with 2,4-D (1.0 mg/L), KT (0.1 mg/L) and MeJA (200 µmol/L, desired concentration) for 48-h culture as the MeJA-treated group. Meanwhile, an equal volume of solvent (ethanol) was dissolved in the experimental controls, which were kept in the same conditions as control group. The total RNA from each group (Con and MeJA) was extracted with TRIzol® Reagent (Invitrogen) following the manufacturer's procedure and then treated with DNase I to remove DNA residue. The mRNA with poly(A) tails was isolated from total RNA using a Dynabeads mRNA DIRECT Kit (Invitrogen), then purified and fragmented into small pieces. Double-stranded cDNA was synthesized from fragmented mRNA with random primers. For Illumina sequencing, the short cDNA fragments were ligated to sequencing adapters. After PCR amplification, 400—500 bp fragments were selected as the cDNA library. These two cDNA libraries were finally sequenced on a HiSeq. 2500 (Illumina) with the PE125 strategy. After trimming the adapters and low-quality reads, 33,145,160 and 28,058,510 clean reads were generated for the two libraries (NCBI accession number SAMN07692812). The FRKM was calculated with the same protocol as used for the other 19 RNA-Seq datasets. C1 and C2 were denoted as the counts of reads mapped to a specific gene obtained from two samples, and then M was defined as (log2C1−log2C2)31. The M-value based on the MA-plot was used to identify DEGs, which were visualized by iTOL together with the phylogenetic tree.
A first-round Pearson's correlation test was performed by the R package “Psych” between the ginsenoside contents and FRKM of PGbHLH-encoding genes in the 9 tissues of the main roots of ginseng27. Coexpression analyses between PGbHLH-encoding genes and triterpene saponin biosynthetic enzyme-encoding genes were performed in all 21 RNA-Seq datasets.
3. Results
3.1. Sequence features and phylogenetic analysis
A total of 169 PGbHLHs containing open reading frame (ORF) sequences were identified (Supplementary Information Table S1). The number of bHLH genes in P. ginseng is similar to that in A. thaliana and D. carota (Table 1). The gene length of PGbHLHs varied from 512 bp (PG38698) to 20,274 bp (PG29796) (Supplementary Information Table S2). Interestingly, although there are a few differences in total number of bHLH genes between O. thomaeum, C. annuum, S. miltiorrhiza, A. ipaensis, P. ginseng, A. thaliana, and D. carota, all 7 species are quite similar in gene length distribution (Fig. 1). The lengths of PGbHLH cDNAs vary from 283 bp (PG15345, PG309998 and PG30999) to 2857 bp (PG12599) (Supplementary Information Table S2), the molecular weights of the predicted proteins range from 10,560.80 Da (PG30998 and PG30999) to 104,842.95 Da (PG26263), and the theoretical isoelectric points are predicted to range from 4.81 (PG29408) to 10.16 (PG38698) (Supplementary Information Table S2).
Table 1.
Number of bHLH proteins and genome feature analysis.
| Species | Number of bHLHs | Genome size |
|---|---|---|
| Arabidopsis thaliana | 153 | 125 Mb |
| Oropetium thomaeum | 108 | 245 Mb |
| Daucus carota | 150 | 473 Mb |
| Salvia miltiorrhiza | 128 | 641 Mb |
| Arachis ipaensis | 124 | 1.35 Gb |
| Panax ginseng | 169 | 3.43 Gb |
| Capsicum annuum | 109 | 3.48 Gb |
Figure 1.

bHLH protein length distribution in P. ginseng and the other six sequenced species.
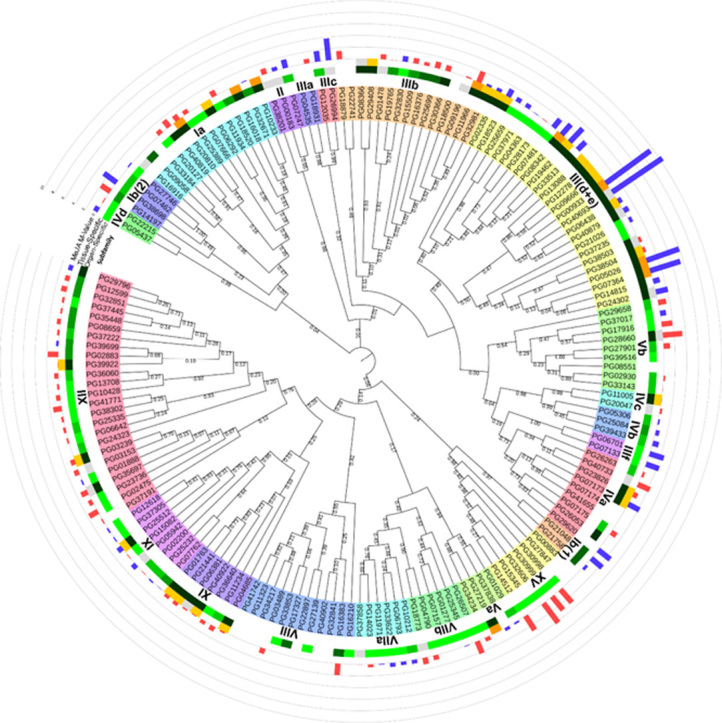
An unrooted phylogenetic tree was constructed using the bHLH gene family in P. ginseng and A. thaliana (Supplementary Information Fig. S1) to determine subfamily designations. A neighbour-joining tree constructed among the 169 bHLH members identified in P. ginseng indicated a total of 24 distinct subfamilies (Fig. 2), with the III(d+e) subfamily having the largest number of members (26 PGbHLHs) and the VIIIc subfamily the fewest (1 PGbHLH). In addition, compared with A. thaliana, the III(d+e) subfamily was dramatically extended (bHLH members from 9 in A. thaliana to 26 in P. ginseng), and this subfamily contains many bHLH proteins involved in JA response and secondary metabolism (Table 2). A similar phenomenon also occurred in the IIIb subfamily (from 5 in A. thaliana to 14 in P. ginseng), which contains many potential activators of cold-responsive genes. However, in the Ib(2) subfamily, which may be involved in Fe uptake regulation, the numbers were decreased (12 in A. thaliana and 3 in P. ginseng).
Figure 2.

Unrooted phylogenetic tree of the bHLH protein family in P. ginseng. Only the bHLH domains were used for phylogenetic tree construction. The JTT model was selected as the best-fitting amino acid substitution model with an estimated proportion of invariable sites (I) and an estimated g-distribution parameter (G). The phylogenetic tree was constructed using the neighbour-joining method with 1000 bootstrap replicates and Poisson correction distance. Roman numerals correspond to the bHLH subfamily. The different colours on the outer two rounds represent organ-specific expression patterns. The blue bars (up-regulated) and red bars (down-regulated) on the outermost round show the changes after MeJA treatment in callus of P. ginseng.
Table 2.
Exon distribution analysis of 169 PGbHLHs in each subfamily.
| Subfamily | Number of PgbHLHs | Number of AtbHLHs | Relative frequency of bHLHs | bHLHs with specific exon number |
AVERAGE | Intron phase |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | >7 | 0 | 1 | 2 | |||||
| Ia | 15 | 10 | 1.50 | 4 | 8 | 3 | 2.93 | 29 | 0 | 0 | ||||
| Ib1 | 2 | 2 | 1.00 | 1 | 1 | 3.50 | 5 | 0 | 0 | |||||
| Ib1 | 3 | 12 | 0.25 | 1 | 1 | 1 | 3.00 | 6 | 0 | 0 | ||||
| II | 3 | 4 | 0.75 | 1 | 1 | 1 | 4.00 | 8 | 0 | 1 | ||||
| IIIa | 2 | 1 | 2.00 | 1 | 1 | 3.50 | 5 | 0 | 0 | |||||
| IIIb | 14 | 5 | 2.80 | 1 | 1 | 7 | 2 | 1 | 2 | 4.43 | 42 | 4 | 1 | |
| IIIc | 2 | 3 | 0.67 | 1 | 1 | 3.00 | 3 | 1 | 0 | |||||
| III(d+e) | 26 | 9 | 2.89 | 20 | 3 | 2 | 1 | 1.46 | 9 | 0 | 3 | |||
| IIIf | 2 | 2 | 1.00 | 2 | 8.00 | 8 | 5 | 0 | ||||||
| IVa | 9 | 4 | 2.25 | 4 | 1 | 2 | 1 | 1 | 4.89 | 31 | 0 | 2 | ||
| IVb | 3 | 2 | 1.50 | 3 | 6.00 | 6 | 1 | 5 | ||||||
| IVc | 2 | 4 | 0.50 | 2 | 6.00 | 4 | 6 | 0 | ||||||
| IVd | 2 | 1 | 2.00 | 1 | 1 | 5.50 | 8 | 1 | 0 | |||||
| Va | 4 | 6 | 0.67 | 1 | 3 | 7.25 | 14 | 6 | 3 | |||||
| Vb | 9 | 3 | 3.00 | 6 | 1 | 1 | 1 | 3.78 | 21 | 1 | 3 | |||
| VIIa | 6 | 6 | 1.00 | 1 | 5 | 9.17 | 35 | 4 | 0 | |||||
| VIIb | 6 | 8 | 0.75 | 2 | 2 | 1 | 1 | 5.67 | 25 | 2 | 1 | |||
| VIIa | 2 | 7 | 0.29 | 1 | 1 | 3.50 | 4 | 1 | 0 | |||||
| VIIIb | 9 | 3 | 3.00 | 6 | 2 | 1 | 1.44 | 3 | 1 | 0 | ||||
| VIIIc | 1 | 5 | 0.20 | 1 | 5.00 | 3 | 0 | 1 | ||||||
| IX | 8 | 7 | 1.14 | 1 | 4 | 3 | 6.63 | 34 | 2 | 8 | ||||
| XI | 7 | 5 | 1.40 | 1 | 1 | 5 | 7.86 | 49 | 1 | 1 | ||||
| XII | 25 | 17 | 1.47 | 1 | 1 | 9 | 14 | 7.96 | 157 | 7 | 5 | |||
| XV | 7 | 6 | 1.17 | 7 | 2.00 | 7 | 0 | 0 | ||||||
| Total | 169 | 132 | 1.28 | 27 | 25 | 22 | 20 | 13 | 24 | 38 | 4.64 | 516 | 43 | 34 |
3.2. Gene structure of PGbHLHs
The 169 PGbHLH genes have a varying number of exons from 1 to 17, and a total of 27 genes are intronless (Supplementary Information Fig. S2). Exon analyses of each subfamily revealed that the average exon number per gene is 2–5, but this number varies in several subfamilies ranging from 1.44 (subfamily VIIIb) to 9.17 (subfamily VIIa). The 27 intronless genes are distributed across 3 subfamilies, particularly subfamily III(d+e) (Fig. 3A), in which 20 genes are intronless. Six of the other 7 intronless genes belong to subfamily VIIIb, while subfamily IIIb only contains 1. Multi-exon genes (exon number ≥7) are mainly distributed in subfamilies VIIa (Fig. 3B), XI and XII . There are several subfamilies where genes with a certain exon number are concentrated, such as subfamily XV, which contains 7 two-exon genes. The same pattern was observed in subfamilies IVb, IVc and IIIf, with 3 six-exon genes, 2 six-exon genes and 2 eight-exon genes, respectively (Table 2).
Figure 3.

The structural features of the III (d+e) and VII a bHLH gene subfamilies. The exons and UTRs are represented by green and pink round-cornered rectangles, respectively, with black connecting lines as the introns. The bHLH domains are emphasized by yellow colour. The numbers above the rectangles correspond to the intron phase.
The intron numbers and phases were also analysed over genomic regions encoding the PGbHLH domains. Among the 593 introns, a great majority (516) are phase 0, whereas only 43 and 34 are phases 1 and 2, respectively. Interestingly, intron pattern distribution shows specificity within some subfamilies: Ia, Ib1, Ib2, IIIa and XV contained only 0-phase-intron genes, while genes in subfamilies IIIf and IVc include several 1-phase introns. Two-phase-intron genes are mainly distributed in subfamilies IVb and IX (Table 2).
3.3. Key amino acid residues in bHLH domain
Based on a bHLH domain alignment of 169 PGbHLHs, 20 amino acid residues were highly conserved with consensus more than 50% (Fig. 4B). Among these, 5 conserved residues were in the basic region, while other 15 were found in the two-helices region.
Figure 4.

Alignment of the bHLH domains of P. ginseng proteins. (A) Alignment of the bHLH domain of PGbHLHs in the III(d+e) subfamily. The shaded boxes above indicate the positions of the DNA-binding basic region, the two α-helixes, and the variable loop region. (B) Highly conserved amino acid residues in the bHLH domain across all PGbHLHs. The overall height of each stack represents the conservation of the sequence at that position. The numbering of the amino acid follows.
There were 14 amino acid residues in the basic region, which is similar to those of other plants. Glu-9 and Arg-10, which are thought to be essential in E-box-binding recognition, were both highly conserved, with 85.12% and 94.65%, respectively, and 83.93% combined. In addition, with the presence of His residues at position 5, 64.88% of PGbHLHs showed G-box specificity to bind to a variation of the E-box hexanucleotide sequence (CACGTG). Interestingly, all 26 III(d+e) members except one (PG22741), contained this G-box binding site (Fig. 4A), which was a higher frequency of H5-E9-R13 structure (96.15%) than the other subfamilies. Because it is a requirement to contact the DNA backbone, most PGbHLHs also showed the critical Arg residues at positions 10 (84.52%) and 12 (95.83%) in the basic region.
The hydrophobic amino acids isoleucine (I), leucine (L), phenylalanine (F), methionine (M) and valine (V) in conserved positions are required for the formation of homo- or heterodimeric complexes between bHLH proteins. These highly conserved positions were ubiquitous in most PGbHLHs. An L residue is present at sites 23, 63 and 73 in 99.40%, 80.95% and 95.83%, while an I residue was found 50.00% and 59.52% of the time at position 16 and 67. When all 5 hydrophobic amino acid residue frequencies were assessed together, 10 sites in the two helices showed high consensus, as shown in Supplementary Information Fig. S4. A conserved proline at position 29 breaks the first helix and starts a loop of variable length.
3.4. Conserved non-bHLH motifs in P. ginseng bHLH proteins
By motif discovery using MEME, 20 conserved motifs were characterized (Supplementary Information Table S4). According to their amino acid sequences, 16 were non-bHLH motifs. In 169 PGbHLHs, these motifs were highly conserved both in combination and relative position. All 9 IVa members contained bHLH domains with motif 12 and motif 4 at the C-terminus; 6 of 7 XI members had their bHLH domain between motif 6 and motif 14, except PG36640, which had lost motif 14. Most motifs were located C-terminal to the bHLH domain, but proteins from subfamilies III(d+e) and IIIf have a stretch of motifs positioned towards the N-terminal of PGbHLHs (Fig. 5 and Supplementary Information Fig. S7). Interestingly, in subfamily III(d+e) there were three subtype motif combinations, with a highly conserved N-terminal region (motifs 8–10-7–11-14), which may be necessary to obtain an active transcription complex. In several subfamilies, such as XV, IVb and IVc, no conserved motifs were present other than the bHLH domain.
Figure 5.

Conserved motifs in each P. ginseng bHLH protein subfamily. A representation of a typical member of each bHLH subfamily is shown, with the bHLH domain and other conserved motifs drawn as coloured boxes. The sequences of each motif in the individual proteins are included in Supplementary Information Table S4.
Commonly motifs only appeared once, but a few special cases were found in P. ginseng. Motif 9 was found on both sides of the bHLH domain in five III(d+e) bHLHs; motif 19 was repeated once in PG37222 and PG37445 (XII subfamily). Several motifs were subfamily-specific, such as motif 15 in XII subfamily, motif 11 in III(d+e) and motif 13 in VIII. However, some motifs also appeared frequently; for instance, motif 4 was shared by subfamilies I , II, III, IV, and V, and motif 6 was distributed across subfamilies IX, XI, and XII.
3.5. Expression profiles of bHLH genes in various organs and tissues
By analysing the expression levels of 169 PGbHLH genes in ten different organ samples of P. ginseng (Fig. 6A), 19 PGbHLH genes were not detected in any organs, and 13 PGbHLH genes were specifically expressed in only one organ. Among these 13 PGbHLHs, fruit, leaflet pedicel, leaf peduncle and stem only had one specific PGHLH (PG32671, PG27139, PG16912 and PG29658 respectively), while seed had three specific PGbHLHs (PG16383, PG17327 and PG09058). There were five specific PGbHLHs in root, whose expression was also associated with growth period (PG29408, PG08366 in 5-year roots; PG26053 in 5- and 12-year roots; PG16018, PG06292, PH10233 in all three periods). Interestingly, 6 of these 13 specific PGbHLHs belonged to the Ia subfamily, which contained many bHLHs controlling cell differentiation. The remaining 137 PGbHLHs also showed altered expression patterns in different organs, 66 of which were detected in ten organs. According to the heatmap, all identified PGbHLHs were clustered into four distinct groups, while ten organs also clustered into four individual groups with specific expression patterns. Forty-five PGbHLHs belonging to 19 subfamilies were differentially high in leaf blade and pedicel. In the root, a total of 44 PGbHLHs including 10 III(d+e) subfamily members and 3 IVa subfamily members showed higher expression patterns (Fig. 2), while 22 PGbHLHs were detected with greater FRKM values in the stem. There were 9 PGbHLHs containing two Ib(1) subfamily members that showed higher expression in the seed.
Figure 6.

Heatmaps representing partial expression profiles of P. ginseng bHLH genes. (A) Clustering and differential expression analysis of 169 PGbHLH genes in various organs. (B) Clustering and differential expression analysis of 44 PGbHLHs genes with root-specific expression in the cortex, periderm and stele. The FPKM values were transformed to log2(value +1). The colour scale is shown at the right, and higher expression levels are shown in red. Genes with FPKM values of 0 in all samples are not shown.
To analyse the ginseng root PGbHLHs further, expression profiles of 44 PGbHLHs highly expressed in the root were also tested in three root tissues (three samples each in cortex, periderm and stele). As shown in Fig. 6B, the 9 samples clustered well in three distinct groups, and the 44 PGbHLHs generated three clusters by their expression pattern. Six PGbHLHs were not detected in any sample, and 38 PGbHLHs were measured in at least one sample. A total of 21 PGbHLHs distributed in 8 subfamilies showed higher expression levels in all three cortex samples, and 8 of these 21 bHLHs belonged to the III(d+e) subfamily (Fig. 2). In the stele, 9 PGbHLHs distributed in 4 subfamilies showed differentially high FRKM, among which 4 were also III(d+e) members. The combined organ-expression profile results indicate that the III(d+e) subfamily may play a role in roots.
The JA signalling network coordinates the production of a broad range of defence-related proteins and secondary metabolites, including ginsenosides and other terpenes. As several groups of bHLH transcription factors were reported to be important for the regulation of these JA-related pathways, a MeJA treatment analysis was performed. After 48 h MeJA treatment, all PGbHLH genes were detected in at least one sample, except 7 and 11 PGbHLH genes in the control and MeJA groups, respectively. For the control group, the FPKM of PGbHLHs ranged from 0.04 to 156.77 with an average of 10.49, while the FPKM for the MeJA group ranged from 0.04 to 451.82 with an average of 17.82. These results indicate that both control and MeJA PGbHLHs showed a wide range of expression levels and that several PGbHLHs were up-regulated after MeJA treatment (Supplementary Information Table S6). Further analysis showed that 14 PGbHLHs were obviously up-regulated (M-value>2.0), among which 9 PGbHLHs had the typical MYC-N structure (Supplementary Information Table S3). Half of these 14 PGbHLHs belonged to the III(d+e) subfamily. Furthermore, all 4 substantially up-regulated PGbHLHs (M-value>5.0) were III(d+e) subfamily members. Eight PGbHLHs were down-regulated by MeJA stimulation (M-value≤2.0). Four of these down-regulated PGbHLHs were distributed in the XV subfamily, which contains many members that act as positive regulators of gibberellin signalling.
3.6. Coexpression analyses of candidate ginsenoside biosynthesis PGbHLHs
To test whether these MeJA-stimulated PGbHLHs were able to induce the accumulation of ginsenosides, Pearson's correlation test was performed between individual or total ginsenoside contents and the expression levels of 14 PGbHLH-encoding genes up-regulated by JA (Supplementary Information Table S7). A total of 6 PGbHLHs, which were 2 III(d+e), 2 IVa, 1 IIIc and 1 IIIf subfamily members, showed strong correlations with ginsenoside accumulation (Cor>0.80; P<0.05). Interestingly, PG40693 and PG07173 showed stronger correlation with PPD-type ginsenosides than with PPT-type ginsenosides (Table 3). Commonly, the transcription factors regulating specialized metabolite biosynthesis are coexpressed with the target genes encoding the pathway enzymes32. Therefore, analyses were conducted to determine the coexpression of these 6 candidate PGbHLHs with target genes in the MEP and MVA pathways (Fig. 7; Supplementary Information Table S8).
Table 3.
Pearson's correlation coefficients of selected bHLH transcription factor expression levels and major ginsenoside contents.
| ID | Rg1 | Re | Rf | Rb1 | Rb2 | Rc | Rd | Total |
|---|---|---|---|---|---|---|---|---|
| PG40693 | 0.8789 | 0.9731 | 0.9209 | 0.9465 | 0.9749 | 0.9825 | 0.9678 | 0.9669 |
| PG26994 | 0.8646 | 0.9112 | 0.9787 | 0.9232 | 0.9280 | 0.8997 | 0.9232 | 0.9249 |
| PG19462 | 0.8203 | 0.8947 | 0.8492 | 0.8797 | 0.8901 | 0.8850 | 0.8501 | 0.8852 |
| PG29620 | 0.8629 | 0.8789 | 0.7424 | 0.8562 | 0.8477 | 0.8620 | 0.7749 | 0.8580 |
| PG06701 | 0.7917 | 0.8762 | 0.8961 | 0.8556 | 0.8773 | 0.8494 | 0.8403 | 0.8708 |
| PG07173 | 0.6911 | 0.8371 | 0.8227 | 0.8061 | 0.8650 | 0.8868 | 0.9309 | 0.8426 |
Figure 7.

Biosynthesis pathway for ginsenosides with the Pearson's correlation coefficients of six candidate bHLH transcription factors. β-AS, β-amyrin synthase; AACT, acetyl-CoA C-acetyltransferase; CAS, cycloartenol synthase; CDP-ME, methylerythritol cytidyl diphosphate; CDP-MEP, 4-diphosphocytidyl-2-C-methyl-d-erythritol-2-phosphate; DDS, dammarenediol synthase; DMAPP, dimethylallyl diphosphate; DXP, 1-deoxy-d-xylulose-5-phosphate; DXR, DXP reductoisomerase; DXS, 1-deoxy-d-xylulose-5-phosphate synthase; FPP, farnesyl diphosphate; FPS, farnesyl diphosphate synthase; HMBPP, 4-hydroxy-3-methyl-butenyl-1-diphosphate; HMG-CoA, 3-hydroxy-3-methylglutaryl-CoA; HMGR, 3-hydroxy-3-methylglutaryl-CoA reductase; HMGS, 3-hydroxy-3-methylglutaryl-CoA synthase; IDI, isopentenyl-diphosphate delta-isomerase; IPP, isopentenyl diphosphate; IspD, CDP-ME synthetase; IspE, 4-diphosphocytidyl-2-C-methyl-d-erythritol kinase; IspF, 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase; IspG, (E)-4-hydroxy-3-methylbut-2-enyl-diphosphate synthase; IspH, 4-hydroxy-3-methylbut-2-en-1-yl diphosphate reductase; LAS, lanosterol synthase; MEcPP, 2-C-methyl-d-erythritol-2,4-cyclodiphosphate; MEP, 2-C-methyl-d-erythritol 4-phosphate; MVD, mevalonate diphosphate decarboxylase; MVK, mevalonate kinase; MVP, mevalonate phosphate; MVPP, diphosphomevalonate; OAS, oleanolic acid synthase; PMK, phosphomevalonate kinase; PPDS, protopanaxadiol synthase; PPTS, protopanaxatriol synthase; SE, squalene epoxidase; SS, squalene synthase.
PG40693 and PG19462, both of which were III(d+e) subfamily members, showed coexpression with genes encoding triterpene saponin biosynthesis enzymes. PG40693, in particular, showed very strong positive correlations with both the MVA and MEP pathways (Cor>0.8). Unlike PG40693 and PG19462, the IIIc subfamily bHLH transcription factor PG26994 only showed moderate positive correlation with 1-deoxy-d-xylulose-5-phosphate synthase (DEX) and β-amyrin synthase (β-AS). Interestingly, two IVa-subfamily bHLH transcription factors showed different patterns. PG29620 showed moderate or even strong correlation with CASs and PPTSs, which is unique among these 6 candidates, while PG07173 showed similar coexpression pattern with 2 III(d+e) subfamily members. Unexpectedly, PG06701, belonging to the IIIf subfamily, had a strong positive correlation with the MEP pathway rather than the MVA pathway. It also showed strong negative correlation with a 3-hydroxy-3-methylglutaryl-CoA reductase (HMGR3) and moderate negative correlations with 3-hydroxy-3-methylglutaryl-CoA synthase (HMGS3), oleanolic acid synthase (OAS1), and 1-deoxy-d-xylulose-5-phosphate synthase (DXS4). Overall, based on expression profiles, the III(d+e) and IVa subfamilies may play roles in ginsenoside biosynthesis regulation.
4. Discussion
As extraordinary progress has been made in genome sequencing technologies, plant genome sequencing and assembly in the last 10 years has increased dramatically33. In modern plant biological research, whole-genome sequencing is one of the most important approaches, which results in an increasing number of sequences deposited in public databases. Therefore, functional annotations for a large number of macromolecules has become one of the current challenges. Assigning protein functions through experimental investigation is well known to be costly and time consuming. Alternatively, using computational approaches to mine and characterize functional gene families in genome-wide datasets is possible due to their significant structural features and conserved domains8, 18, 34, 35. Based on previously used approaches, several bioinformatics methods have been applied to identifying and predicting bHLH transcription factor functions in this study, including BLAST search, sequence alignment, phylogenetic analysis, and domain analysis14, 16, 17. These whole-genome gene-mining approaches have enabled genome-wide identification, phylogenetic analysis, and comparative study of bHLH transcription factors. By combining expression profiles and chemical contents in stimulus-responsive and organ-specific samples, the expression patterns and potential functions of bHLH transcription factors were observed systematically.
In this study, 169 bHLH members in P. ginseng were classified into 24 subfamilies, consistent with other plants14, 18, 36. Furthermore, comparative studies of bHLH gene members in different plant species showed that all 7 species were quite similar in gene length distribution. In contrast to bHLH transcription factors in animals, which are classified into 6 main groups, bHLH classification in plants is on-going. Based on a phylogenetic tree constructed with the bHLHs of A. thaliana, the bHLHs in P. ginseng can be classified into 24 distinct subfamilies (Supplementary Information Fig. S1), with the dramatically extended III(d+e) subfamily having the largest number of members (26 PGbHLHs). Plant bHLH transcription factors of the same subfamily are frequently involved in the same biological process8. As the III(d+e) subfamily contains many bHLH proteins involved in JA response and secondary metabolism, it might be associated with the abundant triterpenoid saponins almost exclusively produced in Panax species. According to the Pfam result, 22 of 26 III(d+e) PGbHLHs and five PGbHLHs from the rest of the III superfamily contained MYC-N structures, which specifically interact with JAZ proteins to repress transcription when JA levels were low. In addition, 7 III(d+e) PGbHLHs showed higher expression levels after MeJA treatment in IR826 ginseng callus, which suggested that the functions of bHLHs belonging same subfamily might be partially or totally redundant. A second significant expansion was observed in the IIIb subfamily (from 5 in A. thaliana to 14 in P. ginseng), which contains many potential activators of cold-responsive genes. It has been reported that 2 MYC-like bHLH transcription factors encoded by inducer of CBF expression genes (ICE1 and its homolog ICE2) in A. thaliana are distributed in the IIIb subfamily37, 38. In addition, in Triticum aestivum, two functional ICE-like bHLH genes were characterized to be candidate regulators of CBF gene expression39. In ginseng, there were only 2 MYC-like bHLH transcription factors (PG08366 and PG29408) in the IIIb subfamily with specific expression activity in roots, whose function will require further study.
The other main objective of our research was to identify the bHLH transcription factors involved in ginsenoside biosynthesis. It has been reported that several JA-stimulated transcription factors specifically modulate plant terpene biosynthesis. Two MYC2 homologs, which are known JA signalling cascade activators, play a role in regulating the biosynthesis of sesquiterpenes in Solanum lycopersicum and Artemisia annua40, 41. Similarly, in P. ginseng, 4 PGbHLHs containing a MYC-N structure and 2 IVa-subfamily members showed expression patterns positively correlated with ginsenoside accumulation. The coexpression phenomenon between transcription factors and regulated genes was also observed. The expression levels of PG40693 and PG19462, distributed in the III(d+e) subfamily, showed very strongly positive correlations with enzyme-encoding genes in both the MVA and MEP pathways. PG06701, belonging to the IIIf subfamily, had a strong positive correlation with the MEP pathway rather than the MVA pathway. Unlike others, a IIIc subfamily bHLH transcription factor PG26994 showed only moderate positive correlative with 1-deoxy-d-xylulose-5-phosphate synthase (DEX) and β-amyrin synthase (β-AS). In addition, four homologous IVa bHLH transcription factors, triterpene saponin biosynthesis activating regulators (TSAR1 and TSAR2) in M. truncatula and TSAR-like (TSARL1 and TSARL2) in C. quinoa were very recently found to regulate the accumulation of triterpenes22, 23. Moreover, there were two bHLH transcription factors reported in other Panax species expect ginseng. One bHLH transcription factor, PnbHLH1 was cloned from P. notoginseng with triterpenoid biosynthesis regulation function7, the other one named PjbHLH was identified in Panax japonicas (ALB38667.1). Multiple protein sequence alignments of bHLH domains of these Panax species bHLH transcription factors were performed using ClustalW. These two bHLH transcription factors showed highly homology, and both are belonged to IVa subfamily. Interestingly, two IVa-subfamily bHLH transcription factors in our study showed strong positive correlations of expression pattern with triterpene accumulation but a different pattern with enzyme-encoding genes. PG29620 showed moderate or even strong correlation with CAS and PPTS, while the expression level of PG07173 showed strong positive correlation with 2-C-methyl-d-erythritol 2,4-cyclodiphosphate synthase (IspF), HMGS and mevalonate kinase (MVK). The similar expression patterns of PGbHLHs from same subfamily indicate their functional redundancy, while synergistic expression of PGbHLHs from different subfamily suggested functional complementation. Further gene-specific overexpression or knockout transgenic analysis may be helpful to unravel their functions.
5. Conclusions
In this study, 169 bHLH transcription factor genes were identified in the genome of P. ginseng, and phylogenetic analysis indicated that these PGbHLHs could be classified into 24 subfamilies consistent with structural similarities. A total of 21 sequencing libraries were constructed for expression pattern analyses using RNA-Seq. Organ/tissue expression patterns and MeJA-responsive patterns suggested complementary and tissue-specific functions of PGbHLHs. Based on gene-specific expression patterns and ginsenoside contents, 6 PGbHLHs were revealed as potentially involved in the regulation of ginsenoside biosynthesis. Some of them might be ideal candidates for further investigation.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Supplementary data associated with this article can be found in the online version at https://doi.org/10.1016/j.apsb.2018.04.004.
Contributor Information
Jiang Xu, Email: jxu@icmm.ac.cn.
Shilin Chen, Email: slchen@icmm.ac.cn.
Appendix A. Supplementary material
Supplementary material
.
Supplementary material
.
References
- 1.Kim Y.J., Zhang D., Yang D.C. Biosynthesis and biotechnological production of ginsenosides. Biotechnol Adv. 2015;33:717–735. doi: 10.1016/j.biotechadv.2015.03.001. [DOI] [PubMed] [Google Scholar]
- 2.Yan X., Fan Y., Wei W. Production of bioactive ginsenoside compound K in metabolically engineered yeast. Cell Res. 2014;24:770–773. doi: 10.1038/cr.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leung K.W., Wong A.S. Pharmacology of ginsenosides: a literature review. Chin Med. 2010;5:20. doi: 10.1186/1749-8546-5-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cao H., Nuruzzaman M., Xiu H. Transcriptome analysis of methyl jasmonate-elicited Panax ginseng adventitious roots to discover putative ginsenoside biosynthesis and transport genes. Int J Mol Sci. 2015;16:3035–3057. doi: 10.3390/ijms16023035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xu J., Chu Y., Liao B. Panax ginseng genome examination for ginsenoside biosynthesis. GigaScience. 2017;6:1–15. doi: 10.1093/gigascience/gix093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang P., Wei Y., Fan Y. Production of bioactive ginsenosides Rh2 and Rg3 by metabolically engineered yeasts. Metab Eng. 2015;29:97–105. doi: 10.1016/j.ymben.2015.03.003. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X., Ge F., Deng B. Molecular cloning and characterization of PnbHLH1 transcription factor in Panax notoginseng. Molecules. 2017;22:1268. doi: 10.3390/molecules22081268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pires N., Dolan L. Origin and diversification of basic-helix-loop-helix proteins in plants. Mol Biol Evol. 2010;27:862–874. doi: 10.1093/molbev/msp288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Massari M.E., Murre C. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 2000;20:429–440. doi: 10.1128/mcb.20.2.429-440.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jones S. An overview of the basic helix-loop-helix proteins. Genome Biol. 2004;5:226. doi: 10.1186/gb-2004-5-6-226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robinson K.A., Lopes J.M. Survey and summary: Saccharomyces cerevisiae basic helix-loop-helix proteins regulate diverse biological processes. Nucleic Acids Res. 2000;28:1449–1505. doi: 10.1093/nar/28.7.1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feller A., Machemer K., Braun E.L., Grotewold E. Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 2011;66:94–116. doi: 10.1111/j.1365-313X.2010.04459.x. [DOI] [PubMed] [Google Scholar]
- 13.Xu W., Dubos C., Lepiniec L. Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 2015;20:176–185. doi: 10.1016/j.tplants.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Gao C., Sun J., Wang C. Genome-wide analysis of basic/helix-loop-helix gene family in peanut and assessment of its roles in pod development. PLoS One. 2017;12:e0181843. doi: 10.1371/journal.pone.0181843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.An J., Hu Z., Che B., Chen H., Yu B., Cai W. Heterologous expression of Panax ginseng PgTIP1 confers enhanced salt tolerance of soybean cotyledon hairy roots, composite, and whole plants. Front Plant Sci. 2017;8:1232. doi: 10.3389/fpls.2017.01232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hickman R., Van Verk M.C., Van Dijken A.J. Architecture and dynamics of the jasmonic acid gene regulatory network. Plant Cell. 2017;29:2086–2105. doi: 10.1105/tpc.16.00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ge F., Luo X., Huang X. Genome-wide analysis of transcription factors involved in maize embryonic callus formation. Physiol Plant. 2016;158:452–462. doi: 10.1111/ppl.12470. [DOI] [PubMed] [Google Scholar]
- 18.Carretero-Paulet L., Galstyan A., Roig-Villanova I., Martínez-García J.F., Bilbao-Castro J.R., Robertson D.L. Genome-wide classification and evolutionary analysis of the bHLH family of transcription factors in Arabidopsis, poplar, rice, moss, and algae. Plant Physiol. 2010;153:1398–1412. doi: 10.1104/pp.110.153593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X., Luo H., Xu Z. Genome-wide characterisation and analysis of bHLH transcription factors related to tanshinone biosynthesis in Salvia miltiorrhiza. Sci Rep. 2015;5:11244. doi: 10.1038/srep11244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ramsay N.A., Glover B.J. MYB-bHLH-WD40 protein complex and the evolution of cellular diversity. Trends Plant Sci. 2005;10:63–70. doi: 10.1016/j.tplants.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 21.Shang Y., Ma Y., Zhou Y. Biosynthesis, regulation, and domestication of bitterness in cucumber. Science. 2014;346:1084–1088. doi: 10.1126/science.1259215. [DOI] [PubMed] [Google Scholar]
- 22.Mertens J., Pollier J., Bossche R.V., Lopez-Vidriero I., Franco-Zorrilla J.M., Goossens A. The bHLH transcription factors TSAR1 and TSAR2 regulate triterpene saponin biosynthesis in Medicago truncatula. Plant Physiol. 2016;170:194–210. doi: 10.1104/pp.15.01645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jarvis D.E., Ho YS, Lightfoot DJ. The genome of Chenopodium quinoa. Nature. 2017;542:307–312. doi: 10.1038/nature21370. [DOI] [PubMed] [Google Scholar]
- 24.Jin J., Tian F., Yang D.C. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic Acids Res. 2017;45:D1040–D1045. doi: 10.1093/nar/gkw982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bailey T.L., Gribskov M. Combining evidence using p-values: application to sequence homology searches. Bioinformatics. 1998;14:48–54. doi: 10.1093/bioinformatics/14.1.48. [DOI] [PubMed] [Google Scholar]
- 26.Wang K., Jiang S., Sun C. The spatial and temporal transcriptomic landscapes of ginseng, Panax ginseng C.A.Meyer. Sci Rep. 2015;5:18283. doi: 10.1038/srep18283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang J.J., Su H., Zhang L. Comprehensive characterization for ginsenosides biosynthesis in ginseng root by integration analysis of chemical and transcriptome. Molecules. 2017;22:889. doi: 10.3390/molecules22060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trapnell C., Roberts A., Goff L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc. 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Trapnell C., Hendrickson D.G., Sauvageau M., Goff L., Rinn J.L., Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang L., Feng Z., Wang X., Wang X., Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 32.De Geyter N., Gholami A., Goormachtig S., Goossens A. Transcriptional machineries in jasmonate-elicited plant secondary metabolism. Trends Plant Sci. 2012;17:349–359. doi: 10.1016/j.tplants.2012.03.001. [DOI] [PubMed] [Google Scholar]
- 33.Goodwin S., McPherson J.D., McCombie W.R. Coming of age: ten years of next-generation sequencing technologies. Nat Rev Genet. 2016;17:333–351. doi: 10.1038/nrg.2016.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Peng W., Wang J., Cai J., Chen L., Li M., Wu F.X. Improving protein function prediction using domain and protein complexes in PPI networks. BMC Syst Biol. 2014;8:35. doi: 10.1186/1752-0509-8-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y., Xu Z., Ji A., Luo H., Song J. Genomic survey of bZIP transcription factor genes related to tanshinone biosynthesis in Salvia miltiorrhiza. Acta Pharm Sin B. 2018;8:295–305. doi: 10.1016/j.apsb.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu Z., Ji A., Song J., Chen S. Genome-wide analysis of auxin response factor gene family members in medicinal model plant Salvia miltiorrhiza. Biol Open. 2016;5:848–857. doi: 10.1242/bio.017178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chinnusamy V., Ohta M., Kanrar S. ICE1: a regulator of cold-induced transcriptome and freezing tolerance in Arabidopsis. Genes Dev. 2003;17:1043–1054. doi: 10.1101/gad.1077503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fursova O.V., Pogorelko G.V., Tarasov V.A. Identification of ICE2, a gene involved in cold acclimation which determines freezing tolerance in Arabidopsis thaliana. Gene. 2009;429:98–103. doi: 10.1016/j.gene.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 39.Badawi M., Reddy Y.V., Agharbaoui Z. Structure and functional analysis of wheat ICE (inducer of CBF expression) genes. Plant Cell Physiol. 2008;49:1237–1249. doi: 10.1093/pcp/pcn100. [DOI] [PubMed] [Google Scholar]
- 40.Spyropoulou E.A., Haring M.A., Schuurink R.C. RNA sequencing on Solanum lycopersicum trichomes identifies transcription factors that activate terpene synthase promoters. BMC Genom. 2014;15:402. doi: 10.1186/1471-2164-15-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ji Y., Xiao J., Shen Y. Cloning and characterization of AabHLH1, a bHLH transcription factor that positively regulates artemisinin biosynthesis in Artemisia annua. Plant Cell Physiol. 2014;55:1592–1604. doi: 10.1093/pcp/pcu090. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material
Supplementary material