

Abstract
The aim of the present study was to examine the effects of atorvastatinon p38 phosphorylation and cardiac remodeling after myocardial infarction in rats. A total of 43 rats were randomly divided into the control, sham operation, post-modeling medication (medication) and post-modeling non-medication (non-medication) groups. The control group did not receive any treatment. Anterior descending arteries of the rats in the medication and non-medication groups were ligated, and threading at the anterior descending arteries was conducted for the rats in the sham operation group. Atorvastatin (10 mg/kg) was given daily to the rats in the medication group, and an equivalent amount of normal saline was given daily to the rats in the sham operation group. Four weeks later, the cardiac function, morphological changes in the myocardial cells, and the expression of tumor necrosis factor-α (TNF-α) and p38 in each group was detected. At 4 weeks after treatment, the myocardial infarction size, fibrosis and myocardial necrosis of the rats in the medication group was examined compared with those in the non-medication group (P<0.05). The cardiac function of the rats in the non-medication group was significantly lower than that of the rats in the control and sham groups (P<0.05), while it was obviously elevated in the medication group compared with that in the non-medication group (P<0.05). The expression of TNF-α and phosphorylated p38 of the left ventricle in the non-medication group was higher than that in the control group (P<0.05), while it was obviously reduced in the non-medication group compared with that in the control group (P<0.05). Atorvastatin can improve cardiac remodeling after myocardial infarction in rats, which may be associated with its inhibition of p38 phosphorylation and its decrease of TNF-α expression.
Keywords: myocardial infarction, rats, atorvastatin, p38, cardiac remodeling
Introduction
Myocardial infarction is one of the leading causes of death in patients (1). The tissues release a variety of chemokines and cytokines after infarction, which leads to the accumulation of inflammatory cells and induces inflammation, resulting in fibrosis of myocardial tissues and cardiac remodeling (2). This causes heart failure due to a reduction in the cardiac function (3–5). Regulating post-infarction inflammatory response can improve cardiac remodeling after myocardial infarction. This enhances cardiac function and reduces mortality.
Atorvastatin is currently one of the most commonly used drugs in clinic for the treatment of coronary heart disease and is mainly used for the control of risk factors in coronary heart disease as well as in the improvement of prognosis. As a result, the incidence of myocardial infarction is reduced (6). Recent findings have shown that atorvastatin can improve cardiac function after myocardial infarction (7), but its specific mechanism remains to be determined. The effects of atorvastatin on myocardial remodeling after myocardial infarction in rats were discussed in this study mainly from the aspect of inflammation.
Materials and methods
Reagents and instruments
Chloral hydrate (Wuhan Yuancheng Technology Development Co., Ltd., Hubei, China), small animal ventilator and BL-420 biological signal collection system (both from Chengdu TME Technology Co., Ltd., Sichuan, China), radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime Institute of Biotechnology, Haimen, China) were used in the present study. Hematoxylin and eosin (H&E), and triphenyl tetrazolium chloride (TTC) staining was carried out (both purchased from Nanjing Jiancheng Bioengineering Institute, Jiangsu, China). Masson's trichrome staining was performed using a Masson Stain kit (D026) (Nanjing Jiancheng Bioengineering Institute). Color Doppler ultrasonic diagnostic apparatus (Mindray North America, Mahwah, NJ, USA), as well as rabbit anti-rat p38 mitogen-activated protein kinase and phosphorylated p38 polyclonal antibodies (all purchased from Cell Signaling Technology, Danvers, MA, USA) were used in the study. Horseradish peroxidase-labeled goat anti-rat antibodies and diaminobenzidine (DAB) coloration reagent kits were used (both from Beyotime Institute of Biotechnology). In addition, we used atorvastatin calcium tablets (Pfizer, Inc., New York, NY, USA), total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), triglyceride (TG) and high-density lipoprotein cholesterol (HDL-C) test kits (all from Nanjing Jiancheng Bioengineering Institute). Further reagents and instruments used were: Protease inhibitor (Roche Diagnostics, Basel, Switzerland), protein electrophoresis apparatus (Bio-Rad Laboratories, Inc., Hercules, CA, USA), rapid tissue cell cracker (Wuxi Voshin Instruments Co., Ltd., Jiangsu, China), protein loading buffer (Beyotime Institute of Biotechnology), pre-stained protein marker (Thermo Fisher Scientific, Inc., Waltham, MA, USA), and fluorescence microscope (IX73; Olympus Corporation, Tokyo, Japan).
Modeling
Chloral hydrate (300–350 mg/kg) was administered by intraperitoneal injection. The rats were connected with the ventilator and BL-420 bioinformation collection system was used after anesthesia. The thoracic cavity was opened from the 3rd and 4th intercostal space of the left thoracic cavity in rats. Anterior descending arteries of the rats in the medication and myocardial infarction groups were ligated with a 6-0 suture. The rats in the sham operation group were threaded but not ligated. Following surgery, the chest was closed and erythromycin ointment was applied. The intratracheal catheter was removed after the rats woke up from anesthesia, and they were returned to the rearing cage for a 4-week rearing. Signs of successful myocardial infarction modeling included: i) Electrocardiogram showed elevated ST segment and ii) the ligation site and the heart tissue at the cardiac apex were whitened. The modeling was considered successful when the aforementioned two requirements were met (8).
Laboratory animals and grouping
A total of 43 male Sprague-Dawley rats (~200 g) were provided by the Laboratory Animal Center of Hubei Medical University (cat. no. SCXK E 2016–0008). The study was approved by the Ethics Committee of Xiangyang No. 1 People's Hospital (Xiangyang, China). The rats were kept in cage at temperature of 22–25°C and humidity 53–60%. The animals had access to food and water ad libitum. The rats that were modeled successfully were divided into four groups using a random number table: i) The control group (10 rats), rats were fed normally; ii) the sham group (11 rats), rats were given 2 ml normal saline by gavage every 24 h continuously for 4 weeks; iii) the non-medication group (11 rats), rats were given 2 ml normal saline by gavage every 24 h continuously for 4 weeks and iv) the medication group (11 rats), atorvastatin was dissolved in the normal saline, and 2 ml atorvastatin (drug dose: 10 mg/kg) (9–11) was given to the rats by gavage every 24 h continuously for 4 weeks after successful modeling.
Measurement of cardiac function via echocardiography
Left ventricular ejection fraction (LVEF), shortening fraction (SF), left ventricular end-systolic diameter (LVESD) and left ventricular end-diastolic diameter (LVEDD) of the rats in each group were measured via color Doppler ultrasonic diagnostic apparatus before and at 4 weeks after modeling.
Measurement of blood lipids in each group
Four weeks later, the rats were anesthetized using chloral hydrate (300–350 mg/kg) via intraperitoneal injection. Blood (4 ml) was drawn from the abdominal aorta and the plasma was frozen at −80°C. The concentrations of TC, TG, HDL-C and LDL-C were detected using the reagent kits.
Left ventricular H&E staining
The anterior wall of the left ventricle of the rats in each group was sampled and fixed in 4% paraformaldehyde. The samples were embedded with paraffin the next day to produce paraffin sections. After H&E staining, the samples were placed under the fluorescence microscope to observe the morphological changes of the cells.
Masson staining
The hearts of the rats were removed and washed repeatedly with normal saline, then fixed in 4% paraformaldehyde. Serial sectioning (5 times in total) was conducted for the hearts of the rats in each group to produce paraffin sections. Masson staining was performed according to the kit instructions after dewaxing. The fibrosis area of each group was analyzed using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
TTC staining
The hearts were removed, washed repeatedly and placed in the heart cutting slot (RWD Life Science Co., Ltd., Shenzhen, China). Then, they were placed in the refrigerator at −20°C for 2 h. Heart sectioning was conducted in the unit of 2 mm and placed in TTC dye liquor for incubation at 37°C for 30 min. The results were observed, and the myocardial infarction size was measured using ImageJ software (National Institutes of Health).
Western blot analysis
The left ventricles of the rats were excised and cut into pieces in the size of rice (12). An appropriate amount of RIPA lysis buffer and protease inhibitors were added. The tissues were pulverized using a rapid tissue cell cracker and centrifuged at 13,500 × g for 5 min at 4°C. The supernatant was removed and an appropriate amount of protein loading buffer was added. Then, it was boiled for 5 min and frozen at −20°C. SDS-PAGE with 5% concentration and 12% separation gel was used. Pre-stained protein marker was added to the first well, and 30 µg protein samples were added to the remaining wells. Electrophoresis was stopped when the bromophenol blue reached the bottom of the gel. Membrane transfer was performed at 4°C for 2 h at 200 mA. The wells were eluted with Tris-buffered saline containing Tween-20 (TBST) and blocked for 2 h with 5% skimmed milk powder (4% bovine serum albumin was used for phosphorylated antibodies). Rabbit anti-rat α-tubulin, p38, phospho-p38 primary polyclonal antibodies (1:500; cat. nos. 2144, 9212 and 9211; Cell Signaling Technology, Danvers, MA, USA) and rabbit anti-rat TNF-α primary polyclonal antibody (1:500; cat. no. ARC3012; Thermo Fisher Scientific, Inc.) was added and incubated overnight at 4°C. Then they were eluted with TBST. Goat anti-rabbit secondary polyclonal antibody (1:1,000; cat. no. 7074; Cell Signaling Technology) was added and incubated for 1 h at room temperature. Finally, they were eluted with TBST, and DAB coloration reagent kits were used for color development for several minutes. They were scanned and the images were saved. Gray value was measured using ImageJ software (National Institutes of Health).
Statistical analysis
SPSS 17.0 (SPSS, Inc., Chicago, IL, USA) was used for result processing. A t-test was used for the comparison of enumeration data between two groups, and analysis of variance with the Bonferroni's post hoc test was used for comparison among multiple groups. The data were expressed as mean ± standard deviation (SD). P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of atorvastatin on improving the cardiac function of the rats
LVEDD, LVESD, LVEF and SF in each group did not show significant differences between groups before modeling (p>0.05, Table I). The ventricular wall movement of the rats in the non-medication group was obviously reduced in comparison with that of the rats in the medication group. Four weeks after modeling LVEF and SF in the medication group were significantly higher than those in the non-medication group (P<0.05). LVESD and LVEDD in the medication group were obviously lower than those in the non-medication group (P<0.05) (Figs. 1 and 2, Tables I and II).
Table I.
Cardiac function in each group before modeling (mean ± SD).
| Groups | LVEDD (cm) | LVESD (cm) | LVEF (%) | SF (%) |
|---|---|---|---|---|
| Control | 0.62±0.08 | 0.32±0.08 | 85.75±7.85 | 57.60±7.98 |
| Sham operation | 0.62±0.11 | 0.32±0.10 | 85.33±8.90 | 57.00±8.78 |
| Non-medication | 0.61±0.11 | 0.31±0.11 | 86.75±7.96 | 58.11±7.88 |
| Medication | 0.62±0.07 | 0.32±0.08 | 83.88±8.56 | 60.19±8.13 |
The remaining quantity of animals in each group: In the sham operation, 9; control, 10; non-medication, 8; and medication groups, 9. LVEDD, LVESD, LVEF and SF in each group show that P>0.05. LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; LVESD, left ventricular end-systolic diameter; SF, shortening fraction; SD, standard deviation.
Figure 1.

Heart echocardiography of the rats in each group before operation. (A) Control, (B) sham operation, (C) non-medication and (D) medication groups.
Figure 2.

Echocardiogram of the rats in each group 4 weeks later. (A) Control, (B) sham operation, (C) non-medication and (D) medication groups.
Table II.
Cardiac function in each group at 4 weeks after modeling (mean ± SD).
| Groups | LVEDD (cm) | LVESD (cm) | LVEF (%) | SF (%) |
|---|---|---|---|---|
| Control | 0.60±0.08 | 0.30±0.75 | 82.20±7.54 | 56.10±9.40 |
| Sham operation | 0.72±0.10 | 0.45±0.70 | 85.75±7.85 | 51.44±9.15 |
| Non-medication | 1.07±0.12a | 0.76±0.11a | 49.13±9.03a | 32.88±9.16a |
| Medication | 0.69±0.09b | 0.39±0.09b | 64.00±8.37b | 49.89±7.51b |
Comparison between the non-medication and control groups shows that P<0.05.
Comparison between the medication and non-medication groups shows that P<0.05. LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; LVESD, left ventricular end-systolic diameter; SF, shortening fraction; SD, standard deviation.
Measurement of blood lipids in each group 4 weeks later
HDL-C and TG of each group had no statistical differences (P>0.05). The contents of TC and LDL-C in the blood of the rats in the medication group were obviously decreased (P<0.05) (Table III).
Table III.
Blood lipids in each group 4 weeks later (mean ± SD) (mmol/l).
| Groups | TC | TG | HDL-C | LDL-C |
|---|---|---|---|---|
| Control | 2.49±0.18 | 1.35±0.14 | 1.46±0.17 | 0.71±0.15 |
| Sham operation | 2.49±0.19 | 1.48±0.14 | 1.53±0.18 | 0.67±0.17 |
| Non-medication | 2.80±0.28 | 1.35±0.18 | 1.40±0.22 | 0.70±0.13 |
| Medication | 1.78±0.26a | 1.33±0.18 | 1.52±0.19 | 0.43±0.09a |
Comparison between the medication and non-medication groups shows that P<0.05. TC, total cholesterol; TG, triglyceride; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; SD, standard deviation.
Comparison of cell morphology of the rats in each group
The non-medication group showed a large area of fibrous tissue hyperplasia. There were only a small number of normal cardiac tissues with incomplete capsule, nuclear fragmentation and dissolution. Cardiac tissues with clear texture could be seen in the sections of the rats in the control group. The capsule was complete, the cell nucleus was blue and the cytoplasm was red. In addition, the medication group showed a small amount of fibrous tissue hyperplasia. Necrotic myocardial cells were significantly reduced, and the texture of the myocardial cells was still visible. The sham operation group showed a small amount of fibrous tissue hyperplasia. Necrotic myocardial cells were rarely observed, and the texture of the myocardial cells was still evident (Fig. 3).
Figure 3.

H&E staining of the anterior myocardial wall in each group (×400). (A) Control, (B) sham operation, (C) non-medication and (D) medication groups. H&E, hematoxylin and eosin.
Comparison of the myocardial fibrosis area of the rats in each group after medication
The proportion of fibrosis in each group was significantly different (P<0.05), and the myocardial fibrosis area in the medication group was significantly lower than that in the non-medication group (P<0.05) at 4 weeks after myocardial infarction in rats (Fig. 4 and Table IV).
Figure 4.

Myocardial Masson staining in each group 4 weeks later. (A) Control, (B) sham operation, (C) non-medication and (D) medication group. The blue part is the fibrosis area.
Table IV.
The area of infarction and fibrosis 4 weeks later (mean ± SD).
| Groups | Proportion of myocardial infarction size | Proportion of cardiac fibrosis area |
|---|---|---|
| Control | 0±0 | 0.01±0.004 |
| Sham operation | 0.11±0.05 | 0.06±0.007 |
| Non-medication | 0.32±0.05a | 0.20±0.019a |
| Medication | 0.22±0.09b | 0.11±0.016b |
Comparison between the non-medication and normal groups shows that P<0.05
Comparison between the medication and non-medication group shows that P<0.05; SD, standard deviation.
Effect of atorvastatin on reducing the myocardial infarction size
The proportion of fibrosis in each group was significantly different (P<0.05), and the myocardial infarction size of the rats in the medication group was significantly lower than that of the rats in the non-medication group (P<0.05) after the 4-week experiment (Fig. 5 and Table IV).
Figure 5.

TTC staining of the hearts of the rats 4 weeks later. (A) Control, (B) sham operation, (C) non-medication and (D) medication groups. The white part is the infarction area and the red part is the non-infarction area. Heart sections were incubated with 2% TTC at 37°C for 30 min. TTC, triphenyltetrazolium chloride.
Protein expression in each group
The cardiac protein expression of the rats in each group was significantly different 4 weeks after successful modeling. Phosphorylated p38 and TNF-α in the non-medication group were obviously increased compared to those in the control group (P<0.05), while they were obviously reduced in the medication group compared with those in the non-medication group (P<0.05) (Figs. 6 and 7).
Figure 6.

Protein expression in each group 4 weeks later. (1) The control, (2) sham operation, (3) non-medication and (4) medication groups.
Figure 7.
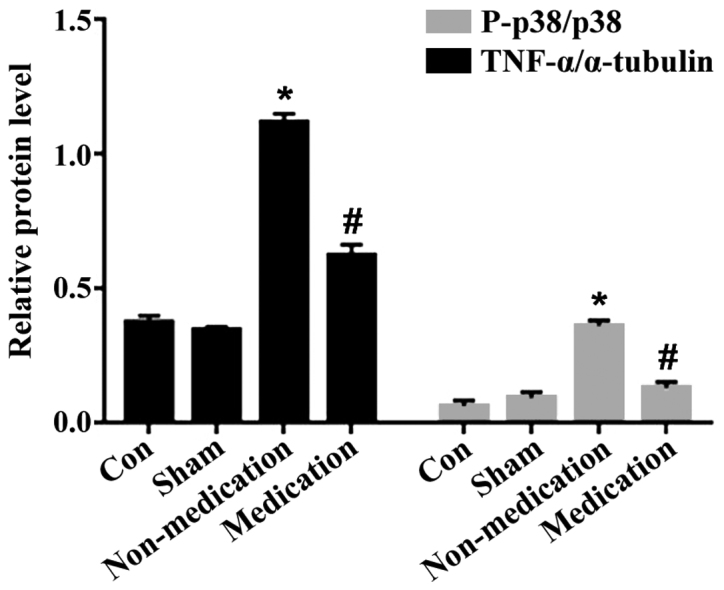
Comparison of protein expression in each group. P-p38, Phosphorylated p38, p38, total p38. *P<0.05, comparison between the non-medication and normal groups. #P<0.05, comparison between the medication and non-medication groups.
Discussion
Myocardial infarction is the necrosis of myocardial cells in the heart caused by ischemia and hypoxia (13). An inflammatory reaction is caused by the accumulation of various inflammatory cells after infarction. The accumulation of inflammatory cells and release of inflammatory cytokines lead to post-infarction cardiac remodeling (2). The expansion of the central cavity, decrease of cardiac function, apoptosis of the myocardial cells and proliferation of fibrous tissues are observed during cardiac remodeling (14). Early remodeling changes the adaptation of the heart. Long-term staying in this state leads to increased cardiac load, decreased cardiac function and heart failure (15). Therefore, the study of the treatment method for cardiac modeling after myocardial infarction is of great importance.
Atorvastatin has the function of regulating lipids. Some scholars believe that atorvastatin can resist inflammation, oxidation and arrhythmia, and improve cardiac remodeling. The long-term administration of atorvastatin can improve patient prognosis and reduce the incidence of adverse events (7,16). However, the exact mechanism is unknown. In the present study, the left ventricular cardiac function, myocardial fibrosis, morphological changes of the myocardial cells and expression of phosphorylated p38 and TNF-α in the rats were detected at 4 weeks after myocardial infarction in order to study the possible mechanism of atorvastatin in improving cardiac remodeling. The results of the experiment showed that the 4-week continuous administration of atorvastatin could improve the left ventricular function of the rats. LVEF and SF were increased compared with those in the non-medication group (P<0.05 for both items), while LVESD and LVEDD were decreased compared with those in the non-medication group (P<0.05 for both items). The myocardial infarction size and fibrosis area in the medication group were significantly reduced compared with those in the non-medication group (P<0.05 for both items). The morphological structure of the myocardial cells was more complete. Necrotic cells were decreased, and the outline of the myocardial cells was clearly visible. The contents of phosphorylated p38 and TNF-α in the left ventricle were significantly decreased (P<0.05 for both items). It indicated that atorvastatin can improve cardiac remodeling after myocardial infarction in rats. Therefore, its action of mechanism may be related to its inhibition of p38 phosphorylation and its decrease of TNF-α expression.
p38 includes 4 subtypes, namely p38α, p38β, p38γ and p38δ (17). p38β, p38γ and p38δ are expressed in the brain, skeletal muscle and pancreas, respectively (17). However, p38α is expressed in any tissue. Its molecular weight is 38 kDa. Endogenous response can be caused by tyrosine site phosphorylation (18). Heat shock protein (HSP) 27 is a target of action at the downstream of p38α. Upon oxidative stress, the activation of HSP27 can convert the actin into a fibrous tissue, in which the p38/MK2/HSP29 pathway plays an important role (18). TNF-α is mainly secreted by macrophages (19), which can cause inflammatory immune response and induce the secretion of various cytokines. It also plays an important role in ischemic cardiomyopathy (20). TNF-α is expressed in large numbers after myocardial infarction in rats (21) and acts on the extracellular matrix (22), leading to myocardial remodeling due to an increased expression of matrix metalloproteinases (23). On the one hand, inflammatory cytokines can activate inducible nitric oxide synthase and increase its activity. On the other hand, it can activate renin-angiotensin-aldosterone system to participate in myocardial remodeling. TNF-α can promote apoptosis (14). The interaction of apoptosis, necrosis and proliferation of fibrous tissues further aggravates myocardial remodeling, which further deteriorates cardiac function. Therefore, reduction of the level of inflammatory cytokines is essential for improving cardiac remodeling after myocardial infarction.
In summary, the findings have shown that the function of atorvastatin on improving cardiac remodeling after myocardial infarction in rats may be associated with its inhibition of p38 phosphorylation and its reduction of TNF-α expression, which provides a basis for clarifying the mechanism of atorvastatin in cardiac remodeling after the treatment of myocardial infarction and lays a foundation for the discovery of subsequent new therapeutic targets.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.
Authors' contributions
ML was responsible for the implementation of the project, the establishment of animal models, the test of indexes, and the wrote the study. FL, the chief executive of the project, provided ideas of selecting topics, conducted experiments and statistical methods and revised the paper. MS collected the data, modificated the pictures and statistical analysis. XS was responsible for the whole experimental methodology. LL was responsible for the production of animal models, animal husbandry and gavage. XW collected and stored the animal specimens. All authors read and approved the final manuscript.
Ethics approval and consent to participate
The study was approved by the Ethics Committee of Xiangyang No.1 People's Hospital (Xiangyang, China).
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Chen J, Cao W, Asare PF, Lv M, Zhu Y, Li L, Wei J, Gao H, Zhang H, et al. Amelioration of cardiac dysfunction and ventricular remodeling after myocardial infarction by danhong injection are critically contributed by anti-TGF-β-mediated fibrosis and angiogenesis mechanisms. J Ethnopharmacol. 2016;194:559–570. doi: 10.1016/j.jep.2016.10.025. [DOI] [PubMed] [Google Scholar]
- 2.Kumagai S, Nakayama H, Fujimoto M, Honda H, Serada S, Ishibashi-Ueda H, Kasai A, Obana M1, Sakata Y, et al. Myeloid cell-derived LRG attenuates adverse cardiac remodelling after myocardial infarction. Cardiovasc Res. 2016;109:272–282. doi: 10.1093/cvr/cvv273. [DOI] [PubMed] [Google Scholar]
- 3.Velasquez LS, Sutherland LB, Liu Z, Grinnell F, Kamm KE, Schneider JW, Olson EN, Small EM. Activation of MRTF-A-dependent gene expression with a small molecule promotes myofibroblast differentiation and wound healing. Proc Natl Acad Sci U S A. 2013;110:16850–16855. doi: 10.1073/pnas.1316764110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sobirin MA, Kinugawa S, Akahashi M, Fukushima A, Homma T, Ono T, Hirabayashi K, Suga T, Azalia P, et al. Activation of natural killer T cells ameliorates postinfarct cardiac remodeling and failure in mice. Circ Res: Aug 10, 2012 (Epub ahead of print) https://doi.org/10.1161/CIRCRESAHA.112.270132. https://doi.org/10.1161/CIRCRESAHA.112.270132 [DOI] [PubMed]
- 5.Qi HP, Wang Y, Zhang QH, Guo J, Li L, Cao YG, Li SZ, Li XL, Shi MM, et al. Activation of peroxisome proliferator-activated receptor γ (PPARγ) through NF-κB/Brg1 and TGF-β1 pathways attenuates cardiac remodeling in pressure-overloaded rat hearts. Cell Physiol Biochem. 2015;35:899–912. doi: 10.1159/000369747. [DOI] [PubMed] [Google Scholar]
- 6.Berwanger O, de Barros E, Silva PG, Barbosa RR, Precoma DB, Figueiredo EL, Hajjar LA, Kruel CD, Alboim C, Almeida AP, Dracoulakis MD, et al. Atorvastatin for high-risk statin-naïvepatients undergoing noncardiac surgery: The lowering the risk of operative complications using atorvastatin loading dose (LOAD) randomized trial. Am Heart J. 2017;184:88–96. doi: 10.1016/j.ahj.2016.11.001. [DOI] [PubMed] [Google Scholar]
- 7.Reichert K, Pereira do Carmo HR, Galluce Torina A, Diógenes de Carvalho D, Carvalho Sposito A, de Souza Vilarinho KA, da Mota Silveira-Filho L, Martins de Oliveira PP, Petrucci O. Atorvastatin improves ventricular remodeling after myocardial infarction by interfering with collagen metabolism. PLoS One. 2016;11:e0166845. doi: 10.1371/journal.pone.0166845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Puhl SL, Müller A, Wagner M, Devaux Y, Böhm M, Wagner DR, Maack C. Exercise attenuates inflammation and limits scar thinning after myocardial infarction in mice. https://doi.org/10.1152/ajpheart.00683.2014. Am J Physiol Heart and Circ Physiol. 2015;309:345–359. doi: 10.1152/ajpheart.00683.2014. [DOI] [PubMed] [Google Scholar]
- 9.Song XJ, Yang CY, Liu B, Wei Q, Korkor MT, Liu JY, Yang P. Atorvastatin inhibits myocardial cell apoptosis in a rat model with post-myocardial infarction heart failure by downregulating ER stress response. Int J Med Sci. 2011;8:564–572. doi: 10.7150/ijms.8.564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.An Z, Yang G, He YQ, Dong N, Ge LL, Li SM, Zhang WQ. Atorvastatin reduces myocardial fibrosis in a rat model with post-myocardial infarction heart failure by increasing the matrix metalloproteinase-2/tissue matrix metalloproteinase inhibitor-2 ratio. Chin Med J (Engl) 2013;126:2149–2156. [PubMed] [Google Scholar]
- 11.Tang XL, Sanganalmath SK, Sato H, Bi Q, Hunt G, Vincent RJ, Peng Y, Shirk G, Dawn B, Bolli R. Atorvastatin therapy during the peri-infarct period attenuates left ventricular dysfunction and remodeling after myocardial infarction. PLoS One. 2011;6:e25320. doi: 10.1371/journal.pone.0025320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song MA, Dasgupta C, Zhang L. Chronic losartan treatment up-regulates AT1R and increases the heart vulnerability to acute onset of ischemia and reperfusion injury in male rats. PLoS One. 2015;10:e0132712. doi: 10.1371/journal.pone.0132712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thygesen K, Alpert JS, White HD, Joint ESC/ACCF/AHA/WHFTask Force for the redefinition of myocardial infarction Universal definition of myocardial infarction. J Am Coll Cardiol. 2007;50:2173–2195. doi: 10.1016/j.jacc.2007.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Fan Z, Fu M, Xu Z, Zhang B, Li Z, Li H, Zhou X, Liu X, Duan Y, Lin PH, et al. Sustained release of a peptide-based MatrixMetalloproteinase-2 inhibitor to attenuate adverse cardiac remodeling and improve cardiac function following myocardial infarction. Biomacromolecules. 2017;18:2820–2829. doi: 10.1021/acs.biomac.7b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heeger CH, Jaquet K, Thiele H, Zulkarnaen Y, Cuneo A, Haller D, Kivelitz D, Schmidt T, Krause K, Metzner A, et al. Percutaneous, transendocardial injection of bone marrow-derived mononuclear cells in heart failure patients following acute ST-elevation myocardial infarction: ALSTER-Stem Cell trial. EuroIntervention. 2012;8:732–742. doi: 10.4244/EIJV8I6A113. [DOI] [PubMed] [Google Scholar]
- 16.Ma H, Liu Y, Xie H, Zhang G, Zhan H, Liu Z, Wang P, Geng Q, Guo L. The renoprotective effects of simvastatin and atorvastatin in patients with acute coronary syndrome undergoing percutaneous coronary intervention: An observational study. Medicine. 2017;96:e7351. doi: 10.1097/MD.0000000000007351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Remy G, Risco AM, Iñesta-Vaquera FA, González-Terán B, Sabio G, Davis RJ, Cuenda A. Differential activation of p38MAPK isoforms by MKK6 and MKK3. Cell Signal. 2010;22:660–667. doi: 10.1016/j.cellsig.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 18.Corre I, Paris F, Huot J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget. 2017;8:55684–55714. doi: 10.18632/oncotarget.18264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Udalova I, Monaco C, Nanchahal J, Feldmann M. Anti-TNF therapy. Microbiol Spectr. 2016;4 doi: 10.1128/microbiolspec.MCHD-0022-2015. doi: 10.1128/microbiolspec. MCHD-0022-2015. [DOI] [PubMed] [Google Scholar]
- 20.Zhang P, Wu X, Li G, He Q, Dai H, Ai C, Shi J. Tumor necrosis factor-alpha gene polymorphisms and susceptibility to ischemic heart disease: A systematic review and meta-analysis. Medicine (Baltimore) 2017;96:e6569. doi: 10.1097/MD.0000000000006569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen H, Xu Y, Wang J, Zhao W, Ruan H. Baicalin ameliorates isoproterenol-induced acute myocardial infarction through iNOS, inflammation and oxidative stress in rat. Int J Clin Exp Pathol. 2015;8:10139–10147. [PMC free article] [PubMed] [Google Scholar]
- 22.Chen WL, Sheu JR, Chen RJ, Hsiao SH, Hsiao CJ, Chou YC, Chung CL, Hsiao G. Mycobacterium tuberculosis upregulates TNF-α expression via TLR2/ERK signaling and induces MMP-1 and MMP-9 production in human pleural mesothelial cells. PLoS One. 2015;10:e0137979. doi: 10.1371/journal.pone.0137979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sato F, Kohsaka A, Takahashi K, Otao S, Kitada Y, Iwasaki Y, Muragaki Y. Smad3 and Bmal1 regulate p21 and S100A4 expression in myocardial stromal fibroblasts via TNF-α. Histochem Cell Biol. 2017;148:617–624. doi: 10.1007/s00418-017-1597-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the present study are available from the corresponding author on reasonable request.