

Ataxia telangiectasia (AT) is an autosomal‐recessive, multisystem disease characterized by progressive neurologic decline, oculocutaneous telangiectasias, immunodeficiency, susceptibility to sinopulmonary infections, autoimmune or other chronic inflammatory diseases, radiation sensitivity (x‐rays and γ‐rays), and malignancies.1, 2 It is caused by a mutation in the ataxia telangiectasia mutated (ATM) gene located on chromosome 11q22‐23.3 In typical cases, progressive ataxia starts in the first year of life, leads to a wheelchair‐bound state around the second decade, and it is variably accompanied by other movement disorders like chorea, dystonia, or myoclonus.1 However, there is increasing evidence of atypical forms or variants in which the clinical picture is different, in that it is less severe, with dystonic‐predominant symptoms and without the typical clinical of AT.1, 2, 4
Case Report
A 27‐year‐old woman (Patient II‐2) was referred for a rapid onset of involuntary head movements starting at the age of 12 years. On examination, she presented with mobile cervical dystonia with patterned dystonic head movements, risus sardonicus‐like orofacial dystonia, tremor of the eyelids, and spasmodic dysphonia with the characteristic of a strangled quality of the voice. The remainder of her examination was unremarkable (Video S1). Her dystonic symptoms tended to be accentuated in stress. The course was without obvious progression or marked fluctuations, except for a few episodes of action‐induced dystonia of the right leg, which appeared during normal gait and resolved while running. There were no obvious psychiatric symptoms. Her family history was unremarkable. In the past, she was treated for pneumonia, recurrent bronchitis, pharyngitis, and oligoarthritis with exanthema in the lower extremities of possible reactive or autoimmune etiology.
Routine blood screening, repeated brain magnetic resonance imaging, abdominal ultrasonography, panels for systemic and rheumatic diseases, thyroid hormones, uric acid, ceruloplasmin, and a peripheral smear for acanthocytes were negative or normal. Slit lamp examination did not reveal a Kayser‐Fleischer ring. A mild axonal polyneuropathy of unknown origin was detected. Treatment with sodium valproate, risperidone, levodopa as well as botulinum toxin injections for cervical dystonia were not helpful. Because a genetic origin was strongly suspected, genetic tests for DYT1, DYT6, DYT11, DYT18, DYT23, DYT24, and DYT25 were conducted, and all produced negative results.
Finally, case‐parent trio exome sequencing was performed using the SureSelect All Exon system (Agilent Technologies, Santa Clara, CA) for capture of exome‐coding DNA and a HiSeq2500 (Illumina Inc., San Diego, CA) for sequence determination (Table S1). Data analysis using a laboratory‐established bioinformatics pipeline (Helmholtz Center, Munich) revealed 2 rare pathogenic variants in the ATM gene, which were confirmed by Sanger sequencing (Fig. 1): c.5573G>A (p.Trp1858*) and c.6154G>A (p.Glu2052Lys); whereas c.5573G>A (p.Trp1858*) was a novel nonsense alteration predicted to cause a truncated protein, c.6154G>A (p.Glu2052Lys) was a low‐frequency missense variant (frequency in the Exome Aggregation Consortium data set, 8 of 121,210 alleles) previously associated with AT.5 The nonsense variant was carried by the patient's unaffected father (I‐1), and the missense variant was carried by the healthy mother (I‐2), indicating a compound heterozygous status in the patient. A variant of AT with predominant craniocervical dystonia was diagnosed. Subsequently, an increased level of α‐fetoprotein, which is a laboratory hallmark of the disease, was found (25.8 ng/mL; normal range, <8.1 ng/mL). Serum protein electrophoresis, lymphocytes, and immunoglobulins (Igs) with all IgG subclasses were normal. Interestingly, a detailed inspection of the patient did not reveal any features of ataxia or telangiectasias on the conjunctivae (Fig. 2A), earlobes (Fig. 2B), skin, or oral cavity. The patient is now medication‐free but is under strict surveillance regarding any tumor detection.
Figure 1.
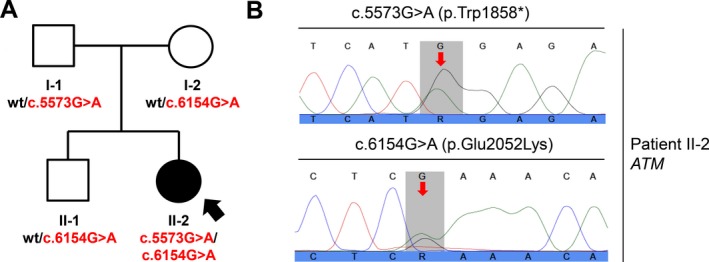
Compound heterozygous ataxia telangiectasia mutated (ATM) mutations in isolated dystonia. Sanger sequencing confirmed that the patient (II‐2) carried the 2 exome‐identified ATM mutations c.5573G>A (p.Trp1858*) and c.6154G>A (p.Glu2052Lys), whereas her family members were heterozygous carriers for either the c.5573G>A (p.Trp1858*) allele (Father I‐1) or the c.6154G>A (p.Glu2052Lys) allele (mother, I‐2; brother, II‐1). A: This is the family pedigree. Symbols are as follows: circles, females; squares, males; solid, dystonia‐affected; open, healthy. Patient II‐2 is indicated by an arrow, and mutational status is shown. Wt indicates wild type. B: Sanger electropherograms demonstrate the ATM mutations in Patient II‐2 (altered nucleotides are highlighted with red arrows).
Figure 2.
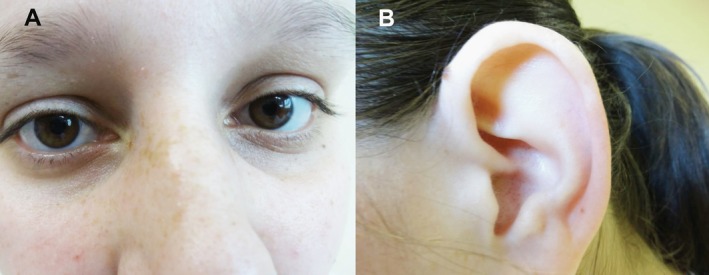
The patient's (A) conjunctivas and (B) earlobes are shown with no visible telangiectasias.
Discussion
Typical AT is associated with ataxia and telangiectasias, immunodeficiency (mainly lower Ig levels or lymphopenia), frequent pulmonary infections, increased sensitivity to ionizing radiation, and an elevated risk of cancer (typically lymphoid tumors in younger patients).1 In contrast, AT variants are characterized by a generally milder course of the disease, usually with a later onset. Immunodeficiency and pulmonary manifestations are less common, and there is a higher risk of malignancy (nonhematopoetic) in later stages.1 Contrary to the classic form of AT, a milder phenotype of the disease is explained by a residual expression of the ATM gene with low but sufficient AT kinase activity, which is involved in the DNA repair processes.6 Unfortunately, ATM kinase activity detection in our patient was not available.
In the clinical picture of variant forms, various hyperkinesias, including dystonia‐dominant cases (e.g., primary torsion dystonia7, 8 or myoclonus‐dystonia8) with a late onset of ataxia, have been reported.1 Although cases of AT without ataxia or telangiectasias were previously reported,2, 4, 7, 8, 9 our case is rather unique because of the long‐term manifestation as a nonprogressive, isolated segmental dystonia in the absence of features typical for AT. Moreover, although the c.6154G>A mutation has been reported in patients who had AT with dopa‐responsive cervical dystonia,5 the pathogenic variant c.5573G>A has not been reported until now.
Because variant cases of AT without clinical manifestation of classical symptoms were previously included under an AT diagnosis, the designation of the term ATM syndrome was recently proposed to cover typical and atypical forms.10 Our case report, along with others,5, 7, 8 highlights the necessity to consider α‐fetoprotein testing and a subsequent genetic examination for AT syndrome in all patients with otherwise unexplained, young‐onset, isolated dystonia.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
J.N.: 1A, 1B, 1C, 3A, 3B
M.Z.: 1B, 1C, 3B
M.Š.: 1A, 1C, 3B
P.H.: 1A, 1B, 1C, 3B
A.F.: 1A, 1B, 1C, 3B
J.W.: 1C, 3B
R.J.: 1A, 1B, 1C, 3B
Disclosures
Ethical Compliance Statement: We confirm that we have read the journal's position on issues involved in ethical publication and affirm that this work is consistent with those guidelines.
Funding Sources and Conflict of Interest: This work was supported by the Czech Science Foundation, Czech Republic (grant GACR16‐13323S); by Charles University, Czech Republic (project Progres Q27/LF1); and by in‐house institutional funding from Technische Universität München, Munich, Germany, and Helmholtz Zentrum München, Munich, Germany. The authors report no conflicts of interest.
Financial Disclosures for the previous 12 months: Dr. Necpál reports speakers’ honoraria from AbbVie, TEVA, and UCB. Dr. Zech reports intramural funding from the Langmatz‐Stiftung, research support from the Else Kröner‐Fresenius‐Stitung, and travel expenses from Pharm‐Allergan GmbH. Dr. Skorvanek has been a member of the AbbVie and Medtronic Consulting and Advisory Boards; he reports research grants from Slovak Research and Development Agency and the Slovak Scientific Grant Agency and honoraria from International Parkinson and Movement Disorders Society, AbbVie, Egis, Krka, Lundbeck, Medtronic, Sandoz, TEVA and UCB. He receives salary from the Safarik University, Kosice, Slovakia; University Hospital of L. Pasteur, Kosice, Slovakia. Dr. Havránková reports honoraria from AbbVie. Dr. Fečíková was supported in part by the Michael J. Fox Foundation (grant: MJFF11362). Dr. Winkelmann reports speaker's honoraria from UCB. Dr. Jech reports honoraria or personal fees from AbbVie, Medtronic, Cardion, Ipsen, and Allergan; he was supported by the Czech Science Foundation (grant GACR 16‐13323S) and Charles University, Prague, Czech Republic (project Progres Q27/LF1).
Supporting information
A video accompanying this article is available in the supporting information here.
Table S1. Whole‐exome sequencing statistics for the dystonia case‐parent trio
Video S1. A 27‐year‐old woman with a 15‐year history of involuntary head movements is shown. On examination, there is a dystonia affecting the cervical and orofacial muscles and the voice. No ataxia on finger‐to‐nose testing, repeated hand movements, or gait is observed.
Acknowledgments
We thank the patient for allowing us to write this article and Drs. Michal Patarák and Jozef Graňák for technical support.
Supporting information may be found in the online version of this article
Relevant disclosures and conflicts of interest are listed at the end of this article.
References
- 1. Rothblum‐Oviatt C, Wright J, Lefton‐Greif MA, McGrath‐Morrow SA, Crawford TO, Lederman HM. Ataxia telangiectasia: a review [serial online]. Orphanet J Rare Dis 2016;11:159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lohmann E, Krüger S, Hauser AK, et al. Clinical variability in ataxia‐telangiectasia. J Neurol 2015;262:1724–1727. [DOI] [PubMed] [Google Scholar]
- 3. Savitsky K, Bar‐Shira A, Gilad S, et al. A single ataxia telangiectasia gene with a product similar to PI‐3 kinase. Science 1995;268:1749–1753. [DOI] [PubMed] [Google Scholar]
- 4. Friedman JH, Weitberg A. Ataxia without telangiectasia. Mov Disord 1993;8:223–226. [DOI] [PubMed] [Google Scholar]
- 5. Charlesworth G, Mohire MD, Schneider SA, Stamelou M, Wood NW, Bhatia KP. Ataxia telangiectasia presenting as dopa‐responsive cervical dystonia. Neurology 2013;81:1148–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Verhagen MM, Last JI, Hogervorst FB, et al. Presence of ATM protein and residual kinase activity correlates with the phenotype in ataxia‐telangiectasia: a genotype‐phenotype study. Hum Mutat 2012;33:561–571. [DOI] [PubMed] [Google Scholar]
- 7. Meissner WG, Fernet M, Couturier J, et al. Isolated generalized dystonia in biallelic missense mutations of the ATM gene. Mov Disord 2013;28:1897–1899. [DOI] [PubMed] [Google Scholar]
- 8. Saunders‐Pullman R, Raymond D, Stoessl AJ, et al. Variant ataxia‐telangiectasia presenting as primary‐appearing dystonia in Canadian Mennonites. Neurology 2012;78:649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Saunders‐Pullman R. Ataxia‐telangiectasia: without ataxia or telangiectasia? Neurology 2009;73:414–415. [DOI] [PubMed] [Google Scholar]
- 10. Teive HA, Moro A, Moscovich M, Arruda WO, Munhoz RP, Raskin S, Ashizawa T. Ataxia‐telangiectasia–a historical review and a proposal for a new designation: ATM syndrome. J Neurol Sci 2015;355:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A video accompanying this article is available in the supporting information here.
Table S1. Whole‐exome sequencing statistics for the dystonia case‐parent trio
Video S1. A 27‐year‐old woman with a 15‐year history of involuntary head movements is shown. On examination, there is a dystonia affecting the cervical and orofacial muscles and the voice. No ataxia on finger‐to‐nose testing, repeated hand movements, or gait is observed.