

Abstract
Mycobacterium tuberculosis is a causative agent of tuberculosis that causes deaths across the world. The pathogen apart from causing disease manifestations can also enter into a phase of latency to re-emerge later. Among the various factors associated with the virulence of pathogen, the lipids composing the cell wall of the bacillus have drawn much interest among. The unique composition of the cell wall composed of mycolic acid, glycolipids such as diacyltrehaloses, polyacyltrehalose, lipomannan, lipoarabinomannan (LAM), mannose-capped-LAM, sulfolipids, and trehalose-6,6’-dimycolate, all have been implicated in providing the pathogen an advantage in the host. The pathogen also alters its metabolism of fatty acids to survive the conditions in the host that is reflected in an altered cell wall composition in terms of lipids. In addition, the lipid profile of the cell wall has been shown to modulate the immune responses launched by the host, especially in the suppression, or production of inflammatory factors, cytokines, and phagocytic cells, such as dendritic cells and macrophages. Apart from M. tuberculosis, the paper also briefly looks at the role of Mycobacterium bovis and its role in tuberculosis in humans along with its lipid profile of its cell wall. This review aims to summarize the various lipids of the cell wall of M. tuberculosis along with their roles in enabling the pathogen to maintain its virulence to infect further humans and its persistence inside the host.
Keywords: Cell wall lipids of Mycobacterium bovis, glycolipids and pathogenicity, lipids of cell wall, Mycobacterium tuberculosis, persistence, role of lipids in virulence
INTRODUCTION
An important reason of death due to lung diseases includes tuberculosis.[1] One of the major causative agents of lung disease is Mycobacterium tuberculosis that has the tag of one of the world's most successful human pathogens because nearly 10 million new cases are reported every year[2] and more than 2 billion people possess a latent infection.[3] The disease affects one-third of the global population causing death of 3 million people in a year.[4] There are several outcomes to an exposure to the bacterium: the pathogen is cleared instantly, or a latent infection is established, or the disease can be manifested quickly, or the symptoms may be reactivated. Looking at the statistics, >90% of healthy people do not manifest an active disease. On the other hand, six out of 10 individuals do not clear the pathogen that could cause a lifelong infection.[5]
The pathogen is a slow-growing bacillus that is associated with transmission through respiration. The bacterium is inhaled in the form of droplets to pass through the respiratory tract and deposited in the alveoli.[3,6] Although it mainly targets the pulmonary tissue, it can also infect other parts of the body. Following infection, the innate immunity springs into action to phagocytose the bacterium. A slew of cells can ingest these pathogens including neutrophils,[7,8] macrophages, and dendritic cells.[8] This is followed by a cell-mediated response by CD4+ and CD8+ cells with the formation of granulomas. The latent infection is caused by the presence of the pathogen within the granulomas, but such immune competent patients do not clear the bacillus.[6]
FORMATION OF GRANULOMA: A CHARACTERISTIC FEATURE OF MYCOBACTERIAL PATHOGENESIS
A characteristic feature of mycobacterial pathogenesis involves the formation of granulomas. These granulomas called tuberculoid granulomas are composed of infected phagocytes in the form of a “nidus” with a layer of active giant cells and macrophages surrounding them. The outermost layer comprises lymphocytes and fibrosis.[7,9]
The formation of granuloma is linked to the limitation of the disease as various cells associated with inflammation protect by surrounding the pathogens to quell the spread and subsequent transmission of the bacterium. In addition, it is seen that the arrangement of granuloma allows for the living bacteria to be kept away from immune cells such as macrophages that activate cytotoxic T-cells. This also facilitates the constant development and evolution of the pathogens within such granulomas.[10]
LIPID PROFILE OF MYCOBACTERIUM TUBERCULOSIS CELL WALL
A characteristic feature of mycobacteria is the presence of a cell envelope that has a high content of lipids and comprises 40% of the cell envelope weight[11] [Figure 1]. All the species of mycobacteria produce a class of α-alkyl, β-hydroxy fatty acids that have a high molecular weight (60–90 carbons).[12] Mycolic acids, 2-alkyl, 3-hydroxy long-chain fatty acids, are the distinctive feature of the M. tuberculosis cell envelope. The mycolic acids are arranged in the form of a bilayer in association with cell wall lipids to serve as a low fluid permeability barrier. The properties of the mycolic acids is reflected in the fluidity and flexibility of the envelope. The chemical nature of the mycolic acids includes α-mycolic acid that shows the presence of unsaturation as well as cyclopropane rings. In addition, mycolic acids can also present in the oxygenated form in majority of the bacteria. Apart from the attenuated species, while Mycobacterium bovis bacillus Calmette–Guérin (BCG) Pasteur possesses α-mycolic and ketomycolic acids, the other species produce methoxy mycolic acids in addition to the mentioned two acids.[12]
Figure 1.
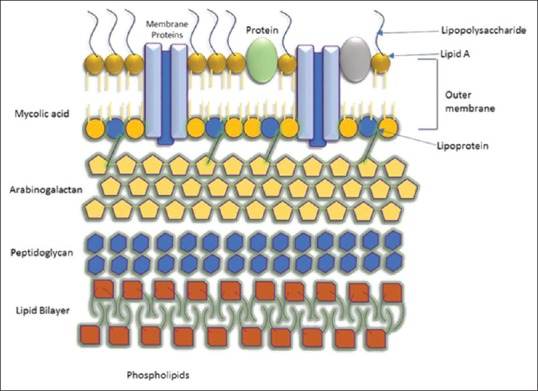
The lipid-rich cell wall of Mycobacterium tuberculosis
Figure 1 shows the lipid-rich cell wall of M. tuberculosis.[1] There is no true outer membrane in M. tuberculosis. The cell wall complex contains peptidoglycan, arabinogalactan, mycolic acid, and lipoarabinomannan (LAM). Over 60% of the mycobacterial cell wall is lipid mycolic acid and 50% of the dry weight of the cell wall. It is strongly hydrophobic and is observed to be important determinant of virulence.
MYCOBACTERIUM TUBERCULOSIS PERSISTENCE IN HOST AND COGNATE LIPID METABOLISM
The persistence of M. tuberculosis inside a host has been linked to its beta-oxidation and glyoxylate metabolic pathways.[13] If the metabolism of the pathogen is studied, the use of lipid metabolism has been documented. For instance, in mice, the source of carbon for the pathogen is fatty acids. This encompasses the use of beta-oxidation where fatty acids with an even number of carbon atoms are processed into acetyl-coenzyme A (CoA) while fatty acids with an odd number of carbon atoms are metabolized into acetyl-CoA and propionyl-CoA.[14]
Further research has shown that the pathogen converts propionyl-CoA into methyl malonyl-CoA-lipids such as sulfolipid-1 (SL-1), polyacyltrehalose (PAT), and phthiocerol dimycocerosate (PDIM).[15] It was shown that acetyl-CoA produced is either converted into intermediates of tricarboxylic acid (TCA) cycle or to mycolic acid.[16] Thus, either of the pathways accordingly enables the lipid metabolism of lipids to help the mycobacteria persist in a host.
MYCOBACTERIAL CELL MEMBRANE LIPIDS AND MODULATION OF RESISTANCE
The role of lipids of the cell envelope and modulation of immune responses has been demonstrated in several studies. This is of utmost importance as the lipids of the cell wall play an important role in the development of resistance of the pathogen. Multidrug-resistant strains show resistance to at least two first-line tuberculosis drugs (isoniazid and rifampicin), while there are extensively drug-resistant (XDR) strains that show resistance to fluoroquinolones and to one or more injectable second-line drugs that include capreomycin, kanamycin, and amikacin. XDR cases have been reported in all the continents, which is a disturbing news.[17]
Another study showed that a polyketide synthase-derived phenolic glycolipid (PGL) produced by Beijing family moderates the clinical outcome of the disease in a mouse model. The lipids specific to the Beijing family were abolished when pks1–15 locus, which encodes a polyketide synthase, was disrupted. Genotyping studies reveal that the locus is mutated in non-Beijing strains but intact in the Beijing strains. Apart from the loss of hypervirulent phenotype, the disruption of PGL causes increased production of pro-inflammatory cytokines tumor necrosis factor (TNF)-alpha and interleukin (IL)-6 and IL-12 in vitro.[18,19]
MYCOBACTERIAL CELL MEMBRANE LIPIDS AND PATHOGENESIS
Trehalose-6,6’-dimycolate
Trehalose-6,6’-dimycolate (TDM) is a mycolic acid-containing glycolipid that is known as cord factor associated with virulence. It is composed of trehalose sugar linked to two mycolic acid residues via an ester bond. The number of residues differs across species to species with 20–80 carbon atoms being most common.[20] Bekierkunst, in 1968, showed for the first time that injection of TDM caused an acute granulomatous reaction[21] and a C-type lectin called minacle on macrophage serves as a recognition receptor for TDM. The activation of the receptor causes production of nitric oxide that in turn induces the production of TNF to cause granulomas.[22]
TDM has been found in Gram-positive and Gram-negative bacteria where it has been implicated in several functions such as resistance toward infectious agents and promotion of angiogenesis and anti-cancer activities.[6] The role of mycolic acids in the pathogenesis of M. tuberculosis has been demonstrated in several studies. The levels of cholesterol inside the macrophages of the peritoneum and alveoli are increased by mycolic acid. Such derivatives of the cells are visualized in the granulomas associated with the pathogen.[23]
TDM has several functions associated such as inhibition of fusion of phospho-bilipid layers by the introduction of stearic hindrance to the fusion. The lipid has also been implicated to possess ability to break down nicotinamide adenine dinucleotide (NAD), hence lowering its concentration in the cell. Such an activity brings down a dip in the activities of NAD enzymes, especially in lung, liver, and spleen.[24] Certain studies have shown that cyclopropane ring of mycolic acids has an important role in virulence and persistence of the bacterium.[25,26] In mice models, it has been shown that oxygenated mycolic acids are essential for virulence of the pathogen.[12]
Other studies have reported the association of mycolic acid in its free form with a biofilm formation.[27] At the stage of infection when free mycolic acid is generated from TDM, the pathogen reduces its ability to cause induction of pro-inflammatory responses along with an increase in the uptake of nutrients to overcome starvation induced by the host and also displays resistance to drugs.[12,27]
Phthiocerol dimycocerosate
Another lipid implicated in the pathogenesis of Mycobacterium is PDIM. A 1974 study suggested a role of this lipid in the pathogen survival in the host. It was observed that an M. tuberculosis H37Rv mutant strain that lacked PDIM could not replicate successfully and caused lesser number of tubercule formation.[28] The lipid has also been associated with permeability of the cell wall. Certain mutants where generated with an inability to generate PDIM were found to be more permeable compared to the wild type.[29] Another study examined the presence or absence of PDIM in the pathogen on the manifestations of the disease. PDIM-deficient M. tuberculosis showed a reduction of bacterial load in the lungs over a period of 4 months when compared to that of the wild-type bacterium used for infecting mice. It was also seen that the PDIM of the bacterial cell envelope does not affect the formation of phagosomes. Instead, they confer protection to the pathogen from reactive nitrogen intermediates produced as part of immune response as the PDIM mutants caused increase in the activity of cytokines by dendritic cells and macrophages.[30]
Sulfolipids
Apart from the above-mentioned lipids, another notable candidate is sulfolipids, theacylated trehaloses that are present in the mycobacterial cell wall.[3] SL-1 is the most 165 abundant lipid on the surface of M. tuberculosis and is not present in nonpathogenic mycobacterium species. SL-1 and its precursor, Ac2SGL, can serve as candidates in biomarker studies[1] Although these lipids were initially associated with inhibiting phagosome–lysosome fusion, subsequent work showed that they inhibit phagosome and enhance the survival of the pathogen in an intracellular milieu.[6] Sulfolipid was suggested as an inhibitor of phagosomes in the cultured mouse peritoneal macrophages by affecting the membranes.[28] The lipid was implied in the inhibition of macrophages, thereby preventing the release of superoxides and affecting phagocytosis and cytokines along with interfering with the signaling pathways of neutrophils.[31]
However, subsequent work suggested that SL-1 affects the mitochondria along with TDM. When murine models were administered SL-1 alone, there was no adverse reaction or toxicity observed. Subsequent work on mmpL8 and pks2 mutants cast a light on the functions of SL-1. MmpL8 is a candidate that is associated with a family of transporters of lipids. A mutant of M. tuberculosis deficient or lacking MmpL8 could not synthesize SL-1 and showed accumulation of its acylated precursor in the cytoplasm. Pks2 is highly similar to mycocerosic acid synthase (mas) gene that codes for an enzyme involved in the synthesis of multiple methyl-branched fatty acids. Mutants of M. tuberculosis for Pks2 did not produce either SL-1 or its acylated precursor however showed growth rates comparable with that of wild-type bacterium.[1]
Despite studies on this lipid, the results have been controversial. Apart from its above effects, genetic manipulation studies suggest the role of this molecule in host–pathogen interactions, but a more lucid explanation of its role is yet to be established.[32]
MYCOBACTERIAL CELL WALL GLYCOLIPIDS AND THEIR ROLES IN PATHOGEN PERSISTENCE AND VIRULENCE
Lipomannan
Other lipids of the cell wall having a role in the host immune system modulation include lipomannan (LM): a glycolipid of the cell wall that causes an induction of IL-12 and ultimately cell death of macrophages.[33] An opposite effect is shown by LAM, another glycolipid of the cell wall that hampers the production of IL-12 by dendritic cells to control the apoptosis induced in macrophages[34] [Figure 2].
Figure 2.
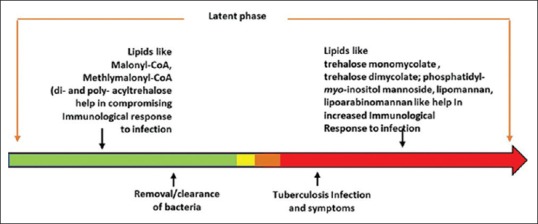
The timeline model of bacterial lipid-regulated host immunological response to infection
Lipoarabinomannan
LAM has been implicated as a virulence factor of M. tuberculosis by occupying a major portion of the cell wall. The glycolipid has also been observed to possess modulatory functions of the bacterial resistance and the host immune systems.[35] LAM also causes dephosphorylation of tyrosine kinase and inactivates the kinase activity and eventually the functions of T-cells and phagocytic cells such as macrophages.[36] A 2003 study reviewed the functions of LAM in altering signaling activities due to its insertion in the membrane and association with Toll-like receptors.[11] A study in 2000 showed LAM in blocking of calcium signaling to enhance the survival of M. tuberculosis inside the host cells. The pathways blocked by the inhibition of calcium signaling include calmodulin and phosphatidyl–inositol-3-kinase hsp34.[37] The virulence of M. tuberculosis involves a role of LAM as it blocks the activity of phosphatidyl-inositol-3-kinase to limit the maturation of phagosomes. LAM also hampers the association of early endosome autoantigen that plays a key role in phagosome maturation.[24]
Diacyltrehaloses
Diacyltrehaloses (DAT) have been implicated in the inhibition of T-cells in a murine model. A 2001 study found that DAT purified from two strains of mycobacteria M. tuberculosis H37Rv and M. fortuitum ATCC 6841 exerted a negative effect on T-cells.[38] Another derivative of glycolipid called mannose-capped LAM (Man-LAM) is associated with host defense. These glycolipids show the presence of mannosyl moieties on D-arabinan. These lipids were shown to possess anti-inflammatory activities following the attachment of mannose to inhibit TNF or IL-12 to facilitate survival of the pathogen inside the host.[24] In a mammalian cell, the lipid derivative serves to mimic a receptor of mannose. This dampens the production of cytokines such as TNF and IL-12, while on the other hand, this increases the production of IL-10 by monocytes or dendritic cells to cause an inhibition of phagosome.[39]
Figure 2 shows the timeline model of bacterial lipid-regulated host immunological response to infection. In the latent phase, a proper homeostasis of cell wall lipid allows M. tuberculosis and thrives in the host. When this homeostasis is upset, it either leads to tuberculosis due to the unregulated expression of pro-inflammatory lipids (the red region) or removal/clearance of bacteria (the green region) due to continued subdued response, which happens mainly when the drugs against M. tuberculosis are administered The role of the ratio of LM and LAM in the cell envelope of M. tuberculosis has also been examined. The former activates co-stimulatory molecules such as CD40 and CD86 to activate macrophages and the inflammation pathways. This glycolipid was also implicated in a Toll-like receptor 2 based-pathway to activate signaling of cells.[33]
ROLE OF CHOLESTEROL AND HOST RESPONSE
It has been reported that cholesterol is associated as a nutrient for M. tuberculosis and also aids in the response of the host.[40] The role of cholesterol includes the providing precursors such as propionyl-CoA for PDIM or methyl malonyl-CoA for sulfolipids. M. tuberculosis can also utilize the molecule as a central pathway as it can supply precursors such as pyruvate. The signaling occurring inside the cell and trafficking of components have been associated with the catabolism of cholesterol. A 2015 study showed the essential role of cholesterol metabolism in the infection of dendritic cells by M. tuberculosis. The study revealed low fitness costs for mutants in the catabolism of cholesterol, mutations in the transporters of cholesterol, and enzymes associated with breaking down the cholesterol side chains and suggested an important role of metabolism of cholesterol and the generation of precursors such as propionyl-CoA for the generation of PDIM and SL-1 [Table 1].[3]
Table 1.
The effects of the various lipids in the mycobacterium cell wall

STUDIES ON MCE: A PUTATIVE TRANSPORTER
Mce is a putative mycobacterial transporter that is associated with the quantity of mycolic acid in the cell wall. It has been indicated that there are four mce operons: Mce1–4 that code for ATP-binding cassette transporters.[41] The levels of free mycolic acids show an increase in mce mutants of M. tuberculosis.[42] A 2015 study examined the quantities of lipids that were differentially expressed in wild-type bacterium versus mce mutants. Twenty-six acylated forms of lipids were identified whose levels were up/downregulated. The forms of 260 lipids that were decreased in the mce mutants were DAT, diacylated sulfoglycolipids (Ac2SGL), PDIM, and phosphatidylethanolamine, while the levels of alpha-, keto-, and methoxy mycolic acids (MA) and hydroxyphthioceranic acid increased in the mutants compared to that in the wild type. These observations suggest that mutations in putative lipid transporters can be linked to lipid content of the cell wall of the pathogen which in turn affects its pathogenicity.[43]
MYCOBACTERIUM BOVIS AND HUMAN TUBERCULOSIS
The pathogen M. bovis causes tuberculosis in bovine species. However, it can be a causative agent of zoonosis whereby, it can induce the disease in humans too. Although the incorporation of certain practices such as pasteurization of milk, sequestering of infected or contaminated cows, and management of infected animals has reduced the incidence of the disease, it has not been eradicated completely.[13] A case study has been presented in a 2010 paper where the misdiagnosis of tuberculosis disease caused a fatal outcome in a patient. A patient from Belgium was diagnosed with TB in May 2001 observing all the tests and was administered tritherapy: isoniazid, rifampicin, and pyrazinamide. Although the sputum became negative, the patient was readmitted with lesions in the lung and positive sputum. The causative agent was identified as M. bovis that resulted from an outbreak of the disease in a farm that spreads to the human as in this case.[13]
LIPIDS OF MYCOBACTERIUM BOVIS CELL WALL
Research has shown that glycolipids of M. bovis BCG are capable of maneuvering inside a host macrophage and infect other macrophages in the vicinity of S281.[44] A subsequent study showed the glycolipids involved to be phosphatidylinositol dimannosides (PIM2), phosphatidylglycerol, cardiolipin (CL), phosphatidylethanolamine, trehalose monomycolate (TMM), TDM, and mycoside B (MycB).[45] It has also been observed that these molecules and their effects are similar in both M. tuberculosis and M. bovis. A research team in 2005 examined the nature of the glycolipids such as PIM2, CL, phosphatidylglycerol, phosphatidylethanolamine, TMM, TDM, and MycB associated with the responses in a mouse model using lipid-coated beads.
They observed that a slew of molecules of the immune system was elicited by M. bovis BCG that include TNF-α, IL-1α, IL-1 β, IL-6, macrophage chemotactic protein-1, and IFN-γ-inducible protein-10 that were also associated with the recruitment of polymorphonuclear and mononuclear leukocytes and lymphocytes. They observed that among the molecules checked, TDM caused the highest production of cytokines such as IL-1 β, IL-6, and TNF-α and also caused greatest recruitment of neutrophils in matrices, suggesting the role of these glycolipids in the formation of granulomas.[10]
GENES AND MUTATIONS INVOLVED IN MYCOBACTERIAL VIRULENCE
The pathogenic strains M. tuberculosis, Mycobacterium africanum, M. bovis, and Mycobacterium microti of the M. tuberculosis complex comprise similar combinations of mycolic acids. These pathogenic species are composed of α-mycolic acids, ketomycolic acids, and methoxy mycolic acids. However, the attenuated variety is M. bovis BCG that lacks the methoxy mycolic acids. Several pathogens inclusive of the mycobacterial family depend on cell wall for their virulence [Table 2]. For instance, the role of lipids in pathogenicity of the bacterium has been demonstrated by inactivating the genes coding for cell wall lipid, resulting in the loss of replication ability of the bacillus.[46]
Table 2.
List of cell wall lipid deletion experiments and their effect on Mycobacterium tuberculosis virulence

The genes fbpA, fbpB, and fbpC are associated with mycolyl transferase property demonstrated in vitro. Certain experiments that involved inactivation of the gene fbpC caused a dip of nearly 40% in the content of the mycolic acid in the cell wall. This altered cell wall mycolate level caused an elevation in the permeability of the cell wall. A subsequent study showed that these bacilli with mutated fbpC gene did not show diminished activities or attenuation in macrophages. A similar experiment with fbpB where the gene was mutated did not affect the properties of the bacterium in macrophage cell lines of both murine and human origin. However, there is an argument in favor of alteration of the cell wall mycolic acid content when compared to that of the parent though actual measurements in the altered cell wall composition were not studied.[12]
It is interesting to note that a study in 2000 showed that complementation studies could restore the levels of altered permeability of cell walls. The team worked on a gene csp1 of Corynebacterium glutamicum that is similar to M. tuberculosis phylogenetically. The gene codes for PS1, a secreted protein, whose N-terminal portion showed similarity with the fbp products and was demonstrated to transfer mycolic acid moieties to trehalose. On inactivating the csp1 gene, the resulting mutants showed a decrease of cell wall mycolate content to nearly half. These mutants also showed an altered permeability of the cell wall envelope and a dip in levels of trehalose dicorynomycolate with an increase in trehalose monocorynomycolate. The authors also showed that when these mutants were complemented with csp1 gene, there was a restoration of the wild-type function. When the csp-1 mutants were given a copy of fbpA, fbpB, or fbpC, it was seen that the cell wall regained its content of mycolic acid and its permeability was restored.[47]
These studies indicate the role of lipids of the cell wall that functions to regulate the permeability which in turn contributes to the survival of the bacterium inside the cell. The role of mycolic acid in the maintenance of the permeability of the cell wall is suggestive that such lipids can be changed by the cell to control the permeability and hence structure and functions of the cell wall.
METABOLISM OF LIPIDS AND MYCOBACTERIUM TUBERCULOSIS VIRULENCE
The composition of the lipids of the cell envelope in M. tuberculosis is regulated by the pathogen to exert an effect on the immune system of the host. The examination of various components of the beta-oxidation pathway reveals the role of the immune system in recognizing several lipids. The role of several lipids such as SL-1, PDIM, and DAT/PAT has been implicated in the suppression of the immune reaction launched at the pathogen.[1]
The role of beta-oxidation of fatty acids over the TCA cycle during the phase of infection is suggested by the research done in 2007. There is a metabolic coupling of PDIM and SL-1 both implicated in virulence of the pathogen using methyl malonyl-CoA that serves as a common precursor for both the lipids. The levels of methyl malonyl-CoA mutated which converts succinyl-CoA of the TCA cycle to methyl malonyl-CoA were increased by upregulation. This caused a dip in the levels of PDIM and SL-1 in vivo that points out to the role of methyl malonyl-CoA.[14] Lipids such as SL-1, DAT, or PDIM possess an odd number of chains. The beta-oxidation pathway has two outcomes; the metabolism of odd chain fatty acids causes the production of both acetyl-CoA and propionyl-CoA while a run of even chain fatty acids yields only acetyl-CoA. A 2015 study revealed that M. tuberculosis is able to survive in dendritic cells by utilizing several genes such as those associated with the catabolism of cholesterol and the synthesis of cell envelope lipids such as SL-1 and PDIM.[3] Thus, quelling of immune responses is linked to the effects of these lipids that are synthesized by the beta-oxidation pathway to generate these odd-chain fatty acids. Such alterations in the pathways associated with lipids and cholesterol play an important role in encouraging the persistence and virulence of the pathogen in intracellular conditions.
Studies performed on putative transporters of lipids have suggested the change in composition of cell wall in M. tuberculosis. A 1998 study has previously shown that the cell wall of the pathogen shows a thickening during the stationary phase of growth.[48] Certain studies performed showed the increase in the levels of free mycolic acids in the cell wall when a putative transporter of lipid was mutated. Mutations in the mce loci that are associated with transport of lipids reported an increased level of fatty acids and associated thickness.[42,49] This increase in thickness of the cell wall due to changes in composition of lipids can have a role in conferring protection to the pathogen from the immune system. A study in 2007 examined the role of a regulator that affects mce1 operon. A repressor: Mce1R shuts down the transcription of the mce1 operon of M. tuberculosis in a mouse model. This suppression of the putative transporter was observed in the initial 2 months of infection that shows an effect akin to that in a mce1-mutant model.[50] It was suggested that the synthesis of SL-1, DAT, and PAT was decreased due to repression of the pathway associated with its precursors either propionyl-CoA or methyl malonyl-CoA. This downregulation is combined with an upregulation of mycolic acids to limit the level of inflammation markers such as TLR-2 generated by the host.[51] The role of mycolic acid transport for the mce1 locus is also suggested. During a phase of limited nutrient availability experienced by the pathogen, the cell wall increases in thickness which in turn causes a downregulation of the mce1 operon to allow the cell to transport free mycolic acids for use as a source of energy.[52] Alternatively, mycolic acid may be produced by the pathways involving acetyl-CoA triggered by stimuli such as starvation.[53] A comparison of metabolic profiles of mce1 operon mutant against that of wild-type M. tuberculosis revealed a decrease in the levels of precursors of SL-1, DAT, and PAT while the levels of mycolic acids showed an elevation. This study is indicative of the role of the 385 mce locus in the maintenance of the pathogen and encouraging its persistence under unfavorable conditions in the cell by exerting an effect on the levels of various lipids of the cell envelope, especially mycolic acids.[43]
QUESTIONS YET TO BE ADDRESSED
The regulation of the composition of the cell wall in response to various requirements as the pathogen progresses its infection requires further elucidation and work. The role of the dynamic change in content of cell wall in response to time to meet stimuli may involve the role of other molecules that are associated with signaling. For instance, a study in 2001 evaluated the role of a bacterial signal transduction system called Pho. This system comprising two components has been linked to response systems toward various external signals. Mutations or damage to the PhoP and PhoR system can damage M. tuberculosis to multiply and maintain itself in in vitro and in vivo conditions.[54] The lipids of the cell envelope such as SL-1, DAT, and PAT are increased by the PhoP component.[55] Another role for the PhoP component came from a study in 2013 where a model was proposed that PhoP is involved in the regulation of the lipids of the cell wall of M. tuberculosis, especially during hypoxic conditions. The network comprises PhoP that regulates another component called WhiB3, a regulator of fatty acid metabolism, which in turn can exert an effect on SL-1, DAT, and PAT.[56]
CELL WALL LIPIDS AND THEIR INHIBITORY ACTIVITY ON SURFACTANTS ON THE LUNG
M. tuberculosis transmits itself by destruction of the lung tissue so that the pathogen can reach the respiratory tract to facilitate its release from the system. A study in 2008 examined the effect of several cell wall lipids that were extracted from M. tuberculosis on lavaged bovine lung surfactant (LS) and a clinically relevant calf LS extract. The authors reported that TMM and TDM (cord factor) both exert inhibition of the surface activity of the tested LSs. Moreover, the total lipid content of the bacterium caused a dip in the activity of the bovine surfactant.[57] Such studies point out to an additional role of the lipids of M. tuberculosis in hampering the activity of the surfactants of the lung. During the course of infection, the lungs already show a decrease in activity of the surfactants due to the activities of the slew of molecules associated with inflammation. The activity of the lipids of cell wall could further exacerbate and diminish the activity of the surfactants to facilitate the release of the bacterium and its transmission.[58]
FUTURE RESEARCH
The above studies on the roles of various lipids of the cell wall of M. tuberculosis along with the gene deletion experiments pave the way for more research as to the exact roles of lipids in pathogenesis of the organism. An important branch is the modulation of immune response by the bacterium as well as the role of lipids in altering surfactant levels in the lungs. With further research into these angles, the virulence can be discussed more in detail to arrive at a potential solution of looking at a complete picture of the organism's virulence. The role of lipids in pathogenesis of a deadly organism with a potential of latency opens up new avenues of research and possible drug targets in the future.
CONCLUSION
According to the WHO, tuberculosis is one of the top ten killer diseases that was diagnosed in a million children and caused the death of 170,000 children.[59] In addition, the ability of the pathogen to form granulomas or establish latent infections gives it an added advantage. This review examined the role of various lipids and their derivatives in the maintenance of structure and virulence of M. tuberculosis. Several lipids such as mycolic acids, TMD, PMIDs, DATs, SLs, and cholesterol contribute to the survival of the pathogen or the suppression of immune responses of the host. Glycolipids such as LM, LAM, and Man-LAM modulate the host immune system and enhance the survival of the pathogen. The bacterium is also thought to regulate the composition of its cell wall lipids in a temporal fashion to facilitate its respond to various stimuli.
Of the several factors of virulence the bacterium has developed, the role of lipids of its cell envelope in the survival, persistence, and virulence has proof. Further detailed elaboration of the roles of the lipids can serve to use them as potential drug targets in the control of this destructive disease.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
REFERENCES
- 1.Queiroz A, Riley LW. Bacterial immunostat: Mycobacterium tuberculosis lipids and their role in the host immune response. Rev Soc Bras Med Trop. 2017;50:9–18. doi: 10.1590/0037-8682-0230-2016. [DOI] [PubMed] [Google Scholar]
- 2.Dye C, Williams BG. The population dynamics and control of tuberculosis. Science. 2010;328:856–61. doi: 10.1126/science.1185449. [DOI] [PubMed] [Google Scholar]
- 3.Mendum TA, Wu H, Kierzek AM, Stewart GR. Lipid metabolism and type VII secretion systems dominate the genome scale virulence profile of Mycobacterium tuberculosis in human dendritic cells. BMC Genomics. 2015;16:372. doi: 10.1186/s12864-015-1569-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dye C, Scheele S, Dolin P, Pathania V, Raviglione MC Consensus Statement. Global burden of tuberculosis: Estimated incidence, prevalence, and mortality by country. WHO Global Surveillance and Monitoring Project. JAMA. 1999;282:677–86. doi: 10.1001/jama.282.7.677. [DOI] [PubMed] [Google Scholar]
- 5.Parrish NM, Dick JD, Bishai WR. Mechanisms of latency in Mycobacterium tuberculosis. Trends Microbiol. 1998;6:107–12. doi: 10.1016/s0966-842x(98)01216-5. [DOI] [PubMed] [Google Scholar]
- 6.Karakousis PC, Bishai WR, Dorman SE. Mycobacterium tuberculosis cell envelope lipids and the host immune response. Cell Microbiol. 2004;6:105–16. doi: 10.1046/j.1462-5822.2003.00351.x. [DOI] [PubMed] [Google Scholar]
- 7.Eum SY, Kong JH, Hong MS, Lee YJ, Kim JH, Hwang SH, et al. Neutrophils are the predominant infected phagocytic cells in the airways of patients with active pulmonary TB. Chest. 2010;137:122–8. doi: 10.1378/chest.09-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wolf AJ, Linas B, Trevejo-Nuñez GJ, Kincaid E, Tamura T, Takatsu K, et al. Mycobacterium tuberculosis infects dendritic cells with high frequency and impairs their function in vivo . J Immunol. 2007;179:2509–19. doi: 10.4049/jimmunol.179.4.2509. [DOI] [PubMed] [Google Scholar]
- 9.Russell DG. Mycobacterium tuberculosis: Here today, and here tomorrow. Nat Rev Mol Cell Biol. 2001;2:569–77. doi: 10.1038/35085034. [DOI] [PubMed] [Google Scholar]
- 10.Geisel RE, Sakamoto K, Russell DG, Rhoades ER. In vivo activity of released cell wall lipids of Mycobacterium bovis bacillus Calmette-Guérin is due principally to trehalose mycolates. J Immunol. 2005;174:5007–15. doi: 10.4049/jimmunol.174.8.5007. [DOI] [PubMed] [Google Scholar]
- 11.Brennan PJ. Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis (Edinb) 2003;83:91–7. doi: 10.1016/s1472-9792(02)00089-6. [DOI] [PubMed] [Google Scholar]
- 12.Dubnau E, Chan J, Raynaud C, Mohan VP, Lanéelle MA, Yu K, et al. Oxygenated mycolic acids are necessary for virulence of Mycobacterium tuberculosis in mice. Mol Microbiol. 2000;36:630–7. doi: 10.1046/j.1365-2958.2000.01882.x. [DOI] [PubMed] [Google Scholar]
- 13.Allix-Béguec C, Fauville-Dufaux M, Stoffels K, Ommeslag D, Walravens K, Saegerman C, et al. Importance of identifying Mycobacterium bovis as a causative agent of human tuberculosis. Eur Respir J. 2010;35:692–4. doi: 10.1183/09031936.00137309. [DOI] [PubMed] [Google Scholar]
- 14.Jain M, Petzold CJ, Schelle MW, Leavell MD, Mougous JD, Bertozzi CR, et al. Lipidomics reveals control of Mycobacterium tuberculosis virulence lipids via metabolic coupling. Proc Natl Acad Sci U S A. 2007;104:5133–8. doi: 10.1073/pnas.0610634104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muñoz-Elías EJ, Upton AM, Cherian J, McKinney JD. Role of the methylcitrate cycle in Mycobacterium tuberculosis metabolism, intracellular growth, and virulence. Mol Microbiol. 2006;60:1109–22. doi: 10.1111/j.1365-2958.2006.05155.x. [DOI] [PubMed] [Google Scholar]
- 16.McKinney JD, Höner zu Bentrup K, Muñoz-Elías EJ, Miczak A, Chen B, Chan WT, et al. Persistence of Mycobacterium tuberculosis in macrophages and mice requires the glyoxylate shunt enzyme isocitrate lyase. Nature. 2000;406:735–8. doi: 10.1038/35021074. [DOI] [PubMed] [Google Scholar]
- 17.Ehrt S, Schnappinger D. Mycobacterium tuberculosis virulence: Lipids inside and out. Nat Med. 2007;13:284–5. doi: 10.1038/nm0307-284. [DOI] [PubMed] [Google Scholar]
- 18.Manca C, Reed MB, Freeman S, Mathema B, Kreiswirth B, Barry CE, 3rd, et al. Differential monocyte activation underlies strain-specific Mycobacterium tuberculosis pathogenesis. Infect Immun. 2004;72:5511–4. doi: 10.1128/IAI.72.9.5511-5514.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reed MB, Domenech P, Manca C, Su H, Barczak AK, Kreiswirth BN, et al. Aglycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431:84–7. doi: 10.1038/nature02837. [DOI] [PubMed] [Google Scholar]
- 20.Spargo BJ, Crowe LM, Ioneda T, Beaman BL, Crowe JH. Cord factor (alpha, alpha-trehalose 6,6’-dimycolate) inhibits fusion between phospholipid vesicles. Proc Natl Acad Sci U S A. 1991;88:737–40. doi: 10.1073/pnas.88.3.737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bekierkunst A. Acute granulomatous response produced in mice by trehalose-6,6-dimycolate. J Bacteriol. 1968;96:958–61. doi: 10.1128/jb.96.4.958-961.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishikawa E, Ishikawa T, Morita YS, Toyonaga K, Yamada H, Takeuchi O, et al. Direct recognition of the mycobacterial glycolipid, trehalose dimycolate, by C-type lectin mincle. J Exp Med. 2009;206:2879–88. doi: 10.1084/jem.20091750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Korf J, Stoltz A, Verschoor J, De Baetselier P, Grooten J. The Mycobacterium tuberculosis cell wall component mycolic acid elicits pathogen-associated host innate immune responses. Eur J Immunol. 2005;35:890–900. doi: 10.1002/eji.200425332. [DOI] [PubMed] [Google Scholar]
- 24.Goyal R, Rao N, Meena LS. Biosynthesis and virulent behavior of lipids produced by Mycobacterium tuberculosis: LAM and cord factor: An overview. Biotechnol Res Int. 2011;2011:274693. doi: 10.4061/2011/274693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Glickman MS, Cox JS, Jacobs WR., Jr A novel mycolic acid cyclopropane synthetase is required for cording, persistence, and virulence of Mycobacterium tuberculosis. Mol Cell. 2000;5:717–27. doi: 10.1016/s1097-2765(00)80250-6. [DOI] [PubMed] [Google Scholar]
- 26.Rao V, Fujiwara N, Porcelli SA, Glickman MS. Mycobacterium tuberculosis controls host innate immune activation through cyclopropane modification of a glycolipid effector molecule. J Exp Med. 2005;201:535–43. doi: 10.1084/jem.20041668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ojha AK, Baughn AD, Sambandan D, Hsu T, Trivelli X, Guerardel Y, et al. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol Microbiol. 2008;69:164–74. doi: 10.1111/j.1365-2958.2008.06274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goren MB, Brokl O, Schaefer WB. Lipids of putative relevance to virulence in Mycobacterium tuberculosis: Phthiocerol dimycocerosate and the attenuation indicator lipid. Infect Immun. 1974;9:150–8. doi: 10.1128/iai.9.1.150-158.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu J, Barry CE, 3rd, Besra GS, Nikaido H. Mycolic acid structure determines the fluidity of the mycobacterial cell wall. J Biol Chem. 1996;271:29545–51. doi: 10.1074/jbc.271.47.29545. [DOI] [PubMed] [Google Scholar]
- 30.Rousseau C, Winter N, Pivert E, Bordat Y, Neyrolles O, Avé P, et al. Production of phthiocerol dimycocerosates protects Mycobacterium tuberculosis from the cidal activity of reactive nitrogen intermediates produced by macrophages and modulates the early immune response to infection. Cell Microbiol. 2004;6:277–87. doi: 10.1046/j.1462-5822.2004.00368.x. [DOI] [PubMed] [Google Scholar]
- 31.Zhang L, English D, Andersen BR. Activation of human neutrophils by Mycobacterium tuberculosis-derived sulfolipid-1. J Immunol. 1991;146:2730–6. [PubMed] [Google Scholar]
- 32.Forrellad MA, Klepp LI, Gioffré A, Sabio y García J, Morbidoni HR, de la Paz Santangelo M, et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013;4:3–66. doi: 10.4161/viru.22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dao DN, Kremer L, Guérardel Y, Molano A, Jacobs WR, Jr, Porcelli SA. Mycobacterium tuberculosis lipomannan induces apoptosis and interleukin-12 production in macrophages. Infect Immun. 2004;72:2067–74. doi: 10.1128/IAI.72.4.2067-2074.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nigou J, Gilleron M, Rojas M, García LF, Thurnher M, Puzo G, et al. Mycobacterial lipoarabinomannans: Modulators of dendritic cell function and the apoptotic response. Microbes Infect. 2002;4:945–53. doi: 10.1016/s1286-4579(02)01621-0. [DOI] [PubMed] [Google Scholar]
- 35.Guérardel Y, Maes E, Briken V, Chirat F, Leroy Y, Locht C, et al. Lipomannan and lipoarabinomannan from a clinical isolate of Mycobacterium kansasii: Novel structural features and apoptosis-inducing properties. J Biol Chem. 2003;278:36637–51. doi: 10.1074/jbc.M305427200. [DOI] [PubMed] [Google Scholar]
- 36.Knutson KL, Hmama Z, Herrera-Velit P, Rochford R, Reiner NE. Lipoarabinomannan of Mycobacterium tuberculosis promotes protein tyrosine dephosphorylation and inhibition of mitogen-activated protein kinase in human mononuclear phagocytes. Role of the Src homology 2 containing tyrosine phosphatase 1. J Biol Chem. 1998;273:645–52. doi: 10.1074/jbc.273.1.645. [DOI] [PubMed] [Google Scholar]
- 37.Malik ZA, Denning GM, Kusner DJ. Inhibition of Ca(2+) signaling by Mycobacterium tuberculosis is associated with reduced phagosome-lysosome fusion and increased survival within human macrophages. J Exp Med. 2000;191:287–302. doi: 10.1084/jem.191.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saavedra R, Segura E, Leyva R, Esparza LA, López-Marín LM. Mycobacterial di-O-acyl-trehalose inhibits mitogen- and antigen-induced proliferation of murine T cells in vitro . Clin Diagn Lab Immunol. 2001;8:1081–8. doi: 10.1128/CDLI.8.6.1081-1088.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Józefowski S, Sobota A, Kwiatkowska K. How Mycobacterium tuberculosis subverts host immune responses. Bioessays. 2008;30:943–54. doi: 10.1002/bies.20815. [DOI] [PubMed] [Google Scholar]
- 40.Ouellet H, Johnston JB, de Montellano PR. Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis. Trends Microbiol. 2011;19:530–9. doi: 10.1016/j.tim.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pandey AK, Sassetti CM. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci U S A. 2008;105:4376–80. doi: 10.1073/pnas.0711159105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Forrellad MA, McNeil M, Santangelo Mde L, Blanco FC, García E, Klepp LI, et al. Role of the mce1 transporter in the lipid homeostasis of Mycobacterium tuberculosis. Tuberculosis (Edinb) 2014;94:170–7. doi: 10.1016/j.tube.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Queiroz A, Medina-Cleghorn D, Marjanovic O, Nomura DK, Riley LW. Comparative metabolic profiling of mce1 operon mutant vs. wild-type Mycobacterium tuberculosis strains. Pathog Dis. 2015;73:ftv066. doi: 10.1093/femspd/ftv066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Beatty WL, Rhoades ER, Ullrich HJ, Chatterjee D, Heuser JE, Russell DG, et al. Trafficking and release of mycobacterial lipids from infected macrophages. Traffic. 2000;1:235–47. doi: 10.1034/j.1600-0854.2000.010306.x. [DOI] [PubMed] [Google Scholar]
- 45.Rhoades E, Hsu F, Torrelles JB, Turk J, Chatterjee D, Russell DG, et al. Identification and macrophage-activating activity of glycolipids released from intracellular Mycobacterium bovis BCG. Mol Microbiol. 2003;48:875–88. doi: 10.1046/j.1365-2958.2003.03473.x. [DOI] [PubMed] [Google Scholar]
- 46.Camacho LR, Ensergueix D, Perez E, Gicquel B, Guilhot C. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol Microbiol. 1999;34:257–67. doi: 10.1046/j.1365-2958.1999.01593.x. [DOI] [PubMed] [Google Scholar]
- 47.Puech V, Bayan N, Salim K, Leblon G, Daffé M. Characterization of the in vivo acceptors of the mycoloyl residues transferred by the corynebacterial PS1 and the related mycobacterial antigens 85. Mol Microbiol. 2000;35:1026–41. doi: 10.1046/j.1365-2958.2000.01738.x. [DOI] [PubMed] [Google Scholar]
- 48.Cunningham AF, Spreadbury CL. Mycobacterial stationary phase induced by low oxygen tension: Cell wall thickening and localization of the 16-kilodalton alpha-crystallin homolog. J Bacteriol. 1998;180:801–8. doi: 10.1128/jb.180.4.801-808.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cantrell SA, Leavell MD, Marjanovic O, Iavarone AT, Leary JA, Riley LW, et al. Free mycolic acid accumulation in the cell wall of the mce1 operon mutant strain of Mycobacterium tuberculosis. J Microbiol. 2013;51:619–26. doi: 10.1007/s12275-013-3092-y. [DOI] [PubMed] [Google Scholar]
- 50.Uchida Y, Casali N, White A, Morici L, Kendall LV, Riley LW, et al. Accelerated immunopathological response of mice infected with Mycobacterium tuberculosis disrupted in the mce1 operon negative transcriptional regulator. Cell Microbiol. 2007;9:1275–83. doi: 10.1111/j.1462-5822.2006.00870.x. [DOI] [PubMed] [Google Scholar]
- 51.Sequeira PC, Senaratne RH, Riley LW. Inhibition of toll-like receptor 2 (TLR-2)-mediated response in human alveolar epithelial cells by mycolic acids and Mycobacterium tuberculosis mce1 operon mutant. Pathog Dis. 2014;70:132–40. doi: 10.1111/2049-632X.12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dunphy KY, Senaratne RH, Masuzawa M, Kendall LV, Riley LW. Attenuation of Mycobacterium tuberculosis functionally disrupted in a fatty acyl-coenzyme A synthetase gene fadD5. J Infect Dis. 2010;201:1232–9. doi: 10.1086/651452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Takayama K, Wang C, Besra GS. Pathway to synthesis and processing of mycolic acids in Mycobacterium tuberculosis. Clin Microbiol Rev. 2005;18:81–101. doi: 10.1128/CMR.18.1.81-101.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pérez E, Samper S, Bordas Y, Guilhot C, Gicquel B, Martín C, et al. An essential role for phoP in Mycobacterium tuberculosis virulence. Mol Microbiol. 2001;41:179–87. doi: 10.1046/j.1365-2958.2001.02500.x. [DOI] [PubMed] [Google Scholar]
- 55.Gonzalo Asensio J, Maia C, Ferrer NL, Barilone N, Laval F, Soto CY, et al. The virulence-associated two-component PhoP-PhoR system controls the biosynthesis of polyketide-derived lipids in Mycobacterium tuberculosis. J Biol Chem. 2006;281:1313–6. doi: 10.1074/jbc.C500388200. [DOI] [PubMed] [Google Scholar]
- 56.Galagan JE, Minch K, Peterson M, Lyubetskaya A, Azizi E, Sweet L, et al. The Mycobacterium tuberculosis regulatory network and hypoxia. Nature. 2013;499:178–83. doi: 10.1038/nature12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Z, Schwab U, Rhoades E, Chess PR, Russell DG, Notter RH, et al. Peripheral cell wall lipids of Mycobacterium tuberculosis are inhibitory to surfactant function. Tuberculosis (Edinb) 2008;88:178–86. doi: 10.1016/j.tube.2007.11.003. [DOI] [PubMed] [Google Scholar]
- 58.Cox JS, Chen B, McNeil M, Jacobs WR., Jr Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402:79–83. doi: 10.1038/47042. [DOI] [PubMed] [Google Scholar]
- 59.Tuberculosis Fact Sheet. World Health Organization. [Last accessed on 2017 Mar 24]. Available from: http://www.who.int/mediacentre/factsheets/fs104/en/