

Abstract
Two reports show FDA-approved nanoparticles can kill cancer cells through iron- and reactive oxygen species-dependent mechanisms, offering new strategies for cancer treatment.
Iron is found in all mammalian cells and is required for cell growth and division. However, iron levels must be tightly controlled, as iron can catalyse the formation of toxic reactive oxygen species (ROS). Rapidly proliferating cancer cells often contain more iron than normal cells and, perhaps for this reason, are more sensitive to ROS stress1,2. Whether it is possible to exploit this sensitivity to treat cancer by increasing iron levels, ROS accumulation or both processes, is unclear. Now, two reports in Nature Nanotechnology identify two Food and Drug Administration (FDA)-approved nanoparticles that can selectively kill cancer cells by increasing iron and ROS levels3,4. These reports suggest novel uses for existing nanoparticles in the treatment of cancer.
In the first report, Bradbury, Overholtzer and colleagues3 investigated the effects on cancer cells of ultrasmall (<10 nm in diameter) poly(ethylene glycol)-coated silica nanoparticles, called C′ dots, currently used in tumour imaging applications. C′ dot ingestion killed cancer cells, but only when they were deprived of amino acids in the growth medium — a condition mimicking the nutrient stress experienced by tumour cells in vivo. Further investigation revealed that C′ dots bind iron in the extracellular environment and, once ingested, increased the levels of intracellular iron and caused an accumulation of ROS (Fig. 1). Crucially, the growth of human tumour cells implanted in mice (that is, tumour xenografts) was blocked by C′ dot administration, suggesting a possible use for these agents in the treatment of cancer.
Figure 1 |.
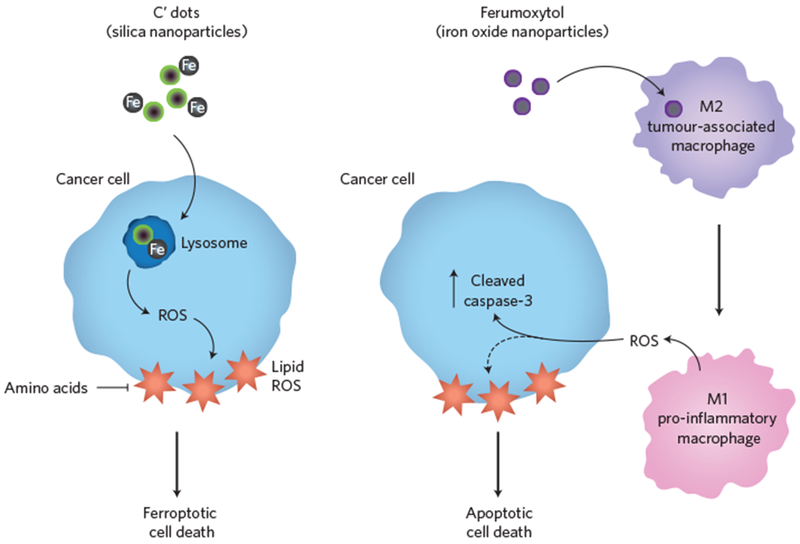
Engineered nanoparticles kill cancer cells by manipulating iron levels and causing an accumulation of reactive oxygen species. Bradbury, Overholtzer and co-workers demonstrated that ultrasmall silica nanoparticles (C′ dots) are selectively lethal to cancer cells (blue) under conditions of amino acid starvation (left). Cell death occurs by ferroptosis — a non-apoptotic form of cell death, resulting from the iron-dependent production of reactive oxygen species (ROS). Daldrup-Link and colleagues showed that the FDA-approved iron oxide nanoparticle drug, ferumoxytol, alters the polarization of tumour-associated macrophages from an anti-inflammatory M2 phenotype to a pro-inflammatory M1 phenotype (right). M1 polarized macrophages potentially release ROS and/or additional diffusible factors, one or more of which may induce apoptotic cell death characterized by an increase in cleaved caspase-3.
The researchers found that C′ dot ingestion does not trigger apoptosis, the well-studied caspase-mediated death process. Rather, C′ dots induce cell death via ferroptosis, a recently described non-apoptotic form of cell death, characterized by the iron-dependent accumulation of ROS and damage to membrane lipids5. Co-administration of a specific inhibitor of ferroptosis, liproxstatin-1, prevented C′ dots from killing implanted tumour cells in vivo. This work demonstrates for the first time that iron-containing nanoparticles can engage the ferroptotic pathway to kill cancer cells in vitro and in vivo. This work complements a recent report describing how ingestion of transferrin complexes, the physiological iron carrier protein in the body, can trigger ferroptosis6. At the biochemical level, it remains unclear how iron accumulation triggers ferroptosis in the context of amino acid deprivation. Iron can act as a catalyst to enhance the production of toxic ROS. Thus, one possibility is that iron-loaded C′ dots release iron into the cell that increases ROS formation directly. Another possibility is that C′ dots, or C′ dot-derived iron, inactivate one or more enzymes that normally restrain ferroptosis. Amino acid deprivation may enhance ROS accumulation by depriving cells of key amino acid building blocks for endogenous antioxidants, such as glutathione.
In a second report, Daldrup-Link and colleagues present a different and equally unexpected mechanism by which the manipulation of iron levels may induce cancer cell death4. The researchers investigated the lethal effects of ferumoxytol, an FDA-approved compound used for the treatment of anaemia resulting from chronic kidney disease. In a variety of cancer cell lines, ferumoxytol treatment triggered marked cell death as detected by increased caspase activation. Furthermore, administration of ferumoxytol with tumour xenografts in mice suppressed tumour growth and inhibited metastatic spread. Mechanistically, ferumoxytol does not appear to act directly on tumour cells, but rather on tumour-associated immune cells, specifically macrophages, causing them to adapt an anti-tumour ‘M1’ phenotype7 (Fig. 1). In a dual-well co-culture system that prevented direct interaction of macrophages and cancer cells, ferumoxytol treatment induced the production of a soluble factor that promotes cancer cell death. While the authors’ suggestion of ROS as this soluble factor is a compelling hypothesis, it would be informative to test the effects of ROS scavengers or inhibitors of ROS formation on cell death in this system. Moreover, genetic and biochemical studies would help to understand how ferumoxytol enhances macrophage ROS production, whether by directly catalysing ROS production, altering the expression of ROS-related enzymes, or some other mechanism.
These two reports describe different ways in which nanoparticles can induce tumour cell death. However, a question that emerges from these studies is whether C′ dots and ferumoxytol could share common mechanisms of action in vivo. For example, Bradbury, Overholtzer and colleagues noted a significant accumulation of macrophages around xenograft tumours following treatment with C′ dots. Could C′ dots promote macrophage recruitment and contribute to cell death in vivo through similar mechanisms as ferumoxytol? Conversely, given that C′ dots were found to induce a non-apoptotic cell death pathway, is it possible that ferumoxytol could do likewise and that this effect was missed because the current work looked exclusively at markers of apoptosis? Looking ahead, nanoparticle-induced non-apoptotic cell death could be especially useful in treating cancers that are incapable of undergoing cell death by apoptosis8.
These reports provide complementary evidence that nanoparticle-induced modulation of iron and ROS levels within cancer cells and the tumour microenvironment could provide a new strategy for cancer treatment. Both C′ dots and ferumoxytol nanoparticles are approved for use in humans for other applications, suggesting intriguing opportunities to repurpose these drugs for cancer therapy. Given the serendipitous nature of these two discoveries, it is possible that other nanoparticles may also induce cancer cell death in unusual ways, perhaps by manipulating intracellular iron or ROS levels, or through as yet undiscovered mechanisms.
References
- 1.Torti SV & Torti FM Nat. Rev. Cancer 13, 342–355 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Trachootham D, Alexandre J & Huang P Nat. Rev. Drug Discov 8, 579–591 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Kim SE et al. Nat. Nanotech 11, 977–985 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zanganeh S et al. Nat. Nanotech 11, 986–994 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dixon SJ et al. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao M et al. Mol. Cell 59, 298–308 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian BZ & Pollard JW Cell 141, 39–51 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sarosiek KA et al. Mol. Cell 51, 751–765 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]