

Abstract
Farming was first introduced to Europe in the mid-7th millennium BCE–associated with migrants from Anatolia who settled in the Southeast before spreading throughout Europe. To understand the dynamics of this process, we analyzed genome-wide ancient DNA data from 225 individuals who lived in southeastern Europe and surrounding regions between 12,000 and 500 BCE. We document a West-East cline of ancestry in indigenous hunter-gatherers and–in far-eastern Europe–early stages in the formation of Bronze Age Steppe ancestry. We show that the first farmers of northern and western Europe passed through southeastern Europe with limited hunter-gatherer admixture, but that some groups that remained mixed extensively, without the male-biased hunter-gatherer admixture that prevailed later in the North and West. Southeastern Europe continued to be a nexus between East and West, with intermittent genetic contact with the Steppe up to 2000 years before the migrations that replaced much of northern Europe’s population.
Introduction
The southeastern quadrant of Europe was the beachhead in the spread of agriculture from its source in the Fertile Crescent of southwestern Asia. After the first appearance of agriculture in the mid-7th millennium BCE 1,2 farming spread westward via a Mediterranean and northwestward via a Danubian route, and was established in both Iberia and Central Europe by 5600 BCE.3,4 Ancient DNA studies have shown that the spread of farming across Europe was accompanied by a massive movement of people5–8 closely related to the farmers of northwestern Anatolia9–11 but nearly all the ancient DNA from Europe’s first farmers is from central and western Europe, with only three individuals reported from the southeast.9 In the millennia following the establishment of agriculture in the Balkan Peninsula, a series of complex societies formed, culminating in sites such as the mid-5th millennium BCE necropolis at Varna, which has some of the earliest evidence of extreme inequality in wealth, with one individual (grave 43) from whom we extracted DNA buried with more gold than is known from any earlier site. By the end of the 6th millennium BCE, agriculture had reached eastern Europe, in the form of the Cucuteni-Trypillian complex in the area of present-day Moldova, Romania and Ukraine, including “mega-sites” that housed hundreds, perhaps thousands, of people.12 After around 4000 BCE, these settlements were largely abandoned, and archaeological evidence documents cultural contacts with peoples of the Eurasian steppe.13 However, the population movements that accompanied these events have been unknown due to the lack of ancient DNA.
Results
We generated genome-wide data from 225 ancient humans (216 reported for the first time), from the Balkan Peninsula, the Carpathian Basin, the North Pontic Steppe and neighboring regions, dated to 12,000-500 BCE (Figure 1, Supplementary Information Table 1, Supplementary Information Note 1). We extracted DNA from skeletal remains in dedicated clean rooms, built DNA libraries and enriched for DNA fragments overlapping 1.24 million single nucleotide polymorphisms (SNPs), then sequenced the product and restricted to libraries with evidence of authentic ancient DNA.7,10,14 We filtered out individuals with fewer than 15,000 SNPs covered by at least one sequence, or that had unexpected ancestry for their archaeological context and were not directly dated. We report, but do not analyze, nine individuals that were first-degree relatives of others in the dataset, resulting in an analysis dataset of 216 individuals. We analyzed these data together with 274 previously reported ancient individuals,9–11,15–27 777 present-day individuals genotyped on the Illumina “Human Origins” array,23 and 300 high coverage genomes from the Simons Genome Diversity Project (SGDP).28 We used principal component analysis (PCA; Figure 1B, Extended Data Figure 1), supervised and unsupervised ADMIXTURE (Figure 1D, Extended Data Figures 2&3),29 D-statistics, qpAdm and qpGraph,30 along with archaeological and chronological information (including 137 newly reported AMS 14C dates) to cluster the individuals into populations and investigate the relationships among them.
Figure 1. Geographic and genetic structure of 216 newly reported individuals.

A: Locations of newly reported individuals. B: Ancient individuals projected onto principal components defined by 777 present-day West Eurasians (shown in Extended Data Figure 1). Includes selected published individuals (faded circles, labeled) and newly reported individuals (other symbols, outliers enclosed in black circles). Colored polygons cover individuals that had cluster memberships fixed at 100% for supervised admixture analysis. C: Date (direct or contextual) for each sample and approximate chronology of southeastern Europe. D: Supervised ADMIXTURE analysis, modeling each ancient individual (one per row), as a mixture of population clusters constrained to contain Anatolian Neolithic (grey), Yamnaya from Samara (yellow), EHG (pink) and WHG (green) populations. Dates in parentheses indicate approximate range of individuals in each population. See Extended Data Figure 2 for individual sample IDs. Map data in A from the R package maps.
We described the individuals in our dataset in terms of their genetic relatedness to a hypothesized set of ancestral populations, which we refer to as their genetic ancestry. It has previously been shown that the great majority of European ancestry derives from three distinct sources.23 First, “hunter-gatherer-related” ancestry that is more closely related to Mesolithic hunter-gatherers from Europe than to any other population, and can be further subdivided into “Eastern” (EHG) and “Western” (WHG) hunter-gatherer-related ancestry.7 Second, “NW Anatolian Neolithic-related” ancestry related to the Neolithic farmers of northwest Anatolia and tightly linked to the appearance of agriculture.9,10 The third source, “steppe-related” ancestry, appears in Western Europe during the Late Neolithic to Bronze Age transition and is ultimately derived from a population related to Yamnaya steppe pastoralists.7,15 Steppe-related ancestry itself can be modeled as a mixture of EHG-related ancestry, and ancestry related to Upper Palaeolithic hunter-gatherers of the Caucasus (CHG) and the first farmers of northern Iran.19,21,22
Hunter-Gatherer substructure and transitions
Of the 216 new individuals we report, 106 from Paleolithic, Mesolithic and eastern European Neolithic contexts have almost entirely hunter-gatherer-related ancestry (in eastern Europe, unlike western Europe, “Neolithic” refers to the presence of pottery,31–33 not necessarily to farming). These individuals form a cline from WHG to EHG that is correlated with geography (Figure 1B), although it is neither geographically nor temporally uniform (Figure 2, Extended Data Figure 4), and contains substructure in phenotypically important variants (Supplementary Information Note 2).
Figure 2. Structure and change in hunter-gatherer-related populations.
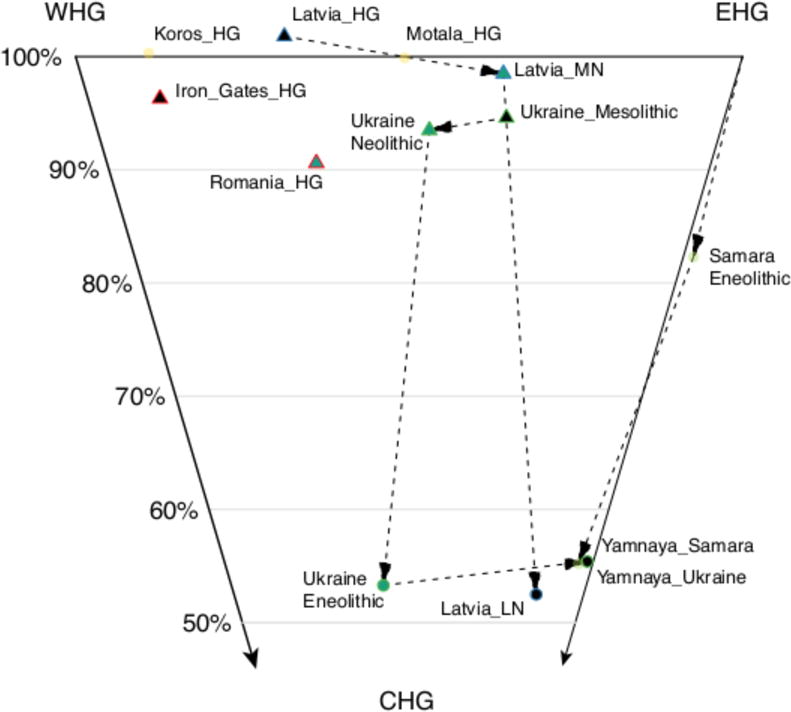
Inferred ancestry proportions for populations modeled as a mixture of WHG, EHG and CHG (Supplementary Table S3.1.3). Dashed lines show populations from the same geographic region. Percentages indicate proportion of WHG+EHG ancestry. Standard errors range from 1.5-8.3% (Supplementary Table S3.1.3).
From present-day Ukraine, our study reports new genome-wide data from seven Mesolithic (~9500-6000 BCE) and 30 Neolithic (~6000-3500 BCE) individuals. On the cline from WHG- to EHG-related ancestry, the Mesolithic individuals fall towards the East, intermediate between EHG and Mesolithic hunter-gatherers from Scandinavia (Figure 1B).7 The Neolithic population has a significant difference in ancestry compared to the Mesolithic (Figures 1B, Figure 2), with a shift towards WHG shown by the statistic D(Mbuti, WHG, Ukraine_Mesolithic, Ukraine_Neolithic); Z=8.5 (Supplementary Information Table 2). Unexpectedly, one Neolithic individual from Dereivka (I3719), which we directly date to 4949-4799 BCE, has entirely NW Anatolian Neolithic-related ancestry.
The pastoralist Bronze Age Yamnaya complex originated on the Eurasian steppe and is a plausible source for the dispersal of steppe-related ancestry into central and western Europe around 2500 BCE.13 All previously reported Yamnaya individuals were from Samara7 and Kalmykia15 in southwest Russia, and had entirely steppe-related ancestry. Here, we report three Yamnaya individuals from further West – Ukraine and Bulgaria – and show that while they all have high levels of steppe-related ancestry, one from Ozera in Ukraine and one from Bulgaria (I1917 and Bul4, both dated to ~3000 BCE) have NW Anatolian Neolithic-related admixture, the first evidence of such ancestry in Yamnaya–associated individuals (Figure 1B&D, Supplementary Data Table 2). Preceding the Yamnaya culture, four Copper Age individuals (I4110, I5882, I5884 and I6561; Ukraine_Eneolithic) from Dereivka and Alexandria dated to ~3600-3400 BCE have ancestry that is a mixture of hunter-gatherer-, steppe- and NW Anatolian Neolithic-related (Figure 1D, Supplementary Data Table 2).
At Zvejnieki in Latvia (17 newly reported individuals, and additional data for 5 first reported in Ref. 34) we observe a transition in hunter-gatherer-related ancestry that is opposite to that seen in Ukraine. We find (Supplementary Data Table 3) that Mesolithic and Early Neolithic individuals (Latvia_HG) associated with the Kunda and Narva cultures have ancestry intermediate between WHG (~70%) and EHG (~30%), consistent with previous reports.34–36 We also detect a shift in ancestry between the Early Neolithic and individuals associated with the Middle Neolithic Comb Ware Complex (Latvia_MN), who have more EHG-related ancestry (we estimate 65% EHG, but two of four individuals appear to be 100% EHG in PCA). The most recent individual, associated with the Final Neolithic Corded Ware Complex (I4629, Latvia_LN), attests to another ancestry shift, clustering closely with Yamnaya from Samara,7 Kalmykia15 and Ukraine (Figure 2).
We report new Upper Palaeolithic and Mesolithic data from southern and western Europe.17 Sicilian (I2158) and Croatian (I1875) individuals dating to ~12,000 and 6100 BCE cluster with previously reported western hunter-gatherers (Figure 1B&D), including individuals from Loschbour23 (Luxembourg, 6100 BCE), Bichon19 (Switzerland, 11,700 BCE), and Villabruna17 (Italy 12,000 BCE). These results demonstrate that WHG populations23 were widely distributed from the Atlantic seaboard of Europe in the West, to Sicily in the South, to the Balkan Peninsula in the Southeast, for at least six thousand years.
A particularly important hunter-gatherer population that we report is from the Iron Gates region that straddles the border of present-day Romania and Serbia. This population (Iron_Gates_HG) is represented in our study by 40 individuals from five sites. Modeling Iron Gates hunter-gatherers as a mixture of WHG and EHG (Supplementary Table 3) shows that they are intermediate between WHG (~85%) and EHG (~15%). However, this qpAdm model does not fit well (p=0.0003, Supplementary Table 3) and the Iron Gates hunter-gatherers show an affinity towards Anatolian Neolithic, relative to WHG (Supplementary Table 2). In addition, Iron Gates hunter-gatherers carry mitochondrial haplogroup K1 (7/40) as well as other subclades of haplogroups U (32/40) and H (1/40) in contrast to WHG, EHG and Scandinavian hunter-gatherers who almost all carry haplogroups U5 or U2. One interpretation is that the Iron Gates hunter-gatherers have ancestry that is not present in either WHG or EHG. Possible scenarios include genetic contact between the ancestors of the Iron Gates population and a NW Anatolian-Neolithic-related population, or that the Iron Gates population is related to the source population from which the WHG split during a re-expansion into Europe from the Southeast after the Last Glacial Maximum.17,37
A notable finding from the Iron Gates concerns the four individuals from the site of Lepenski Vir, two of whom (I4665 & I5405, 6200-5600 BCE), have entirely NW Anatolian Neolithic-related ancestry. Strontium and Nitrogen isotope data38 indicate that both these individuals were migrants from outside the Iron Gates, and ate a primarily terrestrial diet (Supplementary Information section 1). A third individual (I4666, 6070 BCE) has a mixture of NW Anatolian Neolithic-related and hunter-gatherer-related ancestry and ate a primarily aquatic diet, while a fourth, probably earlier, individual (I5407) had entirely hunter-gatherer-related ancestry (Figure 1D, Supplementary Information section 1). We also identify one individual from Padina (I5232), dated to 5950 BCE that had a mixture of NW Anatolian Neolithic-related and hunter-gatherer-related ancestry. These results demonstrate that the Iron Gates was a region of interaction between groups distinct in both ancestry and subsistence strategy.
Population transformations in the first farmers
Neolithic populations from present-day Bulgaria, Croatia, Macedonia, Serbia and Romania cluster closely with the NW Anatolian Neolithic (Figure 1), consistent with archaeological evidence.39 Modeling Balkan Neolithic populations as a mixture of NW Anatolian Neolithic and WHG, we estimate that 98% (95% confidence interval [CI]; 97-100%) of their ancestry is NW Anatolian Neolithic-related. A striking exception is evident in 8 out of 9 individuals from Malak Preslavets in present-day Bulgaria.40 These individuals lived in the mid-6th millennium BCE and have significantly more hunter-gatherer-related ancestry than other Balkan Neolithic populations (Figure 1B,D, Extended Data Figures 1–3, Supplementary Tables 2-4); a model of 82% (CI: 77-86%) NW Anatolian Neolithic-related, 15% (CI: 12-17%) WHG-related, and 4% (CI: 0-9%) EHG-related ancestry fits the data. This hunter-gatherer-related ancestry with a ~4:1 WHG:EHG ratio plausibly represents a contribution from local Balkan hunter-gatherers genetically similar to those of the Iron Gates. Late Mesolithic hunter-gatherers in the Balkans were likely concentrated along the coast and major rivers such as the Danube,41 which directly connects the Iron Gates with Malak Preslavets. Thus, early farmer groups with the most hunter-gatherer-related ancestry may have been those that lived close to the highest densities of hunter-gatherers.
In the Balkans, Copper Age populations (Balkans_Chalcolithic) harbor significantly more hunter-gatherer-related ancestry than Neolithic populations as shown, for example, by the statistic D(Mbuti, WHG, Balkans_Neolithic, Balkans_Chalcolithic); Z=4.3 (Supplementary Data Table 2). This is roughly contemporary with the “resurgence” of hunter-gatherer ancestry previously reported in central Europe and Iberia7,10,42 and is consistent with changes in funeral rites, specifically the reappearance around 4500 BCE of the Mesolithic tradition of extended supine burial – in contrast to the Early Neolithic tradition of flexed burial.43 Four individuals associated with the Copper Age Trypillian population have ~80% NW Anatolian-related ancestry (Supplementary Table 3), confirming that the ancestry of the first farmers of present-day Ukraine was largely derived from the same source as the farmers of Anatolia and western Europe. Their ~20% hunter-gatherer ancestry is intermediate between WHG and EHG, consistent with deriving from the Neolithic hunter-gatherers of the region.
We also report the first genetic data associated with the Late Neolithic Globular Amphora Complex. Individuals from two Globular Amphora sites in Poland and Ukraine form a tight cluster, showing high similarity over a large distance (Figure 1B,D). Both groups of Globular Amphora Complex samples had more hunter-gatherer-related ancestry than Middle Neolithic groups from Central Europe7 (we estimate 25% [CI: 22-27%] WHG ancestry, similar to Chalcolithic Iberia, Supplementary Data Table 3). In east-central Europe, the Globular Amphora Complex preceded or abutted the Corded Ware Complex that marks the appearance of steppe-related ancestry,7,15 while in southeastern Europe, the Globular Amphora Complex bordered populations with steppe-influenced material cultures for hundreds of years44 and yet the individuals in our study have no evidence of steppe-related ancestry, supporting the hypothesis that this material cultural frontier was also a barrier to gene flow.
The movements from the Pontic-Caspian steppe of individuals similar to those associated with the Yamnaya Cultural Complex in the 3rd millennium BCE contributed about 75% of the ancestry of individuals associated with the Corded Ware Complex and about 50% of the ancestry of succeeding material cultures such as the Bell Beaker Complex in central Europe.7,15 In two directly dated individuals from southeastern Europe, one (ANI163) from the Varna I cemetery dated to 4711-4550 BCE and one (I2181) from nearby Smyadovo dated to 4550-4450 BCE, we find far earlier evidence of steppe-related ancestry (Figure 1B,D). These findings push back the first evidence of steppe-related ancestry this far West in Europe by almost 2,000 years, but it was sporadic as other Copper Age (~5000-4000 BCE) individuals from the Balkans have no evidence of it. Bronze Age (~3400-1100 BCE) individuals do have steppe-related ancestry (we estimate 30%; CI: 26-35%), with the highest proportions in the four latest Balkan Bronze Age individuals in our data (later than ~1700 BCE) and the least in earlier Bronze Age individuals (3400-2500 BCE; Figure 1D).
A new source of ancestry in Neolithic Europe
An important question about the initial spread of farming into Europe is whether the first farmers that brought agriculture to northern Europe and to southern Europe were derived from a single population or instead represent distinct migrations. We confirm that Mediterranean populations, represented in our study by individuals associated with the Epicardial Early Neolithic from Iberia7, are closely related to Danubian populations represented by the Linearbandkeramik (LBK) from central Europe7,45 and that both are closely related to the Balkan Neolithic population. These three populations form a clade with the NW Anatolian Neolithic individuals as an outgroup, consistent with a single migration into the Balkan peninsula, which then split into two (Supplementary Information Note 3).
In contrast, five southern Greek Neolithic individuals (Peloponnese_Neolithic) – three (plus one from Ref. 26) from Diros Cave and one from Franchthi Cave – are not consistent with descending from the same source population as other European farmers. D-statistics (Supplementary Information Table 2) show that in fact, these “Peloponnese Neolithic” individuals dated to ~4000 BCE are shifted away from WHG and towards CHG, relative to Anatolian and Balkan Neolithic individuals. We see the same pattern in a single Neolithic individual from Krepost in present-day Bulgaria (I0679_d, 5718-5626 BCE). An even more dramatic shift towards CHG has been observed in individuals associated with the Bronze Age Minoan and Mycenaean cultures,26 suggesting gene flow into the region from populations with CHG-rich ancestry throughout the Neolithic, Chalcolithic and Bronze Age. Possible sources are related to the Neolithic population from the central Anatolian site of Tepecik Çiftlik,21 or the Aegean site of Kumtepe,11 who are also shifted towards CHG relative to NW Anatolian Neolithic samples, as are later Copper and Bronze Age Anatolians.10,26
Sex-biased admixture between hunter-gatherers and farmers
We provide the first evidence for sex-biased admixture between hunter-gatherers and farmers in Europe, showing that the Middle Neolithic “resurgence” of hunter-gatherer-related ancestry7,42 in central Europe and Iberia was driven more by males than by females (Figure 3B&C, Supplementary Data Table 5, Extended Data Figure 5). To document this we used qpAdm to compute ancestry proportions on the autosomes and the X chromosome; since males always inherit a maternal X chromosome, differences imply sex-biased mixture. In the Balkan Neolithic there is no evidence of sex bias (Z=0.27 where a positive Z-score implies male hunter-gatherer bias), nor in the LBK and Iberian_Early Neolithic (Z=-0.22 and 1.09). In the Copper Age there is clear bias: weak in the Balkans (Z=1.66), but stronger in Iberia (Z=3.08) and Central Europe (Z=2.74). Consistent with this, hunter-gatherer mitochondrial haplogroups (haplogroup U)46 are rare and within the intervals of genome-wide ancestry proportions, but hunter-gatherer-associated Y chromosomes (haplogroups I2, R1 and C1)17 are more common: 7/9 in the Iberian Neolithic/Copper Age and 9/10 in Middle-Late Neolithic Central Europe (Central_MN and Globular_Amphora) (Figure 3C).
Figure 3. Structure and change in NW Anatolian Neolithic-related populations.
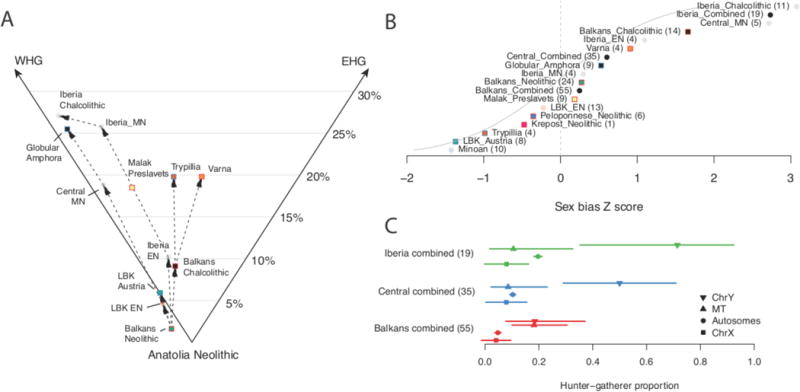
A: Populations modeled as a mixture of NW Anatolia Neolithic, WHG, and EHG. Dashed lines show temporal relationships between populations from the same geographic region. Percentages indicate proportion of WHG+EHG ancestry. Standard errors range from 0.7-6.0% (Supplementary Table S3.2.2). B: Z-scores for the difference in hunter-gatherer-related ancestry on the autosomes compared to the X chromosome when populations are modeled as a mixture of NW Anatolia Neolithic and WHG (N=126 individuals, group sizes in parentheses). Positive values indicate more hunter-gatherer-related ancestry on the autosomes and thus male-biased hunter-gatherer ancestry. “Combined” populations merge all individuals from different times from a geographic area. C: Hunter-gatherer-related ancestry proportions on the autosomes, X chromosome, mitochondrial DNA (i.e. mt haplogroup U), and the Y chromosome (i.e. Y chromosome haplogroups I2, R1 and C1). Points show qpAdm (autosomes and X chromosome) or maximum likelihood (MT and Y chromosome) estimates and bars show approximate 95% confidence intervals (N=109 individuals, group sizes in parentheses).
No evidence that steppe-related ancestry moved through southeast Europe into Anatolia
One version of the Steppe Hypothesis of Indo-European language origins suggests that Proto-Indo-European languages developed north of the Black and Caspian seas, and that the earliest known diverging branch – Anatolian – was spread into Asia Minor by movements of steppe peoples through the Balkan peninsula during the Copper Age around 4000 BCE.47 If this were correct, then one way to detect evidence of it would be the appearance of large amounts of steppe-related ancestry first in the Balkan Peninsula, and then in Anatolia. However, our data show no evidence for this scenario. While we find sporadic steppe-related ancestry in Balkan Copper and Bronze Age individuals, this ancestry is rare until the late Bronze Age. Moreover, while Bronze Age Anatolian individuals have CHG-related ancestry,26 they have neither the EHG-related ancestry characteristic of all steppe populations sampled to date,19 nor the WHG-related ancestry that is ubiquitous in Neolithic southeastern Europe (Extended Data Figure 2&3, Supplementary Data Table 2). An alternative hypothesis is that the ultimate homeland of Proto-Indo-European languages was in the Caucasus or in Iran. In this scenario, westward movement contributed to the dispersal of Anatolian languages, and northward movement and mixture with EHG was responsible for the formation of a “Late Proto-Indo European”-speaking population associated with the Yamnaya Complex.13 While this scenario gains plausibility from our results, it remains possible that Indo-European languages were spread through southeastern Europe into Anatolia without large-scale population movement or admixture.
Discussion
Our study shows that southeastern Europe consistently served as a genetic contact zone. Before the arrival of farming, the region saw interaction between diverged groups of hunter-gatherers, and this interaction continued after farming arrived. While this study has clarified the genomic history of southeastern Europe from the Mesolithic to the Bronze Age, the processes that connected these populations to the ones living today remain largely unknown. An important direction for future research will be to sample populations from the Bronze Age, Iron Age, Roman, and Medieval periods and to compare them to present-day populations to understand how these transitions occurred.
Methods
Ancient DNA Analysis
We extracted DNA and prepared next-generation sequencing libraries in four different dedicated ancient DNA laboratories (Adelaide, Boston, Budapest, and Tuebingen). We also prepared samples for extraction in a fifth laboratory (Dublin), from whence it was sent to Boston for DNA extraction and library preparation (Supplementary Table 1).
Two samples were processed at the Australian Centre for Ancient DNA, Adelaide, Australia, according to previously published methods7 and sent to Boston for subsequent screening, 1240k capture and sequencing.
Seven samples were processed27 at the Institute of Archaeology RCH HAS, Budapest, Hungary, and amplified libraries were sent to Boston for screening, 1240k capture and sequencing.
Seventeen samples were processed at the Institute for Archaeological Sciences of the University of Tuebingen and at the Max Planck Institute for the Science of Human History in Jena, Germany. Extraction48 and library preparation49,50 followed established protocols. We performed in-solution capture as described below (“1240k capture”) and sequenced on an Illumina HiSeq 4000 or NextSeq 500 for 76bp using either single- or paired-end sequencing.
The remaining 199 samples were processed at Harvard Medical School, Boston, USA. From about 75mg of sample powder from each sample (extracted in Boston or University College Dublin, Dublin, Ireland), we extracted DNA following established methods48 replacing the column assembly with the column extenders from a Roche kit.51 We prepared double barcoded libraries with truncated adapters from between one ninth and one third of the DNA extract. Most libraries included in the nuclear genome analysis (90%) were subjected to partial (“half”) Uracil-DNA-glycosylase (UDG) treatment before blunt end repair. This treatment reduces by an order of magnitude the characteristic cytosine-to-thymine errors of ancient DNA data52, but works inefficiently at the 5’ ends,50 thereby leaving a signal of characteristic damage at the terminal ends of ancient sequences. Some libraries were not UDG-treated (“minus”). For some samples we increased coverage by preparing additional libraries from the existing DNA extract using the partial UDG library preparation, but replacing the MinElute column cleanups in between enzymatic reactions with magnetic bead cleanups, and the final PCR cleanup with SPRI bead cleanup.53,54 We screened all libraries from Adelaide, Boston and Budapest by enriching for the mitochondrial genome plus about 3,000 (50 in an earlier, unpublished, version) nuclear SNPs using a bead-capture55 but with the probes replaced by amplified oligonucleotides synthesized by CustomArray Inc. After the capture, we completed the adapter sites using PCR, attaching dual index combinations56 to each enriched library. We sequenced the products of between 100 and 200 libraries together with the non-enriched libraries (shotgun) on an Illumina NextSeq500 using v2 150 cycle kits for 2x76 cycles and 2x7 cycles.
In Boston, we performed two rounds of in-solution enrichment (“1240k capture”) for a targeted set of 1,237,207 SNPs using previously reported protocols.7,14,23 For a total of 34 individuals, we increased coverage by building one to eight additional libraries for the same sample. When we built multiple libraries from the same extract, we often pooled them in equimolar ratios before the capture. We performed all sequencing on an Illumina NextSeq500 using v2 150 cycle kits for 2x76 cycles and 2x7 cycles. We attempted to sequence each enriched library up to the point where we estimated that it was economically inefficient to sequence further. Specifically, we iteratively sequenced more and more from each individual and only stopped when we estimated that the expected increase in the number of targeted SNPs hit at least once would be less than about one for every 100 new read pairs generated. After sequencing, we trimmed two bases from the end of each read and aligned to the human genome (b37/hg19) using bwa.57 We then removed individuals with evidence of contamination based on mitochondrial DNA polymorphism58 or difference in PCA space between damaged and undamaged reads59, a high rate of heterozygosity on chromosome X despite being male59,60, or an atypical ratio of X-to-Y sequences. We also removed individuals that had low coverage (fewer than 15,000 SNPs hit on the autosomes). We report, but do not analyze, data from nine individuals that were first-degree relatives of others in the dataset (determined by comparing rates of allele sharing between pairs of individuals).
After removing a small number of sites that failed to capture, we were left with a total of 1,233,013 sites of which 32,670 were on chromosome X and 49,704 were on chromosome Y, with a median coverage at targeted SNPs on the 216 newly reported individuals of 0.90 (range 0.007-9.2; Supplementary Table 1). We generated “pseudo-haploid” calls by selecting a single read randomly for each individual at each SNP. Thus, there is only a single allele from each individual at each site, but adjacent alleles might come from either of the two haplotypes of the individual. We merged the newly reported data with previously reported data from 274 other ancient individuals9–11,15–27, making pseudo-haploid calls in the same way at the 1240k sites for individuals that were shotgun sequenced rather than captured.
Using the captured mitochondrial sequence from the screening process, we called mitochondrial haplotypes. Using the captured SNPs on the Y chromosome, we called Y chromosome haplogroups for males by restricting to sequences with mapping quality ≥30 and bases with base quality ≥30. We determined the most derived mutation for each individual, using the nomenclature of the International Society of Genetic Genealogy (http://www.isogg.org) version 11.110 (21 April 2016).
Population genetic analysis
To analyze these ancient individuals in the context of present day genetic diversity, we merged them with the following two datasets:
300 high coverage genomes from a diverse worldwide set of 142 populations sequenced as part of the Simons Genome Diversity Project28 (SGDP merge).
777 West Eurasian individuals genotyped on the Human Origins array23, with 597,573 sites in the merged dataset (HO merge).
We computed principal components of the present-day individuals in the HO merge and projected the ancient individuals onto the first two components using the “lsqproject: YES” option in smartpca (v15100)61 (https://www.hsph.harvard.edu/alkes-price/software/).
We ran ADMIXTURE (v1.3.0) in both supervised and unsupervised mode. In supervised mode we used only the ancient individuals, on the full set of SNPs, and the following population labels fixed:
Anatolia_Neolithic
WHG
EHG
Yamnaya
For unsupervised mode we used the HO merge, including 777 present-day individuals. We flagged individuals that were genetic outliers based on PCA and ADMIXTURE, relative to other individuals from the same time period and archaeological culture.
We computed D-statistics using qpDstat (v710). D-statistics of the form D(A,B,X,Y) test the null hypothesis of the unrooted tree topology ((A,B),(X,Y)). A positive value indicates that either A and X, or B and Y, share more drift than expected under the null hypothesis. We quote D-statistics as the Z-score computed using default block jackknife parameters.
We fitted admixture proportions with qpAdm (v610) using the SGDP merge. Given a set of outgroup (“right”) populations, qpAdm models one of a set of source (“left”) populations (the “test” population) as a mixture of the other sources by fitting admixture proportions to match the observed matrix of f4-statistics as closely as possible. We report a p-value for the null hypothesis that the test population does not have ancestry from another source that is differentially related to the right populations. We computed standard errors for the mixture proportions using a block jackknife. Importantly, qpAdm does not require that the source populations are actually the admixing populations, only that they are a clade with the correct admixing populations, relative to the other sources. Infeasible coefficient estimates (i.e. outside [0,1]) are usually a sign of poor model fit, but in the case where the source with a negative coefficient is itself admixed, could be interpreted as implying that the true source is a population with different admixture proportions. We used the following set of seven populations as outgroups or “right populations”:
Mbuti.DG
Ust_Ishim_HG_published.DG
Mota.SG
MA1_HG.SG
Villabruna
Papuan.DG
Onge.DG
Han.DG
For some analyses where we required extra resolution (Supplementary Data Table 4) we used an extended set of 14 right (outgroup) populations, including additional Upper Paleolithic European individuals17:
ElMiron
Mota.SG
Mbuti.DG
Ust_Ishim_HG_published.DG
MA1_HG.SG
AfontovaGora3
GoyetQ116-1_published
Villabruna
Kostenki14
Vestonice16
Karitiana.DG
Papuan.DG
Onge.DG
Han.DG
We also fitted admixture graphs with qpGraph (v6021)30 (https://github.com/DReichLab/ AdmixTools, Supplementary Information Note 3). Like qpAdm, qpGraph also tries to match a matrix of f-statistics, but rather than fitting one population as a mixture of other, specified, populations, it fits the relationship between all tested populations simultaneously, potentially incorporating multiple admixture events. However, qpGraph requires the graph relating populations to be specified in advance. We tested goodness-of-fit by computing the expected D-statistics under the fitted model, finding the largest D-statistic outlier between the fitted and observed model, and computing a Z-score using a block jackknife.
For 114 individuals with hunter-gatherer-related ancestry we estimated an effective migration surface using the software EEMS (https://github.com/dipetkov/eems)62. We computed pairwise differences between individuals using the bed2diffs2 program provided with EEMS. We set the number of demes to 400 and defined the outer boundary of the region by the polygon (in latitude-longitude co-ordinates) [(66,60), (60,10), (45,-15), (35,-10), (35,60)]. We ran the MCMC ten times with different random seeds, each time with one million burn-in and four million regular iterations, thinned to one in ten thousand.
To analyze potential sex bias in admixture, we used qpAdm to estimate admixture proportions on the autosomes (default option) and on the X chromosome (option “chrom: 23”). We computed Z-scores for the difference between the autosomes and the X chromosome as where pA and pX are the hunter-gatherer admixture proportions on the autosomes and the X chromosome, and σA and σX are the corresponding jackknife standard deviations. Thus, a positive Z-score means that there is more hunter-gatherer admixture on the autosomes than on the X chromosome, indicating that the hunter-gatherer admixture was male-biased. Because X chromosome standard errors are high and qpAdm results can be sensitive to which population is first in the list of outgroup populations, we checked that the patterns we observe were robust to cyclic permutation of the outgroups. To compare frequencies of hunter-gatherer uniparental markers, we counted the individuals with mitochondrial haplogroup U and Y chromosome haplogroups C1, I2 and R1, which are all common in Mesolithic hunter-gatherers but rare or absent in Anatolian Neolithic individuals. The Iron Gates hunter-gatherers also carry H and K1 mitochondrial haplogroups so the proportion of haplogroup U represents the minimum maternal hunter-gatherer contribution. We computed binomial confidence intervals for the proportion of haplogroups associated with each ancestry type using the Agresti-Coull method63,64 implemented in the binom package in R.
Given autosomal and X chromosome admixture proportions, we estimated the proportion of male and female hunter-gatherer ancestors by assuming a single-pulse model of admixture. If the proportions of male and female ancestors that are hunter-gatherer-related are given by m and f, respectively, then the proportions of hunter-gatherer-related ancestry on the autosomes and the X chromosome are given by and . We approximated the sampling error in the observed admixture proportions by the estimated jackknife error and computed the likelihood surface for (m,f) over a grid ranging from (0,0) to (1,1).
Direct AMS 14C Bone Dates
We report 137 new direct AMS 14C bone dates for 136 individuals from multiple AMS radiocarbon laboratories. In general, bone samples were manually cleaned and demineralized in weak HCl and, in most cases (PSU, UCIAMS, OxA), soaked in an alkali bath (NaOH) at room temperature to remove contaminating soil humates. Samples were then rinsed to neutrality in Nanopure H2O and gelatinized in HCL.65 The resulting gelatin was lyophilized and weighed to determine percent yield as a measure of collagen preservation (% crude gelatin yield). Collagen was then directly AMS 14C dated (Beta, AA) or further purified using ultrafiltration (PSU, UCIAMS, OxA, Poz, MAMS).66 It is standard in some laboratories (PSU/UCIAMS, OxA) to use stable carbon and nitrogen isotopes as an additional quality control measure. For these samples, the %C, %N and C:N ratios were evaluated before AMS 14C dating.67 C:N ratios for well-preserved samples fall between 2.9 and 3.6, indicating good collagen preservation.68 For 119 of the new dates, we also report δ13C and δ15N values (Supplementary Table 6).
All 14C ages were δ13C-corrected for mass dependent fractionation with measured 13C/12C values69 and calibrated with OxCal version 4.2.370 using the IntCal13 northern hemisphere calibration curve.70 For hunter-gatherers from the Iron Gates, the direct 14C dates tend to be overestimates because of the freshwater reservoir effect (FRE), which arises because of a diet including fish that consumed ancient carbon, and for these individuals we performed a correction (Supplementary Information Note 1),71 assuming that 100% FRE = 545±70 yr, and δ15N values of 8.3% and 17.0% for 100% terrestrial and aquatic diets, respectively.
Data Availability
The aligned sequences are available through the European Nucleotide Archive under accession number PRJEB22652. The pseudo-haploid genotype dataset used in analysis and consensus mitochondrial genomes are available at https://reich.hms.harvard.edu/datasets.
Code Availability
Software used to analyze the data is available from the following sources:
smartpca, qpAdm, qpDstat, qpGraph: https://github.com/DReichLab/AdmixTools/
ADMIXTURE: https://www.genetics.ucla.edu/software/admixture/
Extended Data
Extended Data Figure 1.
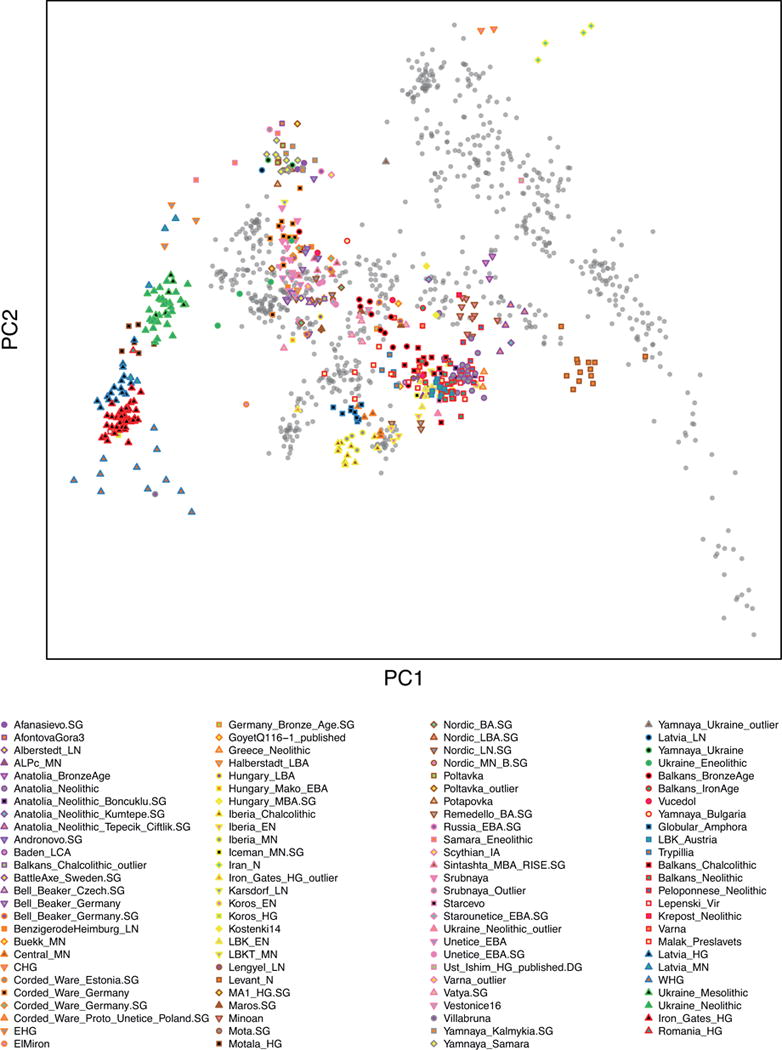
PCA of 486 ancient individuals, projected onto principal components defined by 777 present-day West Eurasian individuals (grey points). This differs from Figure 1B in that the plot is not cropped and the present-day individuals are shown.
Extended Data Figure 2.
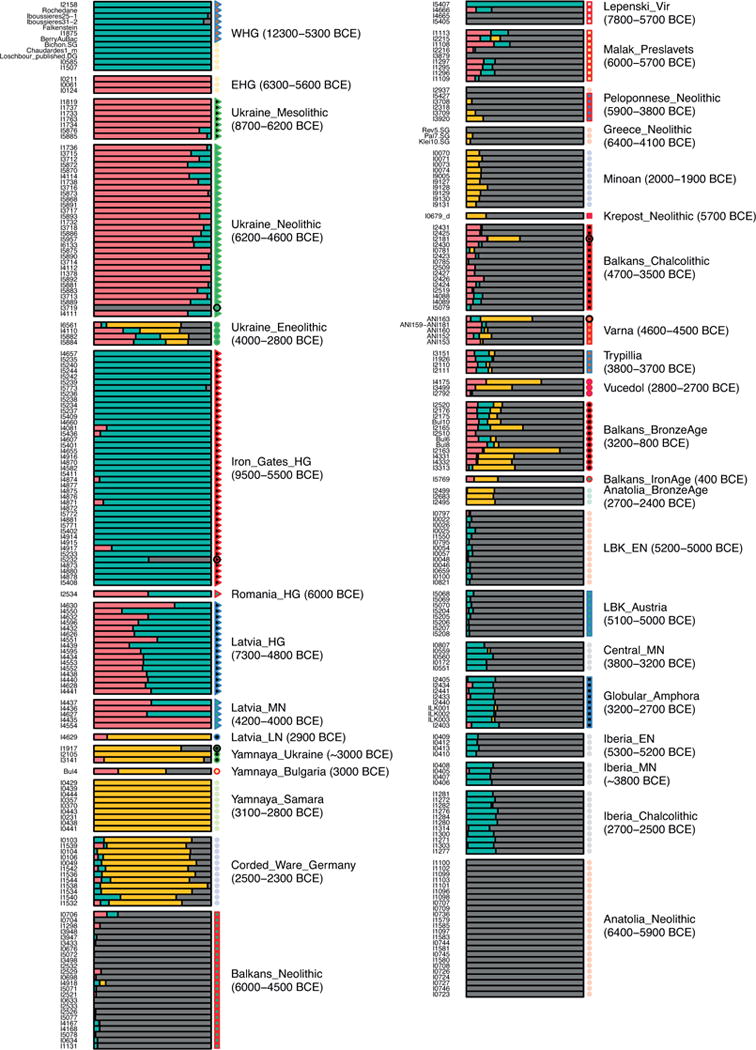
Supervised ADMIXTURE analysis modeling each ancient individual (one per row), as a mixture of populations represented by clusters that are constrained to contain Anatolian Neolithic (grey), Yamnaya from Samara (yellow), EHG (pink) and WHG (green) populations. Dates in parentheses indicate approximate range of individuals in each population. This differs from Figure 1D in that it contains some previously published samples, and includes sample IDs.
Extended Data Figure 3.
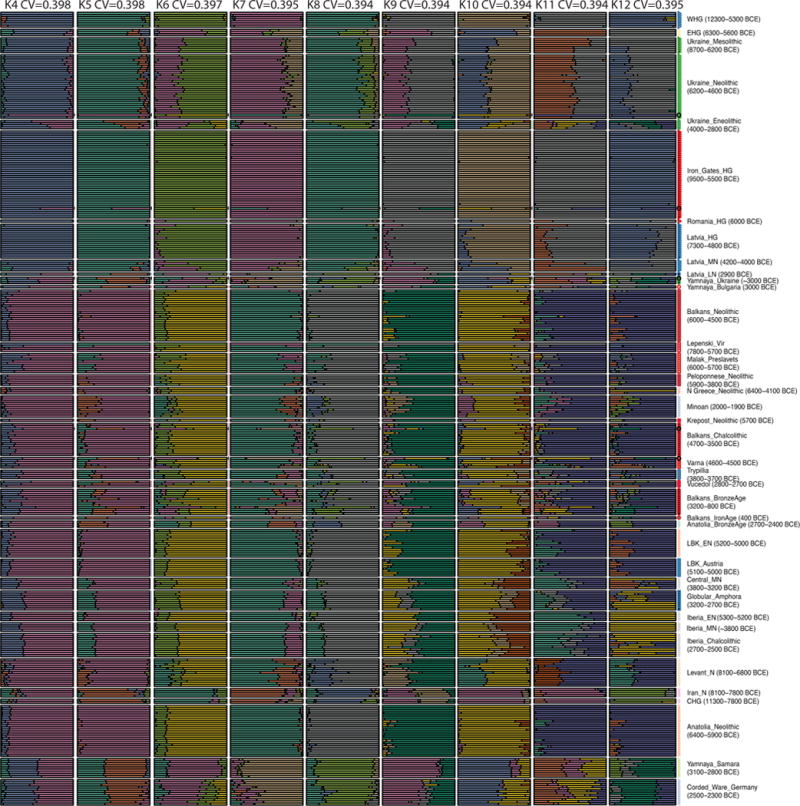
Unsupervised ADMIXTURE plot from k=4 to 12, on a dataset consisting of 1099 present-day individuals and 476 ancient individuals. We show newly reported ancient individuals and some previously published individuals for comparison.
Extended Data Figure 4.
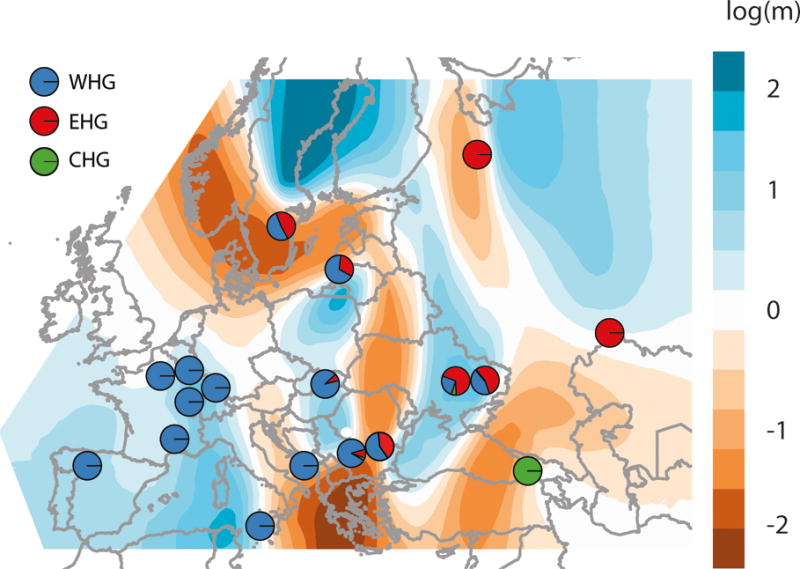
Spatial structure in hunter-gatherers. Estimated effective migration surface (EEMS).62 This fits a model of genetic relatedness where individuals move (in a random direction) from generation to generation on an underlying grid so that genetic relatedness is determined by distance. The migration parameter m defines the local rate of migration, varies on the grid and is inferred. This plot shows log10(m), scaled relative to the average migration rate (which is arbitrary). Thus log10(m)=2, for example, implies that the rate of migration at this point on the grid is 100 times higher than average. To restrict as much as possible to hunter-gatherer structure, the migration surface is inferred using data from 116 individuals that date to earlier than ~5000 BCE and have no NW Anatolian-related ancestry. Though the migration surface is sensitive to sampling, and fine-scale features may not be interpretable, the migration “barrier” (region of low migration) running north-south and separating populations with primarily WHG from primarily EHG ancestry seems to be robust, and consistent with inferred admixture proportions. This analysis suggests that Mesolithic hunter-gatherer population structure was clustered and not smoothly clinal, in the sense that genetic differentiation did not vary consistently with distance. Superimposed on this background, pies show the WHG, EHG and CHG ancestry proportions inferred for populations used to construct the migration surface (another way of visualizing the data in Figure 2, Supplementary Table 3.1.3 – we use two population models if they fit with p>0.01, and three population models otherwise). Pies with only a single color are those that were fixed to be the source populations.
Extended Data Figure 5.
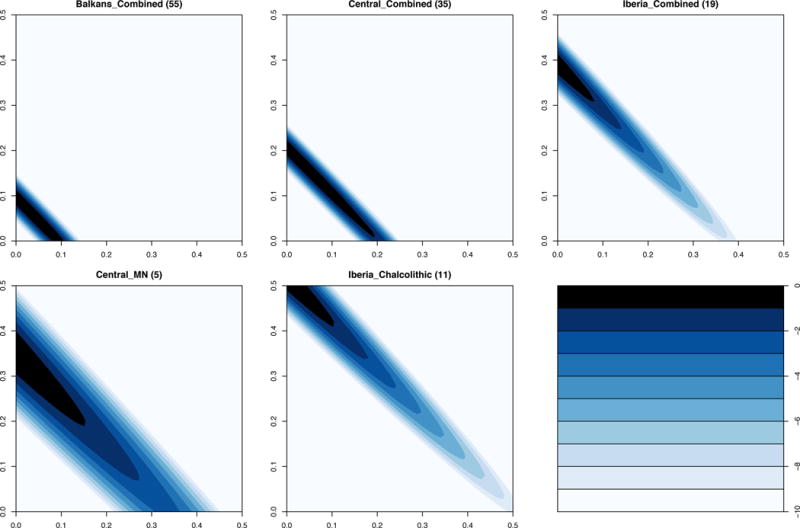
log-likelihood surfaces for the proportion of female (x-axis) and male (y-axis) ancestors that are hunter-gatherer-related for the combined populations analyzed in Figure 3C, and the two populations with the strongest evidence for sex-bias. Numbers in parentheses give the number of individuals in each group. Log-likelihood scale ranges from 0 to -10, where 0 is the feasible point with the highest likelihood.
Supplementary Material
Acknowledgments
We thank David Anthony, Iosif Lazaridis, and Mark Lipson for comments on the manuscript, Bastien Llamas, Alan Cooper and Anja Furtwängler for contributions to laboratory work, Richard Evershed for contributing 14C dates and Friederike Novotny for assistance with samples. Support for this project was provided by the Human Frontier Science Program fellowship LT001095/2014-L to I.M.; by DFG grant AL 287 / 14-1 to K.W.A.; by Irish Research Council grant GOIPG/2013/36 to D.F.; by the NSF Archaeometry program BCS-1460369 to DJK (for AMS 14C work at Penn State); by MEN-UEFISCDI grant, Partnerships in Priority Areas Program – PN II (PN-II-PT-PCCA-2013-4-2302) to C.L.; by Croatian Science Foundation grant IP-2016-06-1450 to M.N.; by European Research Council grant ERC StG 283503 and Deutsche Forschungsgemeinschaft DFG FOR2237 to K.H.; by ERC starting grant ADNABIOARC (263441) to R.P.; and by US National Science Foundation HOMINID grant BCS-1032255, US National Institutes of Health grant GM100233, the Howard Hughes Medical Institute, and an Allen Discovery Center grant from the Paul Allen Foundation to D.R.
Footnotes
Author Contributions
SAR, AS-N, SVai, SA, KWA, RA, DA, AA, NA, KB, MBG, HB, MB, ABo, YB, ABu, JB, SC, NC, RC, MC, CC, DD, NE, MFr, BGal, GG, BGe, THa, VH, KH, THi, SI, IJ, IKa, DKa, AK, DLa, MLa, CL, MLe, KL, DLV, DLo, IL, MMa, FM, KM, HM, MMe, PM, VM, VP, TDP, ASi, LS, MŠ, VS, PS, ASt, TS, MT-N, CT, IV, FVa, SVas, FVe, SV, EV, BV, CV, JZ, SZ, PWS, GC, RK, DC, GZ, BGay, MLi, AGN, IP, AP, DB, CB, JK, RP & DR assembled and interpreted archaeological material. CP, AS-N, NR, NB, FC, OC, DF, MFe, BGam, GGF, WH, EH, EJ, DKe, BK-K, IKu, MMi, AM, KN, MN, JO, SP, KSi, KSt & SVai performed laboratory work. IM, CP, AS-N, SM, IO, NP & DR analyzed data. DJK, ST, DB, CB interpreted 14C dates. JK, RP & DR supervised analysis or laboratory work. IM & DR wrote the paper, with input from all co-authors.
References
- 1.Tringham RE. In: The Transition to Agriculture in Prehistoric Europe. Price D, editor. Cambridge University Press; 2000. pp. 19–56. [Google Scholar]
- 2.Bellwood P. First Farmers: The Origins of Agricultural Societies. 2nd. Wiley-Blackwell; 2004. [Google Scholar]
- 3.Golitko M. In: Ancient Europe, 8000 BC to AD 1000: An Encyclopedia of the Barbarian World. Bogucki P, Crabtree PJ, editors. Charles Scribners & Sons; 2003. pp. 259–266. [Google Scholar]
- 4.Vander Linden M. In: Investigating Archaeological Cultures: Material Culture, Variability, and Transmission. Roberts BW, Linden M Vander, editors. Springer; 2012. pp. 289–319. [Google Scholar]
- 5.Bramanti B, et al. Genetic discontinuity between local hunter-gatherers and central Europe’s first farmers. Science. 2009;326:137–140. doi: 10.1126/science.1176869. [DOI] [PubMed] [Google Scholar]
- 6.Skoglund P, et al. Origins and genetic legacy of Neolithic farmers and hunter-gatherers in Europe. Science. 2012;336:466–469. doi: 10.1126/science.1216304. [DOI] [PubMed] [Google Scholar]
- 7.Haak W, et al. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature. 2015;522:207–211. doi: 10.1038/nature14317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cassidy LM, et al. Neolithic and Bronze Age migration to Ireland and establishment of the insular Atlantic genome. Proc Natl Acad Sci U S A. 2016;113:368–373. doi: 10.1073/pnas.1518445113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hofmanova Z, et al. Early farmers from across Europe directly descended from Neolithic Aegeans. Proc Natl Acad Sci U S A. 2016;113:6886–6891. doi: 10.1073/pnas.1523951113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mathieson I, et al. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015;528:499–503. doi: 10.1038/nature16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Omrak A, et al. Genomic Evidence Establishes Anatolia as the Source of the European Neolithic Gene Pool. Curr Biol. 2016;26:270–275. doi: 10.1016/j.cub.2015.12.019. [DOI] [PubMed] [Google Scholar]
- 12.Müller J, Rassmann K, Videiko M. BCE. Routledge; 2016. Trypillia Mega-Sites and European Prehistory: 4100-3400. [Google Scholar]
- 13.Anthony DW. The horse the wheel and language. Princeton University Press; 2007. [Google Scholar]
- 14.Fu Q, et al. An early modern human from Romania with a recent Neanderthal ancestor. Nature. 2015 doi: 10.1038/nature14558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allentoft ME, et al. Population genomics of Bronze Age Eurasia. Nature. 2015;522:167–172. doi: 10.1038/nature14507. [DOI] [PubMed] [Google Scholar]
- 16.Fu Q, et al. Genome sequence of a 45,000-year-old modern human from western Siberia. Nature. 2014;514:445–449. doi: 10.1038/nature13810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fu Q, et al. The genetic history of Ice Age Europe. Nature. 2016;534:200–205. doi: 10.1038/nature17993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gallego Llorente M, et al. Ancient Ethiopian genome reveals extensive Eurasian admixture in Eastern Africa. Science. 2015;350:820–822. doi: 10.1126/science.aad2879. [DOI] [PubMed] [Google Scholar]
- 19.Jones ER, et al. Upper Palaeolithic genomes reveal deep roots of modern Eurasians. Nature communications. 2015;6:8912. doi: 10.1038/ncomms9912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keller A, et al. New insights into the Tyrolean Iceman’s origin and phenotype as inferred by whole-genome sequencing. Nature communications. 2012;3:698. doi: 10.1038/ncomms1701. [DOI] [PubMed] [Google Scholar]
- 21.Kilinc GM, et al. The Demographic Development of the First Farmers in Anatolia. Curr Biol. 2016 doi: 10.1016/j.cub.2016.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lazaridis I, et al. Genomic insights into the origin of farming in the ancient Near East. Nature. 2016;536:419–424. doi: 10.1038/nature19310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lazaridis I, et al. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014;513:409–413. doi: 10.1038/nature13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Olalde I, et al. Derived immune and ancestral pigmentation alleles in a 7,000-year-old Mesolithic European. Nature. 2014;507:225–228. doi: 10.1038/nature12960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Raghavan M, et al. Upper Palaeolithic Siberian genome reveals dual ancestry of Native Americans. Nature. 2014;505:87–91. doi: 10.1038/nature12736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lazaridis I, et al. Genetic origins of the Minoans and Mycenaeans. Nature. 2017;548:214–218. doi: 10.1038/nature23310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lipson M, et al. Parallel ancient genomic transects reveal complex population history of early European farmers. Nature. 2017;551:368–372. doi: 10.1038/nature24476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mallick S, et al. The Simons Genome Diversity Project: 300 genomes from 142 diverse populations. Nature. 2016;538:201–206. doi: 10.1038/nature18964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Patterson N, et al. Ancient admixture in human history. Genetics. 2012;192:1065–1093. doi: 10.1534/genetics.112.145037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gronenborn D, Dolukhanov P. In: The Oxford Handbook of Neolithic Europe. Fowler C, Harding J, Hofmann D, editors. Oxford University Press; 2015. pp. 195–214. [Google Scholar]
- 32.Telegin D Ya. Neolithic cultures of Ukraine and their chronology. Journal of World Prehistory. 1987;I:307–331. [Google Scholar]
- 33.Telegin D Ya, Potekhina ID. Neolithic cemeteries and populations in the Dnieper Basin. British Archaeological Reports: 1987. [Google Scholar]
- 34.Jones ER, et al. The Neolithic Transition in the Baltic Was Not Driven by Admixture with Early European Farmers. Curr Biol. 2017:2185–2193. doi: 10.1016/j.cub.2016.12.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mittnik A, et al. The Genetic History of Northern Europe. bioRxiv. 2017 doi: 10.1101/113241. [DOI]
- 36.Saag L, et al. Extensive farming in Estonia started through a sex-biased migration from the Steppe. Curr Biol. 2017;27:2185–2193. doi: 10.1016/j.cub.2017.06.022. [DOI] [PubMed] [Google Scholar]
- 37.Maier A. The Central European Magdalenian: Regional Diversity and Internal Variability. Springer; 2015. [Google Scholar]
- 38.Borić D, Price TD. Strontium isotopes document greater human mobility at the start of the Balkan Neolithic. Proc Natl Acad Sci U S A. 2013;110:3298–3303. doi: 10.1073/pnas.1211474110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krauß R, Marinova E, De Brue H, Weninger B. The rapid spread of early farming from the Aegean into the Balkans via the Sub-Mediterranean-Aegean Vegetation Zone. Quaternary International. 2017;XXX:1–18. [Google Scholar]
- 40.Bacvarov K. In: Moments in time: Papers Presented to Pál Raczky on His 60th Birthday. Anders A, Kulcsár G, editors. L’Harmattan; 2013. pp. 29–34. [Google Scholar]
- 41.Gurova M, Bonsall C. ‘Pre-Neolithic’ in Southeast Europe: a Bulgarian perspective. Documenta Praehistorica. 2014;XLI:95–109. [Google Scholar]
- 42.Brandt G, et al. Ancient DNA reveals key stages in the formation of central European mitochondrial genetic diversity. Science. 2013;342:257–261. doi: 10.1126/science.1241844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Borić D. In: The Oxford Handbook of Neolithic Europe. Fowler C, Harding J, Hofmann D, editors. Oxford University Press; 2015. pp. 927–957. [Google Scholar]
- 44.Szmyt M. In: Transition to the Bronze Age (Archaeolingua 30) Heyd V, Kulcsár G, Szeverényi V, editors. Archaeolingua; 2013. pp. 93–111. [Google Scholar]
- 45.Olalde I, et al. A Common Genetic Origin for Early Farmers from Mediterranean Cardial and Central European LBK Cultures. Mol Biol Evol. 2015;32:3132–3142. doi: 10.1093/molbev/msv181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Posth C, et al. Pleistocene Mitochondrial Genomes Suggest a Single Major Dispersal of Non-Africans and a Late Glacial Population Turnover in Europe. Curr Biol. 2016;26:827–833. doi: 10.1016/j.cub.2016.01.037. [DOI] [PubMed] [Google Scholar]
- 47.Anthony DW, Ringe D. The Indo-European Homeland from Linguistic and Archaeological Perspectives. Annual Review of Linguistics. 2015;1:199–219. [Google Scholar]
References
- 48.Dabney J, et al. Complete mitochondrial genome sequence of a Middle Pleistocene cave bear reconstructed from ultrashort DNA fragments. Proc Natl Acad Sci U S A. 2013;110:15758–15763. doi: 10.1073/pnas.1314445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harb Protoc. 2010 doi: 10.1101/pdb.prot5448. pdb prot5448 2010. [DOI] [PubMed] [Google Scholar]
- 50.Rohland N, Harney E, Mallick S, Nordenfelt S, Reich D. Partial uracil-DNA-glycosylase treatment for screening of ancient DNA. Philos Trans R Soc Lond B Biol Sci. 2015;370:20130624. doi: 10.1098/rstb.2013.0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Korlevic P, et al. Reducing microbial and human contamination in DNA extractions from ancient bones and teeth. BioTechniques. 2015;59:87–93. doi: 10.2144/000114320. [DOI] [PubMed] [Google Scholar]
- 52.Briggs AW, et al. Removal of deaminated cytosines and detection of in vivo methylation in ancient DNA. Nucleic Acids Res. 2010;38:e87. doi: 10.1093/nar/gkp1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeAngelis MM, Wang DG, Hawkins TL. Solid-phase reversible immobilization for the isolation of PCR products. Nucleic Acids Res. 1995;23:4742–4743. doi: 10.1093/nar/23.22.4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rohland N, Reich D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 2012;22:939–946. doi: 10.1101/gr.128124.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maricic T, Whitten M, Pääbo S. Multiplexed DNA sequence capture of mitochondrial genomes using PCR products. PLoS One. 2010;5:e14004. doi: 10.1371/journal.pone.0014004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kircher M, Sawyer S, Meyer M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic Acids Res. 2012;40:e3. doi: 10.1093/nar/gkr771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Fu Q, et al. A revised timescale for human evolution based on ancient mitochondrial genomes. Curr Biol. 2013;23:553–559. doi: 10.1016/j.cub.2013.02.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Skoglund P, et al. Separating endogenous ancient DNA from modern day contamination in a Siberian Neandertal. Proc Natl Acad Sci U S A. 2014;111:2229–2234. doi: 10.1073/pnas.1318934111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Korneliussen TS, Albrechtsen A, Nielsen R. ANGSD: Analysis of Next Generation Sequencing Data. BMC Bioinformatics. 2014;15:356. doi: 10.1186/s12859-014-0356-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Price AL, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 62.Petkova D, Novembre J, Stephens M. Visualizing spatial population structure with estimated effective migration surfaces. Nat Genet. 2016;48:94–100. doi: 10.1038/ng.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Brown L, Cai T, DasGupta A. Interval Estimation for a Binomial Proportion. Statistical Science. 2001;16:101–133. [Google Scholar]
- 64.Agresti A, Coull B. Approximate is better than “exact” for interval estimation of binomial proportions. The American Statistician. 1998;52:119–126. [Google Scholar]
- 65.Longin R. New method of collagen extraction for radiocarbon analysis. Nature. 1971;230:241–242. doi: 10.1038/230241a0. [DOI] [PubMed] [Google Scholar]
- 66.Brown TA, Nelson DE, Vogel JS, Southon JR. Improved collagen extraction by modified longin method. Radiocarbon. 1988;30:171–177. [Google Scholar]
- 67.Kennett DJ, et al. Archaeogenomic evidence reveals prehistoric matrilineal dynasty. Nature communications. 2017;8:14115. doi: 10.1038/ncomms14115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Klinken GJ. Bone collagen quality indicators for paleodietary and radiocarbon measurements. JAS. 1999;26:687–695. [Google Scholar]
- 69.Stuiver M, Polach HA. Discussion: Reporting of 14C data. Radiocarbon. 1977;19:355–363. [Google Scholar]
- 70.Bronk Ramsey C. OxCal 4.23 Online Manual. 2013 https://c14.arch.ox.ac.uk/oxcalhelp/hlp_contents.html.
- 71.Cook GT, et al. A freshwater diet-derived 14C reservoir effect at the Stone Age sites in the Iron Gates gorge. Radiocarbon. 2001;43:453–460. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The aligned sequences are available through the European Nucleotide Archive under accession number PRJEB22652. The pseudo-haploid genotype dataset used in analysis and consensus mitochondrial genomes are available at https://reich.hms.harvard.edu/datasets.