

Abstract
Dravet Syndrome is a devastating childhood epilepsy disorder with high incidence of premature death plus comorbidities of ataxia, circadian rhythm disorder, impaired sleep quality, autistic-like social-interaction deficits and severe cognitive impairment. It is primarily caused by heterozygous loss-of-function mutations in the SCN1A gene that encodes brain voltage-gated sodium channel type-1, termed NaV1.1. Here I review experiments on mouse genetic models that implicate specific loss of sodium currents and action potential firing in GABAergic inhibitory interneurons as the fundamental cause of Dravet Syndrome. The resulting imbalance of excitatory to inhibitory neurotransmission in neural circuits causes both epilepsy and co-morbidities. Promising therapeutic approaches involving atypical sodium channel blockers, novel drug combinations, and cannabidiol give hope for improved outcomes for Dravet Syndrome patients.
Keywords: Epilepsy, autism, cognitive deficit, sodium channels, interneurons
Introduction
Voltage-gated sodium (NaV) channels initiate action potentials in neurons and other excitable cells [1]. They are composed of a large central pore-forming α subunit in complex with one or two auxiliary β subunits [2]. In response to depolarizing stimuli, brain NaV channels rapidly activate, open, and then inactivate with 1–2 msec [2,3]. A further slow inactivation process is engaged by long trains of stimuli or prolonged depolarizations in the range of 100 msec [4]. The kinetics and voltage dependence of sodium channel activation and inactivation strongly influence the threshold for action potential firing and the initiation, firing frequency, and durations of trains of action potentials [4]. Because information is encoded in the frequency and pattern of trains of action potentials, sodium channels play critical roles in information processing in neural circuits as well as in information transmission throughout the brain.
Dravet Syndrome
Dravet Syndrome is a devastating childhood epilepsy disorder with a high incidence of premature death plus co-morbidities of developmental delay, severe cognitive impairment, ataxia, circadian rhythm disorder, impaired sleep quality, and autistic-like social interaction deficits [5]. It is primarily caused by heterozygous loss-of-function mutations in the SCN1A gene that encodes the brain voltage-gated sodium channel type-1, termed NaV1.1 [5]. Approximately 80% of patients with a clinical diagnosis of Dravet Syndrome have identified mutations in exons encoding NaV1.1 [5]. As the exons of SCN1A only comprise 6 kb of the gene, which is over 100 kb in size, many of the remaining patients may have loss-of-function mutations in the non-coding portion of the gene that prevent NaV1.1 protein expression.
Dravet Syndrome typically begins at 6 to 9 months of age with seizures induced by elevation in core body temperature due to a fever, a hot day, or a hot bath [5]. Spontaneous seizures follow within weeks, and they become progressively more frequent and severe throughout childhood. After six years of age, the frequency and severity of seizures are reduced, but seizures continue into adulthood [5]. Seizures in Dravet Syndrome are resistant to standard pharmacotherapy, even with mixtures of multiple antiepileptic drugs [6]. Historically, up to 30% of children with Dravet Syndrome died before their teenage years. Even with modern diagnosis and care, up to 15% of affected children die prematurely.
Children with Dravet Syndrome also have debilitating co-morbidities. As their seizures reach peak frequency and severity during years 1–5, developmental milestones are lost, and cognitive achievements regress, resulting in a typical IQ of 50 in teenage years [5]. This severe cognitive deficit is as problematic as seizures for older patients because they require constant care. Most Dravet Syndrome patients also have autistic-like social interaction deficits, and ataxia, circadian rhythm defect, and sleep impairment are common [5]. In this article, I review research on mouse genetic models that has provided important insights into the pathophysiology and therapy of this devastating disease
Mouse Genetic Models of Epilepsy and Premature Death in Dravet Syndrome
It is a paradox that loss-of-function mutations in a NaV channel cause epilepsy. Two mouse genetic models of Dravet Syndrome based on different disease mutations both showed spontaneous seizures (Fig. 1a). Surprisingly, these loss-of-function mutations have a specific effect to reduce the sodium currents and electrical excitability of GABAergic interneurons [7,8], which would imbalance the ratio of excitation and inhibition in neural circuits throughout the brain and lead to general hyperexcitability [9]. By postnatal day (P) 21, mice with heterozygous mutations in NaV1.1 become susceptible to thermal induction of seizures by slowly raising their core body temperature to 38.5°C or higher (Fig. 1c) [10]. One day later, at P22, they begin to have spontaneous seizures, which increase to peak frequency and severity at P24-P27 [7,11]. The frequency of spontaneous seizure declines with increasing age, but these mice continue to have seizures through adulthood [11–13]. The temporal evolution of Dravet Syndrome is similar in humans and mice, when compared to the developmental time courses of the two species, with seizures beginning near the time of weaning (P20 vs. 6–9 months), reaching peak during early development (P24–27 vs. 2–5 years), and declining to a steady state at puberty (P60 vs.13 years) [14]. The onset of Dravet Syndrome in humans and mice is approximately correlated with the normal developmental loss of embryonic NaV1.3 channels paired with the failure of normal development of NaV1.1 channels to replace them [14].
Figure 1. Spontaneous and thermally induced seizures in DS mice.
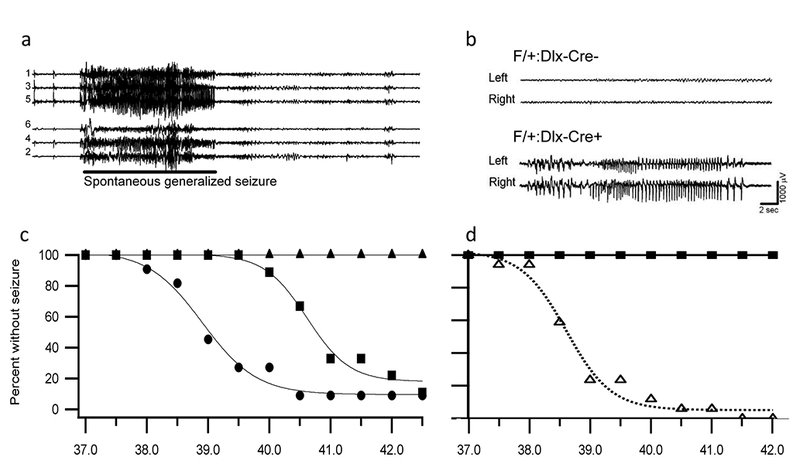
The body core temperature of mice was raised to the indicated levels by a thermal feedback circuit and heat lamp in steps of 0.5°C per two minutes, and seizures were recorded by video camera and implanted EEG electrodes. (a) Representative EEG traces during a spontaneous seizure in a DS mouse. (b) Representative EEG traces of F/+:Dlx-Cre- and F/+:Dlx-Cre+ mice during thermal induction of seizures at P24. Top, F/+:Dlx-Cre- mouse at 39.5°C. Bottom, F/+:Dlx-Cre+ mouse during a seizure at 39.5°C. (c) Temperature dependence of thermal induction of seizures in DS mice. Thermally-induced seizures were evoked in DS mice at P30–46 (circles, mean temperature of 39°C) and P20–22 (squares, mean temperature of induction 40.2°C), but no seizures were induced in P17–18 mice (triangles). (d) Temperature dependence of thermal induction of seizures in Dlx-Cre+ mice. Thermally-induced seizures were evoked in all F/+:Dlx-Cre+ animals with a mean temperature of 39°C. No F/+:Dlx-Cre- mice had a seizure.
As expected from the deficit in action potential firing in GABAergic interneurons, inhibitory GABAergic neurotransmission is reduced. Recordings of spontaneous, action potential-driven synaptic activity in hippocampal slices show an increase in the time interval between inhibitory postsynaptic currents (IPSCs) and a reduction in the frequency of these synaptic events (Fig. 2a). In response to the reduced inhibitory neurotransmission, excitatory neurotransmission is increased (Fig. 2b). These results reveal an increase in the ratio of excitatory to inhibitory neurotransmission in neural circuits in Dravet Syndrome.
Figure 2. Spontaneous inhibitory and excitatory synaptic activity in WT and DS mice.
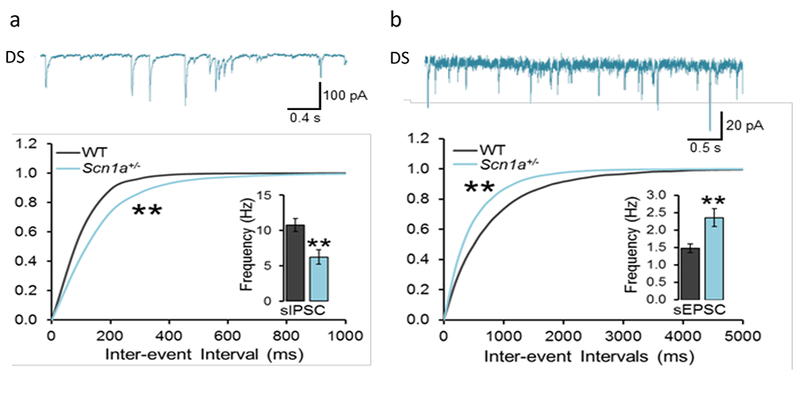
Hippocampal slices were prepared from WT and DS mice, and spontaneous, AP-dependent synaptic events were recorded by whole-cell voltage clamp of CA1 neurons. (a) Top. Representative recording of inhibitory postsynaptic currents (IPSCs) from a CA1 neuron from a DS mouse. Bottom. Histogram of inter-event intervals between IPSCs. Inset. Mean frequency of IPSCs. (b) Top. Representative recording of excitatory postsynaptic currents (EPSCs) from a CA1 neuron from a DS mouse. Bottom. Histogram of inter-event intervals between EPSCs. Inset. Mean frequency of EPSCs.
The key role of sodium channel mutations in GABAergic interneurons as the fundamental genetic cause of Dravet Syndrome was further supported by specific Cre-Lox deletion of NaV1.1 in forebrain GABAergic interneurons, which precisely recapitulates the thermally induced seizures, spontaneous seizures, and premature death of the disease [15]. Thermal induction of seizures is half-maximal at 38.5°C, just as for mice with global mutation of NaV1.1 (Fig. 1b, d), suggesting that mutation in forebrain GABAergic interneurons quantitatively recapitulates the epilepsy syndrome [15]. In contrast, specific deletion in excitatory neurons ameliorates the effects of Dravet Syndrome in mice [16]. Remarkably, these mouse genetic models also recapitulate each of the co-morbidities of Dravet Syndrome, and failure of firing of GABAergic inhibitory neurons is implicated in each case.
Co-Morbidities in Mouse Genetic Models of Dravet Syndrome
Ataxia.
The first co-morbidity to be analyzed in a mouse model of Dravet Syndrome was ataxia. A mild ataxia phenotype was observed in digital video recordings [17]. Electrophysiological studies revealed a defect in action potential firing in cerebellar Purkinje neurons [17]. This defect is sufficient to cause ataxia, because deficits of similar magnitude in Purkinje cell function cause ataxia in other contexts (Table 1).
Table 1.
| Co-Morbidity | Symptoms in DS Mice | Physiological Correlates | Causal Evidence for Mechanism |
|---|---|---|---|
| Ataxia | Abnormal foot placement in walking | Failure of AP firing in GABAergic cerebellar Purkinje neurons | |
| Circadian Rhythm | Long circadian cycle; weak light-induced phase shift; increased negative masking | Failure of AP firing in GABAergic neurons in the suprachiasmatic nucleus | Not observed in forebrain-specific knockout mouse; reversed by clonazepam |
| Sleep Impairment | Reduced non-REM sleep, delta wave power, sleep spindles | Failure of rebound AP firing in GABAergic neurons in the RNT | Observed in forebrain-specific knockout mouse |
| Cognitive Deficit | Failure of spatial learning and memory | Failure of AP firing by GABAergicneurons in hippocampus and cerebral cortex | Observed in forebrain-specific knockout mouse and PV+SST-specific knockout; reversed by clonazepam |
| Autistic-like Behavior | Impaired social interaction; repetitive behaviors | Failure of AP firing by PV-expressing GABAergicneurons in hippocampus and cerebral cortex | Observed in forebrain-specific and PV-specific knockout mouse; reversed by clonazepam |
AP, action potential; SCN, suprachiasmatic nucleus of the hypothalamus; RNT, reticular nucleus of the thalamus; REM, rapid-eye-movement.
Circadian rhythm.
Dravet Syndrome children have a circadian rhythm defect, which prevents them from establishing a normal sleep-wake cycle [18, 19]. Dravet Syndrome mice have an abnormally long circadian cycle length, defects in phase shift after change of their light-dark cycle, analogous to exaggerated jet lag in humans, and impaired light-induced shifts of their sleep-wake cycle [19]. GABAergic neurotransmission is impaired in the suprachiasmatic nucleus of the hypothalamus, the primary site of the circadian clock [19]. These deficits were substantially reversed by treatment with the benzodiazepine clonazepam, a positive allosteric modulator of the postsynaptic response of GABAA receptors to synaptically released GABA [19]. These results add further support for the conclusion that co-morbidities of Dravet Syndrome are caused by failure of action potential firing of GABAergic interneurons (Table 1).
Sleep quality.
Children with Dravet Syndrome wake frequently, independent of their pattern of seizures during sleep [18,19]. Dravet Syndrome mice also have a defect in sleep quality [21], including excess brief waking episodes. This sleep defect is observed in mice with specific deletion of NaV1.1 in GABAergic interneurons in the forebrain, confirming that it is independent of the impairment of circadian rhythms in the suprachiasmatic nucleus [21]. Amplitudes of delta waves and sleep spindles in NREM sleep are decreased more than 60%. Sleep spindles and behavioral aspects of sleep are driven by a tri-synaptic pathway of excitatory neurons in the basal nucleus of the thalamus, pyramidal cells in the cerebral cortex, and GABAergic inhibitory neurons in the reticular nucleus of the thalamus, which control this circuit by firing rapid trains of action potentials upon release from a hyperpolarizing stimulus [22]. This rebound firing is reduced by 60% in Dravet Syndrome mice [21], consistent with the conclusion that impaired action potential firing of these GABAergic interneurons is responsible for the defect in sleep quality (Table 1).
Autistic-like behaviors.
Children with Dravet Syndrome also exhibit autistic-like behaviors [23–25]. Dravet Syndrome mice have a substantial deficit in social interactions (Fig. 3) [26,27]. In the three-chamber test, Dravet Syndrome mice (Fig. 3a) show no preference for interaction with a stranger mouse in preference to a novel object. In the open field, they interact less with a stranger mouse, and they exhibit rapid defensive escapes from social interactions [26,27]. These deficits in social interaction are also present in mice with specific deletion of NaV1.1 channels in forebrain inhibitory neurons (Dlx, Fig. 3c) [26], indicating that this autistic-like behavior is caused by reduced action potential firing in inhibitory neurons (Table 1). Consistent with this mechanism, autistic-like behaviors are rescued by treatment of Dravet Syndrome mice with low doses of the benzodiazepine clonazepam [26].
Figure 3. Autism and cognitive deficit in DS mice.
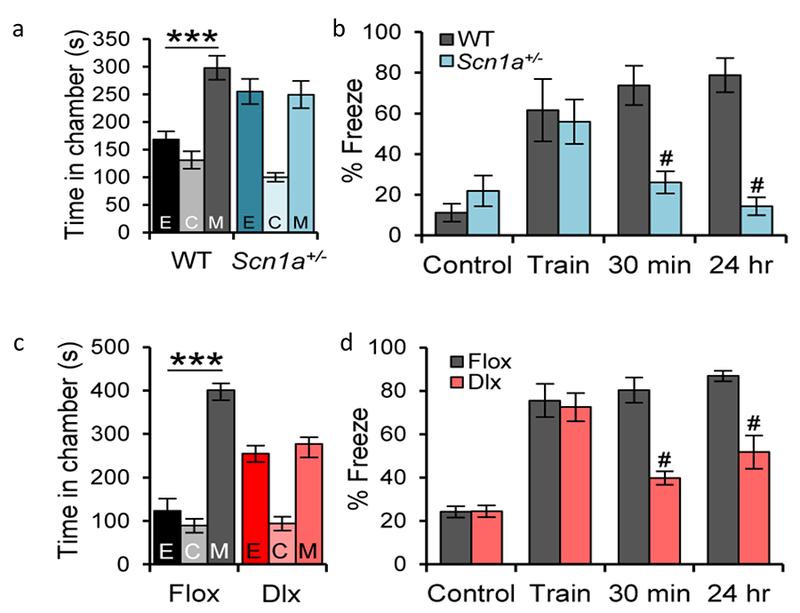
(a) A test mouse was placed in the center chamber (C) of a three-chamber apparatus with connecting passageways, and its movement among the three chambers was recorded and digitally analyzed. WT mice spend more time in the side chamber containing a stranger mouse in a small inverted wire cup (M) compared to the side chamber containing an empty wire cup (E). In contrast, DS mice spend equal time in the E and M chambers. (b) In the contextual fear-conditioning test, a mouse was placed in a well-marked cage with an electric grid as its floor, and the movement of the mouse was recorded and digitally analyzed. The % of time that the mouse displays fear-induced freezing behavior was quantified. WT and DS test mice freeze infrequently while exploring the cage (Control), but they freeze in fear during and immediately after a mild foot shock (Train, 2s, 0.5 mA). Test mice were transported to their home cage. Upon return to the context of the fearful shock after 30 min and 24 h, WT mice freeze in anticipation of a shock, even though none is administered. In contrast, DS mice display a profound deficit in short-term (30 min) and long term (24 hr) memory of the spatial context of the foot shock (0.5 mA), (c) In the 3-chamber test, Flox control mice prefer to interact with the stranger mouse (M) compared to the empty wire cup, whereas Dlx1/2-Scn1a+/− mice have no preference for the stranger mouse. (d) In the contextual fear-conditioning test, Dlx1/2-Scn1a+/− mice have a normal fear response during and immediately after the foot shock (Train) but display a profound deficit in short-term (30 min) and long-term (24 hr) memory of the spatial context of the foot shock compared to Flox control mice.
Spatial learning and memory.
According to parental reports, children with Dravet Syndrome have exceptional difficulty learning and remembering the location of dangerous situations, and therefore have an unusual number of accidents from falling on stairs, jumping into swimming pools, running into the street, etc. [19]. Dravet Syndrome mice also have a major deficit in spatial learning and memory [26]. In the context-dependent fear conditioning test, Dravet Syndrome mice exhibit a normal acute ‘freezing’ response to a fearful foot shock, but they do not remember the spatial context of that fearful event when later returned to the same location (Fig. 3b) [26]. They also are impaired in the Barnes maze test, in which mice learn to find a dark refuge hole on the periphery of a brightly lighted field [26]. These impairments in spatial learning and memory are also observed when the NaV1.1 channel is specifically deleted in forebrain inhibitory neurons (Fig. 3d), and they are rescued by a single low dose of clonazepam [26]. Thus, this defect in fear-associated spatial learning is characteristic of mice and humans with Dravet Syndrome, and it is caused by loss of electrical excitability in forebrain GABAergic interneurons (Table 1).
Interneuron Types
In the cerebral cortex, interneurons can be divided into three non-overlapping classes, recognizable by their expression of the marker proteins parvalbumin (PV), somatostatin (SST), and serotonin receptor 3a (5-HT3aR) [28]. PV interneurons make synapses on the cell bodies and axon initial segments of pyramidal neurons, where their fast-spiking discharges exert potent inhibition of action potential firing by their postsynaptic target [28]. SST interneurons make synapses on distal synapses of pyramidal neurons. Their activity opposes incoming EPSCs from synapses on the distal dendrites and exerts disynaptic inhibition of the activity of neighboring excitatory neurons [28]. 5-HT3aR neurons form synapses on both inhibitory neurons and excitatory neurons, which may provide opposing influences on pyramidal neuron excitability [28]. In Dravet Syndrome mice, action potential firing is impaired in some subtypes of 5HT3aR inhibitory neurons in layers M/MI [29] and in both PV and SST interneurons in Layer V (Fig. 4) [30]. Disynaptic inhibition between pairs of pyramidal neurons, which is mediated by SST interneurons [31], was also markedly impaired [30]. No impairment of action potential firing was observed in pyramidal neurons in these cortical brain areas [29,30].
Figure 4. Impaired action potential generation in PV and SST interneurons from from Cre+ mice.
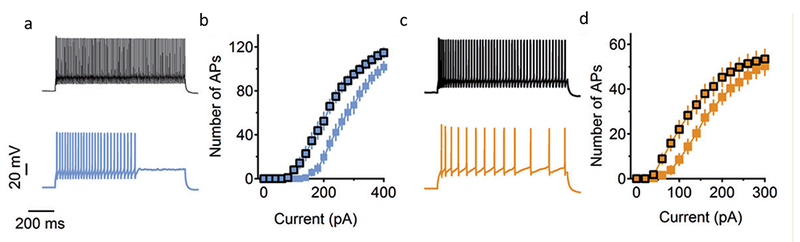
DS Flox mice were bred with mice expressing PV-Cre (PV mice) or with mice expressing SST-Cre (SST mice). Cerebral cortical slices were prepared, PV and SST interneurons were identified by specific labeling, and APs were recorded in whole-cell current clamp mode in response to injection of the indicated amounts of depolarizing current. (a) Representative trains of action potentials recorded in whole-cell current clamp during injection of 240 pA of depolarizing current. WT, black; PV, blue. (b) Action potential number plotted vs. the level of depolarizing current. (c) Representative trains of action potentials recorded in whole-cell current clamp during injection of 160 pA of depolarizing current. WT, black; SST, yellow. (d) Action potential number plotted vs. the level of depolarizing current.
We used the Cre-Lox method to delete the NaV1.1 channel in each of these classes of interneurons [32]. Action potential firing was impaired in all thee classes of interneurons [29,32]. Deletion in PV interneurons gave autistic-like social interaction deficits and pro-epileptic effects (Table 1) [32]. Deletion in SST interneurons gave hyperactivity and pro-epileptic effects that were not as strong as deletion in PV interneurons (Table 1) [32]. Combined deletion in PV and SST interneurons gave synergistic pro-epileptic effects, cognitive impairments, and premature death (Table 1) [32]. In contrast, deletion in 5-HT3aR neurons caused comparatively mild effects, with no evident pro-epileptic effects or premature death, no cognitive deficit, and relatively mild social interaction impairment [29]. Evidently, Dravet Syndrome is caused primarily by defects in action potential firing in PV and SST interneurons. These deficits create an imbalance of excitatory glutamatergic neurotransmission over inhibitory GABAergic neurotransmission, which is a key piece in the puzzle of Dravet Syndrome (Fig. 5). This imbalance of electrical signaling in brain circuits leads to the pleiotropic effects of this disease on ataxia, circadian defect, sleep impairment, autistic-like behaviors, and cognitive deficit (Table 1).
Figure 5. Imbalance of excitatory vs. inhibitory neurotransmission in DS.

Failure of AP firing in GABAergic interneurons in DS causes a reduction in GABA release and an imbalance of excitatory vs. inhibitory neurotransmission in circuits in the brain. This imbalance leads to epilepsy, cognitive deficit, and autistic-like behaviors plus other co-morbidities in DS.
Genetic Background Effects
Children with apparently complete loss-of-function mutations in NaV1.1 have different time course and severity of Dravet Syndrome symptoms, implicating strong effects of genetic background in determining disease severity [23]. All of our studies cited above were carried out with NaV1.1 mutations expressed in homozygous C57BL/6J mice, which recapitulate all of the phenotypes of human Dravet Syndrome [12,26]. With this genetic background, all of the effects of these mutations are caused by failure of action potential firing by GABAergic interneurons [12,15,26,33]. However, in our initial studies, we found that the effect of deletion of NaV1.1 channels was much more severe in the C57BL/6J genetic background than in the 129SvJ genetic background, because the ~70% of C57BL/6 mice died by P80 whereas 129SvJ mice died no more frequently than WT [7].
Mistry et al. [34] compared the Dravet Syndrome NaV1.1+/− genotype in 50:50 C57BL/6:J:129S6/SvEvTac genetic background to homozygous 129S6/SvEvTac genetic background. As in our previous work [7], they found that mice in a 50:50 C57BL/6J genetic background had spontaneous seizures and premature death, whereas mice in the homozygous 129S6/SvEvTac genetic background had both spontaneous seizures and premature death [34]. Electrophysiological studies revealed reduced sodium currents in hippocampal GABAergic interneurons in 50:50 C57BL/6:J:129S6/SvEvTac but not in homozygous 129S6/SvEvTac genetic background [34], consistent with a causal role for impaired action potential generation in GABAergic interneurons in Dravet Syndrome. They also observed increased sodium currents in pyramidal neurons of P21 mice that may contribute to disease phenotypes in these genetic backgrounds.
Further studies of genetic background effects in mice with homozygous genetic backgrounds showed that NaV1.1+/− mice in the 129SvJ genetic background have a less severe phenotype, with no evident spontaneous seizures, premature death, or cognitive deficit [35]. This less severe phenotype resembles Generalized Seizures with Febrile Seizures Plus (GEFS+) [12], which is a milder NaV1.1 genetic epilepsy syndrome. The mild phenotype of Dravet Syndrome in 129SvJ mice was correlated with less reduction of action potential firing in GABAergic interneurons and less impairment of postsynaptic boosting of synaptic inputs in pyramidal neurons [35]. No changes in pyramidal cell excitability were observed in these genetic backgrounds [35]. Overall, these results indicate that genetic background effects on the phenotypes of NaV1.1 mutations are potentially strong enough to change the clinical diagnosis from Dravet Syndrome to GEFS+, consistent with the model that mutations with a spectrum of severity and genetic background can account for the spectrum of clinical diagnoses from familial febrile seizures to GEFS+ to Dravet Syndrome [12].
Gene mapping studies are in progress to identify potential modifier genes for Dravet Syndrome. The initial phase of this work revealed sites of significant genetic background effects on chromosomes 5, 7, 8, and 11 [36]. Follow-up studies of chromosome 5 identified a 9 Mb region containing candidate genes encoding GABAA receptor subunits [37]. Genetic background effects of polymorphisms in GABAA receptor subunits would be expected in a disease caused by failure of action potential firing in GABAergic interneurons. It will be of great interest to discover the specific polymorphisms in these genes and analyze their effects on GABA receptor function.
Novel Therapeutic Approaches
Current therapy.
Current treatment of Dravet Syndrome is not sufficient to prevent the storm of seizures and debilitating co-morbidities that are characteristic of this disease, even though combinations of antiepileptic drugs are used [6]. One standard treatment includes four antiepileptic drugs: valproate, clobazam, topiramate, and stiripentol [6]. The nontraditional antiepileptic drug leviteracetam is also frequently used as an add-on medication [38]. Unfortunately, even with these complex drug cocktails, control of seizures and prevention of co-morbidities are usually not achieved.
Sodium channel-blocking antiepileptic drugs.
Drugs that block sodium channels are a mainstay of antiepileptic therapy, including phenytoin and lamotrigine. However, as would be expected in a disease caused by loss-of-function mutations in a sodium channel, these traditional sodium channel blockers actually exacerbate seizures in Dravet Syndrome in both children and mice [39,40]. Although traditional sodium channel blocking antiepileptic drugs are not effective, the atypical sodium channel blocker GS967 has beneficial effects on seizure frequency and premature death in Dravet Syndrome mice [41]. This atypical compound preferentially inhibits sustained sodium currents compared to peak sodium currents [41]. This mechanism of action may allow GS967 to prevent the synchronous repetitive action potential firing characteristic of epileptic seizures while leaving normal action potential generation and propagation in inhibitory neurons relatively unaffected.
Enhancing GABAergic neurotransmission.
Because the primary effect of mutation of NaV1.1 is failure of action potential generation in inhibitory interneurons, the most direct approach to restoring normal nervous system function would be to enhance GABAergic neurotransmission. Our evidence that epilepsy, premature death, and all co-morbidities of Dravet Syndrome are caused by failure of action potential firing in inhibitory neurons suggests that both epilepsy and co-morbidities could potentially be prevented by appropriate enhancement of phasic GABAergic neurotransmission. An ideal therapeutic approach would be to both increase the level of GABA in the synaptic cleft and enhance its postsynaptic effects. Tiagabine and related drugs inhibit the uptake of GABA from the synaptic cleft by inhibiting the GAT1 transporter [42,43]. Benzodiazepines like clonazepam enhance the postsynaptic response to GABA by serving as positive allosteric modulators [44]. We found synergistic effects of tiagabine and clonazepam on thermally induced seizures in Dravet Syndrome mice and reduced side effects for equi-effective combined doses compared to the single drugs [45]. Unfortunately, sedative side effects of clonazepam and increased incidence of myoclonic seizures caused by tiagabine were still noticeable with the optimum drug combination [45].
Cannabidiol.
Parental reports indicate that the nonpsychotropic cannabis component cannabidiol is effective in reducing the frequency and severity of seizures in Dravet Syndrome [46]. We found that treatment with a single dose of cannabidiol decreases the duration and severity of thermally induced seizures (Fig. 6), reduces the frequency of spontaneous seizures, and ameliorates the social interaction deficits in Dravet Syndrome mice [47], and a small clinical trial reported efficacy in reducing the frequency of spontaneous seizures in Dravet Syndrome children [48]. The beneficial effects of cannabidiol in mice were correlated with increased GABAergic inhibitory neurotransmission, reduced ratio of excitatory to inhibitory neurotransmission, enhanced action potential firing in GABAergic interneurons in the dentate gyrus, and, at high stimulus intensity, inhibition of action potential firing of excitatory dentate granule neurons [47]. These electrophysiological effects were mimicked and occluded by inhibition of the lipid-activated G protein-coupled receptor GPR55 [47], suggesting a key role for this signaling pathway in mediating the effects of cannabidiol. Drugs that act directly on the GPR55 pathway may also be efficacious in Dravet Syndrome.
Figure 6. CBD reduced seizure duration and severity in DS mice.
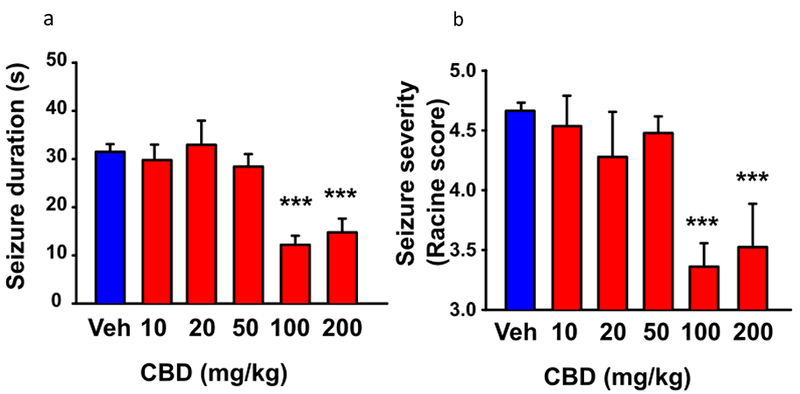
The core body temperature of mice was warmed to 38.5°C as in Fig. 1, and the duration and severity of the resulting seizures were video recorded. (a) Dose dependence of CBD reduction in duration of thermally induced seizures. (b) Dose dependence of CBD reduction in seizure severity based on Racine Score.
Conclusion
Studies of multiple mouse genetic models of Dravet Syndrome all lead to the conclusion that the primary pathogenic event is loss of action firing in GABAergic interneurons. This loss of electrical excitability in GABAergic interneurons leads to an imbalance of excitation over inhibition in many neural circuits. This imbalance leads directly to the severe epilepsy, premature death, and many co-morbidities of Dravet Syndrome. Genetic dissection of the phenotypes of Dravet Syndrome indicates different roles for specific classes of interneurons in the disease. Secondary up-regulation of sodium channel expression and function may also contribute to the disease phenotypes, and genetic background effects have a major impact on disease severity. Recent pharmacological studies in mice and humans give a more optimistic outlook for future effective therapies for this devastating disease.
Highlights.
Dravet Syndrome results from heterozygous loss-of-function mutation of Nav1.1 channel
Mouse genetic models recapitulate all aspects of the disease
Epilepsy is caused by loss of action potential firing in GABAergic inhibitory neurons
Co-morbidities also correlate with loss of excitability of inhibitory neurons
Novel therapeutic approaches show promise for improved treatment
Acknowledgements
Research on Dravet Syndrome in the author’s laboratory was supported by the National Institute of Neurological Disorders & Stroke of the National Institutes of Health (Research Grant R01 25470) and by a grant from the Simons Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The author declares no conflicts of interest.
References
- 1.Hille B: Ionic Channels of Excitable Membranes edn 3rd Ed Sunderland, MA: Sinauer Associates Inc; 2001. [Google Scholar]
- 2.Catterall WA: From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 2000, 26:13–25. [DOI] [PubMed] [Google Scholar]
- 3.Hille B: Ionic Channels of Excitable Membranes, 3rd Ed Sunderland, MA: Sinauer Associates Inc; 2001. [Google Scholar]
- 4.Vilin YY, Ruben PC: Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem Biophys 2001, 35:171–190. [DOI] [PubMed] [Google Scholar]
- 5.Dravet C: The core Dravet syndrome phenotype. Epilepsia 2011, 52 Suppl 2:3–9. [DOI] [PubMed] [Google Scholar]
- 6.Chiron C, Dulac O: The pharmacologic treatment of Dravet syndrome. Epilepsia 2011, 52 Suppl 2:72–75. [DOI] [PubMed] [Google Scholar]
- 7.Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA: Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 2006, 9:1142–1149. [DOI] [PubMed] [Google Scholar]
- 8.Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, et al. : NaV1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scnla gene mutation. J Neurosci 2007, 27:5903–5914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liautard C, Scalmani P, Carriero G, de Curtis M, Franceschetti S, Mantegazza M: Hippocampal hyperexcitability and specific epileptiform activity in a mouse model of Dravet syndrome.Epilepsia 2013, 54:1251–1261. [DOI] [PubMed] [Google Scholar]
- 10.Oakley JC, Kalume F, Yu FH, Scheuer T, Catterall WA: Temperature- and age-dependent seizures in a mouse model of severe myoclonic epilepsy in infancy. Proc Natl Acad Sci U S A 2009, 106:3994–3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kalume F, Westenbroek RE, Cheah CS, Yu FH, Oakley JC, Scheuer T, Catterall WA: Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Invest 2013, 123:1798–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Catterall WA, Kalume F, Oakley JC: NaV1.1 channels and epilepsy. Journal of Physiology 2010, 588:1849–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mantegazza M: Dravet syndrome: insights from in vitro experimental models. Epilepsia 2011, 52 Suppl 2:62–69. [DOI] [PubMed] [Google Scholar]
- 14.Cheah CS, Westenbroek RE, Roden WH, Kalume F, Oakley JC, Jansen LA, Catterall WA: Correlations in timing of sodium channel expression, epilepsy, and sudden death in Dravet syndrome. Channels (Austin) 2013, 7:468–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheah CS, Yu FH, Westenbroek RE, Kalume FK, Oakley JC, Potter GB, Rubenstein JL, Catterall WA: Specific deletion of NaV1.1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A 2012, 109:14646–14651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ogiwara I, Iwasato T, Miyamoto H, Iwata R, Yamagata T, Mazaki E, Yanagawa Y, Tamamaki N, Hensch TK, Itohara S, et al. : Nav1.1 haploinsufficiency in excitatory neurons ameliorates seizure-associated sudden death in a mouse model of Dravet syndrome. Hum Mol Genet 2013, 22:4784–4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kalume F, Yu FH, Westenbroek RE, Scheuer T, Catterall WA: Reduced sodium current in Purkinje neurons from NaV1.1 mutant mice: implications for ataxia in severe myoclonic epilepsy in infancy. J Neurosci 2007, 27:11065–11074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nolan KJ, Camfield CS, Camfield PR: Coping with Dravet syndrome: parental experiences with a catastrophic epilepsy. Dev Med Child Neurol 2006, 48:761–765. [DOI] [PubMed] [Google Scholar]
- 19.Nolan K, Camfield CS, Camfield PR: Coping with a child with Dravet syndrome: insights from families. J Child Neurol 2008, 23:690–694. [DOI] [PubMed] [Google Scholar]
- 20.Han S, Yu FH, Schwartz MD, Linton JD, Bosma MM, Hurley JB, Catterall WA, de la Iglesia HO: NaV1.1 channels are critical for intercellular communication in the suprachiasmatic nucleus and for normal circadian rhythms. Proc Natl Acad Sci U S A 2012, 109:E368–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kalume F, Oakley JC, Westenbroek RE, Gile J, de la Iglesia HO, Scheuer T, Catterall WA: Sleep impairment and reduced interneuron excitability in a mouse model of Dravet Syndrome. NeurobiolDis 2015, 77:141–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuentealba P, Steriade M: The reticular nucleus revisited: intrinsic and network properties of a thalamic pacemaker. Prog Neurobiol 2005, 75:125–141. [DOI] [PubMed] [Google Scholar]
- 23.Genton P, Velizarova R, Dravet C: Dravet syndrome: the long-term outcome. Epilepsia 2011, 52 Suppl 2:44–49. [DOI] [PubMed] [Google Scholar]
- 24.Besag F, Gobbi G, Aldenkamp A, Caplan R, Dunn DW, Sillanpaa M: Psychiatric and Behavioural Disorders in Children with Epilepsy (ILAE Task Force Report): Behavioural and psychiatric disorders associated with childhood epilepsy syndromes. Epileptic Disord 2016. [DOI] [PubMed] [Google Scholar]
- 25.Li BM, Liu XR, Yi YH, Deng YH, Su T, Zou X, Liao WP: Autism in Dravet syndrome: prevalence, features, and relationship to the clinical characteristics of epilepsy and mental retardation. Epilepsy Behav 2011, 21:291–295. [DOI] [PubMed] [Google Scholar]
- 26.Han S, Tai C, Westenbroek RE, Yu FH, Cheah CS, Potter GB, Rubenstein JL, Scheuer T, de la Iglesia HO, Catterall WA: Autistic-like behaviour in Scn1a+/− mice and rescue by enhanced GABA-mediated neurotransmission. Nature 2012, 489:385–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito S, Ogiwara I, Yamada K, Miyamoto H, Hensch TK, Osawa M, Yamakawa K: Mouse with NaV1.1 haploinsufficiency, a model for Dravet syndrome, exhibits lowered sociability and learning impairment. Neurobiol Dis 2013, 49:29–40. [DOI] [PubMed] [Google Scholar]
- 28.Rudy B, Fishell G, Lee S, Hjerling-Leffler J: Three groups of interneurons account for nearly 100% of neocortical GABAergic neurons. Dev Neurobiol 2011, 71:45–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams AD, Cheah CS, Catterall WA, Oakley JC: Diminished excitability of 5-HT3aR-expressing GABAergic interneurons but no pro-epileptic effects caused by selective deletion of NaV1.1 channels in a mouse model of Dravet Syndrome Soc. Neurosci. Abst 2017, In press. [Google Scholar]
- 30.Tai C, Abe Y, Westenbroek RE, Scheuer T, Catterall WA: Impaired excitability of somatostatin- and parvalbumin-expressing cortical interneurons in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A 2014, 111:E3139–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Silberberg G, Markram H: Disynaptic inhibition between neocortical pyramidal cells mediated by Martinotti cells. Neuron 2007, 53:735–746. [DOI] [PubMed] [Google Scholar]
- *32.Rubinstein M, Han S, Tai C, Westenbroek RE, Hunker A, Scheuer T, Catterall WA: Dissecting the phenotypes of Dravet syndrome by gene deletion. Brain 2015, 138:2219–2233.This article shows that mutation of NaV1.1 channels in specific classes of interneurons causes different aspects Dravet Syndrome. Deletion in PV interneurons causes pro-epileptic effects and autistic-like behaviors, deletion in SST interneurons causes milder pro-epileptic effects and hyperactivity, and deletion in both of these classes of interneurons causes synergistic effects on epilepsy and cognitive impairment.
- 33.Catterall WA: Sodium channels, inherited epilepsy, and antiepileptic drugs. Annu Rev Pharmacol Toxicol 2014, 54:317–338. [DOI] [PubMed] [Google Scholar]
- 34.Mistry AM, Thompson CH, Miller AR, Vanoye CG, George AL Jr., Kearney JA: Strain- and age-dependent hippocampal neuron sodium currents correlate with epilepsy severity in Dravet syndrome mice. Neurobiol Dis 2014, 65:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rubinstein M, Westenbroek RE, Yu FH, Jones CJ, Scheuer T, Catterall WA: Genetic background modulates impaired excitability of inhibitory neurons in a mouse model of Dravet syndrome.NeurobiolDis 2015, 73:106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Miller AR, Hawkins NA, McCollom CE, Kearney JA: Mapping genetic modifiers of survival in a mouse model of Dravet syndrome. Genes Brain Behav 2014, 13:163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *37.Hawkins NA, Zachwieja NJ, Miller AR, Anderson LL, Kearney JA: Fine mapping of a Dravet Syndrome modifier locus on mouse chromosome 5 and candidate gene analysis by RNA-Seq. PLoS Genet 2016, 12:e1006398.This article presents the first fine mapping of chromosomal regions that cause genetic background effects in Dravet Syndrome and identifies genes encoding subunits of GABAA receptors on chromosome 5 as prime candidates for modifier genes.
- 38.Striano P, Coppola A, Pezzella M, Ciampa C, Specchio N, Ragona F, Mancardi MM, Gennaro E, Beccaria F, Capovilla G, et al. : An open-label trial of levetiracetam in severe myoclonic epilepsy of infancy. Neurology 2007, 69:250–254. [DOI] [PubMed] [Google Scholar]
- 39.Guerrini R, Dravet C, Genton P, Belmonte A, Kaminska A, Dulac O: Lamotrigine and seizure aggravation in severe myoclonic epilepsy. Epilepsia 1998, 39:508–512. [DOI] [PubMed] [Google Scholar]
- 40.Hawkins NA, Anderson LL, Gertler TS, Laux L, George AL Jr., Kearney JA: Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Ann Clin TranslNeurol 2017, 4:326–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *41.Anderson LL, Hawkins NA, Thompson CH, Kearney JA, George AL Jr.: Unexpected efficacy of a novel sodium channel modulator in Dravet Syndrome. Sci Rep 2017, 7:1682.This article revealed unexpected efficacy of an atypical sodium channel inhibitor in a mouse genetic model of Dravet Syndrome. In contrast to traditional sodium channel inhibitors, GS967 was effective in reducing seizure frequency and severity and in reducing premature death.
- 42.Meldrum BS, Chapman AG: Basic mechanisms of gabitril (tiagabine) and future potential developments. Epilepsia 1999, 40 Suppl 9:S2–6. [DOI] [PubMed] [Google Scholar]
- 43.Bauer J, Cooper-Mahkorn D: Tiagabine: efficacy and safety in partial seizures - current status. Neuropsychiatr Dis Treat 2008, 4:731–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rudolph U, Mohler H: Analysis of GABAA receptor function and dissection of the pharmacology of benzodiazepines and general anesthetics through mouse genetics. Annu Rev Pharmacol Toxicol 2004, 44:475–498. [DOI] [PubMed] [Google Scholar]
- 45.Oakley JC, Cho AR, Cheah CS, Scheuer T, Catterall WA: Synergistic GABA-enhancing therapy against seizures in a mouse model of Dravet syndrome. J Pharmacol Exp Ther 2013, 345:215–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Porter BE, Jacobson C: Report of a parent survey of cannabidiol-enriched cannabis use in pediatric treatment-resistant epilepsy. Epilepsy Behav 2013, 29:574–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *47.Kaplan JS, Stella N, Catterall WA, Westenbroek RE: Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc Natl Acad Sci U S A 2017.This article showed that cannabidiol, the most abundant nonpsychotropic component of cannabis extracts, reduces the frequency, severity, and duration of seizures and improves social interaction behaviors in a mouse genetic model of Dravet Syndrome. These effects are prevented by an inhibitor of the lipid-activated G protein-coupled receptor GPR55, implicating this regulatory pathway in the beneficial effects of cannabidiol.
- *48.Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, Scheffer IE, Thiele EA, Wright S, Cannabidiol in Dravet Syndrome Study G: Trial of cannabidiol for drug-resistant seizures in the Dravet Syndrome. N Engl J Med 2017, 376:2011–2020.This article presents the first results from a placebo-controlled clinical trial of the effecacy of cannabidiol in Dravet Syndrome. Highly significant reduction in the frequency of seizures in Dravet Syndrome patients was achieved with cannabidiol treatment.