

Abstract
The rational design of anti-cancer agents includes a new approach based on ruthenium complexes that can act as nitric oxide (NO) donor agents against specific cellular targets. One of the most studied classes of those compounds is based on bis(bipyridine) ruthenium fragment and its derivative species. In this work, we present the chemical and cytotoxicity properties against the liver hepatocellular carcinoma cell line HepG2 of cis-[RuII(NO+)Cl(dcbpy)2]2− conjugated to a polyclonal antibody IgG (anti-VDAC) recognizing a cell surface marker. UV–visible bands of the ruthenium complex were assigned with the aid of density functional theory, which also allowed estimation of the structures that explain the biological effects of the ruthenium complex–IgG conjugate. The interaction of cis-[RuII(NO+)Cl(dcbpy)2]3− with mitochondria was evaluated due to the potential of these organelles as anti-cancer targets, and considering they interact with the anti-VDAC antibody. The cytotoxicity of cis-[RuII(NO+)Cl(dcbpy)2]3−-anti-VDAC antibody was up to 80% greater in comparison to the free cis-[RuII(NO+)Cl(dcbpy)2]3− complex. We suggest that this effect is due to site-specific interaction of the complex followed by NO release.
Keywords: Nitrosyl ruthenium complexes, Conjugated ruthenium-antibody complex, Nitric oxide delivery agent
Introduction
In the last decade, there has been growing interest in the physiology of nitric oxide (NO) which has led to the development of a variety of new NO donors [1–7]. Metal-based compounds are one of the most promising classes of NO donor agents. Among these compounds, nitrosyl ruthenium complexes are the most studied and best understood species [4, 5, 8–21].
In general, these complexes have the structure {Ru–NO}3+, and they can act as NO delivery agents via (a) a reduction process [12, 15–17], (b) via direct light irradiation on the metal–ligand charge transfer band dπ(RuII)–π*(NO+) [16, 17], or (c) by photoinduced electron transfer [20, 21]. Depending on the pH, {Ru–NO}3+ can be fully converted to {Ru–NO2}+, which could act as an NO delivery agent either by photolysis or by an oxygen transfer reaction, as described in the literature [16, 22]. The dπ(RuII)–π*(NO+) backbonding strongly influences the formation of the nitro compound [22].
For example, in the case of the complex cis-[Ru(NO2) (bpy)2L]n+/0 (L = Cl−, pyridine, isonicotinamide, 4-picoline, or 4-acetylpyridine), and where bpy is bipyridine (a strong π-acceptor ligand), cis-[RuII(NO+)(bpy)2L]n+1 is formed when the complex is dissolved in aqueous solution at pH ≤ 4.0 [5]. This may pave the way for the application of this kind of system in bioassays, which are usually conducted in physiological pH. Regarding in vitro and in vivo assays, NO delivery agents based on ruthenium complexes have been used for a number of biological purposes [4] including experiments on vasorelaxation [22], Chagas disease [23–25], and cytotoxicity [12–15]. Maybe the most appealing feature is the possibility to control the amount of NO release based on the uptake of ruthenium species. For example, in cancer cells, NO has been suggested to modulate several different events [26, 27]. It seems to show dual functionality: NO can be anti- or pro-tumorigenic depending on its local concentration [26]. NO levels between 50 and 100 nM activate signal transduction pathways that elicit tumorigenic effects; above this level, NO exerts antitumoral functions [26]. The key to maximize the efficacy and/or reduce the side effects associated with NO, is better targeted delivery of these agents to tumor sites. Several strategies have been used in this way mainly focus in drug delivery system [28–34]. Although targeting NO delivery agents can promote an increase in cellular uptake, it may reduce overall cytotoxicity since certain threshold levels of NO over large areas may be required for activation of signaling pathways. Effective targeted NO delivery agents based on ruthenium complexes may improve local NO concentration, which could increase the cytotoxic effect of the nitric oxide derivative ruthenium complex. Compounds containing ruthenium(II) and some unsaturated ligands “L” show good stability mainly due to existing backbonding dπ(RuII)–π*(L), and have been used as models in kinetic and photochemical studies [35, 36]. One class of compounds, for example, [RuII(bpy)2L(NO+)]n+ has been studied due to its thermal stability, and also its ability to release NO under external stimulation [4, 9, 10, 13, 14, 37–40]. This suggests these compounds might find applications as NO delivery agents and possible anti-cancer agents. One possible target within cancer cells is mitochondria. In addition to providing cellular energy, these organelles play an important role in cell signaling events, including those involving cell death [41]. The voltage-dependent anion channel (VDAC) is a protein in the mitochondrial outer membrane, controlling cross-talk between mitochondria and the remainder of cell. VDAC plays a central role in mitochondria-mediated apoptosis by means of reactive oxygen species (ROS) generation, and it should be noted that NO can increase ROS levels [41]. Therefore, VDAC is a potential target for NO donor agents based on ruthenium complexes.
To achieve this goal, one approach to improve the molecular specificity of the ruthenium complex is by attaching it to a ligand that recognizes a surface marker that is preferentially expressed in cancer cells. Such strategy has been used for some ruthenium complexes [42, 43]. Hence, this work aimed to evaluate the cytotoxic effect of polyclonal antibody IgG, anti-VDAC conjugated to the nitrosyl ruthenium complex cis-[Ru(NO)Cl(dcbpy)2]3− where dcbpy is 2,2′-bipyridine-4,4′-dicarboxylic acid as a NO delivery agent against the hepatocellular carcinoma cell line (HepG2).
Materials and methods
Chemical and reagents
RuCl3·nH2O, sodium nitrite, triethanolamine, N,N′-dicyclohexylcarbodiimide (DCC), and N-hydroxysuccinimide (NHS) were purchased from Sigma-Aldrich Chemicals (Brazil) as high purity reagents. VDAC 1/2/3, FL-238, and rabbit polyclonal IgG (200 μg mL−1) were acquired from Santa Cruz Biotechnology (Santa Cruz, USA). Doubly distilled water was used in all experiments. Nitric oxide was prepared by the reaction of nitric oxide with copper turnings and was purified according to the literature method [37].
Apparatus
The ultraviolet–visible (UV–Vis) spectra were recorded on an Agilent 8453 spectrophotometer; the samples were placed in a quartz cell with an optical path length of 1 cm. The infrared spectra were acquired on a Prestige 21 Schimadzu FTIR spectrometer; the samples were pressed into KBr pellets.
The pH was measured with a DM 20 pH meter (Digimed). The photochemical experiments were carried out in aqueous solution containing the ruthenium complex at 1.0 × 10−4 M. A 447-nm Diode Laser Colibri (Quantum Tec®) was employed. The electrochemical studies were performed in aqueous solution using an AUTOLAB PGSTAT 30 potentiostat/galvanostat. A conventional three-electrode cell arrangement consisting of a glassy carbon disk working electrode, a platinum wire auxiliary electrode, and an Ag/AgCl reference electrode was employed. The supporting electrolyte was KCl at 0.1 mol L−1.
NO was measured on a Dual Channel Nitric Oxide Measuring system inNO-T equipped with a selective amperometric sensor amiNO-600 from Innovative Instruments, Inc. Mass spectrometry was accomplished on an ULTROTOFQESI-TOF Mass Spectrometer (Bruker Daltonics, Billerica, USA) operating in the positive mode.
Computational methodology
Density functional theory (DFT) computations were employed to fully optimize the ground state geometries of all the species described in this work. The calculations were done with Gaussian 09 using Becke’s three-parameter hybrid functional with the correlation functional of Lee, Yang and Parr formalized as the B3LYP hybrid functional [44–47] and the effective core potential basis set LanL2DZ [48–51], which proved to be suitable for geometry predictions in coordination compounds containing metals of the second row of the transition elements in the periodic table. Different degrees of protonation of the carboxylate groups in the dcbpy ligand were explored. The non-equivalency of the –CO2− groups of the molecule renders four different possibilities for monoprotonation and six possible combinations for diprotonation. In addition, the carboxylato groups can be engaged in solute–solvent hydrogen-bonding specific interactions, which have to be taken into account to avoid artifacts in the computations arising from the excess of negative charge on the CO2− substituents. For this purpose, four explicit water molecules were incorporated in the geometry optimizations and all subsequent computations. All the different cases of mono- and diprotonated structures plus the fully deprotonated molecule were fully optimized in vacuo. Tight SCF convergence criteria and default setting were used along the geometry optimizations. In all cases, vibrational analyses prove that the stationary points reached along the optimizations correspond to local minima in the potential energy surface. The computations suggest that whereas the monoprotonated stage is dominated by one single species, several isomers might contribute in significant proportions to the diprotonated species. The analysis of the electronic structure was complemented with (TD)DFT computations including at least 90 states of the same multiplicity than the ground state. The spectra were computed at the gas phase geometry. Apart from the explicit water molecules solvation effects in aqueous solution were incorporated in the computations employing the PCM approximation, as implemented in Gaussian 09.
Synthesis of cis-[Ru(NO)Cl(dcbpy-H)2]
The complex cis-[Ru(NO)Cl(dcbpy-H)2] (Ru-DCBPY) was synthesized according to a previously similar published procedure [21]. The complex cis-[RuCl2(dcbpy-H2)2] was the starting material; it was prepared in the same way as cis-[RuCl2(bpy)2]·2H2O, described in [38]. Briefly, cis-[RuCl2(dcbpy-H2)2]·2H2O (0.100 g, 0.14 mmol) was dissolved in 25 mL of milli-Q H2O and degassed with argon for 30 min. Gaseous NO was then generated by reaction between nitric acid solution (50% V/V) and metallic copper. The produced gaseous mixture was passed through a concentrated NaOH solution to eliminate NO2 traces. The nitric oxide was then bubbled through the cis-[RuCl2(dcbpy-H2)2]·2H2O complex solution for 40 min to give a reddish-brown solution. This procedure was repeated three times for 24 h under constant stirring. The resulting brown precipitate was collected by filtration, washed with 30 mL of diethyl ether, and stored under vacuum in the dark. Yield: 59% (Anal. calc. for C24H22N5O13ClRu: C, 39.76; H, 3.06; N, 9.66. Found: C, 39.31; H, 2.96; N, 9.51%). m/z = 652.95 ({[Ru(NO)Cl(dcbpy-H)2]}+).
Isolation of rat liver mitochondria
Mitochondria were isolated by standard differential centrifugation [52]. Male Wistar rats weighing approximately 200 g were killed by decapitation, according to research protocols approved by CEUA-USP, Rib. Preto. Livers (10–15 g) were immediately removed, sliced in medium (50 mL) consisting of 250 mM sucrose, 1 mM ethyleneglycolbis(β-aminoethyl)-N,N,N′,N′-tetraacetic acid (EGTA), and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)–KOH, pH 7.2, and homogenized three times for 15 s, at 1-min intervals, with a Potter–Elvehjem homogenizer. Homogenate was centrifuged (580g, 5 min), and the resulting supernatant was centrifuged further (10,300g, 10 min). Pellets were then suspended in medium (10 mL) consisting of 250 mmol L−1 sucrose, 0.3 mM EGTA, and 10 mM HEPES–KOH, pH 7.2, and centrifuged (3400g, 15 min). The final mitochondrial pellet was suspended in medium (1 mL) consisting of 250 mM sucrose and 10 mM HEPES–KOH, pH 7.2, and used within 3 h. Mitochondrial protein content was determined by the Biuret reaction. For the assays, mitochondria (0.5 or 1 mg protein mL−1) were energized with 5 mM potassium succinate (plus 2.5 μM rotenone) in a standard medium consisting of 125 mM sucrose, 65 mM KCl, and 10 mM HEPES–KOH, pH 7.4, at 30 °C.
Direct detection of NO release from complex (Ru-DCBPY)
NO release from complex (Ru-DCBPY) (100 μM) after incubation with succinate-energized mitochondria (1 mg protein mL−1) in standard medium was assessed with a Selective Sensor Electrode (AmiNO 700) in the dark.
Mitochondrial assays
Mitochondrial oxygen consumption was monitored in the standard medium, in an oxygraph equipped with a Clark-type oxygen electrode (Hansatech instruments, oxytherm electrode unit, UK). Mitochondrial membrane potential was estimated spectrofluorimetrically; 10 μM of cationic dye safranine O was used as probe and fluorescence was measured at 495/586 nm excitation/emission wavelength pair [53]. ROS/RNS levels were evaluated by monitoring oxidation of dichlorodihydrofluorescein diacetate (H2DCFDA) (Molecular Probes, Eugene) at the 503/528 nm excitation/emission wavelength pair [54]. The fluorimetric experiments were accomplished on a Hitachi Model F-4500 spectrofluorimeter (Tokyo, Japan). Mitochondrial swelling was estimated from the decrease in the apparent absorbance at 540 nm [55].
Synthesis of cis-[RuCl(dcbpy)2(NO2)]4−-anti-VDAC
The porin antibody anti-VDAC was bound to the (Ru-DCBPY) forming cis-[RuCl (dcbpy)2(NO2)]4−-anti-VDAC via amide bond formation using a procedure similar to the procedure described by Terpetsching et al. [56]. In this protocol, DCC (0.230 g, 1.11 mM) and NHS (0.119 g, 1.030 mM) were dissolved in dimethylformamide (3.0 mL), under stirring, and cooled in an ice bath. Next, (Ru-DCBPY) (0.200 g, 2.53 mM) was added, and the mixture was stirred for 2 h. The precipitate was removed by filtration through a syringe filter (Millipore-Millex 0.22 μm). The filtrate (500 μL) containing the active Ru complex was added to an IgG solution in 0.2 M carbonate buffer pH 8.3 (1.0 mL, 1% IgG) and stirred for 12 h. Purification of the labeled protein was performed by gel filtration chromatography on Sephadex G-25 coupled to a peristaltic pump (GILSON, miniplus 3) at 1 mL min−1 flow; 0.1 M phosphate buffer pH 7.2 was used. The fractions were collected in a Fraction Collector LKB, BROMA.
Antibody detection (western blotting) and (UV–Vis) assessment of fractions
IgG (anti-VDAC) was determined in (aliquot) fractions by western blotting. The mitochondria harvested from aliquots obtained from the Sephadex column were re-suspended in CelLytic™M (Sigma, Brazil) lysis buffer for 2 min, on ice, and centrifuged at 10,000g for 5 min. The resulting supernatants were subjected to 12% SDS-PAGE, transferred to a nitrocellulose membrane, and probed with antibodies against cytochrome c (BD Pharmigen, purified mouse anticytochrome c) followed by horseradish peroxidase-coupled detection.
HepG2 cell culture
The HepG2 cells were obtained from the American Type Culture Collection. The cell line was cultured in Dulbecco’s medium supplemented with 10% fetal bovine fetal serum, 100 IU mL−1 penicillin G, 100 mg mL−1 streptomycin, and 1 μg mL−1 amphotericin. The cells were plated in 6, 12, 24 and 96-well plates and cultured until 75–90% confluence was achieved (depending on the assay) in a humidified incubator at 37.0 °C with 5.0% CO2.
MTT cell viability assay
The viability of the HepG2 cells was assessed by means of the [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide] (MTT) colorimetric assay. The cells were plated on 96-well microtiter cell culture plates at 2 × 104 cells/well for 24 h. After the indicated treatment, the cells were incubated at 37.0 °C for 3 h in a culture medium containing 10 mol L−1 MTT in phosphate-buffered saline (PBS). The blue MTT formazan precipitate was then dissolved in 50 L of DMSO, and the absorbance was measured at 570 nm with a multi-well plate reader [57]. The cell viability was expressed as the percentage of the absorption values in the treated cells relative to the non-treated (control) cells. The data are presented as the means ± the standard error of three independent experiments.
Statistical analysis
Statistical analysis was performed by two-way ANOVA. Equality of variance was assumed with Bonferroni’s post hoc test for pairwise comparisons. Results with P < 0.05 were considered statistically significant.
Results and discussion
NO bubbling through an aqueous cis-[RuCl2(dcbpy-H2)2] solution afforded the nitrosyl ruthenium complex (Ru-DCBPY) in high yield. Elemental analyses of complex (Ru-DCBPY) as well as mass spectrum analysis (Supplementary material: Figure 1) agreed with the proposed molecular formula. Complex (Ru-DCBPY) displayed electronic absorption spectrum with poor defined bands at 323 nm (log ε = 3.97) and 305 nm (log ε = 4.13), which was assigned based on theoretical calculations. Figure 1 provides a schematic representation of the (Ru-DCBPY) complex, with the carboxylato groups labeled to assist in the discussion that follows. Table 1 compiles the most relevant DFT-computed structural parameters (Ru–N and N–O distances, Ru–N–O angle and IR frequencies) for the different cis-[RuII(NO+) Cl(dcbpy-Hx)2]n (n vary of 2+ to −2) species explored in this work.
Fig. 1.
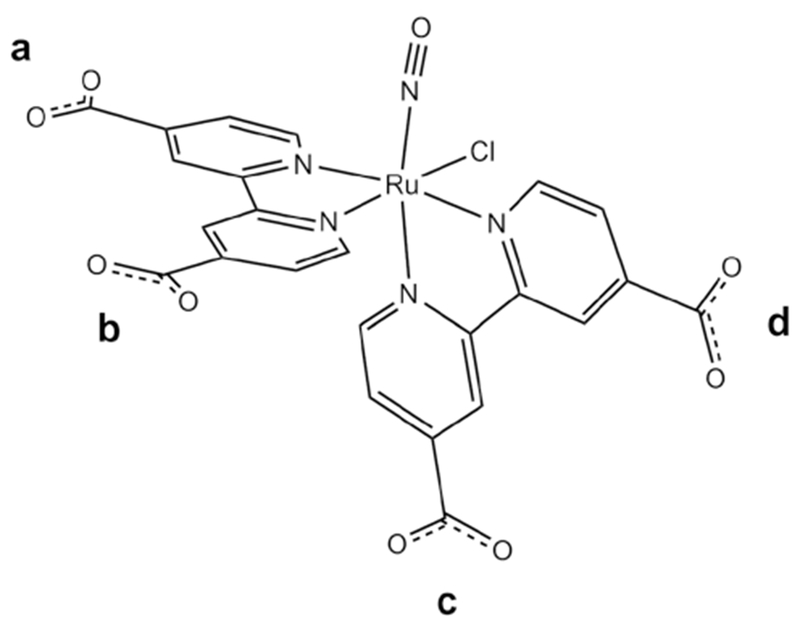
Schematic representation of the molecular structure of cis-[Ru(NO)Cl(dcbpy)2]2−
Table 1.
Structural parameters, IR frequencies and relative energies for the different cis-[Ru(NO)Cl(dcbpy)2Hn]n−2 species computed in this computations. The labels employed to identify the protonation sites correspond to the ones in Fig. 1
| N | Species | Protonation pattern | dRu–N(O) (Å) | dN–O (Å) | Ru–N–O | νNOa | Fractionb |
|---|---|---|---|---|---|---|---|
| Isolated system | |||||||
| 0 | 1 | – | 1.771 | 1.192 | 173.2 | 1808 | 100 |
| 1 | 2 | a | 1.790 | 1.194 | 174.3 | 1790 | 78.8 |
| 3 | b | 1.786 | 1.193 | 175.2 | 1797 | 2.5 | |
| 4 | c | 1.774 | 1.190 | 175.2 | 1811 | 0.8 | |
| 5 | d | 1.783 | 1.193 | 174.0 | 1795 | 18.0 | |
| 2 | 6 | a–b | 1.802 | 1.194 | 173.4 | 1785 | 80.1 |
| 7 | a–c | 1.787 | 1.189 | 175.3 | 1808 | 0.4 | |
| 8 | a–d | 1.794 | 1.192 | 174.1 | 1798 | 1.1 | |
| 9 | b–c | 1.783 | 1.188 | 174.6 | 1815 | 0.0 | |
| 10 | b–d | 1.792 | 1.192 | 175.4 | 1797 | 0.5 | |
| 11 | c–d | 1.790 | 1.192 | 175.0 | 1799 | 17.9 | |
| With explicit water molecules | |||||||
| 0 | 1 | – | 1.771 | 1.189 | 173.2 | 1819 | 100 |
| 1 | 2 | a | 1.780 | 1.189 | 173.4 | 1814 | 54.0 |
| 3 | b | 1.777 | 1.188 | 175.0 | 1820 | 9.4 | |
| 4 | c | 1.768 | 1.187 | 175.1 | 1829 | 4.1 | |
| 5 | d | 1.774 | 1.188 | 173.7 | 1819 | 32.5 | |
| 2 | 6 | a–b | 1.793 | 1.191 | 175.3 | 1801 | 53.0 |
| 7 | a–c | 1.782 | 1.187 | 175.1 | 1819 | 4.5 | |
| 8 | a–d | 1.786 | 1.189 | 174.1 | 1812 | 8.4 | |
| 9 | b–c | 1.779 | 1.186 | 174.6 | 1826 | 1.5 | |
| 10 | b–d | 1.785 | 1.189 | 175.5 | 1813 | 11.0 | |
| 11 | c–d | 1.780 | 1.189 | 175.9 | 1816 | 21.6 | |
Computed for the optimized geometries in vacuo, values in cm−1
Computed on the basis of the zero-point corrected energies arising from the PCM computations, neglecting entropic contributions
The metric parameters are within the expectations for this kind of compounds, with slight differences depending on the presence or absence of explicit water molecules along the optimization procedure. In all cases, the N–O bond length at ca. 1.19 Å and the almost linear Ru–N–O moiety is consistent with a predominant RuII–NO+ character. The computations predict NO stretching frequencies (νNO) be insensitive to the protonation degree of the complex. Although the trans NO ligand has offered effect on the nitrosyl stretching frequency [58, 59], it seems to be not the case for nitrosyl bipyridine ruthenium complexes, in which a net effect for the σ-donor and π-acceptor character of different forms of carboxybipyridine ligand does not show significant change on the νNO stretching frequency. Bipyridine nitrosyl ruthenium(II) complexes present νNO stretching bands in the 1800–2000 cm−1. The peak intensity varies from medium to strong [4, 5, 10, 12–16, 20, 23, 37–40, 58–60]. In the FTIR spectrum of (Ru-DCBPY) complex, νNO was found in 1945 cm−1 indicating that the complex is of {RuII-NO+}6 type. In addition, the spectrum exhibits peaks at 1610, 1560 and 1460 cm−1, which is characteristic of the pyridine rings. The carboxylate group is evidenced by a peak at 1690 cm−1. The calculated νNO stretching frequencies for (Ru-DCBPY) in this work were 1800 cm−1, which are lower than the experimental ones. It is in part due to a tendency of DFT methods with standard functionals to provide too delocalized molecular and electronic structures due to inherent self-interaction errors [61–63]. However, the inclusion of explicit water molecules at hydrogen-bonding distance of the carboxylato groups brings the νNO values closer to the experimental ones. In the absence of these specific interactions, the high charge located on the –CO2− groups leads to several anomalies, not only in the stretching frequencies but also in the electronic structure picture and the predicted electronic spectroscopy. Even when isolated molecules have been included in the table for completeness, the following discussion will involve only hydrogen-bonded complexes.
There is no easy way to obtain by DFT reliable acidity constant values, which could assist in the determination of the degree of protonation in solution. The literature related to other species holding the {Ru(dcbpy)2}2+ core suggests that, at the pH employed in our experiments, the nitrosyl species is probably best described as holding an average of two protonated carboxylato sites. Simple enthalpic arguments allow establishing that three out of the six possible diprotonated species present in solution (labeled 6–11 in Table 1) should predominate in acidic aqueous solution, with still non-negligible amounts of the remaining ones. The electronic structure and the (TD)DFT predicted spectra for all these species are in fact very similar, because the Ru–NO interaction dominates the orbital ordering. Figure 2 displays a MO diagram highlighting some relevant orbitals computed for isomer 6, expected to be the most abundant in solution.
Fig. 2.
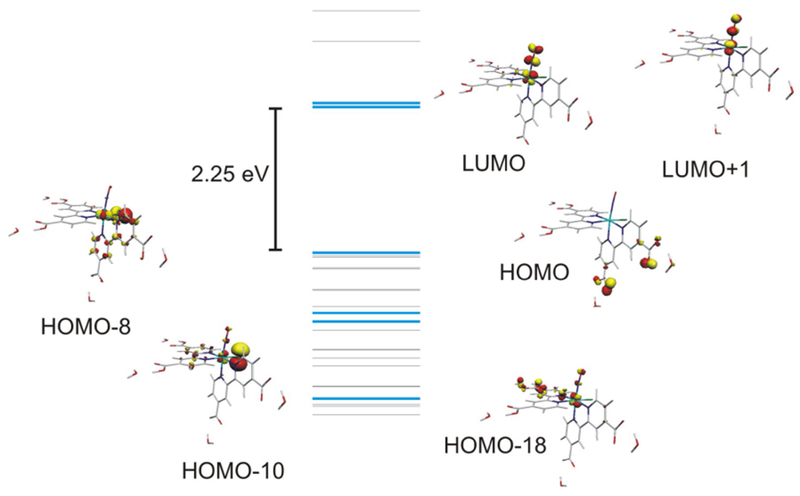
Molecular orbital diagram for species number 6
The antibonding LUMO and LUMO + 1 orbitals have 22 and 25% contribution of metal-centered atomic orbitals, reflecting extensive mixing between the Ru dπ and the NO π* orbitals. This composition is comparable to other {RuNO}6 species and is consistent with a well-preserved nitrosonium character of the coordinated NO fragment. The high covalency of the Ru–NO bond results in a noticeable stabilization of the dπ orbitals, which leads to a HOMO that is mostly centered on the non-protonated carboxylato groups of the dcbp ligand. The occupied MOs immediately below the HOMO are also dcbpy, dcpyH2 or Cl− centered and show little contribution from the Ru fragment. The HOMO-8 is the first MO with measurable dπ Ru-centered contribution, and it is necessary to go as deep as HOMO-10 and HOMO-18 to find the bonding MOs that result from the interaction between the dπ set and the nitrosyl ligand. This electronic picture results in an electronic spectrum, which is shifted to the high energy end of the visible region of the spectrum. In aqueous acidic medium, the Ru-DCBPY was found to be light yellow in color exhibiting band at 305 nm. The spectrum feature was similar to the data reported in the literature for nitrosyl ruthenium bipyridine [5, 14, 17, 21, 22, 37]. Figure 3 compares the experimental and computed electronic spectra.
Fig. 3.
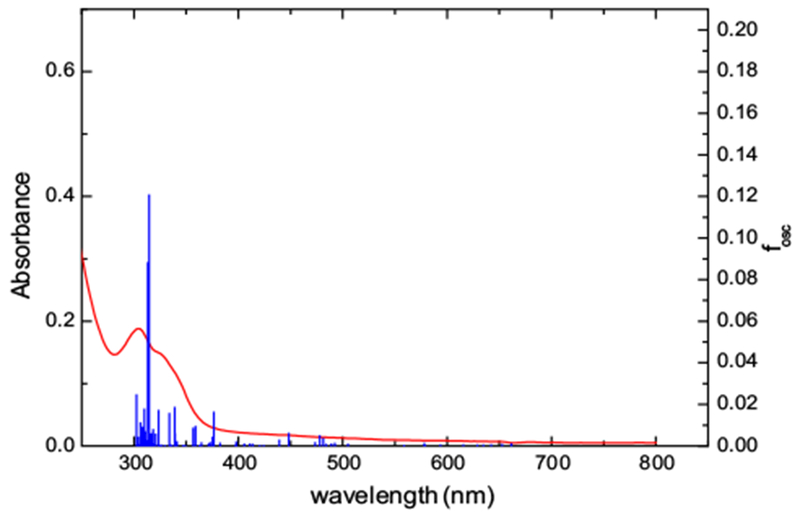
Experimental and (TD)DFT-computed spectra for [Ru(NO) Cl(dcbpy-H)2]. The computed spectrum corresponds to species number 7
The individual six possible diprotonated species spectra show very subtle variations. Nevertheless, the differences in the absorption maxima for the six cases are within the error for the computational methodology, allowing to focus the analysis on one particular example without loss of generality. For the species number 6 (TD)DFT predicts the most intense absorption in the 250–800 nm to be centered at 315 nm. This transition, which is mostly a ligand-to-ligand charge transfer transition between dcbpyH2 and NO+ centered MO’s, accounts for the experimental absorption maximum at 305 nm. The shoulder at lower energies originates in a series of closely spaced low intensity absorptions of similar origin involving any of the three dcbpy, dcpyH2 or Cl− co-ligands. These transitions overlap with the usual π–π* absorptions due to the bipyridinic moieties. Finally, the experimental weak absorption band in the 450–500 nm region seems to involve transitions between orbitals in the range HOMO-7 to HOMO-14 and the NO-centered LUMO and LUMO + 1 orbitals and have mixed ligand to ligand and metal to ligand charge transfer character, revealing the existence of low lying excited states with Ru(III)–NO(0) character.
In physiological pH, Ru-DCBPY was slowly converted to cis-[RuCl(dcbpy)2(NO2)]4− by nucleophilic attack of hydroxide ion (Scheme 2); kobs was 5.35 × 10−4 ± 2.47 × 10−5 s−1 at 25.0 ± 0.1 °C (Supplementary Material: Figure 2). In pH ≤ 2.0, a precipitate starts to form, probably due protonation of all carboxylate substituent. In that condition, we are not being able to measure the pKa of bonded 4,4′-dicarboxilic acid. As judged by published pKa for some cis-[Ru(dcbpy-H2)2L2]n+ [64], it is reasonable to assume three equilibriums depending on pH for cis-[Ru(NO)Cl(dcbpy-H2)2]2+ complex (Scheme 1). In physiological pH, the best assumption for the molecular structure of the nitrosyl ruthenium complex discussed in this work is the existence of the equilibrium constant described in Eq. 3.
Scheme 2.
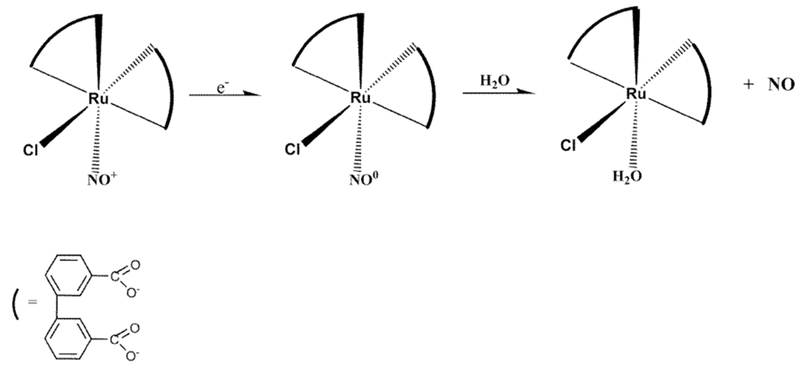
Control reduction potential electrolysis of (Ru-DCBPY) specie at 0.1 V vs. Ag/AgCl in pH 3.0
Scheme 1.
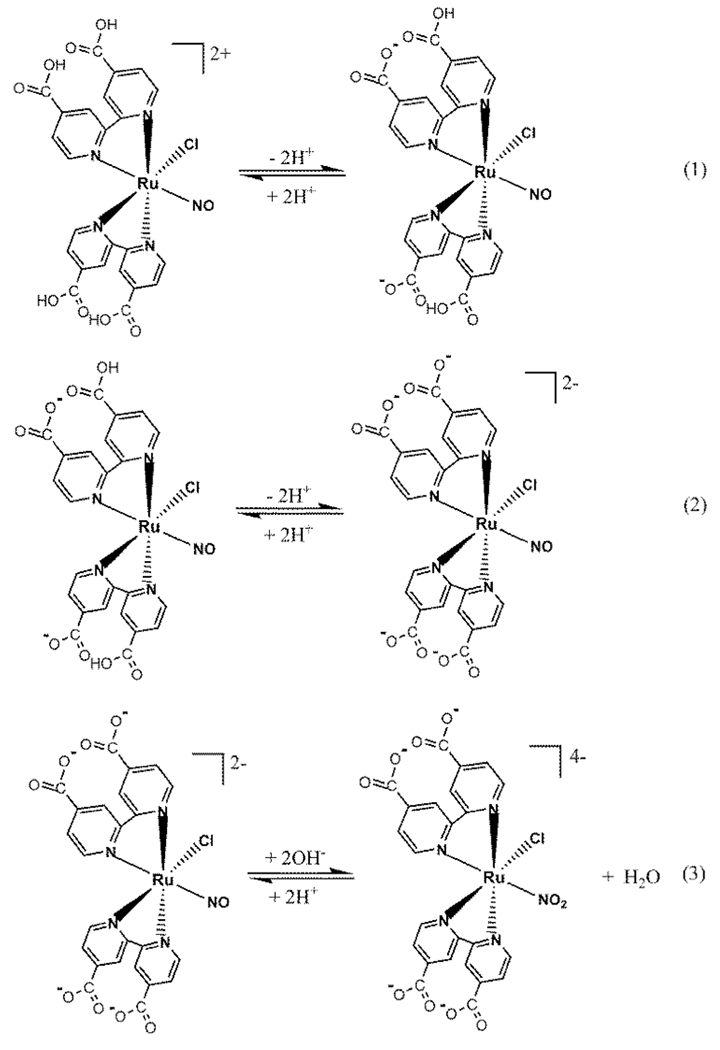
Effect of pH on the chemical composition of cis-[Ru(NO)Cl(dcbpy-H2)2]2+ complex
The electrochemistry of nitrosyl ruthenium complexes has been extensively studied in both aqueous and nonaqueous media [37, 39, 65, 66]. In general, these complexes undergo two one-electron processes between − 1.0 and 1.0 V vs. Ag/AgCl centered on the nitrogen oxide derivative ligand [58].
Cyclic voltammogram (CV) measurements of (Ru-DCBPY) in aqueous solution KCl 0.1 and 0.01 M acetate buffer solution was made separately in two different scan windows to isolate redox events of interest. The first redox couple has E1/2 = 0.14 V (vs. Ag/AgCl) and is attributed to the redox process centered on the nitrosyl group (NO+/0) when the CV was performed in a potential window of + 0.4 to − 0.2 V for the negative scan at the scan rate of 100 mV s−1 at 25 °C. The ratio of the anodic (Epa) and cathodic (Epc) current peak heights decreases in slow scan rate consistent with a coupled chemical reaction. The results are in line with the loss of NO following reduction process of (Ru-DCBPY) as generally observed for nitrosyl ruthenium complexes [4, 5, 10–16, 22, 58, 65, 66]. A second peak attributed as irreversible process was observed at E1/2 = − 0.42 V (vs. Ag/AgCl), due to NO0/−, when the CV was performed in a potential window of + 0.4 to − 0.8 V. In aqueous solution, E1/2 for (Ru-DCBPY) couples shifts 0.02 V to a higher potential in comparison to cis-[Ru(NO) Cl(bpy)2]2+ due different σ-donor, π-acceptor character of co-ligands. It is consistent with the cis-[Ru(NO)L(bpy)2]3+ series where the highest π-acceptor character of L changes the electrochemical process centered on nitrosyl ligand to higher values [67].
Chemical reduction of (Ru-DCBPY) in 0.015 M ascorbic acid produced cis-[RuCl(dcbpy-H)2(H2O)]− as judged by NO-sensor measurements (inset Fig. 4). Conducted spectroelectrochemistry at 0.1 V vs. Ag/AgCl in 0.01 M KCl pH = 3.0 controlled by the addition of HCl solution (Fig. 4) helped us to infer the electro-reduction process of the (Ru-DCBPY) species (Scheme 2).
Fig. 4.
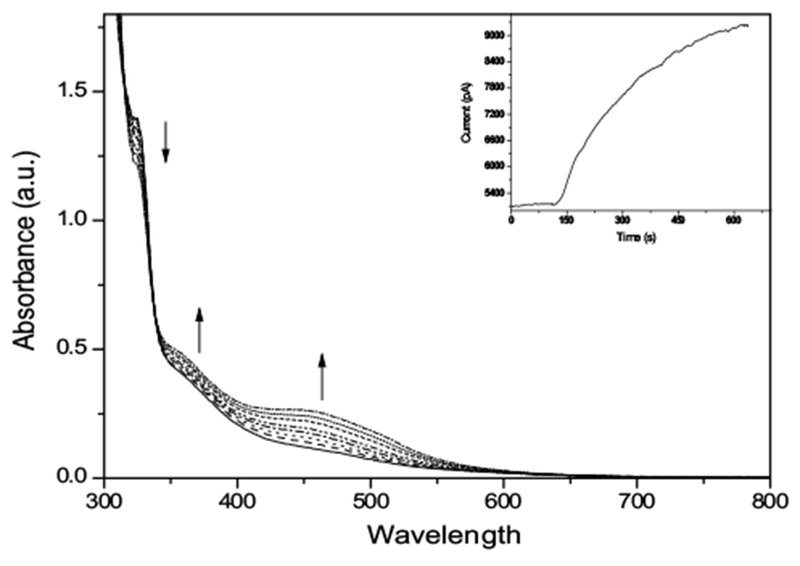
Spectroelectrochemistry measurements of (Ru-DCBPY) (7.4 × 10−5 M) in aqueous 0.01 M KCl solution, pH = 3.0. Inset: Chronoamperometry with NO sensor of aqueous (Ru-DCBPY) (7.4 × 10−5 M) containing ascorbic acid (0.015 M)
The well-established electrochemical mechanism of (Ru-DCBPY) in aqueous solution may help to explain some biological results related to its use as NO delivery agent. Interaction sites for (Ru-DCBPY) in biological medium are cell/organelles membranes, which are also considered potential targets of anti-cancer drugs [16, 29, 41]. Among them, mitochondrial plays a critical role in numerous biochemical processes, particularly oxidative phosphorylation, process in which ATP synthesis is driven by the transfer of electrons from NADH and FADH2 along the respiratory chain in the inner mitochondrial membrane (IMM), to molecular oxygen. This process generates electrochemical proton gradient across the IMM used to drive the ATP synthesis via F1F0-ATP synthase. During this process, mitochondrial reactive oxygen species (ROS) are produced and may elicit the mitochondrial permeability transition (MPT) in association with Ca2+. MPT is a cyclosporine-(CsA)-sensitive well-documented phenomenon mediated by the opening mitochondrial transition pores, which is related with cellular necrosis and cellular apoptosis [68–71]. Mitochondrial parameters, oxidative and bioenergetics provide perspectives for anti-cancer strategies. In this context, we have explored the effects of NO generated from ruthenium nitrosyl (Ru-DCBPY) on mitochondria.
First, we have determined the capability of succinate-energized isolated mitochondria induce NO release from (RU-DCBPY) using a selective electrode. Figure 5, in the gray area, shows time-course of direct determination of NO release suggesting that ruthenium complex underwent one-electron reduction centered on nitrogen oxide derivative ligand site, followed by NO release in the presence of mitochondrial NADH coenzyme. Previously, we demonstrated two similar mechanisms of NO release from ruthenium complexes of this same class, exposed to NADH [12, 16]. NADH would bind directly to metal centers of ruthenium and ruthenium complexes, which suggests applicability of coenzyme.
Fig. 5.
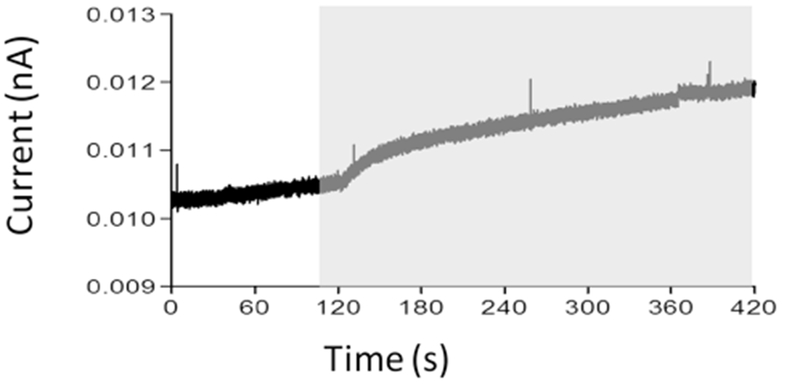
Release of NO from (Ru-DCBPY), evaluated by mitochondria-dependent oxy-reduction systems, detected directly through selective electrode. (Ru-DCBPY) (100 μM) was incubated with mitochondria in standard medium. The plot, representative of three independent experiments, shows NO release. Gray area shows time course after mitochondria addition
Effects of complex (Ru-DCBPY) on mitochondrial reactive oxygen/nitrogen species (ROS/RNS) levels and mitochondrial swelling
The production of ROS/RNS by HepG2 cells after incubation with (Ru-DCBPY) was examined to provide evidence on cell death mechanisms involving mitochondria. Figure 6 shows increased ROS/RNS levels in succinate-energized mitochondria, incubated as indicated, through oxidation of the nonspecific probe H2DFDA [54]. The mitochondrial permeability transition inhibitor cyclosporine A (CsA) partially inhibited the effect of NO released from complex (Ru-DCBPY) at 100 μM (higher concentration).
Fig. 6.
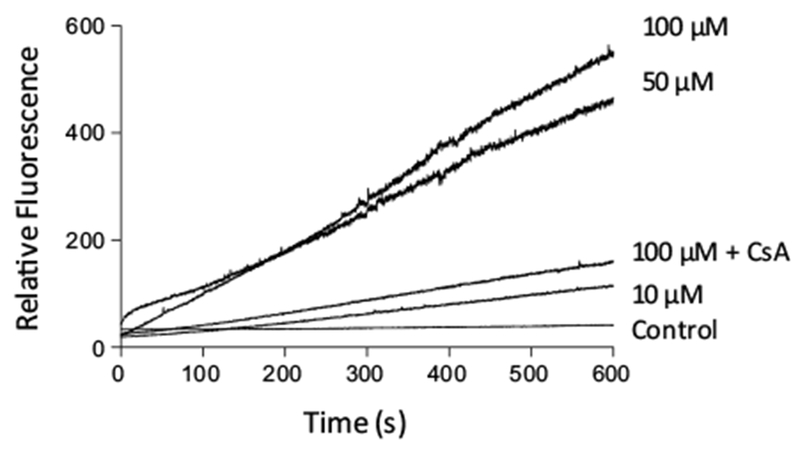
Time course of mitochondrial ROS generation in the presence of NO released from complex (Ru-DCBPY), as assessed with H2DCFDA. Mitochondria (0.5 mg protein mL−1) were incubated in the standard medium described in Fig. 1, at 30 °C, in the presence of 5 μM succinate + 2.5 μM rotenone. Plots are representative of at least three experiments with different mitochondrial preparations
NO released from complex (Ru-DCBPY) induced Ca2+-dependent/CsA-sensitive mitochondrial swelling at 10–100 μM, which attested to NO involvement in the classic mitochondrial permeability transition (MPT) (Fig. 7), a process usually associated with ROS/RNS generation [71–74]. CsA almost completely inhibited swelling induced by complex (Ru-DCBPY) at 50 μM. The inhibition of swelling by 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (cPTIO), a NO scavenger, suggests that NO was the MPT inducer. We propose that in the presence of superoxide anion (O·−2) from mitochondria, NO generates peroxynitrite, which in turn, inhibits respiratory chain and elicits MPT [15, 16].
Fig. 7.

Time course of mitochondrial swelling induced by NO released from complex (Ru-DCBPY). Mitochondria (0.5 mg protein mL−1) were incubated in the standard medium described in Fig. 1, at 30 °C, in the presence of 5 μM succinate + 2.5 μmol L−1 rotenone. Data are representative of at least three experiments with different mitochondrial preparations
Effects of NO released from complex (Ru-DCBPY) on mitochondrial membrane potential and mitochondrial respiration
Respiratory control ratio (RCR: state 3/state 4 respiration rates) indicates the degree of mitochondrial coupling. Table 2 shows that complex (Ru-DCBPY) at 10 and 50 μM decreased RCR values, whilst at 100 μM it increased the values. High NO concentrations inhibited respiration probably because reaction between NO and superoxide generated peroxynitrite, an irreversible respiratory chain inhibitor [72–75]. In agreement with the RCR results, complex (Ru-DCBPY) decreased the mitochondrial membrane potential in a dose–response manner (10–100 μM) (Fig. 8). Mitochondria efficiently induced NO release from complex (Ru-DCBPY) in a controlled way, via an oxidation–reduction mechanism. We propose that, once released, NO inhibited respiratory chain and raised ROS levels inducing MPT; ROS levels could be even increased by MPT. According to Brown and Borutaite, NO-induced cell necrosis occurs via respiratory chain inhibition at site I and/or IV and via RNS- or oxidant-induced signal transduction. DNA damage may activate mitochondrial pathway to apoptosis. RNS-induced MPT may induce both apoptosis and necrosis. Various mechanisms by which NO acts to kill different types of cancer, such as breast, cervical, gastric, colorectal, and head and neck, have been addressed. However, local NO concentration is the main factor involved.
Table 2.
Effects of NO released from complex (Ru-DCBPY) on respiratory parameters of succinate-energized isolated rat liver mitochondria (1 mg protein mL−1) in standard medium
| Mitochondrial parameters | dcbpy (μM) |
|||
|---|---|---|---|---|
| Control | 10 | 50 | 100 | |
| V3 (ng of O atoms min−1) | 231.5 ± 6.3 | 197.0 ± 11.3 | 202.2 ± 1.4 | 123.6 ± 7.7 |
| V4 (ng of O atoms min−1) | 31.3 ± 2.2 | 34.5 ± 2.1 | 48.3 ± 8.1 | 120. ± 8.3* |
| VCCCP | 245.1 ± 7.4 | 189.1 ± 5.3* | 250.4 ± 8.9 | 255.1 ± 4.6 |
| RCR | 6.4 ± 1.4 | 5.8 ± 1.5 | 4.3 ± 0.5 | 1.3 ± 0.9* |
| ADP/O | 2.0 ± 0.2 | 1.7 ± 0.2 | 1.7 ± 0.3 | 1.3 ± 0.4* |
Values are presented as mean ± SEM (n = 3), P < 0.05 vs. control
Fig. 8.
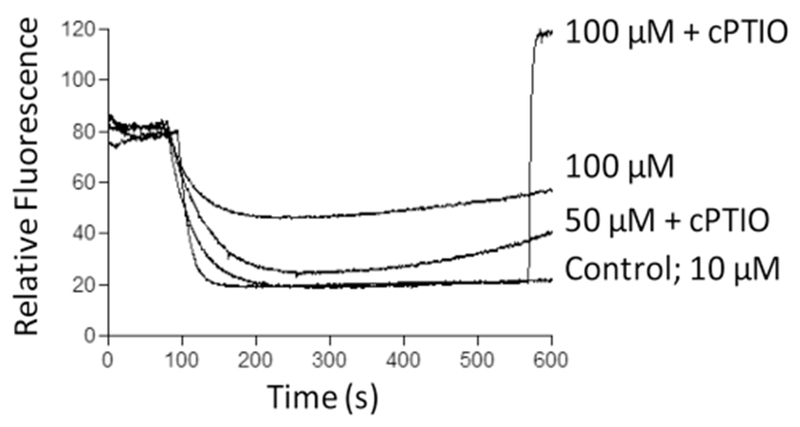
Time course of membrane potential induced by NO released from complex (Ru-DCBPY) in succinate-energized mitochondria. Mitochondria (1 mg of protein mL−1) were incubated with 10–100 μM of (Ru-DCBPY) in the standard medium, in the presence of 5 μM succinate (plus 2.5 μmol L−1 rotenone), at 30 °C. Plots are representative of at least three experiments with different mitochondrial preparations
(Ru-DCBPY)–IgG conjugate
Nitrosyl ruthenium complexes may constitute useful NO delivery agents in anti-cancer therapy because they allow for control of the local NO concentration. We hypothesized that conjugating the nitrosyl ruthenium complex (Ru-DCBPY) with antibody, in basic medium, might improve the specificity of the complex for the target action site. Antibody–drug conjugates (ADCs) have been extensively studied as a promising class of anti-cancer agents. Cell recognition by drugs and consequent binding of the drug to target cells potentially increase drug accumulation in the desired tissues and organs. In this proof-of-concept study, we considered a novel system involving polyclonal antibody IgG anti-VDAC, the mitochondrial voltage-dependent anion-selective channel, conjugated to complex (Ru-DCBPY). The aim is targeting drugs to mitochondria, an organelle with high levels of the reducer agent NADH. Complex (Ru-DCBPY) covalently bound to the IgG immunoglobulin through a coupling reaction illustrated in Scheme 3; a previously published procedure was employed [56].
Scheme 3.
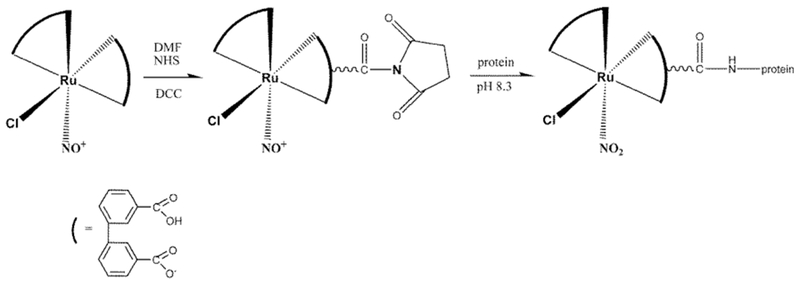
Chemical synthesis of the NHS-ester of (Ru-DCBPY) ligation, in basic medium, with IgG anti-VDAC antibody (protein)
The resulting complex (Ru-DCBPY)–IgG conjugate was purified in a Sephadex G-25 column. UV–Vis spectroscopy helped to identify the separated fractions containing the complex (Ru-DCBPY)–IgG conjugate. The eluted conjugates were analyzed by Western blotting. Figure 9 depicts the comparative cytotoxicity on the Hepg2 cell line induced by free VDAC, complex (Ru-DCBPY) and complex (Ru-DCBPY)–IgG conjugate. The viability of HepG2 cell decreased in the presence of selected fractions from the column separation, containing the conjugates (fractions 14–17) as compared to the control (cell culture medium only), free VDAC, cis-[RuCl2(dcbpy-H)2] and free (Ru-DCBPY). Cellular uptake measured for aqueous complex (Ru-DCBPY) was in the order of 10−8 mol L−1, which contained the (Ru-DCBPY)–IgG conjugate. Cytotoxicity was probably a consequence of interactions between the anti-VDAC antibody conjugate and the mitochondrial VDAC protein [72, 73].
Fig. 9.
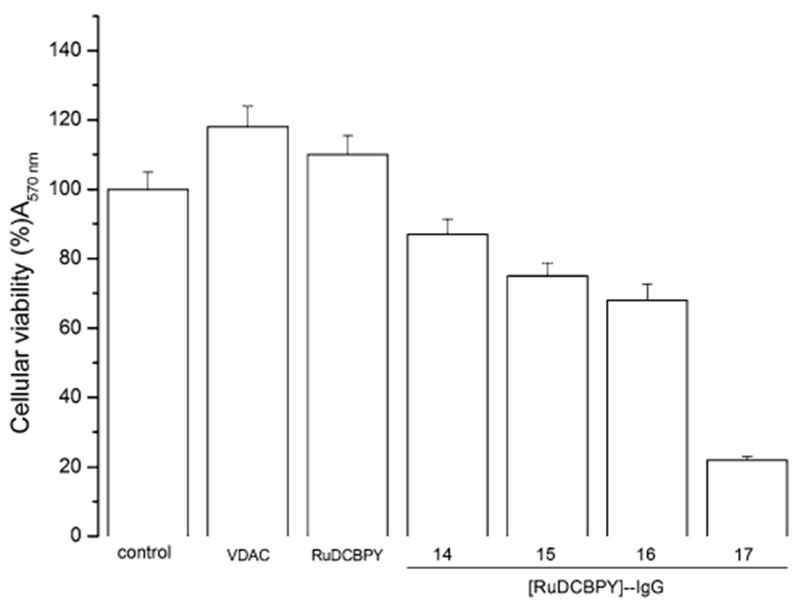
MTT cell viability assay. Effects on HepG2 cell viability of follow treatments: control, DMEM cell culture medium; VDAC (IgG anti-VDAC antibody solubilized in DMEM medium); complex (I), ruthenium nitrosyl complex Ru-DCBPY not conjugated with IgG anti-VDAC antibody, e.g., Ru-DCBPY; Fractions 14, 15, 16, and 17 were from the conjugate purification on a Sephadex column, as described in “Materials and methods”, (Ru-DCBPY)–IgG. The experiments were performed in triplicate, independently, and expressed as the mean and standard deviation
In principle, the remaining complex (Ru-DCBPY) in the complex (Ru-DCBPY)–IgG conjugate underwent chemical reaction in the cancer cell membrane. This process could be attributed to the reduction of conjugated (Ru-DCBPY) or oxygen transfer reaction of cis-[RuCl(dcbpy)2(NO2)]4− generating NO, which may have reacted with superoxide to give peroxynitrite, a species that inhibits the mitochondrial respiration process by several biochemical pathways [16]. Typically, these mitochondrial events are involved in cell death and can potentially kill tumor cells.
Conclusions
The use of an antibody–nitrosyl ruthenium complex conjugate was shown to be able to kill the HepG-2 cancer cell line. Local NO release accounted for the observed cytotoxicity, which could be explained by the (Ru-DCBPY) complex targeting the mitochondrial VDAC where it undergoes reduction and NO release. Alterations of the NAD+/NADH redox couple have also been described as a new mechanism for anti-cancer drug activity, and may be involved in the cytotoxicity of the (Ru-DCBPY)–IgG conjugate.
Supplementary Material
Acknowledgements
This work was supported by CNPq, CAPES, FAPESP Grant 2016/08851-0 and scholarship 2009/11471-9 (Loyanne Ramos). MRH was supported by US NIH Grants R01AI050875 and R21AI121700, Air Force Office of Scientific Research Grant FA9550-13-1-0068, by US Army Medical Research Acquisition Activity Grant W81XWH-09-1-0514, and by US Army Medical Research and Materiel Command Grant W81XWH-13-2-0067.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00775-018-1589-x) contains supplementary material, which is available to authorized users.
References
- 1.Miller MR, Megson IL (2007) Recent developments in nitric oxide donor drugs. Br J Pharmacol 151(3):305–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonavida B (ed) (2010) Nitric oxide (NO) and cancer: prognosis, prevention, and therapy. Springer Science and Business Media, New York [Google Scholar]
- 3.Serafim RA, Primi MC, Trossini GH, Ferreira EI (2012) Nitric oxide: state of the art in drug design. Curr Med Chem 19(3):386–405 [DOI] [PubMed] [Google Scholar]
- 4.Tfouni E, Truzzi DR, Tavares A, Gomes AJ, Figueiredo LE, Franco DW (2012) Biological activity of ruthenium nitrosyl complexes. Nitric Oxide 26(1):38–53 [DOI] [PubMed] [Google Scholar]
- 5.de Lima RG, Silva BR, da Silva RS, Bendhack LM (2014) Ruthenium complexes as NO donors for vascular relaxation induction. Molecules 19(7):9628–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szabo C (2016) Gasotransmitters in cancer: from pathophysiology to experimental therapy. Nat Rev Drug Discovery 15:185–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lundberg JO, Gladwin MT, Weitzberg E (2015) Strategies to increase nitric oxide signalling in cardiovascular disease. Nat Rev Drug Discov 14(9):623–641 [DOI] [PubMed] [Google Scholar]
- 8.Paulo M, Banin TM, de Andrade FA, Bendhack LM (2014) Enhancing vascular relaxing effects of nitric oxide-donor ruthenium complexes. Future Med Chem 6(7):825–838 [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Duan Q, Wang J, Song Z, Qiao X, Wang H (2015) Photocontrolled nitric oxide release from two nitrosylruthenium isomer complexes and their potential biomedical applications. J Biomed Opt 20(1):015004. [DOI] [PubMed] [Google Scholar]
- 10.Carneiro ZA, Biazzotto JC, Alexiou AD, Nikolaou S (2014) Nitric oxide photorelease from a trinuclear ruthenium nitrosyl complex and its in vitro cytotoxicity against melanoma cells. J Inorg Biochem 134:36–38 [DOI] [PubMed] [Google Scholar]
- 11.Castro PFDS, Andrade DL, Reis CDF, Costa SHN, Batista AC, Silva RS, Rocha ML (2016) Relaxing effect of a new ruthenium complex nitric oxide donor on airway smooth muscle of an experimental model of asthma in rats. Clin Exp Pharmacol Physiol 43(2):221–229 [DOI] [PubMed] [Google Scholar]
- 12.Rodrigues FP, Carneiro ZA, Mascharak P, Curti C, da Silva RS (2016) Incorporation of a ruthenium nitrosyl complex into liposomes, the nitric oxide released from these liposomes and HepG2 cell death mechanism. Coord Chem Rev 306:701–707 [Google Scholar]
- 13.Ramos LCB, Marchesi MSP, Callejon D, Baruffi MD, Lunardi CN, Slep LD, Bendhack LM, da Silva RS (2016) Enhanced antitumor activity against melanoma cancer cells by nitric oxide release and photosensitized generation of singlet oxygen from ruthenium complexes. Eur J Inorg Chem 22:3592–3597 [Google Scholar]
- 14.Negri LB, Martins TJ, Ramos LCB, da Silva RS (2017) Nitric oxide derivative ruthenium compounds as NO-based chemotherapeutic and phototherapeutic agents In: Sebara AB (ed) Nitric oxide donors: novel biomedical applications and perspectives, 1st edn Elsevier, New York, pp 1–24 [Google Scholar]
- 15.Toledo JC, de França Lopes LG, Alves AA, da Silva LP, Franco DW (2002) Release of NO by a nitrosyl complex upon activation by the mitochondrial reducing power. J Inorg Biochem 89(3):267–271 [DOI] [PubMed] [Google Scholar]
- 16.Rodrigues FP, Pestana CR, Polizello AC, Pardo-Andreu GL, Uyemura SA, Santos AC, Alberici LC, da Silva RS, Curti C (2012) Release of NO from a nitrosyl ruthenium complex through oxidation of mitochondrial NADH and effects on mitochondria. Nitric Oxide 26(3):174–181 [DOI] [PubMed] [Google Scholar]
- 17.Pereira ADC, Ford PC, da Silva RS, Bendhack LM (2011) Ruthenium-nitrite complex as pro-drug releases NO in a tissue and enzyme-dependent way. Nitric Oxide 24(4):192–198 [DOI] [PubMed] [Google Scholar]
- 18.Fry NL, Mascharak PK (2011) Photoactive ruthenium nitrosyls as NO donors: how to sensitize them toward visible light. Acc Chem Res 44(4):289–298 [DOI] [PubMed] [Google Scholar]
- 19.Smith NA, Sadler PJ (2013) Photoactivatable metal complexes: from theory to applications in biotechnology and medicine. Philos Trans R Soc A 371:20120519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.da Silva RS, Marchesi MS, Khin C, Lunardi CN, Bendhack LM, Ford PC (2007) Photoinduced electron transfer between the cationic complexes [Ru(NH3)5pz]2+ and trans-[RuCl([15]aneN4)NO]+ mediated by phosphate ion: visible light generation of nitric oxide for biological targets. J Phys Chem B 111(24):6962–6968 [DOI] [PubMed] [Google Scholar]
- 21.Cicillini SA, Prazias ACL, Tedesco AC, Serra OA, da Silva RS (2009) Nitric oxide and singlet oxygen photo-generation by light irradiation in the phototherapeutic window of a nitrosyl ruthenium conjugated with a phthalocyanine rare earth complex. Polyhedron 28(13):2766–2770 [Google Scholar]
- 22.Da Rocha ZN, Marchesi MSP, Molin JC, Lunardi CN, Miranda KM, Bendhack LM, Ford PC, da Silva RS (2008) The inducing NO-vasodilation by chemical reduction of coordinated nitrite ion in cis-[Ru(NO2)L(bpy)2]2+ complex. Dalton Trans 32:4282–4287 [DOI] [PubMed] [Google Scholar]
- 23.Sarniguet C, Toloza J, Cipriani M, Lapier M, Vieites M, Toledano-Magaña Y, García-Ramos JC, Ruiz-Azuara L, Moreno V, Maya JD, Azar CO, Gambino D, Otero L (2014) Water-soluble ruthenium complexes bearing activity against protozoan parasites. Biol Trace Elem Res 159(1–3):379–392 [DOI] [PubMed] [Google Scholar]
- 24.Sesti-Costa R, Carneiro ZA, Silva MC, Santos M, Silva GK, Milanezi C, da Silva RS, Silva JS (2014) Ruthenium complex with benznidazole and nitric oxide as a new candidate for the treatment of chagas disease. PLoS Negl Trop Dis 8(10):3207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Iniguez E, Sánchez A, Vasquez MA, Martínez A, Olivas J, Sattler A, Sánchez-Delgado RA, Maldonado RA (2013) Metal–drug synergy: new ruthenium (II) complexes of ketoconazole are highly active against Leishmania major and Trypanosoma cruzi and nontoxic to human or murine normal cells. J Biol Inorg Chem 18(7):779–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridnour LA, Thomas DD, Switzer C, Flores-Santana W, Isenberg JS, Ambs S, Roberts DD, Wink DA (2008) Molecular mechanisms for discrete nitric oxide levels in cancer. Nitric Oxide 19(2):73–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomas DD, Ridnour LA, Isenberg JS, Flores-Santana W, Switzer CH, Donzelli S, Colton C, Harris CC, Roberts DD, Wink DA (2008) The chemical biology of nitric oxide: implications in cellular signaling. Free Radic Biol Med 45(1):18–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stevens EV, Wells A, Shin JH, Liu J, Der CJ, Schoenfisch MH (2010) Nitric oxide-releasing silica nanoparticle inhibition of ovarian cancer cell growth. Mol Pharm 7(3):775–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hirst D, Robson T (2007) Targeting nitric oxide for cancer therapy. J Pharm Pharmacol 59(1):3–13 [DOI] [PubMed] [Google Scholar]
- 30.Xiang HJ, Deng Q, An L, Guo M, Yang SP, Liu JG (2016) Tumor cell specific and lysosome-targeted delivery of nitric oxide for enhanced photodynamic therapy triggered by 808 nm near-infrared light. Chem Commun 52(1):148–151 [DOI] [PubMed] [Google Scholar]
- 31.Park J, Pramanick S, Kim J, Leeab J, Kim WJ (2017) Nitric oxide-activatable gold nanoparticles for specific targeting and photo-thermal ablation of macrophages. Chem Commun 53(81):11229–11232 [DOI] [PubMed] [Google Scholar]
- 32.Safdar S, Payne CA, Tu NH, Taite LJ (2013) Targeted nitric oxide delivery preferentially induces glioma cell chemosensitivity via altered p53 and O6-methylguanine-DNA methyltransferase activity. Biotechnol Bioeng 110(4):1211–1220 [DOI] [PubMed] [Google Scholar]
- 33.Li YH, Guo M, Shi SW, Zhang QL, Yang SP, Liu JG (2017) A ruthenium-nitrosyl-functionalized nanoplatform for the targeting of liver cancer cells and NIR-light-controlled delivery of nitric oxide combined with photothermal therapy. J Mater Chem B 5(38):7831–7838 [DOI] [PubMed] [Google Scholar]
- 34.Deng Q, Xiang HJ, Tang WW, An L, Yang SP, Zhang QL, Liu JG (2016) Ruthenium nitrosyl grafted carbon dots as a fluorescence-trackable nanoplatform for visible light-controlled nitric oxide release and targeted intracellular delivery. J Inorg Biochem 165:152–158 [DOI] [PubMed] [Google Scholar]
- 35.Wyland KR, Hoffman EE, Jain A (2017) DNA interaction studies and photoinduced ligand exchange kinetics of a sterically strained Ruthenium(II) complex. Inorg Chim Acta 454:62–66 [Google Scholar]
- 36.Campagna S, Puntoriero F, Nastasi F, Bergamini G, Balzani V (2007) Photochemistry and photophysics of coordination compounds: ruthenium In: Balzani V, Campagna S (eds) Photochemistry and photophysics of coordination compounds I. Springer, Berlin, pp 117–214 [Google Scholar]
- 37.Sauaia MG, de Lima RG, Tedesco AC, da Silva RS (2005) Nitric oxide production by visible light irradiation of aqueous solution of nitrosyl ruthenium complexes. Inorg Chem 44(26):9946–9951 [DOI] [PubMed] [Google Scholar]
- 38.Meyer TJ, Godwin JB (1971) Preparation of ruthenium nitrosyl complexes containing 2, 2′-bipyridine and 1, 10-phenanthroline. Inorg Chem 10(3):471–474 [Google Scholar]
- 39.Sauaia MG, Oliveira FS, Tedesco AC, da Silva RS (2003) Control of NO release by light irradiation from nitrosyl-ruthenium complexes containing polypyridyl ligands. Inorg Chim Acta 355:191–196 [Google Scholar]
- 40.Ghosh K, Kumar S, Kumar R (2011) Synthesis and characterization of a novel ruthenium nitrosyl complex and studies on photolability of coordinated NO. Inorg Chem Commun 14(1):146 [Google Scholar]
- 41.Elmore S (2007) Apoptosis: a review of programmed cell death. Toxicol Pathol 35(4):495–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grau-Campistany A, Massaguer A, Carrion-Salip D, Barragán F, Artigas G, López-Senín P, Moreno V, Marchán V (2013) Conjugation of a Ru(II) arene complex to neomycin or to guanidinoneomycin leads to compounds with differential cytotoxicities and accumulation between cancer and normal cells. Mol Pharm 10(5):1964–1976 [DOI] [PubMed] [Google Scholar]
- 43.Wang T, Zabarska N, Wu Y, Lamla M, Fischer S, Monczak K, Ng DYW, Sven Rau S, Weil T (2015) Receptor selective ruthenium-somatostatin photosensitizer for cancer targeted photodynamic applications. Chem Commun 51:12552–12555 [DOI] [PubMed] [Google Scholar]
- 44.Becke ADJ (1988) Density functional calculations of molecular bond energies. J Chem Phys 84:4524–4529 [Google Scholar]
- 45.Becke ADJ (1993) Density-functional thermochemistry. III. The role of exact exchange. J Chem Phys 98:5648–5652 [Google Scholar]
- 46.Lee C, Yang W, Parr RG (1988) Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys Rev B 37(2):785–789 [DOI] [PubMed] [Google Scholar]
- 47.Perdew JP (1986) Density-functional approximation for the correlation energy of the inhomogeneous electron gas. Phys Rev B 33:8822–8824 [DOI] [PubMed] [Google Scholar]
- 48.Dunning TH Jr, Hay PJ (1976) Modern theoretical chemistry In: Schaefer HF III (ed) Modern theoretical chemistry. Plenum, New York, pp 1–28 [Google Scholar]
- 49.Hay PJ, Wadt WRJ (1985) Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J Chem Phys 82:270–283 [Google Scholar]
- 50.Hay PJ, Wadt WRJ (1985) Ab initio effective core potentials for molecular calculations. Potentials for K to Au including the outermost core orbitals. J Chem Phys 82:299–310 [Google Scholar]
- 51.Wadt WR, Hay PJJ (1985) Ab initio effective core potentials for molecular calculations. Potentials for main group elements Na to Bi. J Chem Phys 82:284–298 [Google Scholar]
- 52.Pedersen PL, Greenawalt JW, Reynafarje B, Hullihen J, Decker GL, Soper JW, Bustamente E (1978) Preparation and characterization of mitochondria and submitochondrial particles of rat liver and liver-derived tissues. Methods Cell Biol 20:411–481 [DOI] [PubMed] [Google Scholar]
- 53.Åkerman KE, Wikström MK (1976) Safranine as a probe of the mitochondrial membrane potential. FEBS Lett 68(2):191–197 [DOI] [PubMed] [Google Scholar]
- 54.Wrona M, Patel K, Wardman P (2005) Reactivity of 2′, 7′-dichlorodihydrofluorescein and dihydrorhodamine 123 and their oxidized forms toward carbonate, nitrogen dioxide, and hydroxyl radicals. Free Radic Biol Med 38(2):262–270 [DOI] [PubMed] [Google Scholar]
- 55.Blondin GA, Green DE (1967) The mechanism of mitochondrial swelling. Proc Natl Acad Sci USA 58(2):612–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Terpetschnig E, Szmacinski H, Malak H, Lakowicz JR (1995) Metal–ligand complexes as a new class of long-lived fluorophores for protein hydrodynamics. Biophys J 68(1):342–350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mosmann T (1983) Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays. J Immunol Methods 65:55–63 [DOI] [PubMed] [Google Scholar]
- 58.Tfouni E, Krieger M, McGarvey BR, Franco DW (2003) Structure, chemical and photochemical reactivity and biological activity of some ruthenium amine nitrosyl complexes. Coord Chem Rev 236(1–2):57–69 [Google Scholar]
- 59.Caramori GF, Frenking G (2007) The nature of the Ru–NO bond in ruthenium tetraammine nitrosyl complexes. Organometallics 26(24):5815–5825 [Google Scholar]
- 60.Videla M, Jacinto JS, Baggio R, Garland MT, Singh P, Kaim W, Slep LD, Olabe JA (2006) Structure, spectroscopy, and electrophilic and nucleophilic reactivities of bound nitrosyl. Inorg Chem 45:8608–8617 [DOI] [PubMed] [Google Scholar]
- 61.Cohen AJ, Mori-Sánchez P, Yang W (2008) Insights into current limitations of density functional theory. Science 321(5890):792–794 [DOI] [PubMed] [Google Scholar]
- 62.Coropceanu V, Malagoli M, André JM, Brédas JL (2002) Charge-transfer transitions in triarylamine mixed-valence systems: a joint density functional theory and vibronic coupling study. J Am Chem Soc 124(35):10519–10530 [DOI] [PubMed] [Google Scholar]
- 63.Renz M, Kaupp M (2012) Predicting the localized/delocalized character of mixed-valence diquinone radical anions. Toward the right answer for the right reason. J Phys Chem A 116(43):10629–10637 [DOI] [PubMed] [Google Scholar]
- 64.Shimidzu T, Iyoda T, Izaki K (1985) Photoelectrochemical properties of bis (2,2′-bipyridine)(4,4′-dicarboxy-2,2′-bipyridine) ruthenium(II) chloride. J Phys Chem 89(4):642–645 [Google Scholar]
- 65.Borges SDS, Davanzo CU, Castellano EE, Zukerman-Schpector J, Silva SC, Franco DW (1998) Ruthenium nitrosyl complexes with N-heterocyclic ligands. Inorg Chem 37(11):2670–2677 [DOI] [PubMed] [Google Scholar]
- 66.Calandreli I, Oliveira FD, Liang GG, da Rocha ZN, Tfouni E (2009) Synthesis and characterization of trans-[Ru(NO)Cl(L)4] (PF6)2(L = isonicotinamide; 4-acetylpyridine) and related species. Inorg Chem Commun 12(7):591–595 [Google Scholar]
- 67.Sauaia MG, da Silva RS (2003) The reactivity of nitrosyl ruthenium complexes containing polypyridyl ligands. Transit Met Chem 28(3):254–259 [Google Scholar]
- 68.Zoratti M, Szabò I (1995) The mitochondrial permeability transition. Biochim Biophys Acta 1241(2):139–176 [DOI] [PubMed] [Google Scholar]
- 69.Halestrap AP (2009) What is the mitochondrial permeability transition pore. J Mol Cell Cardiol 46(6):821–831 [DOI] [PubMed] [Google Scholar]
- 70.Petronilli V, Cola C, Massari S, Colonna R, Bernardi P (1993) Physiological effectors modify voltage sensing by the cyclosporin A-sensitive permeability transition pore of mitochondria. J Biol Chem 268(29):21939–21945 [PubMed] [Google Scholar]
- 71.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P (1994) The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols. Increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem 269(24):16638–16642 [PubMed] [Google Scholar]
- 72.Brown GC (2007) Nitric oxide and mitochondria. Front Biosci 12(6):1024–1033 [DOI] [PubMed] [Google Scholar]
- 73.Brown GC, Borutaite V (2001) Nitric oxide, mitochondria, and cell death. IUBMB Life 52(3–5):189–195 [DOI] [PubMed] [Google Scholar]
- 74.Kroemer G, Galluzzi L, Brenner C (2007) Mitochondrial membrane permeabilization in cell death. Physiol Rev 87(1):99–163 [DOI] [PubMed] [Google Scholar]
- 75.Gogvadze V, Orrenius S, Zhivotovsky B (2009) Mitochondria as targets for chemotherapy. Apoptosis 14(4):624–640 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.