

Abstract
Background
The APOE4 allele is the strongest genetic risk factor for Alzheimer’s disease (AD). It has been associated with an accumulation of amyloid-β (Aβ) in the brain, which is produced through the sequential cleavage of the amyloid-β precursor protein (AβPP) by β- and γ-secretases. Alternatively, AβPP is also cleaved by α-secretases such as A Disintegrin and Metalloproteinase Domain-containing Protein 10 (ADAM10).
Objective
While several studies have investigated the impact of apoE on β- and γ-secretase, interactions between apoE and α-secretases have not been fully examined. We investigated the effect of each apoE isoform on ADAM10 in vitro and in human cortex samples.
Method
ADAM10 activity and kinetics was assessed in cell-free assays and the biological activity of ADAM10 further investigated in 7WCHO cells over-expressing wild type AβPP through ELISA. Finally, ADAM10 expression and activity was observed in the soluble fraction of both control and Alzheimer’s Disease human cortex samples through ELISA.
Results
In a cell free assay, ADAM10 activity was found to be significantly lower in apoE4 samples compared to apoE2. 7WCHO cells over expressing wild type AβPP exposed to apoE4 demonstrated reduced formation of sAβPPα compared to other apoE isoforms. We also identified APOE and AD dependent changes in ADAM10 activity and expression in the soluble brain fraction of human brain cortex.
Conclusion
Overall, our data demonstrates an apoE isoform-dependent effect on ADAM10 function and AβPP processing which may describe the elevated amyloid levels in the brains of AD subjects carrying the APOE4 allele.
Keywords: Alzheimer’s Disease, ADAM10, α-secretase, apolipoprotein E, APOE, amyloid
1. INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia affecting an estimated 44 million individuals worldwide. One of the pathological hallmarks of AD includes the extracellular deposition of amyloid-β peptides (Aβ) in the brain. The accumulation of Aβ is thought to be one of the primary drivers of AD pathology as brain Aβ levels strongly correlate with the severity of neurodegeneration and cognitive impairment [1–4]. In addition, the elevation of Aβ appears to precede the neuroinflammatory and neurotoxic events associated with AD, indicating Aβ accumulation has a role in the early stages of AD [5].
Aβ is produced via proteolysis of the type I transmembrane protein amyloid-β precursor protein (AβPP) through the β secretase pathway [6]. Sequential cleavage by beta-site AβPP cleaving enzyme 1 (BACE) and γ-secretase results in the production of Aβ40 as well as the pathogenic Aβ42. While the β- and γ-secretase enzymes have been extensively studied in AD [7, 8], treatments targeting these enzymes in clinical trials have largely failed to improve the AD phenotype and generally exhibit poor toxicological profiles [8–11]. In addition to cleavage via β-secretase, AβPP can also be proteolysed through the α-secretase pathway resulting in the formation of soluble AβPPα (sAβPPα) which has been shown to rescue synaptic function and promote neurite growth [12, 13]. A prominent enzyme in the α-secretase pathway is A Disintegrin and Metalloproteinase Domain-containing Protein 10 (ADAM10), the expression of which is controlled by the retinoic acid receptor (RAR) and the retinoid X receptor (RXR) [14]. It exists as both a membraneassociated protein and soluble extracellular protein (sADAM10), formed via ectodomain shedding at the cell surface. The resulting intracellular domain (ICD) is sequentially processed by γ-secretase and translocates to the areas of the nucleus involved in active transcription [15]. Recently, several ADAM10 mutations were identified that resulted in decreased ADAM10 activity. The reduced activity was associated with elevated Aβ levels in vitro, suggesting that diminished α-secretase activity may be a contributing factor in AD pathogenesis [16]. As such, promoting the α-secretase processing of AβPP may be a viable therapeutic strategy to abrogate Aβ brain pathology and the development of AD.
ApoE is a 34kDa glycoprotein that exists as three isoforms in humans (apoE2, apoE3 and apoE4), with the APOE4 allele representing the strongest genetic risk factor for AD [17]. Possession of one or two APOE4 alleles increases the chance of developing AD by 4- and 15-fold, respectively when compared to APOE3 homozygous individuals [18]. In addition, AD patients and transgenic AD animals homozygous for APOE3 show reduced brain Aβ deposition compared to subjects homozygous for APOE4 [19, 20]. Despite the well established link between APOE genotype and AD pathology, the exact mechanism by which apoE contributes to the AD phenotype remains unclear. Prior studies have identified apoE-dependent differences in the processing of AβPP, the majority of this work has been focused on the β-secretase pathway [21, 22]. While it has been suggested that inadequate processing through the α-secretase pathway may lead to increased levels of pathological Aβ [23], there has been little investigation into the role of apoE in α-secretase function. Therefore, the purpose of this study is to assess the influence of APOE genotype on the activity and expression of the α-secretase ADAM10 in the context of AD.
2. MATERIALS AND METHOD
2.1. Human Cortex Samples
Human cortex samples (inferior frontal gyrus) were obtained from the autopsied brains of non-demented (ND) and AD subjects with different APOE genotypes as summarized in (Table 1). The origins of the human samples are listed in the acknowledgements.
Table 1.
Demographic details of human brain cortex samples.
| APOE | Non-Demented | Alzheimer’s Disease | ||
|---|---|---|---|---|
| Mean age ± StDev | N (M/F) | Mean age ± StDev | N (M/F) | |
| 2/2 | 90.0 ± 24.00 | 10(3/7) | 79.1 ± 19.30 | 2(0/2) |
| 2/3 | 85.5 ± 10.05 | 10(4/6) | 79.8 ± 10.84 | 10(7/3) |
| 3/3 | 86.3 ± 6.16 | 10(9/1) | 79.8 ± 15.53 | 10(2/8) |
| 3/4 | 81.0 ± 10.23 | 10(7/3) | 82.5 ± 8.68 | 10(5/5) |
| 4/4 | 80.8 ± 8.41 | 10 (5/5) | 51.7 ± 16.20 | 10 (5/5) |
Abbreviations: apoE, apolipoprotein E genotype; StDev, Standard deviation; M, male; F, female.
2.2. Isolation of Brain Fractions
Brain fractions were isolated from the human cortex samples using a modified technique as previously described by our group [24]. Briefly, using a Dounce homogenizer, the tissue was homogenized in HBSS on ice. A sample was collected representing the whole brain homogenate and diluted 1:1 in lysis buffer (M-PER + 1%EDTA +0.2%PMSF (Thermo Scientific, IL, USA)) supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Scientific, IL, USA). The remaining homogenized tissue was diluted 1:1 with 40% dextran followed by centrifugation at 6000g for 15 minutes at 4°C. The soluble fraction of the cortex sample was obtained from the supernatant. All samples were immediately stored at −80°C until further analysis.
2.3. Collection and Enrichment of Human Lipidated ApoE
Glial cells expressing human apoE2, 3 or 4 isolated from neonatal mice were kindly provided by Dr. Mary Jo LaDu (University of Illinois at Chicago) [25]. Lipidated apoE particles were collected and enriched from the mixed glial cultures as described in our previous work [26]. Briefly, human apoE expressing mixed glial cells were plated in 150cm2 flasks (≈1.5 brains/flask) in DMEM/F12 (+10% FBS, L-glutamine (2mM), and 1% penicillin/streptomycin). Upon confluency, cells were washed and incubated with serum-free media for 72 hours. Glial-conditioned media was collected and centrifuged at 1000g for 3 min to remove any residual debris before concentrating (≈10x) using the Vivaspin 15R centrifugal concentrator with a molecular weight cut-off of 10 000 Da (Sartorius Stedim Biotech, USA). The resulting concentrate was analysed for apoE content using a human apoE ELISA (MBL International Corporation, MA, USA) and stored at −80°C until further analysis.
2.4. ApoE and AD Mediated ADAM10 Activity
The effect of apoE on ADAM10 activity was assessed in a cell free paradigm utilising a fluorescent substrate. Recombinant ADAM10 (1μM) (R&D Systems, MN, Canada) was incubated in the presence of lipidated apoE2, 3 and 4 (0.5–250ng/ml) and the fluorescent substrate PEPDAB010 (5nM) (BioZyme Inc., NC, USA) for 1 hour at 37°C before detection of fluorescence. Additional kinetic activity analysis of ADAM10 (1μM) was carried out in the presence of apoE (0.1 – 300ng/ml) and a fixed concentration of PEPDAB010 (5nM). ADAM10 and apoE were pre-incubated at 37°C for 1 hour before addition of PEPDAB010. Enzymatic activity was then measured by fluorescence every 35 seconds for 30 minutes at 37°C. The rate of reaction for each concentration of apoE calculated by taking the initial gradient in the linear region and plotting it against the concentration of apoE. In all the cell free activity assays, to determine whether the substrate was metabolised by apoE itself, fluorescent substrate was incubated with various concentrations of apoE alone in the absence of ADAM10. These values were used as background controls for each apoE isoform-ADAM10 treatment combination. For all cell free activity paradigms, fluorescence was measured at 485/528 Ex/Em using BioTek Synergy HT microplate reader.
2.5. Human Cortex ADAM10 Activity and Expression
To evaluate ADAM10 activity in the soluble fraction of human brain tissue, 50μl of the soluble fraction was added to PEPDAB010 at a final concentration of 5nM before incubation at 37°C for 1 hour. Activity was analysed as previously described in section 3.4. After completion of the original study, an ADAM10 specific substrate, PEPDAB063 (Biozyme Inc., NC, USA), was made commercially available. Therefore to validate our previous results, a panel of 10 samples (one of each genotype both the AD and the ND cortex samples) was randomly selected and tested against the values obtained with the original substrate (PEPDAB010). An identical method to the PEPDAB010 protocol was used with the exception of the fluorescence being measured at 480/530 Ex/Em. Percentage variation in the same sample between each substrate was calculated after being standardised to a reference sample. This analysis was repeated using each of the samples being used as the reference sample and an average variance obtained. Expression of ADAM10 in the cortex samples (both homogenate and soluble fraction) were obtained via ADAM10 ELISA (Cloud-Clone Corp., USA) as per the manufacturer’s instructions. Expression and activity data was standardized by total protein content obtained though bicinchoninic acid (BCA) assay (Thermo Scientific, IL, USA) of the homogenate.
2.6. ApoE Dependent sAβPPα Production
7WCHO cells over-expressing wild type AβPP were incubated at 37°C in DMEM F-12 (10% FBS, 5% Pen/Strep, 5% Glutamax, 5% Gentamycin). Upon confluency, cells were detached with trypsin (Invitrogen, CA, USA) and seeded into precoated poly-D-lysine 24-well plates. When approximately 90% confluent, cells were treated with lipidated apoE at 50ng/ml alone and in combination with the ADAM10 specific inhibitor GI254023X (1μm) (Tocris Bioscience, UK) in FBS-free media for 24 hours. Following treatment, media was collected and the total protein content in the media assessed using BCA assay (Thermo Scientific, IL, USA). sAβPPα expression was quantified using a human sAβPPα ELISA as per the manufacturer’s instructions (IBL, Japan) and levels standarized by total protein content in the media.
2.7. Statistical Analysis
Statistical significance of cell free, in vitro and ex vivo experiments were assessed by one- or two-way ANOVA and post-hoc analyses where appropriate (JMPv11.0.0, SAS Institute Inc., NC, USA; GraphPad Prism 5, GraphPad Software, Inc., CA, USA). When necessarily, data was normalized prior to statistical analysis. EC50 values were generated using a nonlinear dose-response regression of the data set (Michaelis Menten curve fit, GraphPad Prism 5). Normalisation of data for correlation analysis used the Johnson SU correction (GraphPad Prism 5). In addition, to assess the relationship between the enzymatic efficiency and expression, Pearson product-moment correlation coefficients were computed (GraphPad Prism5).
3. RESULTS
3.1. Cell-free ApoE Isoform Dependent ADAM10 Activity
To determine the effect of the apoE isoforms on the activity of ADAM10, a cell free activity assay was utilised (Fig. 1A). Following exposure of apoE isoforms and an ADAM10 fluorescent substrate to active recombinant ADAM10, apoE isoform- and concentration-dependent changes in the activity of ADAM10 were detected with a rank order of apoE2>apoE3>apoE4. At 25ng/ml apoE, ADAM10 activity was significantly lower in the presence of apoE4 compared to apoE2 (≈40% reduction). Although it did not reach statistical significance, apoE3 showed consistently higher activity than apoE4 at all concentrations. In addition we also investigated the influence of apoE isoforms on ADAM10 activity via Michaelis Menten kinetics (Fig. 1B). This revealed that apoE2 demonstrated the greatest impact on ADAM10 activity (EC50 = 0.69ng/ml) followed by apoE3 and apoE4 (EC50 = 16.81ng/ml and 27.17ng/ml, respectively).
Fig. 1.

ADAM10 activity is apoE-isoform dependent. (A) ADAM10 activity in a cell free paradigm was significantly modulated by apoE in an isoform and dose dependent manner (apoE2>apoE3>apoE4), (F (11, 24) = 25.77; p<0.0001). Differences between apoE2 and apoE4 were statistically significant at concentrations ≥25ng/ml. For all apoE concentrations, ADAM10 activity was lower in the presence of apoE4 compared to the other isoforms. (B) ApoE isoform-specific influence on ADAM10 activity. ApoE2 demonstrated the greatest influence on ADAM10 activity (EC50 = 0.69ng/ml ± 0.18) in comparison to apoE3 (EC50 = 24.27ng/ml ± 8.04) or apoE4 (52.24ng/ml ± 14.01). Activity data are presented as ADAM10 activity (n=3) ±SEM. Statistical significance was determined by two-way ANOVA followed by Sidak post-hoc analysis. EC50 values were generated using nonlinear regression analyses (EC50 ± SEM (n=3)). *p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001.
3.2. ApoE Isoform Dependent sAβPPα Production In Vitro
The effect of apoE on AβPP processing in vitro was evaluated using wild type AβPP over-expressing 7WCHO cells treated with exogenous apoE. We observed that apoE4 exposure resulted in a 50% reduction sAβPPα production compared to apoE2 treated cells (Fig. 2). Treatment with the ADAM10-specific inhibitor GI254023X in combination with apoE ablated the production of sAβPPα and removed any apoE isoform effect.
Fig. 2.
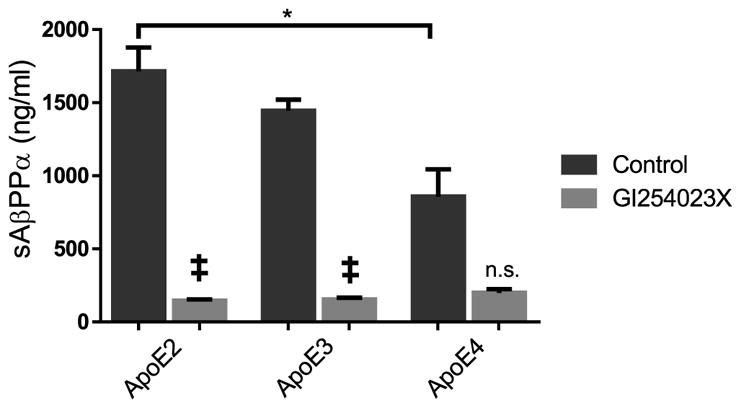
ADAM10 mediated processing of AβPP in cell culture is apoE isoform dependent. In 7WCHO cells over expressing AβPP, apoE treatment (50ng/ml) for 24 hours resulted in significant differences in the expression of sAβPPα as measured by ELISA (n=3) (F (7, 16) = 22.41; p < 0.0001). ApoE4 demonstrated significantly less detectable sAβPPα compared to the apoE2-treated cells (p<0.05). Treatment with the ADAM10 specific inhibitor GI254023X significantly reduced sAβPPα formation compared to apoE2 or apoE3 alone (p<0.001) but not apoE4 alone. Data are presented as mean±SEM. Statistical significance was determined by Two-Way ANOVA followed by Sidak post-hoc analysis. *p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001; ‡ comparison to non-inhibitor p<0.005.
3.3. ADAM10 Expression and Activity in Human Brain Fractions
ADAM10 analyses in brain homogenate from human cortical tissue revealed a small but insignificant small but insignificant decrease in the expression of ADAM10. In addition, when stratified by APOE genotype there was no observable trend and no significance was found (data not shown). In the soluble brain fraction, levels of the proteolytically cleaved sADAM10 were found to be significantly lower in the AD samples compared to ND samples (Fig. 3A). In addition, these changes in sADAM10 levels were also found be both APOE genotype and disease dependent (Fig. 3C). In the AD samples, no significant changes in sADAM10 levels were detected across APOE genotypes. In the ND samples, there was a significant increases in sADAM10 levels in the APOE4/4 genotype compared to all other genotypes. In addition, the activity of soluble ADAM10 was also found to be significantly lower in the AD samples compared to the ND samples (Fig. 3B) and was dependent on APOE genotype and disease state. ND APOE4/4 samples demonstrated significantly more activity than both AD APOE4/4 and ND APOE2/3 samples (Fig. 3D). In addition, no significant correlations were found in the human cortex samples with either activity or expression when compared to age. ADAM10 activity in the panel of random samples utilising the ADAM10 substrate PEPDAB063, as detailed in section 3.5, showed no differences compared to the original substrate PEPDAB010 with an average percentage change of −0.001% ± 6.39%. As such, the results of our enzyme activity studies appear to be specific for ADAM10.
Fig. 3.

ADAM10 expression and activity is altered by APOE genotype and AD in human brain cortex. sADAM10 expression (A) and activity (B) was reduced in AD compared to the ND controls (t=2.770 df=86 p<0.01 and t=3.089 df=87 p<0.01). (C) When stratified by APOE genotype, sADAM10 expression was found to be dependent on APOE genotype (F (4,78) = 7.571 P < 0.0001), disease (F (1,78) = 10.77 P < 0.005) and dependent on a interaction between disease state and APOE (F (4, 78) = 9.192 P < 0.0001). (D) A significant difference in sADAM10 expression in the APOE4/4 genotype was detected compared to all other apoE genotypes in the ND samples (p<0.0001 for all). In addition, sADAM10 expression in the ND APOE4/4 genotype was significantly higher than the AD APOE4/4 (p < 0.0001). There was no significant difference in the levels of sADAM10 between the AD samples. (D)When sADAM10 activity was stratified by APOE and disease, it was found to be disease state dependent (F (1, 61) = 10.00; p<0.005) and dependent on an interaction between disease and apoE (F (3, 61) = 8.816; p<0.0001). In AD samples, a significant reduction ADAM10 activity in the APOE2/3 genotype compared to APOE4/4 was observed (p<0.05). In ND samples, sADAM10 activity in the APOE4/4 genotype was significantly higher than the APOE2/3 (p<0.05). There was also a significant difference between the disease states in the APOE4/4 samples (p<0.001). Data are presented as mean sADAM10 activity (fluorescent units) and mean sADAM10 expression (pg) normalised to total protein content in the whole brain homogenate (μg) ±SEM. Statistical significance was determined by Two-Way ANOVA followed by Sidak post-hoc analysis. *p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001.
3.4. Enzymatic Efficiency of ADAM10
In addition to raw activity or expression levels, we assessed the enzymatic efficiency of ADAM10 for each sample by calculating the activity of ADAM10 per unit of expression (activity/pg). This revealed both disease and APOE genotype differences in the efficiency of sADAM10 (Fig. 4A). In the AD samples, enzymatic efficiency appeared to be unaltered by the APOE genotype. However, in the ND samples, the APOE2/2 genotype showed the highest enzymatic efficiency demonstrating a 10-fold increase over the ND APOE4/4 genotype. In addition, in the ND soluble fraction, a significant inverse correlation was apparent between the enzymatic efficiency of sADAM10 and the sADAM10 levels (r = −0.93, p<0.0001). This relationship was not evident in the AD samples (r = −0.30, p>0.05) (Fig. 4B).
Fig. 4.

The enzymatic efficiency of sADAM10 is APOE dependent (F (4, 77) = 4.816; p<0.005) and dependent on an interaction between disease state and APOE genotype (F (4, 77) = 4.551; p<0.005) (A). In the ND samples, the APOE2/2 genotype demonstrated significantly higher sADAM10 enzymatic efficiency compared to all other ND APOE genotypes as well as the APOE2/2 AD samples (p<0.01). (B) Enzymatic efficiency of sADAM10 against sADAM10 expression demonstrated highly significant inverse correlation in the ND group, while no such correlation was observed in the AD group (ND r =−0.93, n =47, p<0.0001; AD r =−0.31, n =37, p > 0.05). Enzymatic efficiency data presented as enzymatic efficiency of ADAM10 (ADAM10 activity/ sADAM10 expression). Statistical significance was determined by Two-Way ANOVA followed by Sidak post-hoc analysis. Correlation data presented as normalised enzymatic efficiency of ADAM10 against normalised ADAM10 expression. *p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001.
4. DISCUSSION
Alzheimer’s disease is the primary cause of dementia in the elderly and is characterized by an excessive accumulation of Aβ peptides in the brain and cerebrovasculature. Aβ is formed through sequential proteolysis of AβPP by the β- and γ-secretase enzymes. In addition, α-secretases such as ADAM10 compete with β-secretases for AβPP resulting in the production the neurotropic sAβPPα fragment [12]. As the strongest genetic risk factor for AD, APOE has previously been shown to influence the processing of AβPP [21], but the impact of APOE genotype on α-secretase function has yet to be fully explored. The purpose of this study was to examine the effect of apoE on ADAM10 function and advance our understanding of the association between APOE genotype and the development of AD. Our results indicate the expression and activity of ADAM10 is altered in AD and influenced by APOE genotype, which may help describe the propensity of individuals carrying the APOE4 allele to exhibit AD pathology.
We found that ADAM10 activity was reduced in the presence of apoE4 compared to the other apoE isoforms in a cell free paradigm (apoE2>apoE3>apoE4). Although prior work failed to detect apoE-dependent differences in sAβPPα levels in vitro [27], our in vitro studies utilized lipidated apoE [25], which is likely a better representation of apoE under normal physiological conditions than de-lipidated recombinant apoE. This is highly relevant as recent studies have indicated that apoE function is dependent on the lipidation status of apoE; with poorly lipidated apoE demonstrating altered binding affinities to Aβ and increased amyloidogenesis in mouse models of AD [28–30]. The lowered ADAM10 activity in the presence of apoE4 compared to apoE2 resulted in reduced sAβPPα formation which could potentially increase the pool of AβPP available for Aβ synthesis and, at the same time, diminish the neurotropic effects of sAβPPα.
We initially investigated the expression of ADAM10 in human brain homogenate but identified no significant differences in AD versus control subjects or between APOE genotypes. This is in agreement with previous studies by Bernstein et al. which demonstrated a slight but insignificant reduction in ADAM10 expression in AD compared to ND brain specimens using stereological analyses [31]. Based on information from studies conducted by Parkin et al. and Tousseyn et al., which identified a soluble form of ADAM10, we interrogated the activity of ADAM10 in the soluble brain fraction isolated from human cortical tissue [15, 32]. Here we observed a significant reduction in the activity of sADAM10 in AD versus ND samples. Moreover, these changes were both APOE genotype and disease dependent with APOE4 AD samples displaying the lowest activity. Although alterations in ADAM10 activity have been observed by other groups [33], changes due to APOE genotype have not been investigated. When combined with our in vitro data, these studies implicate apoE as a modulator of AβPP processing by altering ADAM10 activity.
In terms of expression, reduced ADAM10 levels have only been reported in the platelets of AD subjects [34], although they did appear to correlate with reduced mini-mental state exam scores [35]. In the current study, while we found no difference in the expression of total ADAM10 between the ND and AD subjects in cortical brain homogenate. However a significant decrease in the expression of the extracellular proteolytically shed sADAM10 was identified in the AD samples compared to the ND control group. Furthermore, differences in sADAM10 levels were also evident when stratified by APOE genotype, with the highest levels of sADAM10 detected in the APOE4/4 ND samples. Interestingly, there were no changes in the expression of sADAM10 in the AD samples between APOE genotypes. Altogether, these data suggests that APOE genotype influences the ectodomain shedding of ADAM10. Previously, it was demonstrated that ADAM10 shedding is mediated by ADAM9 and ADAM15 followed by cleavage of the intracellular domain (ICD) fragment by γ-secretase. Currently, the extent to which these other ADAM family members are involved in the etiology of AD is not known and their activity may also be influenced by the APOE genotype. However, the role of γ-secretase in AD has been well documented and was the focus of several high profile clinical trials targeting γ-secretase activity which ultimately failed to reach their clinical endpoints [9, 10]. If γ-secretases are implicit in the ectodomain shedding of ADAM10 and this mechanism proves to be protective against AD, it may provide rationale for the disappointing results of these prior AD clinical trials, although further investigation is certainly warranted.
To ascertain whether changes in the activity of sADAM10 were independent of changes in the overall expression profile of ADAM10, we assessed the enzymatic efficiency of sADAM10 by examining activity as a function of expression. sADAM10 enzymatic efficiency was APOE-dependent for the ND samples and coincided with the results we observed in the cell free activity assay (APOE2/2> APOE3/3>APOE4/4) while, on the other hand, the AD samples showed no significant changes in sADAM10 efficiency across APOE genotypes. Previously, it was demonstrated that differences in lipidation states between apoE isoforms confers altered binding affinities to Aβ (apoE2>apoE3> apoE4) [30]. Therefore, it is possible that the altered influence of apoE on ADAM10 activity we observed in our cell free assay and the APOE-dependent enzymatic efficiency in the ND human soluble samples may be attributed to lipidation status. Alternatively, in the AD samples, the influence of APOE on sADAM10 enzymatic efficiency was absent. Currently, it is not known whether there are any differences in the lipidation status of apoE in the brains of AD patients versus ND subjects, although it is a burgeoning area of interest in this field. Interestingly, the ADAM10 transcription factors RAR and RXR, which are known to be defective in AD [36], are involved in the regulation of apoE expression and their lipidation status [14, 37]. Therefore, these elements may play a key role in the effect of apoE on ADAM10 activity as well as ADAM10 expression.
In addition, to changes in the expression and enzymatic efficiency of sADAM10, a strong inverse correlation was identified in the ND control samples between these two variables. We propose that, under ND conditions, the increased shedding of ADAM10 observed in the APOE4/4 genotype may be part of a feedback mechanism that compensates for the lower enzymatic efficiency of sADAM10 due to the presence of apoE4 (Fig. 5). Previous studies have demonstrated that upon proteolytic cleavage of ADAM10 at the cellular membrane and subsequent γ-secretase processing, the ADAM10-ICD translocates to areas of the nucleus involved in active transcription and contain the ADAM10 transcription factors RAR and RXR [14, 15, 38]. We propose that ectodomain shedding of ADAM10 prompts the release of the ADAM10-ICD which may have a role in regulating ADAM10 expression as well as other ADAM10 family members in a manner similar to other protein ICDs [39–41]. However, in AD, the RAR/RXR transcription processes are known to be defective and, as a result, the ability to regulate ADAM10 levels may be compromised [36]. In support of this, we observed no correlation between sADAM10 expression and enzymatic efficiency in the AD samples. Importantly, therapies aimed at restoring the normal function of these transcriptional areas [42] have been shown to result in increased expression of ADAM10 [14], suggesting this mechanism may be a viable therapeutic target to restore the normal feedback mechanism and boost sADAM10 levels. Our data also suggests that if the efficiency of the ADAM10 transcription mechanism is reduced, subjects with an APOE4 genotype may be affected to a greater degree as sADAM10 activity is inherently reduced in these individuals due to the presence of the apoE4 isoform.
Fig. 5.
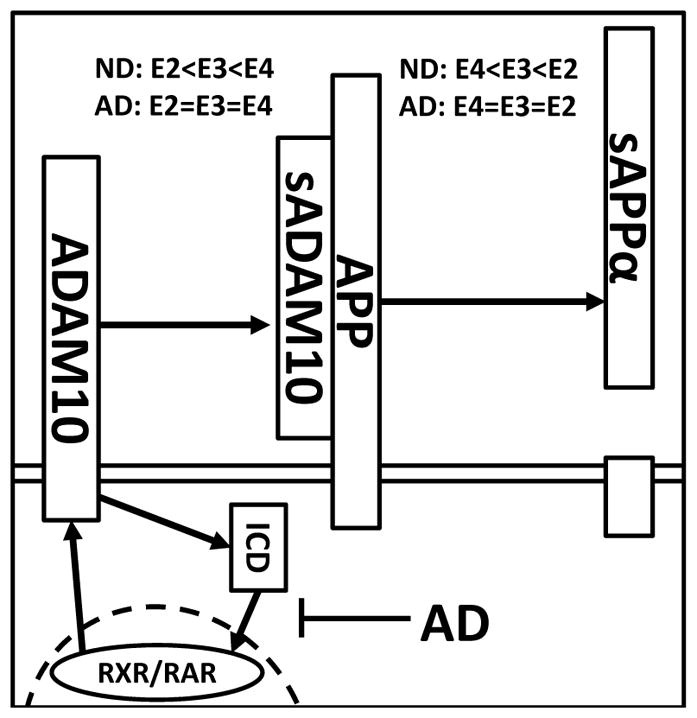
Proposed schematic of one potential mechanism for sADAM10 regulation in AD and the role of apoE genotype. ADAM10 expression is driven by RAR/RXR transcription. At the cell surface, full-length ADAM10 undergoes proteolysis in an apoE genotype dependent manner (apoE2<apoE3<apoE4) resulting in soluble extracellular fragment (sADAM10) and an intracellular domain fragment (ICD). sADAM10 metabolizes full length AβPP to produce sAβPPα, a process that is apoE dependent (apoE2>apoe3>apoE4). Under normal conditions, the ICD fragment of ADAM10 transitions to the nucleus where it induces ADAM10 expression. In APOE4 carriers, sADAM10 activity is less efficient than in other APOE genotypes and, in response, the ADAM10 protein population is increased to compensate for the lack of ADAM10 efficiency. However, in AD individuals the RAR/RXR machinery is defective and, as a result, ADAM10 levels cannot be sufficiently upregulated to overcome the diminished efficiency of ADAM10 in APOE4 carriers. Consequently, there is less processing of AβPP by sADAM10, which can lead to increased Aβ synthesis and the accumulation of Aβ peptides in the brains of AD subjects with an APOE4 genotype.
Overall, our studies demonstrate an isoform-dependent effect of apoE on the activity of the α-secretase, ADAM10, which ultimately impacts the processing of sAβPPα in the AD brain. Specifically, the enzymatic efficiency of ADAM10 is diminished in subjects with an APOE4 genotype. Furthermore, we found that ND subjects are able to compensate for the reduced enzymatic efficiency of sADAM10, imposed by apoE4 isoform, by increasing sADAM10 expression levels. This compensatory mechanism is dysfunctional or absent in individuals with AD, which may describe the elevated Aβ burden found in the brains of AD patients carrying the APOE4 allele. While the contribution of apoE to Aβ-related pathology is likely mediated through several pathways (Aβ synthesis, degradation, and BBB clearance, etc.), these findings provide new insight into apoE function and the development of AD. As such, restoration of the enzymatic efficiency of sADAM10 or increasing the expression of sADAM10 may be a novel therapeutic strategy to mitigate Aβ pathology in the AD brain.
Acknowledgments
We are grateful to the Banner Sun Health Research Institute Brain and Body Donation Program of Sun City, Arizona for the provision of the human brain tissue specimens. The Brain and Body Donation Program is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. Human brain tissue specimens were also obtained from the University of Maryland Brain and Tissue Bank which is a Brain and Tissue Repository of the NIH NeuroBioBank (Baltimore, MD) and the Mount Sinai NIH Brain and Tissue Repository (New York, NY). This research was supported by the Roskamp Foundation and the National Institute on Aging of the National Institutes of Health under award number R01AG041971. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Send Orders for Reprints to reprints@benthamscience.ae
CONFLICT OF INTEREST
The authors confirm that this article content has no conflict of interest.
References
- 1.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, et al. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann Neurol. 1999;46:860–6. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 2.Koyama A, Okereke OI, Yang T, Blacker D, Selkoe DJ, Grodstein F. Plasma Amyloid β as a predictor of dementia and cognitive decline: a systematic review and meta-analysis. Arch Neurol. 2012;69:824–31. doi: 10.1001/archneurol.2011.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Querfurth HW, LaFerla FM. Alzheimer’s Disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 4.Ghiso J, Frangione B. Amyloidosis and Alzheimer’s disease. Adv Drug Deliv Rev. 2002;54:1539–51. doi: 10.1016/s0169-409x(02)00149-7. [DOI] [PubMed] [Google Scholar]
- 5.LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer’s disease. Nat Rev Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 6.Turner PR, O’Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70:1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- 7.Selkoe DJ. Resolving controversies on the path to Alzheimer’s therapeutics. Nat Med. 2011;17:1060–5. doi: 10.1038/nm.2460. [DOI] [PubMed] [Google Scholar]
- 8.Vassar R, Kandalepas PC. The β-secretase enzyme BACE1 as a therapeutic target for Alzheimer’s disease. Alzheimers Res Ther. 2011;3:20. doi: 10.1186/alzrt82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369:341–50. doi: 10.1056/NEJMoa1210951. [DOI] [PubMed] [Google Scholar]
- 10.Imbimbo BP. Why did tarenflurbil fail in Alzheimer’s disease? J Alzheimers Dis. 2009;17:757–60. doi: 10.3233/JAD-2009-1092. [DOI] [PubMed] [Google Scholar]
- 11. [accessed April 14, 2016];Lilly Halts Phase 2 Trial of BACE Inhibitor Due to Liver Toxicity | ALZFORUM. n.d http://www.alzforum.org/news/research-news/lilly-halts-phase-2-trial-bace-inhibitor-due-liver-toxicity.
- 12.Bell KFS, Zheng L, Fahrenholz F, Cuello AC. ADAM-10 overexpression increases cortical synaptogenesis. Neurobiol Aging. 2008;29:554–65. doi: 10.1016/j.neurobiolaging.2006.11.004. [DOI] [PubMed] [Google Scholar]
- 13.Hasebe N, Fujita Y, Ueno M, Yoshimura K, Fujino Y, Yamashita T. Soluble β-amyloid precursor protein alpha binds to p75 neurotrophin receptor to promote neurite outgrowth. PloS One. 2013;8:e82321. doi: 10.1371/journal.pone.0082321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tippmann F, Hundt J, Schneider A, Endres K, Fahrenholz F. Upregulation of the α-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 2009;23:1643–54. doi: 10.1096/fj.08-121392. [DOI] [PubMed] [Google Scholar]
- 15.Tousseyn T, Thathiah A, Jorissen E, Raemaekers T, Konietzko U, Reiss K, et al. ADAM10, the rate-limiting protease of regulated Intramembrane proteolysis of notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the γ-secretase. J Biol Chem. 2009;284:11738–47. doi: 10.1074/jbc.M805894200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim M, Suh J, Romano D, Truong MH, Mullin K, Hooli B, et al. Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate α-secretase activity. Hum Mol Genet. 2009;18:3987–96. doi: 10.1093/hmg/ddp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chartier-Harlin MC, Parfitt M, Legrain S, Pérez-Tur J, Brousseau T, Evans A, et al. Apolipoprotein E, epsilon 4 allele as a major risk factor for sporadic early and late-onset forms of Alzheimer’s disease: analysis of the 19q13.2 chromosomal region. Hum Mol Genet. 1994;3:569–74. doi: 10.1093/hmg/3.4.569. [DOI] [PubMed] [Google Scholar]
- 18.Ashford JW. APOE genotype effects on Alzheimer’s disease onset and epidemiology. J Mol Neurosci. 2004;23:157–65. doi: 10.1385/JMN:23:3:157. [DOI] [PubMed] [Google Scholar]
- 19.Schmechel DE, Saunders AM, Strittmatter WJ, Crain BJ, Hulette CM, Joo SH, et al. Increased amyloid beta-peptide deposition in cerebral cortex as a consequence of apolipoprotein E genotype in late-onset Alzheimer disease. Proc Nat Acad Sci USA. 1993;90:9649–53. doi: 10.1073/pnas.90.20.9649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, et al. Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2000;97:2892–7. doi: 10.1073/pnas.050004797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dodart J-C, Bales KR, Johnstone EM, Little SP, Paul SM. Apolipoprotein E alters the processing of the β-amyloid precursor protein in APPV717F transgenic mice. Brain Res. 2002;955:191–9. doi: 10.1016/s0006-8993(02)03437-6. [DOI] [PubMed] [Google Scholar]
- 22.Ewers M, Zhong Z, Bürger K, Wallin A, Blennow K, Teipel SJ, et al. Increased CSF-BACE 1 activity is associated with ApoE-epsilon 4 genotype in subjects with mild cognitive impairment and Alzheimer’s disease. Brain J Neurol. 2008;131:1252–8. doi: 10.1093/brain/awn034. [DOI] [PubMed] [Google Scholar]
- 23.Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, et al. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest. 2004;113:1456–64. doi: 10.1172/JCI20864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bachmeier C, Paris D, Beaulieu-Abdelahad D, Mouzon B, Mullan M, Crawford F. A multifaceted role for apoE in the clearance of beta-amyloid across the blood-brain barrier. Neurodegener Dis. 2013;11:13–21. doi: 10.1159/000337231. [DOI] [PubMed] [Google Scholar]
- 25.Manelli AM, Bulfinch LC, Sullivan PM, LaDu MJ. Aβ42 neurotoxicity in primary co-cultures: effect of apoE isoform and Aβ conformation. Neurobiol Aging. 2007;28:1139–47. doi: 10.1016/j.neurobiolaging.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bachmeier C, Shackleton B, Ojo J, Paris D, Mullan M, Crawford F. Apolipoprotein E isoform-specific effects on lipoprotein receptor processing. Neuromol Med. 2014;16:686–96. doi: 10.1007/s12017-014-8318-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Irizarry MC, Deng A, Lleo A, Berezovska O, Arnim CAFV, Martin-Rehrmann M, et al. Apolipoprotein E modulates γ-secretase cleavage of the amyloid precursor protein. J Neurochem. 2004;90:1132–43. doi: 10.1111/j.1471-4159.2004.02581.x. [DOI] [PubMed] [Google Scholar]
- 28.Wahrle SE, Jiang H, Parsadanian M, Hartman RE, Bales KR, Paul SM, et al. Deletion of abca1 increases Aβ deposition in the PDAPP transgenic mouse model of Alzheimer disease. J Biol Chem. 2005;280:43236–42. doi: 10.1074/jbc.M508780200. [DOI] [PubMed] [Google Scholar]
- 29.Wahrle SE, Jiang H, Parsadanian M, Kim J, Li A, Knoten A, et al. Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J Clin Invest. 2008;118:671–82. doi: 10.1172/JCI33622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tokuda T, Calero M, Matsubara E, Vidal R, Kumar A, Permanne B, et al. Lipidation of apolipoprotein E influences its isoformspecific interaction with Alzheimer’s amyloid beta peptides. Biochem J. 2000;348:359–65. [PMC free article] [PubMed] [Google Scholar]
- 31.Bernstein H-G, Bukowska A, Krell D, Bogerts B, Ansorge S, Lendeckel U. Comparative localization of ADAMs 10 and 15 in human cerebral cortex normal aging, Alzheimer disease and Down syndrome. J Neurocytol. 2003;32:153–60. doi: 10.1023/b:neur.0000005600.61844.a6. [DOI] [PubMed] [Google Scholar]
- 32.Parkin E, Harris B. A disintegrin and metalloproteinase (ADAM)-mediated ectodomain shedding of ADAM10. J Neurochem. 2009;108:1464–79. doi: 10.1111/j.1471-4159.2009.05907.x. [DOI] [PubMed] [Google Scholar]
- 33.Tyler SJ, Dawbarn D, Wilcock GK, Allen SJ. α- and β-secretase: profound changes in Alzheimer’s disease. Biochem Biophys Res Commun. 2002;299:373–6. doi: 10.1016/s0006-291x(02)02635-9. [DOI] [PubMed] [Google Scholar]
- 34.Colciaghi F, Borroni B, Pastorino L, Marcello E, Zimmermann M, Cattabeni F, et al. α-Secretase ADAM10 as well as αAPPs is reduced in platelets and CSF of Alzheimer disease patients. Mol Med Camb Mass. 2002;8:67–74. [PMC free article] [PubMed] [Google Scholar]
- 35.Manzine PR, Barham EJ, Vale Fde AC do, Selistre-de-Araújo HS, Iost PSC Cominetti MR. Correlation between mini-mental state examination and platelet ADAM10 expression in Alzheimer’s disease. J Alzheimers Dis. 2013;36:253–60. doi: 10.3233/JAD-130125. [DOI] [PubMed] [Google Scholar]
- 36.Goodman AB, Pardee AB. Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc Nat Acad Sci. 2003;100:2901–5. doi: 10.1073/pnas.0437937100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao J, Fu Y, Liu C-C, Shinohara M, Nielsen HM, Dong Q, et al. Retinoic acid isomers facilitate apolipoprotein E production and lipidation in astrocytes through the retinoid X receptor/retinoic acid receptor pathway. J Biol Chem. 2014;289:11282–92. doi: 10.1074/jbc.M113.526095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lallemand-Breitenbach V, de Thé H. PML Nuclear Bodies. Cold Spring Harb Perspect Biol. 2010;2:a000661. doi: 10.1101/cshperspect.a000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perrot V, Rechler MM. Characterization of insulin inhibition of transactivation by a C-terminal fragment of the forkhead transcription factor Foxo1 in rat hepatoma cells. J Biol Chem. 2003;278:26111–9. doi: 10.1074/jbc.M212750200. [DOI] [PubMed] [Google Scholar]
- 40.El Fiky A, Arch AE, Krolewski JJ. Intracellular domain of the IFNaR2 interferon receptor subunit mediates transcription via Stat2. J Cell Physiol. 2005;204:567–73. doi: 10.1002/jcp.20305. [DOI] [PubMed] [Google Scholar]
- 41.Schöler J, Ferralli J, Thiry S, Chiquet-Ehrismann R. The intracellular domain of teneurin-1 induces the activity of microphthalmia-associated transcription factor (MITF) by binding to transcriptional repressor HINT1. J Biol Chem. 2015;290:8154–65. doi: 10.1074/jbc.M114.615922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu JH, Nakajima A, Nakajima H, Diller LR, Bloch KD, Bloch DB. Restoration of promyelocytic leukemia protein-nuclear bodies in neuroblastoma cells enhances retinoic acid responsiveness. Cancer Res. 2004;64:928–33. doi: 10.1158/0008-5472.can-03-1199. [DOI] [PubMed] [Google Scholar]