

Abstract
Despite the recent identification of over 40 missense heterozygous RELN mutations in ASD, none of these has been functionally characterized. Reelin is an integral signaling ligand for proper brain development and postnatal synapse function – properties likely disrupted in ASD patients. We find that the R2290C mutation, which arose de novo in an affected ASD proband, and other analogous mutations in RXR domains reduce protein secretion. Closer analysis of RELN R2290C heterozygous neurospheres reveals upregulation of Protein Disulfide Isomerase A1, best known as an ER-chaperone protein, which has been linked to neuronal pathology. This effect is recapitulated in a heterozygous RELN mouse mutant that is characterized by defective Reelin secretion. These findings suggest that both a deficiency in Reelin signaling and pathologic impairment of Reelin secretion may contribute to ASD risk.
Keywords: AUTS1, RELN Orleans, Dab1, autism, PDI, eIF2α
Introduction
Autism spectrum disorder (ASD; OMIM 209850) is characterized by deficits in verbal and nonverbal communication, as well as socialization and behavioral deficits (American Psychiatric Association & American Psychiatric Association. DSM-5 Task Force. 2013). According to the most recent estimates, as many as 1 in 45 children were diagnosed with ASD in 2014 in the United States, which represents a 10-fold increase in prevalence over the past 40 years (Zablotsky et al. 2015). Despite this prevalence, at best, only 20% of cases are caused by defined monogenetic disturbances such as fragile X syndrome or tuberous sclerosis (Persico & Napolioni 2013). Early linkage analysis studies to identify genetic causes of idiopathic ASD identified 7q22 as the first autism susceptibility locus (AUTS1) (IMGSAC 2001, IMGSAC 1998, Autism Genome Project et al. 2007). Since then over 15 linkage studies examining RELN as a risk factor for ASD have been published with both positive and negative findings, consistent with locus heterogeneity (Anney et al. 2012). A recent meta-analysis did, however, find evidence that at least one RELN SNP likely segregates with ASD (r362691) (Wang et al. 2014b).
More compelling suggestions that RELN has a role in the disease have come from exome sequencing of ASD families. These studies have identified several inherited and de novo RELN mutations in patients (Bonora et al. 2003, Iossifov et al. 2014, Neale et al. 2012, De Rubeis et al. 2014, Koshimizu et al. 2013, Yuen et al. 2015, Zhang et al. 2015, Lammert & Howell 2016). Based on the RELN Q416X nonsense allele, the false discovery rate (FDR) for RELN in ASD is estimated at <0.05 (De Rubeis et al. 2014). Further evidence for a role in ASD is the decreased Reelin protein expression in the blood and brain of ASD patients (Fatemi et al. 2005, Fatemi et al. 2002, Fatemi et al. 2001). Consistent with a role in ASD, RELN is expressed and functionally important for the developing cerebral cortex and cerebellum, which are two areas with anatomical findings in ASD patients (Fatemi et al. 2012, Wang et al. 2014a, Tsai et al. 2012, Butts et al. 2014, Stoner et al. 2014).
The Reelin protein is a ligand for LDL-superfamily receptors ApoER2 and VLDLR. Its binding to the receptors leads to recruitment and tyrosine phosphorylation of the cytoplasmic adaptor protein Dab1, which ultimately regulates cellular behavior to influence neuron positioning in the developing brain and n-methyl-d-aspartate receptor (NMDAR) activity (Bock et al. 2004, Howell et al. 1997, Rice & Curran 2001, Hiesberger et al. 1999, D'Arcangelo et al. 1999, Jossin & Cooper 2011, Beffert et al. 2002, Hoe et al. 2006). The intersection of the Reelin-Dab1 signaling pathway with both NMDAR function and mTOR signaling positions it at the intersection of two major ASD protein networks and emphasizes the importance of understanding how RELN participates in the etiology of ASD (Lammert & Howell 2016).
Despite mounting evidence linking RELN and ASD, there is no experimental evidence supporting a mechanism for the role of Reelin in ASD. Homozygotic deficiency in RELN is uncommon and is associated with lissencephaly with cerebellar hypoplasia (Hong et al. 2000, Zaki et al. 2007). The heterozygous mutations that are identified in ASD probands thus far have not been characterized, and only a few mutations are clearly loss-of-function for the canonical pathway, since they would not produce protein that binds the receptors (Lammert & Howell 2016).
Here, we test the hypothesis that the de novo RELN R2290C mutation is not a benign polymorphism and has characteristics that may contribute to ASD. This mutation falls in a class of mutations that map to a conserved arginine-amino acid-arginine (RXR) motif (Lammert & Howell 2016). We find R2290C and the other RXR mutations reduce Reelin secretion. In addition, protein disulfide isomerase A1 (PDIA1) expression is increased in neurospheres that are heterozygous (+/−) for RELN R2290C as well as in the cerebella of RELN Orleans (Orl) +/− mice, which also have reduced Reelin protein secretion. PDIA1 is best known as an ER resident chaperone that assures correct disulfide bond formation in nascent proteins (Parakh & Atkin 2015), but its overexpression may contribute to neuropathology (Perri et al. 2015, Zeeshan et al. 2016). These results suggest that at least a subset of RELN mutations found in ASD patients are not exclusively loss-of-function, but also cause other cellular perturbations.
Materials and Methods
Animals
Animals were used in compliance with approved protocols by the Animal Care and Use Committee for SUNY Upstate Medical University following NIH guidelines. Timed pregnant Swiss Webster dams were purchased from Taconic. RELN Orleans (Orl) mice were kindly provided by Dr. Patricia Phelps (UCLA, USA) and were maintained on the BALB/c background. RELN null-allele mice were maintained on a C57BL6 background and were from Jackson Laboratories.
Alignments and Structures
Clustal Omega was used to generate the alignment of Reelin sub-repeat domains using boundaries specified by NCBI Conserved Domains for GenBank: AACS1105.1. UCSF Chimera was used to generate the rendering of RCSB Protein Data Bank PDB 2E26.
Constructs
To eliminate the confusion of comparing mouse, human, full-length, and fusion protein, the numbering of the residue in the human full-length Reelin protein was used throughout. The pcDNA3 full-length wild-type mouse RELN construct pcRL was graciously provided by Dr. Gabriella D’Arcangelo (Rutgers, USA) (D'Arcangelo et al. 1997). To generate the point mutations in full-length RELN the region of interest was subcloned in pBS and the mutations were introduced by QuikChange mutagenesis (Agilent Technologies) and then re-cloned into pCRL (detailed method available upon request). Wild-type version of the RRfp and Reln56SD fusion constructs were generated by gene synthesis (GeneArt), cloned into pCDNA, and mutations were introduced as above. All mutant constructs were validated by bidirectional sequencing. The PA-tagged WT RELN construct was a generous gift from Dr. Junichi Takagi (Osaka University, Japan) (Fujii et al. 2014).
Antibodies
Anti-Reelin (G10; Millipore; AB_565117), anti-Flag (Sigma-Aldrich; AB_262044), anti-phosphotyrosine (4G10; Millipore; AB_916370), anti-β-actin (Sigma-Aldrich; AB_476744), anti-PA tag (AB_10920577; WAKO), anti-BIP (AB_732737; Abcam), anti-PERK (AB_10831515), anti-PDI (AB_2156433; against PDIA1), anti-Ero1-Lα (AB_823684), anti-phospho-eIF2α (AB_10692650), anti-total-eIF2α (AB_330951; Cell Signaling), and anti-V5 (AB_2556564; Invitrogen). Anti-Dab H1 antibody was a generous gift from Dr. Andre Goffinet (University of Louvain, Belgium).
Cell Culture
HeLa cells and HEK293 cells were grown in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher) supplemented with 10% Fetal Bovine Serum and 1% Penicillin-Streptomycin. All cell cultures were maintained at 37°C at 5% CO2. Stable HEK293 wild-type (WT) Reelin-expressing cells were the kind gift of Dr. Eckart Förster (Zelluläre Neurobiologie, Hamburg) (Forster et al. 2002). Stable cell lines of HEK293 cells secreting full-length R2290C or R2290H Reelin were generated in the same manner. Transfections were performed using X-tremeGENE HP following the manufacturer’s protocols (Sigma). Cortical neuronal cultures were isolated from embryonic (E) day 15 or 16 and grown in neurobasal media supplemented with 2% B27 (Invitrogen) and 1% Pen-Strep. Cells were plated at a density of 3×105/cm2 on poly-L-lysine coated polystyrene dishes. One quarter of culture media was replaced every 2-3 days.
Genomic Editing
P19 embryonal carcinoma cells (McBurney et al. 1982) were edited using CRISPR/CAS9 and a single-stranded template (ssODN) with nucleotide sequence to alter Arg 2290 (R2291 in mouse) to Cys as previously described (Ran et al. 2013). Guide (sg) RNA sequences proximal to the codon for R2290 were generated based on the Zhang lab CRISPR Design tool (http://crispr.mit.edu/). Each guide sequence was then inserted into a vector designed to express the guide and wild-type Cas9 [pSpCas9(BB)-2A-Puro (PX459); Addgene; (Ran et al. 2013)]. One guide (F1; CAGATGGCTGCCACCAGCGG) was selected based on its effectiveness at generating indels in P19 cells (not shown). P19 cells were transfected with F1 sgRNA-containing Cas9 vector and a single-stranded oligonucleotide (ssODN) template using X-tremeGENE HP (Sigma). The ssODN template extended 50 bp on either side of the R2290 codon and had at its core (TCGACAtGtCTCaGa) mutations (lower case) to change the R2290 codon, introduce a PciI site and prevent the F1 guide from functioning (complete sequence available on request). Twenty-four hours after transfection, cells were split and selected in puromycin (1μg/mL) for 48 hours. Edited P19 cell colonies were identified by PCR amplification of the target area followed by PciI digest and forward and reverse strand sequencing. Heterozygous line 21 was propagated from a single cell to ensure clonality and the wild-type control line was processed identically.
P19 Cell Differentiation
We found that P19 cells grown as aggregates and treated with retinoic acid (RA) (10μM; Sigma) for 48 h in 50% MED-II media and 50% DMEM, 10% FBS, 1% Penicillin-streptomycin, 0.5% GlutaMAX (Gibco) differentiated into neuron-specific class III β-tubulin positive, Reelin-producing cells by 2-4 DIV and had robust Reelin expression by 8 days of differentiation (Figure 2B and data not shown). Cells differentiated with this protocol do not augment Dab1 phosphorylation in response to Reelin stimulation (data not shown). MEDII media, previously used to influence the differentiation of stem cells, refers to HepG2 cell-conditioned media (Rathjen et al. 1999). After the first 48 h, media was replaced every 2 days with no RA.
Reelin Stimulation Assay
Reelin stimulation was done essentially as described with modifications (Howell et al. 1999). Reelin-conditioned media (RCM) was collected from stably transfected cells in neurobasal media with 1% penicillin-streptomycin (Thermo Fisher) for three days and pooled, after which it was concentrated 10-fold with a 100kDa Amplicon (Millipore) filter (Matsuki et al. 2010). Control-conditioned media (CCM) was generated in the same manner by collecting media from untransfected 293 cells. Concentration of Reelin was normalized based on Western blot densitometry. Cortical neuron cultures (DIV 5 or 6) were rinsed with PBS and treated for 20 minutes with wild type, R2290C, or R2290H RCM, neurobasal (NB) media, or CCM. To stop the reaction, media was removed, cells were washed (1x with PBS) and then lysed in RIPA (0.15M NaCl, 1% NP40, 1% sodium deoxycholate, 0.1% SDS, 50mM NaF, 2mM EDTA, 1 mM phenylarsinine oxide, protease inhibitor [complete mini, EDTA-free; Roche], phosphatase inhibitor cocktail 1 [Sigma]) by sonication. Western blots were imaged by near-infrared fluorescence (Odyssey CLx, LI-COR Biosciences).
Western Blotting
Lysates and media were collected at 48 hours post transfection, cleared by centrifugation at 15,000g × 20 minutes and resolved by SDS PAGE, prior to immunoblotting with indicated antibodies. Blots were developed with ECL chemiluminescence substrate (Thermo Scientific) and visualized on the chemidoc (Biorad). Protein bands were analyzed by densitometry using ImageJ (NCBI). Lysate values are normalized to actin. For mouse experiments, cerebella from both male and female mice were collected and frozen. Samples were homogenized in RIPA buffer and processed as above for Western blotting. The extracellular Reelin ratio is reported as the fraction of Reelin media signal to Reelin lysate signal. Graphing and statistics were generated using Prism Graphpad software and Microsoft Excel (n≥3 for all experiments). Groups were compared using t-test or 1-way ANOVA with Dunnett’s posthoc test, and significance reported as p<0.05 or lower.
Immunocytochemistry
For differentiated P19 cells, spheres were fixed in 4% PFA in PBS for 5 minutes, washed 3 times in PBS, then stored in 20% sucrose (Sigma) in PBS overnight. Cells were stained with methylene-blue then mounted in OCT, cryosectioned at 12μm thickness, and melted directly onto glass slides. Slides were allowed to dry, washed with PBS, then cold absolute methanol for 10 minutes, followed by a PBS wash. Slides were blocked for 1 hour at room temperature in PBST (2% w/v BSA, .5% Triton X 100, PBS). Primary and secondary antibody stains were carried out in PBST. Coverslips were prepared with Vectashield (Vector Laboratories) containing DAPI. Images were taken on a Zeiss Imager.A2 with Nikon Elements Software package. Images were formatted and compiled with Adobe Photoshop.
Pulse-Chase Assay
For experiments with full-length Reelin, HEK239 stable cells lines (WT, R2290C, or R2290H) were plated at 2×106 cells per well in collagen-coated 12-well polystyrene dishes. On the labeling day, cells were methionine-starved for 30 min in pulse-labeling (P-L) media (DMEM without methionine, 10% FBS, 50mM HEPES, 200mM glutamine, 1% pen-strep) then treated with P-L media (500μL) containing 0.2mCi/mL 35S L-methionine (EasyTag; Perkin Elmer) for 1 hour. The cold chase was commenced by removing radioactive media, washing with warmed PBS, and then incubating in P-L media containing cold methionine [1× Methionine (Sigma)]. Culture media and lysates were collected at 5 minutes (0 h) and every hour thereafter. Lysates were collected in RIPA; DNA was sheared using a 22G needle and clarified by centrifugation (13,000 g). Samples (200 μL) were immunoprecipitated with anti-Reelin (1 μg; G10) for full-length Reelin, followed by addition of Protein A-G plus Sepharose beads (Santa Cruz) at 4°C for 4 hours and 4 serial washes with RIPA buffer. Samples were resolved by SDS-PAGE as above, dried, and visualized by phosphorimaging (Molecular Imager; Biorad). Reelin counts for lysates (CL) and media (CM) were normalized to the sample’s corresponding 0 hour lysate sample (C0). Averages were plotted and non-linear regression line automatically fitted. Significance was determined using 2-way ANOVA with Dunnett’s posthoc test and significance reported as p<0.05.
Real time (Q)-PCR
P19 neurospheres at 8 days of differentiation were collected and rinsed with PBS. RNA was isolated using RNEasy Mini Kit (Qiagen) and cDNA synthesized using QuantiTect Reverse Transcription Kit (Qiagen). Spliced XBP-1 Q-PCR was performed with primers as previously described (van Schadewijk et al. 2012). Actin and TBP were used as controls. Results were analyzed with Biorad CFX manager and significant differential expression determined using delta delta Cq and t-test.
RNAseq
RNA was extracted from three 6-week old male RELN Orl or wild-type cerebella using the RNeasy Mini Kit (Qiagen). Samples were given a number and all processing was done in a double-blinded fashion. Libraries were prepped using the Stranded Total RNA kit (Illumina) and validated on the 2100 Bioanalyzer (Agilent). The libraries were run on the Illumina NextSeq 500 instrument using the NextSeq 75 cycle High Output Reagent kit. Seventy-five base pair single end reads were performed with libraries loaded at 1.9pM with 1% Phi X control. Reads were aligned using the TopHat2 application in BaseSpace (Illumina). This procedure involved first filtering out mitochondrial and ribosomal sequences, followed by trimming of 2 bases from the 5’ end of each read, and alignment to the Mm10 build of the mouse genome using the Bowtie 1 aligner. After alignment, quantification of transcript abundance was performed using Cufflinks for all annotated reference genes and transcripts that were detected in the samples, with data reported as Fragments Per Kilobase of exon per Million fragments (FPKM) values. Differential expression was then performed with Cuffdiff 2.1.1, with results reported as log2 (Heterozygous/Wildtype), and p values adjusted using the FDR. For this report, we and analyze genes involved in the UPR (unfolded protein response) network or Simons Foundation Autism Database (AutDB) (Supplemental File 1, GEO accession number pending).
Statistical Analysis
Pairwise comparisons were done by Student’s t-test (e.g. Figure 2E, 2H). When more than two samples were compared, we used 1-way or 2-way ANOVA with Dunnett’s post hoc test for comparing one or more measurements, respectively (e.g. Figure 1E vs. 1G). See the relevant methods sections for details. Data analysis was done in Microsoft Excel and GraphPad Prism 7. Due to correction for the multiple testing done in the RNAseq analysis, we may have false negatives or type two errors in our analysis, and therefore may have overlooked an ER stress gene that is differentially expressed in our samples.
Figure 2. Decreased extracellular Reelin expression by R2290C+/− compared to wild-type neurospheres.
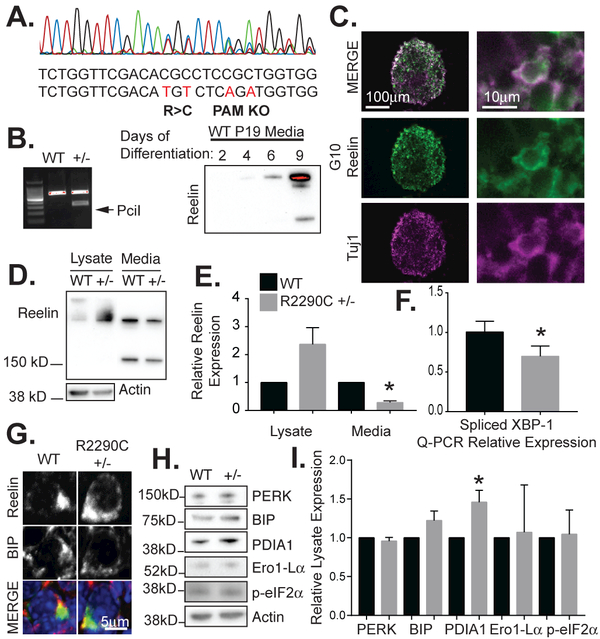
(A) A heterozygous R2290C P19 cell line was generated by CRISPR-Cas9 genome editing and verified by sequencing that showed the expected mutation and silent changes indicated below. Silent mutations inactivate the PAM sequence and prevent further Cas9 cleavage in a P19 cell clone. (B) P19 cells grown as neurospheres in MEDII media produce extracellular Reelin by 8 days of differentiation. Restriction digest by PciI demonstrates the heterozygous R2290C mutation. (C) Neurospheres were cryosectioned and stained for Reelin (anti-Reelin, G10 antibody) or Tuj1, a neural-specific marker. (D) Neurosphere lysates and media were collected and analyzed by Western blot for Reelin (G10). (E) The fraction of Reelin in the media in R2290C+/− neurospheres (media/lysate) was significantly decreased compared to wild-type neurospheres (n=4, error bars = SEM, *p value <0.05 by t-test), while the levels of Reelin in the lysates (lysates/actin) trended higher but were not statistically different. (F) Q-PCR showed decreased XBP-1 splicing in R2290C+/− neurospheres compared to WT. (G) Reelin expression in WT and R2290C+/− neurspheres co-localizes with BIP, an ER protein. (H) Western blot of ER stress markers comparing WT and R2290C+/− neurosphere lysates. (I) PDI was significantly increased in R2290C+/− lysates compared to control (n=4, error bars = SEM, *p value <0.05 by t-test).
Figure 1. The RELN R2290C and other ASD mutations reside in a conserved RXR consensus and impair Reelin secretion.

(A) Rendering of Reelin repeat 5 and 6 crystal structure (PDB 2E26; Chimera). The positions of the R2290C and R2292C mutations in the RXR motif of 5B are shown facing into the hydrophilic pore of RELN repeat 5. The corresponding locations of the RXR motif in an A repeat are also indicated. The position of the receptor-binding loops in 6A are indicated. (B) Alignment of the conserved region of the Reelin sub-repeat domains (Clustal Omega). The RXR consensus element is boxed and indicated below. (C) Schematic of Reelin structure showing an F-spondin homology domain, an N-terminal domain unique to Reelin, and 8 Reelin repeat domains. Each Reelin repeat domain contains an A and B sub-repeat domain (blue ovals) connected by an EGF domain (green ovals). ASD-associated mutations are indicated above their respective repeat. (D) Less Reelin was detected in media of HeLa cells transfected with full-length mutant (R2290C, R2290H, R1742Q, or R1742W) as compared to wild-type (WT) Reelin by Western blotting (anti-Reelin G10), whereas equal or greater amounts were observed in the cell lysates. One full-length (>400 kD), smeared Reelin band was observed in the cell lysates and three products (full length and two cleavage products: 410, 380, and 180 kD) were observed in the media, as expected (Jossin et al. 2004). Reelin is not expressed in vector-alone transfected HeLa cells (not shown). (E) The ratio of Reelin signal in the media (M) to Reelin lysate signal (L) was significantly greater for WT than mutant Reelin (n=4, error bars ± SEM, and * p value < 0.05 by 1-way ANOVA with Dunnett’s post hoc test). (F) WT Reelin metabolically labeled with 35S-methionine was most abundant in 293 cell lysates at the beginning of a cold chase, but by two hours the majority was detected in the cell media. In contrast, the majority of radiolabeled R2290C and R2290H mutant RELN remained in cell lysates over a 4h time course, as detected by phosphor-imaging of immunoprecipitated Reelin (anti-G10). (G) In the 293 cell lysate the Reelin counts (CL) at times indicated relative to counts at time 0 (C0) decreased more rapidly for WT Reelin than the R2290C and R2290H mutants (n=3, error bars ± SEM, * p<0.05 R2290C, # p<0.05 R2290H by 2-way ANOVA with Dunnett’s post hoc test). (H) In the media the Reelin counts (CM) at indicated times relative to lysate counts at time 0 (C0) increased more rapidly for WT than R2290C or R2290H (n=3, error bars ± SEM, * p<0.05 R2290C, # p<0.05 R2290H by 2-way ANOVA with Dunnett’s post hoc test).
Results
The de novo RELN R2290C Mutation Falls in an RXR Protein Consensus Element That Is Mutated in Other ASD Patients
Reelin is a large (440kDa) protein composed of an N-terminal F-spondin homology domain, a unique region, and 8 highly homologous Reelin repeat domains (D'Arcangelo et al. 1995, Panteri et al. 2006, Yasui et al. 2010, Yasui et al. 2007, Nogi et al. 2006). Each repeat is divided further into homologous A and B sub-repeats that are separated by a hinge-like EGF domain. Each sub-repeat contains primarily β-sheets oriented in an 11-stranded jellyroll containing a hydrophilic pore (Figure 1A).
The RELN R2290C mutation maps to an RXR consensus element that is revealed by aligning the 16 Reelin subrepeats (Lammert & Howell 2016). Interestingly, thus far, 9 ASD patients have been found with mutations in the RXR domain and 7 of these are distinct, representing at least 7 founder mutational events (Figure 1C, Table 1). The RXR sequence at the beginning of the β-sheets and the Arg residues are oriented towards the preceding sub-repeat (Figure 1A, 1B). In the B sub-repeats, these Arg residues are positioned on the surface of the jellyroll’s hydrophilic pore, whereas in the A sub-repeats they are oriented towards the previous repeat. No other group of RELN mutations identified in ASD patients align in a consensus element (Lammert & Howell 2016). We reasoned, therefore, that the RXR mutations are the most likely of the annotated mutations to have an effect relevant to ASD.
Table 1.
ASD Patient RELN mutations by sex. Mutations identified in the RXR consensus sequence in ASD probands are listed along with patient gender, either male (M), female (F), or no report available (N/A). Exome Aggregation Consortium (ExAC) database allele count is listed for each variant (exac.broadinstitute.org). This database includes sequence information from 60,706 unrelated individuals from several case-control studies, excluding ASD, but including schizophrenia and bipolar patients.
| Disease Mutation | Sex | Reference | ExAC frequency |
|---|---|---|---|
| R1742Q | M, M | Bonora et al., De Rubeis et al. |
31 |
| R1742W | N/A | Bonora et al. | 2 |
| R2290C | M | Iossifov et al. | 0 |
| R2290H | F, M | Bonora et al. | 0 |
| R2292C | M | De Rubeis et al. | 5 |
| R2639H | M | De Rubeis et al. | 2 |
| R2833S | M | De Rubeis et al. | 0 |
RXR Mutations Reduce Reelin Secretion
To study the de novo RELN R2290C mutation and other patient mutations in the RXR consensus element, we generated mutations at the murine equivalent of R1742 and R2290. These patient mutations represent the first Arg of the RXR motif in sub-repeats 4A and 5B, and were changed to Trp or Gln and Cys or His, respectively. Therefore, mutations in both the A and B sub-repeat domains were represented, and all four of these mutations occur within the Reelin repeat 3-6 fragment, a naturally occurring proteolytic fragment competent for signaling (Nogi et al. 2006, Jossin et al. 2004). These full-length constructs were transfected into cells and Reelin levels in the media and cell lysates were compared by anti-Reelin immunoblotting (Figure 1D, 1E). All three naturally occurring, N-terminal proteolytic fragments were visible by Western blotting the media of wild-type Reelin transfected cells. Reelin was, however, significantly decreased in the media from cells transfected with each of the R1742 and R2290 mutants compared to wild-type. Similar results were obtained with a fusion protein containing the minimal 5 and 6 signaling domain (Reln56SD) (Figure S1A), which had an exogenous nidogen secretion signal. The Reln56SD R2290C and R2290H transfected cells produced barely detectable extracellular Reelin, whereas the wild-type version of the fusion was highly expressed in the media (Figure S1B, S1C).
To better investigate the consequences of an RXR mutation under endogenous Reelin regulation, we used CRISPR-Cas9 gene editing to generate neurons with a heterozygous R2290C mutation (Figure 2A, 2B, and data not shown). Residue R2290 is particularly close to the receptor binding surface in sub-repeat 6A (Figure 1A, residues K2361 and K2468) (Yasui et al. 2007). Furthermore, the R2290C mutation is of particular interest since it arose de novo in an ASD proband (Iossifov et al. 2014) and has not been observed in control samples in ASD sequencing projects or in over 60,000 case-control sequences archived in the ExAC collection (Table 1). The mutation was introduced into pluripotent P19 cells (McBurney 1993) that we found can be differentiated into Reelin-secreting cells by growing them as aggregates in the presence of retinoic acid and HepG2 cell-conditioned media (Figure 2B and data not shown). After 8 days of differentiation, R2290C+/− P19 neurospheres expressed significantly less Reelin into the media and tended to express more in the cellular lysates than their wild-type counterparts (Figure 2D, 2E), demonstrating that under physiologic conditions this mutation selectively compromises Reelin biosynthesis of secreted Reelin protein. The apparent accumulation of intracellular Reelin, though not statistically significant, is consistent with a deficiency in Reelin R2290C secretion (Figure 2D, 2E).
To investigate if reduced extracellular levels of full-length mutant Reelin proteins were due to altered secretion, we performed pulse-chase experiments using stable cell populations expressing Reelin WT, R2290C, or R2290H (Figure 1F) and Reln56SD WT or R2290C (Figure S1D). Transfected cells were exposed to a pulse of 35S-methionine for 1 hour, which was then chased with excess cold methionine up to 4 hours. 35S labeled wild-type full-length Reelin and Reln56SD were found in the media within 1 hour (Figure 1H, S1D). This was accompanied by a corresponding decrease in labeled Reelin in the cell lysates (Figure 1G, S1E). Mutant full-length Reelin (R2290C and R2290H) and mutant Reln56SD (R2290C) were inefficiently secreted into the media (Figure 1H, S1E). Consistent with these results, cell lysate levels for mutant Reelin were relatively unchanged over this time. This suggests that the full-length R2290C and R2290H mutant Reelin are secreted only about 25% as efficiently as wild-type (Figure 1F-H). The R2290C mutant form of the shorter RelnSD56 fusion protein was secreted less efficiently than the full-length Reelin mutants, which may be a consequence of the exogenous secretion signal or the absence of other Reelin domains.
To assay the consequence of the remaining RXR mutations on extracellular accumulation of Reelin, we generated fusion proteins with each of the affected Reelin repeat domains in the wild-type and mutant forms. These Reelin repeat fusion protein (RRfp) constructs were designed to facilitate further biochemical analyses and contained the secretion signal from the nidogen protein, 3xFlag tag, and the respective Reelin repeat domain, followed by a C-terminal HA tag (Figure 3A). The mutant RRfp and the corresponding wild-type fusions were compared for expression in cell lysates and media by Western blotting (Figure 3B, 3C). For each Reelin repeat (4, 5, 6, and 7), the wild-type RRfp was present in both the media and the cell lysate. In media samples, each RRfp showed an upward molecular weight shift as compared to the lysate, consistent with glycosylation known for Reelin. For all mutant RRfps, the ratio of expression in media versus total cell lysate was significantly reduced compared to the respective wild-type control.
Figure 3. Reelin repeat fusion proteins (RRfps) with ASD RXR mutations are less abundant in the media as compared with their wild-type counterparts.
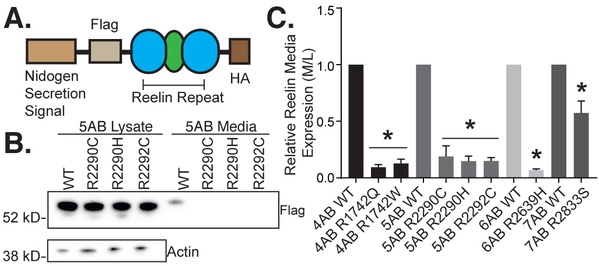
(A) Schematic of RRfps designed to test all ASD-associated RXR mutations identified in genetic studies thus far. The fusion protein is composed of a nidogen secretion signal, an N-terminal 3xFlag tag, a C-terminal HA tag and either wild-type or mutant versions of Reelin repeat 4, 5, 6, or 7. (B) Representative Western blot of RRfp 5AB for wild-type and mutant constructs. All the RXR mutants are expressed and easily detected in the cell lysates of transfected HeLa cells at comparable levels to the WT counterpart. However, in cell media the mutant proteins are under-represented and barely detectable by Western blotting (anti-Flag tag). (C) All ASD mutant RRfps showed decreased extracellular Reelin ratios (media/lysate) than their respective wild-type controls (n≥3, error bars ± SEM, * p value < 0.05 by 1-way ANOVA with Dunnett’s post hoc test).
RELN RXR Mutations Are Not Dominant Negative
RXR mutations in patients are present as heterozygous mutations. Therefore, we examined whether the Reelin generated from the mutant alleles might interfere with WT Reelin. To determine whether secretion-defective Reelin mutants had a dominant-negative effect on extracellular wild-type Reelin, N-terminal PA-tagged wild-type Reelin was co-transfected with either wild-type, R2290C, or R2290H untagged Reelin. Western blot with anti-PA tag antibody showed no effect of mutant RXR RELN expression on secretion of wild-type PA-tagged Reelin (Figure 4A, 4B). These findings indicate that the mutant forms of Reelin do not inhibit secretion of wild-type Reelin. We repeated this experiment using the Reln56SD fusion proteins, which are tagged with a V5 epitope. Wild-type full length Reelin was secreted as efficiently with co-expression of Reln56SD wild-type, R2290C, or R2290H fusion proteins (Figure S1F, S1G).
Figure 4. RXR mutations do not impair wild-type secretion and encoded proteins are capable of inducing Dab1 tyrosine-phosphorylation.
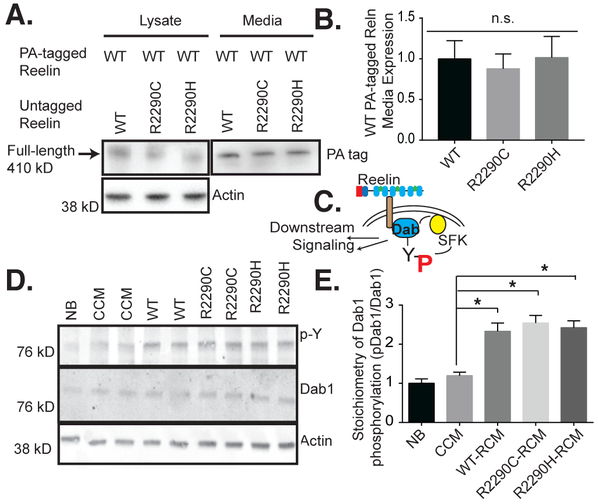
(A) Untagged mutant Reelin (R2290C, H) did not reduce the media accumulation of PA-tagged wild-type Reelin from co-expressing HeLa cells. Western blots show the anti-PA (WT Reelin) and actin levels. (B) Quantification of A (n=3, error bars ± SEM; 1-way ANOVA with Dunnett’s post hoc test, not statistically different, n.s.). (C) Schematic of the canonical Reelin signaling pathway: Reelin interacts with and multimerizes the receptors ApoER2 and VLDLR, recruiting Dab1 to their cytoplasmic domains, and leading to the Dab1-dependent activation of Src, which then phosphorylated Dab1 on tyrosine causing a cascade of downstream events including Dab1 degradation. (D) Dab1 tyrosine phosphorylation (4G10; p-Y) was induced to an equal extent by normalized amounts of WT, R2290C, or R2290H mutant Reelin, in stimulated primary neuronal cultures. (E) Densitometry measurements were quantified by dividing 4G10 signal by total Dab1 for each sample and values reported relative to neurobasal (NB). The stoichiometry of Dab1 tyrosine phosphorylation was augmented by WT and mutant Reelin treatment to approximately the same extent (n=4, error bars ± SEM, * p<0.05, 1-way ANOVA with Dunnett’s post hoc test). There was no significant difference between R2290C, R2290H and wild-type (WT) Reelin-conditioned media (RCM), but all were statistically different from control-conditioned media (CCM) in pairwise comparisons. CCM was not statistically different from NB media.
Since the full-length RXR proteins are secreted, albeit at reduced levels compared to wild-type, it is possible that they interfere with the normal signaling function of Reelin. Residue R2290 is proximal to the receptor-binding domain on 6A (Figure 1A); therefore, we determined whether the R2290 mutations might affect Reelin binding to its receptor and ultimately its signaling function, by monitoring the induction of Dab1 tyrosine phosphorylation (Figure 4C; Howell et al. 1999).
Media collected from wild-type and mutant Reelin-producing cells was concentrated and normalized for total Reelin levels. Primary cortical neuron cultures were then treated with these normalized Reelin-conditioned media (RCM), control-conditioned media (CCM) or neurobasal (NB) for 20 minutes before lysates were collected and analyzed for Dab1 tyrosine phosphorylation, which is a downstream consequence of Reelin signaling (Figure 4C, 4D, 4E). Wild-type and the mutant RCM significantly induced Dab1 tyrosine phosphorylation over CCM and NB (Figure 4D). Importantly neither mutant appeared to be deficient for signaling under these conditions compared to CCM. Therefore, the R2290C and R2290H mutant proteins are effective ligands, and they are unlikely to interfere with wild-type Reelin signaling.
Impaired Reelin Secretion Upregulates Protein Disulfide Isomerase A1
The inability to efficiently secrete mutant Reelin may lead to an increase in intracellular Reelin and ER stress-related effects in the Reelin-producing cell. Therefore, we first examined if Reelin accumulates in intracellular aggregates. Differentiated P19 R2290C+/− or WT neurospheres and HeLa cells transfected with R1742Q, R1742W, R2290C, and R2290H or wild-type Reelin were stained with anti-Reelin and anti-BIP, an ER resident protein (data not shown). In all instances, Reelin co-localized with BIP (Figure 2G), as expected for a secreted protein and consistent with previous reports (Campo et al. 2009, D'Arcangelo et al. 1997). No obvious mislocalization or aggregates were detected in cells producing mutant Reelin.
Build-up of proteins in the ER can initiate an unfolded protein response (UPR). This is initiated by one or more sensor proteins IRE1, PERK, and ATF6, all of which eventually lead to coordinated changes in gene expression to regulate chaperones, ER-associated degradation (ERAD) components, and cell survival and apoptosis signaling molecules (Wang et al. 2009). Therefore we examined by immunoblotting whether RELN RXR mutants caused upregulation of ER stress markers compared to wild-type. R2290C +/− neurospheres did not show significant changes in PERK, BIP, Ero1-Lα or phospho-eIF2α as compared to equivalent wild-type P19 cultures (Figure 2H, 2I). Activation of IRE1 during ER stress leads to the removal of a 26-nucleotide intron from XBP-1 mRNA (Wang et al. 2009). In RELN Orl +/− samples, however, we found decreased XBP-1 splicing as compared to wild-type cerebella (Figure 2F). There was, however, a significant increase in protein disulfide isomerase A1 (PDIA1; also known as prolyl 4-hydrozylase β) (Figure 2H, 2I).
Since a RELN RXR mutant mouse model is not currently available, we instead utilized a spontaneous RELN mutant mouse known as Orleans (RELN Orl) (de Bergeyck et al. 1997). A 220bp deletion in the mRNA generates a truncated protein at the 8th EGF-like domain that fails to be secreted, possibly due to protein misfolding, since the C-terminus is not essential for secretion (Nakano et al. 2007). Therefore, the RELN Orl allele provides an in vivo model of a secretion defect. Since autistic patients with RELN mutations are heterozygous, we used RELN Orl +/− mice to model the disease.
We chose the cerebellum as the focus for our analysis for several reasons. First, the cerebellum has recently been shown to have a large role in cognition and behavior and is a central player in ASD (Wang et al. 2014a, Tsai et al. 2012, Butts et al. 2014). Combined behavioral and neuroimaging studies of children with isolated perinatal cerebellar damage suggest that the occurrence of such damage may be the most significant acquired risk factor for a positive ASD diagnosis yet defined, with affected children showing more than a 35-fold increased risk of ASD-like features (Wang et al. 2014a, Limperopoulos et al. 2007). Furthermore, cerebellar granule cell neurons are the most abundant neuron in the cerebellum and it is the site of highest postnatal RELN expression in the brain (GENSAT.org). Thus, if a failure to secrete mutant Reelin adversely affects the Reelin-producing cell, it would most likely be detectable in the cerebellum.
We examined ER stress markers in the cerebellum as we did in the P19 derived neurospheres to determine if there would be consistent findings between the two models and any indication of ER stress. Similar to the neurosphere results, PDIA1 expression was much higher in the cerebella from RELN Orl +/− mutants compared to wild-type mice (Figure 5A, 5B). Unfortunately, Ero1-Lα expression was below the detection limit of our blots (data not shown). During ER stress, phosphorylation of eIF2α leads to decreased protein translation reducing the load on the ER, downstream of PERK activation (Wang et al. 2009). Interestingly, phospho-eIF2α levels were reduced, which would increase protein translation (Figure 5A, 5B). Total eIF2α protein levels tended to be reduced as well, but this was not significant.
Figure 5. Heterozygous Reeler Orleans (RELN Orl) mice, a model of impaired Reelin secretion, show impaired Reelin signaling and increased PDI expression in the cerebellum.
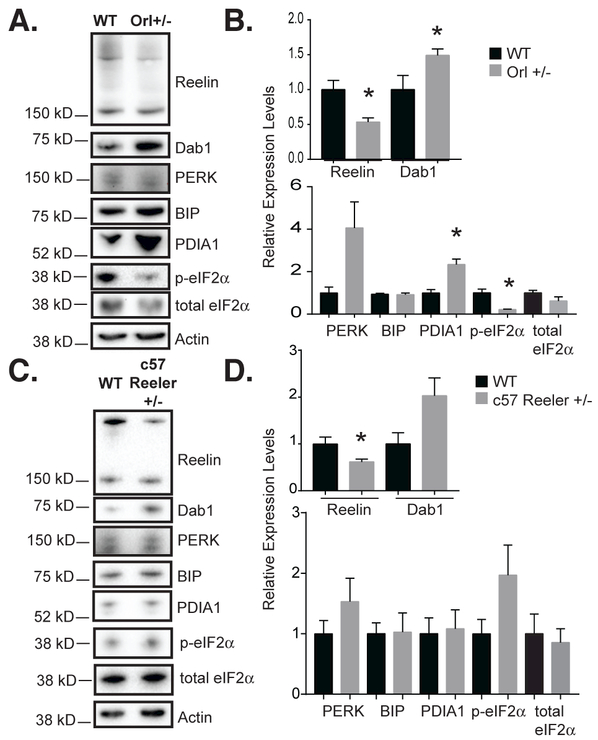
(A) Western blot of 6-week-old male RELN Orl mice cerebella from wild-type and heterozygous (Orl+/−) mice for Reelin, total Dab1, and ER stress markers. Reelin expression (anti-G10) is reduced and total Dab1 expression (anti-DabH1) is augmented in 6-week-old RELN Orl +/− cerebellum as compared to wild-type (+/+). (B) Quantification of A. Reelin was significantly decreased, while Dab1 was significantly increased in Orl+/− cerebella. PDIA1 was significantly increased in Orl+/− cerebella (n=3, error bars ± SEM, * p< 0.05, t-test). (C) RELN +/− null allele mice cerebellar lysates were analyzed by Western blot for ER stress markers. (D) Quantification of C. Except for reduced Reelin levels in the mutant, no statistically significant differences were seen between the wild-type and RELN +/− null allele cerebella. Dab1 levels tended to be higher in the mutant but this was not significant (n=4-5, error bars ± SEM).
To determine if changes in PDIA1 were caused by accumulation of intracellular RELN or were an effect of reduced Reelin protein, we examined expression of PDIA1 and other stress markers in RELN +/− null allele mice. The levels of PDIA1 as well as PERK, BIP, phospho-eIF2α and total eIF2α were unchanged between wild-type and RELN +/− null allele mice (Figure 5C, 5D). This suggests that there are phenotypic differences in the cerebella between mice that carry a RELN allele that fails to produce a protein (null allele) and those that make a protein that fails to be secreted (Orl allele).
Each of the three major ER stress pathways ultimately leads to changes in gene transcription. Thus, we compared wild-type and RELN Orl +/− mice cerebellum by RNAseq. Analysis was performed on 3 heterozygous and 3 wild-type cerebella. For RNAseq, samples averaged 37 million reads each (with 97% alignment and 99.5% strand specificity). Comparison of the expression levels in heterozygous versus wild-type mice indicated a total of 72 annotated genes with FDR q values < 0.15 (Supplemental File 1). No UPR or ER-stress response genes, including the key factors ATF4 and CHOP, were significantly changed at this threshold (Supplemental File 1). Although PDIA1 levels were increased in heterozygous cerebella at the protein level, there was not an increase in the mRNA level between samples, suggesting that the change was not caused by increased transcription in the RELN Orl +/− mutants.
Dab1 Protein Levels Increase in RELN Orl +/− Cerebella
Previously it has been shown that in RELN-deficient animals Dab1 protein levels are increased (Rice et al. 1998, Howell et al. 1999). Once Dab1 is tyrosine phosphorylated in response to Reelin stimulation, it is degraded by the proteasome pathway (Arnaud et al. 2003). In postnatal animals it is more difficult to detect Dab1 tyrosine phosphorylation and its levels are similar between WT and mutant animals in the neocortex (Howell et al. 1999). Here we observe that in the cerebellum there is a significant increase in Dab1 protein level in the RELN Orl +/− animals compared to wild-type (Figure 5A, 5B), suggesting that reduced Reelin signaling has an impact on this tissue.
Discussion
This study provides the first experimental evidence that heterozygous RELN mutations identified in ASD patients, particularly the de novo RELN R2290C mutation, alter biological function. Previously RELN had been implicated in ASD by whole genome linkage analysis, identification of potentially damaging mutations, including de novo mutations in patients, and the observation of reduced RELN gene and protein expression in the general ASD population (IMGSAC 2001, IMGSAC 1998, Wang et al. 2014b, Fatemi et al. 2005, Fatemi et al. 2002, Fatemi et al. 2001, Lammert & Howell 2016). Focusing on de novo mutation R2290C and studying a subset of analogous RELN mutations that are enriched in an RXR consensus sequence of the Reelin sub-repeat domains (Figure 1A-1C), we found that these mutations lead to reduced protein secretion and reduced extracellular accumulation (Figure 1D-1H, 3, S1). Furthermore RELN R2290C +/− neurospheres had higher expression of the endoplasmic reticulum foldase PDIA1 than wild-type controls (Figure 2H, 2I). Similarly, PDIA1 expression was increased in the cerebella of mice with defective Reelin secretion, i.e., RELN Orl +/− mutants, compared to wild-type (Figure 5A, B). These mice also had higher levels of Dab1 expression than controls, suggesting that Reelin-signaling efficacy was reduced (Figure 5A, B). Together it appears that at least a subset of RELN mutations in ASD patients, particularly R2290C, cause reduced Reelin secretion, reduced signaling, and an elevated chaperone protein PDIA1 expression, which may have combinatorial effects in the disorder.
Consistent with our findings that RXR mutant Reelin causes a partial loss-of-function, two RELN mutations (Q416X and E221fs) either truncate the Reelin protein before the receptor-binding surface on repeat 6A or lead to nonsense-mediated RNA decay (Lammert & Howell 2016, De Rubeis et al. 2014, Neale et al. 2012). Furthermore, it has also been shown that Reelin is decreased in the blood and brain of ASD patients (Fatemi et al. 2005, Fatemi et al. 2002, Fatemi et al. 2001), possibly as a result of differential RELN promoter methylation in ASD patients (Lintas et al. 2016, Zhubi et al. 2014). This has also been observed in maternal-stress/infection mouse models of ASD (Palacios-Garcia et al. 2015). Therefore, reduced RELN expression may also play a role in a broader population of ASD individuals who do not carry RELN mutations.
In addition to loss-of-function, PDIA1 protein expression was increased in both R2290C+/− neurospheres and RELN Orl +/− cerebella as compared to controls, suggesting a problem in the Reelin-producing cells. Interestingly, the RELN null allele, which makes no protein, did not elevate PDIA1 expression in heterozygotes as compared to wild-type littermates. PDIA1 functions as an ER chaperone, which, like many other chaperones, is often upregulated in response to ER stress. However, we did not find any other evidence of ER stress (Figure 2H, 2I, 5B, Supplemental Table 1). PDIA1 mRNA was not increased in the RELN Orl +/− cerebella, nor were other known UPR genes upregulated (Supplemental File 1). Contrary to what is expected in ER stress, XBP splicing and eIF2α phosphorylation were decreased in R2290C+/− neurospheres and Reln Orl +/− cerebella, respectively (2F, 5B). These disparate findings might suggest that elevation of PDIA1, and perhaps other chaperones not analyzed here, is sufficient to restore proteostasis such that ER stress is successfully resolved leaving only a few remaining indications of it. Alternatively, PDIA1 might be elevated in response to another pathway; for instance, the reticulon family has been shown to redistribute and activate PDIA1 in an ER stress-independent manner (Bernardoni et al. 2013, Yang et al. 2009).
Although PDIA1 is generally thought to have positive effects in neurons, its increased expression increases redox signaling, generating reactive oxygen species and has been linked to neurodegenerative disorders (Perri et al. 2015, Zeeshan et al. 2016). Thus PDIA1 may have detrimental effects in the Reelin-producing cells that are additive with the loss-of-function effects from reduced secretion. Some detrimental effects may be due to cytosolic rather than ER functions of PDIA1 and may not directly affect protein secretion (Parakh & Atkin 2015, Perri et al. 2015). Therefore, mutations that prevent secretion may have more than one injurious effect, and this may explain the propensity of RELN RXR mutations in ASD patients. For instance, if both the reduction of Reelin signaling and increased expression of PDIA1 had additive effects in the disorder, we would expect individual mutations that caused both to be more likely to appear in the patient population.
These findings make it tenable to determine how RELN mutations might contribute to ASD. At this time, three potential mechanisms are apparent: 1. Neuronal positioning, since this is regulated by RELN (D'Arcangelo et al. 1995, Hong et al. 2000), and recently it has been shown that a subset of ASD patients have focal regions of neuronal lamination defects (Stoner et al. 2014). It would thus be interesting to determine if patients with heterozygous RELN mutations have similar neuronal ectopias. 2. Synaptic function, since Reelin has also been implicated in synapse physiology postnatally with roles in the presynapse to regulate spontaneous neuron transmitter release (Bal et al. 2013) and the postsynapse to regulate NMDAR phosphorylation (Beffert et al. 2005, Chen et al. 2005, Groc et al. 2007, Campo et al. 2009, Ventruti et al. 2011). The synapse has become a central focus in ASD research with the realization that several mutations found in patients map to key regulatory genes (De Rubeis et al. 2014). 3. Regulation of mTOR signaling, since Reelin has been shown to regulate this pathway (Jossin & Goffinet 2007). Furthermore the Reelin pathway is altered by mutations in the upstream regulator of mTOR, TSC2 (Moon et al. 2015). Dysregulation of translation and mTOR are thought to be central to ASD in fragile X and tuberous sclerosis patients, respectively (Laggerbauer et al. 2001, Davis et al. 2015).
In pursuing a mechanism, it is important to keep in mind that in conjunction with RELN mutations, the male genotype is a contributing risk factor, as ASD affects 4 to 5 times as many males as females. Of the 8 ASD patients with RXR mutations and known sex, 7 are male (Table 1) (Bonora et al. 2003, Iossifov et al. 2014, De Rubeis et al. 2014). Other genetic mutations and/or environmental factors likely contribute to an ASD diagnosis in patients carrying RELN mutations, including the RXR mutations, since some of the mutations are found in parents and in the ExAC database (Table 1). This database represents cases and controls from other sequencing projects including psychiatric conditions but not ASD. Thus, at least for some RELN alleles, it appears that other genetic and environmental factors contribute to the presentation of the disorder.
In sum, our findings provide the first evidence that heterozygous mutations identified in ASD probands have functional consequences in neurons and are not benign polymorphisms. The de novo R2290C mutation in RELN leads to decreased Reelin secretion and elevated PDIA1 expression. Since increased PDIA1 expression can be detrimental to neurons, it will be interesting to determine if it has a role in ASD, in patients with missense mutations in RELN, and in other genes encoding proteins that pass through the ER.
Supplementary Material
Acknowledgements
The authors would like to thank Dr. Rick Matthews and Kimberly Wong for discussions, Drs. Andrea Viczian, Richard Wojcikiewicz, Andre Goffinet, Junichi Takagi, and Patricia Phelps for reagents, and Karen Gentile for running the RNAseq assay. Thank you to Bonnie Lee Howell for reading and editing the manuscript. This work was supported by a National Institute for Neurological Disorders and Stroke grant to NS073662 (BWH) and fellowship to DBL (NS086731).
Abbreviations
- +/−
Heterozygous
- ASD
autism spectrum disorder
- AutDB
Simons Foundation Autism Database
- CCM
control-conditioned media
- CR
Cajal-Retzius cell
- DIV
days in vitro
- ER
endoplasmic reticulum
- ERAD
endoplasmic reticulum-associated degradation
- FDR
false discovery rate
- GCN
cerebellar granule cell neuron
- NB
neurobasal media
- PBS
phosphate buffered saline
- RCM
Reelin-conditioned media
- RELN
reelin gene
- RELN Orl
Orleans reeler mouse
- RELN56SD
Reelin repeat 5 and 6 signaling domain fusion protein
- RRfp
Reelin repeat fusion protein
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SNP
single nucleotide polymorphism
- UPR
untranslated protein response
- WT
wild-type
- NMDAR
n-methyl-d-aspartate receptor
- RXR
arginine-amino acid-arginine
- DAVID
Database for Annotation, Visualization, and Integrated Discovery
- ExAC
Exome Aggregation Consortium
Footnotes
Competing interest
The authors have no competing interests to declare.
References
- American Psychiatric Association. and American Psychiatric Association. DSM-5 Task Force. (2013) Diagnostic and statistical manual of mental disorders : DSM-5. American Psychiatric Association, Washington, D.C. [Google Scholar]
- Anney R, Klei L, Pinto D et al. (2012) Individual common variants exert weak effects on the risk for autism spectrum disorders. Human molecular genetics, 21, 4781–4792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnaud L, Ballif BA and Cooper JA (2003) Regulation of protein tyrosine kinase signaling by substrate degradation during brain development. Molecular and cellular biology, 23, 9293–9302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autism Genome Project C, Szatmari P, Paterson AD et al. (2007) Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nature genetics, 39, 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bal M, Leitz J, Reese AL, Ramirez DM, Durakoglugil M, Herz J, Monteggia LM and Kavalali ET (2013) Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron, 80, 934–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U, Morfini G, Bock HH, Reyna H, Brady ST and Herz J (2002) Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. The Journal of biological chemistry, 277, 49958–49964. [DOI] [PubMed] [Google Scholar]
- Beffert U, Weeber EJ, Durudas A et al. (2005) Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron, 47, 567–579. [DOI] [PubMed] [Google Scholar]
- Bernardoni P, Fazi B, Costanzi A, Nardacci R, Montagna C, Filomeni G, Ciriolo MR, Piacentini M and Di Sano F (2013) Reticulon1-C modulates protein disulphide isomerase function. Cell death & disease, 4, e581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock HH, Jossin Y, May P, Bergner O and Herz J (2004) Apolipoprotein E receptors are required for reelin-induced proteasomal degradation of the neuronal adaptor protein Disabled-1. The Journal of biological chemistry, 279, 33471–33479. [DOI] [PubMed] [Google Scholar]
- Bonora E, Beyer KS, Lamb JA et al. (2003) Analysis of reelin as a candidate gene for autism. Molecular psychiatry, 8, 885–892. [DOI] [PubMed] [Google Scholar]
- Butts T, Green MJ and Wingate RJ (2014) Development of the cerebellum: simple steps to make a 'little brain'. Development, 141, 4031–4041. [DOI] [PubMed] [Google Scholar]
- Campo CG, Sinagra M, Verrier D, Manzoni OJ and Chavis P (2009) Reelin secreted by GABAergic neurons regulates glutamate receptor homeostasis. PloS one, 4, e5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Beffert U, Ertunc M, Tang TS, Kavalali ET, Bezprozvanny I and Herz J (2005) Reelin modulates NMDA receptor activity in cortical neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience, 25, 8209–8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo G, Homayouni R, Keshvara L, Rice DS, Sheldon M and Curran T (1999) Reelin is a ligand for lipoprotein receptors. Neuron, 24, 471–479. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI and Curran T (1995) A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature, 374, 719–723. [DOI] [PubMed] [Google Scholar]
- D'Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K and Curran T (1997) Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. The Journal of neuroscience : the official journal of the Society for Neuroscience, 17, 23–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis PE, Peters JM, Krueger DA and Sahin M (2015) Tuberous Sclerosis: A New Frontier in Targeted Treatment of Autism. Neurotherapeutics : the journal of the American Society for Experimental NeuroTherapeutics, 12, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bergeyck V, Nakajima K, Lambert de Rouvroit C, Naerhuyzen B, Goffinet AM, Miyata T, Ogawa M and Mikoshiba K (1997) A truncated Reelin protein is produced but not secreted in the 'Orleans' reeler mutation (Reln[rl-Orl]). Brain research. Molecular brain research, 50, 85–90. [DOI] [PubMed] [Google Scholar]
- De Rubeis S, He X, Goldberg AP et al. (2014) Synaptic, transcriptional and chromatin genes disrupted in autism. Nature, 515, 209–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Aldinger KA, Ashwood P et al. (2012) Consensus paper: pathological role of the cerebellum in autism. Cerebellum, 11, 777–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatemi SH, Snow AV, Stary JM, Araghi-Niknam M, Reutiman TJ, Lee S, Brooks AI and Pearce DA (2005) Reelin signaling is impaired in autism. Biological psychiatry, 57, 777–787. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Stary JM and Egan EA (2002) Reduced blood levels of reelin as a vulnerability factor in pathophysiology of autistic disorder. Cellular and molecular neurobiology, 22, 139–152. [DOI] [PubMed] [Google Scholar]
- Fatemi SH, Stary JM, Halt AR and Realmuto GR (2001) Dysregulation of Reelin and Bcl-2 proteins in autistic cerebellum. Journal of autism and developmental disorders, 31, 529–535. [DOI] [PubMed] [Google Scholar]
- Forster E, Tielsch A, Saum B, Weiss KH, Johanssen C, Graus-Porta D, Muller U and Frotscher M (2002) Reelin, Disabled 1, and beta 1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proceedings of the National Academy of Sciences of the United States of America, 99, 13178–13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii Y, Kaneko M, Neyazaki M, Nogi T, Kato Y and Takagi J (2014) PA tag: a versatile protein tagging system using a super high affinity antibody against a dodecapeptide derived from human podoplanin. Protein expression and purification, 95, 240–247. [DOI] [PubMed] [Google Scholar]
- GENSAT.org The Gene Expression Nervous System Atlas (GENSAT) Project, NINDS Contracts N01NS02331 & HHSN271200723701C to The Rockefeller University; (New York, NY: ). [Google Scholar]
- Groc L, Choquet D, Stephenson FA, Verrier D, Manzoni OJ and Chavis P (2007) NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein Reelin. The Journal of neuroscience : the official journal of the Society for Neuroscience, 27, 10165–10175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA and Herz J (1999) Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron, 24, 481–489. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Tran TS, Matsuoka Y, Howell BW and Rebeck GW (2006) DAB1 and Reelin effects on amyloid precursor protein and ApoE receptor 2 trafficking and processing. The Journal of biological chemistry, 281, 35176–35185. [DOI] [PubMed] [Google Scholar]
- Hong SE, Shugart YY, Huang DT, Shahwan SA, Grant PE, Hourihane JO, Martin ND and Walsh CA (2000) Autosomal recessive lissencephaly with cerebellar hypoplasia is associated with human RELN mutations. Nature genetics, 26, 93–96. [DOI] [PubMed] [Google Scholar]
- Howell BW, Hawkes R, Soriano P and Cooper JA (1997) Neuronal position in the developing brain is regulated by mouse disabled-1. Nature, 389, 733–737. [DOI] [PubMed] [Google Scholar]
- Howell BW, Herrick TM and Cooper JA (1999) Reelin-induced tyrosine [corrected] phosphorylation of disabled 1 during neuronal positioning. Genes & development, 13, 643–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IMGSAC (1998) A full genome screen for autism with evidence for linkage to a region on chromosome 7q. International Molecular Genetic Study of Autism Consortium. Human molecular genetics, 7, 571–578. [DOI] [PubMed] [Google Scholar]
- IMGSAC (2001) Further characterization of the autism susceptibility locus AUTS1 on chromosome 7q. Human molecular genetics, 10, 973–982. [DOI] [PubMed] [Google Scholar]
- Iossifov I, O'Roak BJ, Sanders SJ et al. (2014) The contribution of de novo coding mutations to autism spectrum disorder. Nature, 515, 216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y and Cooper JA (2011) Reelin, Rap1 and N-cadherin orient the migration of multipolar neurons in the developing neocortex. Nature neuroscience, 14, 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y and Goffinet AM (2007) Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Molecular and cellular biology, 27, 7113–7124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y, Ignatova N, Hiesberger T, Herz J, Lambert de Rouvroit C and Goffinet AM (2004) The central fragment of Reelin, generated by proteolytic processing in vivo, is critical to its function during cortical plate development. The Journal of neuroscience : the official journal of the Society for Neuroscience, 24, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshimizu E, Miyatake S, Okamoto N, Nakashima M, Tsurusaki Y, Miyake N, Saitsu H and Matsumoto N (2013) Performance comparison of bench-top next generation sequencers using microdroplet PCR-based enrichment for targeted sequencing in patients with autism spectrum disorder. PloS one, 8, e74167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laggerbauer B, Ostareck D, Keidel EM, Ostareck-Lederer A and Fischer U (2001) Evidence that fragile X mental retardation protein is a negative regulator of translation. Human molecular genetics, 10, 329–338. [DOI] [PubMed] [Google Scholar]
- Lammert DB and Howell BW (2016) RELN Mutations in Autism Spectrum Disorder. Front Cell Neurosci, 10, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limperopoulos C, Bassan H, Gauvreau K et al. (2007) Does cerebellar injury in premature infants contribute to the high prevalence of long-term cognitive, learning, and behavioral disability in survivors? Pediatrics, 120, 584–593. [DOI] [PubMed] [Google Scholar]
- Lintas C, Sacco R and Persico AM (2016) Differential methylation at the RELN gene promoter in temporal cortex from autistic and typically developing post-puberal subjects. Journal of neurodevelopmental disorders, 8, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuki T, Matthews RT, Cooper JA, van der Brug MP, Cookson MR, Hardy JA, Olson EC and Howell BW (2010) Reelin and stk25 have opposing roles in neuronal polarization and dendritic Golgi deployment. Cell, 143, 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBurney MW (1993) P19 embryonal carcinoma cells. The International journal of developmental biology, 37, 135–140. [PubMed] [Google Scholar]
- McBurney MW, Jones-Villeneuve EM, Edwards MK and Anderson PJ (1982) Control of muscle and neuronal differentiation in a cultured embryonal carcinoma cell line. Nature, 299, 165–167. [DOI] [PubMed] [Google Scholar]
- Moon UY, Park JY, Park R et al. (2015) Impaired Reelin-Dab1 Signaling Contributes to Neuronal Migration Deficits of Tuberous Sclerosis Complex. Cell reports, 12, 965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y, Kohno T, Hibi T, Kohno S, Baba A, Mikoshiba K, Nakajima K and Hattori M (2007) The extremely conserved C-terminal region of Reelin is not necessary for secretion but is required for efficient activation of downstream signaling. The Journal of biological chemistry, 282, 20544–20552. [DOI] [PubMed] [Google Scholar]
- Neale BM, Kou Y, Liu L et al. (2012) Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature, 485, 242–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogi T, Yasui N, Hattori M, Iwasaki K and Takagi J (2006) Structure of a signaling-competent reelin fragment revealed by X-ray crystallography and electron tomography. The EMBO journal, 25, 3675–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palacios-Garcia I, Lara-Vasquez A, Montiel JF, Diaz-Veliz GF, Sepulveda H, Utreras E, Montecino M, Gonzalez-Billault C and Aboitiz F (2015) Prenatal stress down-regulates Reelin expression by methylation of its promoter and induces adult behavioral impairments in rats. PloS one, 10, e0117680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panteri R, Paiardini A and Keller F (2006) A 3D model of Reelin subrepeat regions predicts Reelin binding to carbohydrates. Brain research, 1116, 222–230. [DOI] [PubMed] [Google Scholar]
- Parakh S and Atkin JD (2015) Novel roles for protein disulphide isomerase in disease states: a double edged sword? Frontiers in cell and developmental biology, 3, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perri ER, Thomas CJ, Parakh S, Spencer DM and Atkin JD (2015) The Unfolded Protein Response and the Role of Protein Disulfide Isomerase in Neurodegeneration. Frontiers in cell and developmental biology, 3, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persico AM and Napolioni V (2013) Autism genetics. Behavioural brain research, 251, 95–112. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA and Zhang F (2013) Genome engineering using the CRISPR-Cas9 system. Nature protocols, 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathjen J, Lake JA, Bettess MD, Washington JM, Chapman G and Rathjen PD (1999) Formation of a primitive ectoderm like cell population, EPL cells, from ES cells in response to biologically derived factors. Journal of cell science, 112 (Pt 5), 601–612. [DOI] [PubMed] [Google Scholar]
- Rice DS and Curran T (2001) Role of the reelin signaling pathway in central nervous system development. Annual review of neuroscience, 24, 1005–1039. [DOI] [PubMed] [Google Scholar]
- Rice DS, Sheldon M, D'Arcangelo G, Nakajima K, Goldowitz D and Curran T (1998) Disabled-1 acts downstream of Reelin in a signaling pathway that controls laminar organization in the mammalian brain. Development, 125, 3719–3729. [DOI] [PubMed] [Google Scholar]
- Stoner R, Chow ML, Boyle MP et al. (2014) Patches of disorganization in the neocortex of children with autism. The New England journal of medicine, 370, 1209–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai PT, Hull C, Chu Y et al. (2012) Autistic-like behaviour and cerebellar dysfunction in Purkinje cell Tsc1 mutant mice. Nature, 488, 647–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Schadewijk A, van't Wout EF, Stolk J and Hiemstra PS (2012) A quantitative method for detection of spliced X-box binding protein-1 (XBP1) mRNA as a measure of endoplasmic reticulum (ER) stress. Cell stress & chaperones, 17, 275–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ventruti A, Kazdoba TM, Niu S and D'Arcangelo G (2011) Reelin deficiency causes specific defects in the molecular composition of the synapses in the adult brain. Neuroscience, 189, 32–42. [DOI] [PubMed] [Google Scholar]
- Wang M, Wey S, Zhang Y, Ye R and Lee AS (2009) Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxidants & redox signaling, 11, 2307–2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Kloth AD and Badura A (2014a) The cerebellum, sensitive periods, and autism. Neuron, 83, 518–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Hong Y, Zou L et al. (2014b) Reelin gene variants and risk of autism spectrum disorders: an integrated meta-analysis. American journal of medical genetics. Part B, Neuropsychiatric genetics : the official publication of the International Society of Psychiatric Genetics, 165B, 192–200. [DOI] [PubMed] [Google Scholar]
- Yang YS, Harel NY and Strittmatter SM (2009) Reticulon-4A (Nogo-A) redistributes protein disulfide isomerase to protect mice from SOD1-dependent amyotrophic lateral sclerosis. The Journal of neuroscience : the official journal of the Society for Neuroscience, 29, 13850–13859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui N, Nogi T, Kitao T, Nakano Y, Hattori M and Takagi J (2007) Structure of a receptor-binding fragment of reelin and mutational analysis reveal a recognition mechanism similar to endocytic receptors. Proceedings of the National Academy of Sciences of the United States of America, 104, 9988–9993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui N, Nogi T and Takagi J (2010) Structural basis for specific recognition of reelin by its receptors. Structure, 18, 320–331. [DOI] [PubMed] [Google Scholar]
- Yuen RK, Thiruvahindrapuram B, Merico D et al. (2015) Whole-genome sequencing of quartet families with autism spectrum disorder. Nature medicine, 21, 185–191. [DOI] [PubMed] [Google Scholar]
- Zablotsky B, Black LI, Maenner MJ, Schieve LA and Blumberg SJ (2015) Estimated Prevalence of Autism and Other Developmental Disabilities Following Questionnaire Changes in the 2014 National Health Interview Survey. National health statistics reports, 1–20. [PubMed] [Google Scholar]
- Zaki M, Shehab M, El-Aleem AA, Abdel-Salam G, Koeller HB, Ilkin Y, Ross ME, Dobyns WB and Gleeson JG (2007) Identification of a novel recessive RELN mutation using a homozygous balanced reciprocal translocation. American journal of medical genetics. Part A, 143A, 939–944. [DOI] [PubMed] [Google Scholar]
- Zeeshan HM, Lee GH, Kim HR and Chae HJ (2016) Endoplasmic Reticulum Stress and Associated ROS. International journal of molecular sciences, 17, 327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Kong W, Gao Y et al. (2015) Gene Mutation Analysis in 253 Chinese Children with Unexplained Epilepsy and Intellectual/Developmental Disabilities. PloS one, 10, e0141782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhubi A, Chen Y, Dong E, Cook EH, Guidotti A and Grayson DR (2014) Increased binding of MeCP2 to the GAD1 and RELN promoters may be mediated by an enrichment of 5-hmC in autism spectrum disorder (ASD) cerebellum. Translational psychiatry, 4, e349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.