

Abstract
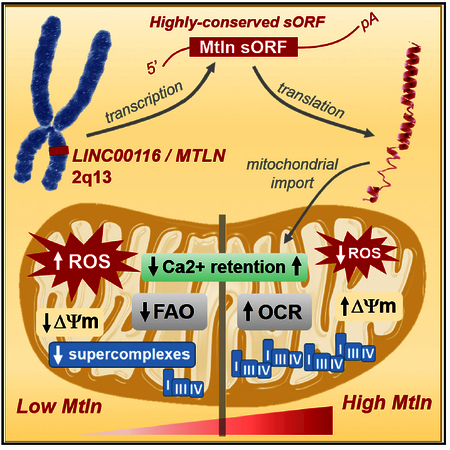
Mitochondria are composed of many small proteins that control protein synthesis, complex assembly, metabolism, and ion and reactive oxygen species (ROS) handling. We show that a skeletal muscle- and heart-enriched long non-coding RNA, LINC00116, encodes a highly conserved 56-amino-acid microprotein that we named mitoregulin (Mtln). Mtln localizes to the inner mitochondrial membrane, where it binds cardiolipin and influences protein complex assembly. In cultured cells, Mtln overexpression increases mitochondrial membrane potential, respiration rates, and Ca2+ retention capacity while decreasing mitochondrial ROS and matrix-free Ca2+. Mtln-knockout mice display perturbations in mitochondrial respiratory (super)complex formation and activity, fatty acid oxidation, tricarboxylic acid (TCA) cycle enzymes, and Ca2+ retention capacity. Blue-native gel electrophoresis revealed that Mtln co-migrates alongside several complexes, including the complex I assembly module, complex V, and supercomplexes. Under denaturing conditions, Mtln remains in high-molecular-weight complexes, supporting its role as a sticky molecular tether that enhances respiratory efficiency by bolstering protein complex assembly and/or stability.
Graphical Abstract
INTRODUCTION
Only ~5% of the human genome encodes for proteins, yet studies show that much of our DNA gives rise to thousands of uncharacterized RNAs (ENCODE Project Consortium, 2012). Although many are annotated as long non-coding RNAs (lncRNAs) that are thought to serve gene regulatory roles, emerging ribosomal profiling data support that several harbor actively translated short open-reading frames (sORFs) that encode previously overlooked microproteins (<100 amino acids, below the traditional cutoff applied for ORF annotation) (Bazzini et al., 2014). Several follow-up investigations have shown that some lncRNA-derived microproteins are highly conserved across species and play important roles in ion channel modulation, cell signaling, and RNA regulation (Anderson et al., 2016; D’Lima et al., 2017; Matsumoto et al., 2017). Although some studies have demonstrated that lncRNAs control cellular bioenergetics through gene regulatory mechanisms (Long et al., 2016), the potential involvement of lncRNA-derived microproteins in cellular metabolism remains underexplored.
Mitochondria generate the bulk of cellular ATP via oxidative phosphorylation (OXPHOS), supported by reducing equivalents generated by the tricarboxylic acid (TCA) cycle and fatty acid β-oxidation (FAO). Also, mitochondria play crucial roles in Ca2+ homeostasis, reactive oxygen species (ROS) generation and removal, redox signaling, and cell death regulation. Mitochondria are composed of >1,000 proteins, many of which are assembled into distinct complexes in inner and outer mitochondrial membranes (IMMs and OMMs), with many subunits deriving from nuclear-encoded genes (Calvo et al., 2016). Indeed, mitochondrial proteins are enriched among the hundreds of currently annotated proteins with <100 amino acids; these include subunits of the mitochondrial ribosome, protein import machinery, and respiratory chain complexes, along with related assembly factors (Amunts et al., 2015; Guerrero-Castillo et al., 2017; Letts et al., 2016).
Mitochondrial assembly factors facilitate the precise construction and arrangement of mitochondrial protein complexes. The OXPHOS machinery is made up of ~100 subunits distributed across five major complexes, including the electron transport chain, which can be further arranged into supercomplexes to optimize electron transfer and energy return while limiting ROS generation. Despite much progress, there remains great interest in understanding the pathways that guide and support these architectural configurations (Cogliati et al., 2016; Guerrero-Castillo et al., 2017; Letts et al., 2016; Ostojić et al., 2013; Stroud et al., 2016). Numerous investigations of mutations in patients with mitochondrial deficiencies have identified specific assembly factors that contribute to respiratory chain dysfunction (Abdulhag et al., 2015; Alston et al., 2016; Fernandez-Vizarra et al., 2007; Heide et al., 2012; Nouws et al., 2010; Ogilvie et al., 2005; Saada et al., 2009; Sugiana et al., 2008). Considering the number of unsolved clinical cases of mitochondrial deficiency, it is believed that many assembly factors remain to be discovered. Here, we describe the identification of a new lncRNA-derived microprotein that enhances mitochondrial functions and present data supporting that this may occur via modulation of assembly and stability of complexes and supercomplexes involved in FAO, the TCA cycle, and electron transport.
RESULTS
LINC00116 Encodes a Highly Conserved Muscle-Enriched Transmembrane Microprotein
While evaluating independent human lncRNA expression datasets (GTEx Consortium, 2013; Gibb et al., 2011; Uhlén et al., 2015; Wu et al., 2009), we identified LINC00116 (NC116) as a skeletal muscle- and heart-enriched lncRNA (Figure 1A). Although genomic annotation suggests that the NC116 gene spans ~11.4 kb and produces longer-transcript isoforms, available RNA sequencing (RNA-seq) data indicate that the shorter ~440-bp transcript is the major isoform (GTEx Consortium, 2013; Michel et al., 2014) (Figure 1B). While inspecting the NC116 genomic architecture, we noted a region of ultraconserved nucleotide base composition within the first exon of the major transcript isoform and hypothesized that this may encode protein. Query of ribosomal profiling datasets support that this region is actively translated in human and mouse cell lines and tissues (Figure 1B) (Bazzini et al., 2014; Michel et al., 2014). In silico translation across this region in various species indicated a common sORF that is predicted to encode for a highly conserved 56-amino-acid single-pass transmembrane microprotein (Phobius and TMHMM algorithms; Figure 1C). We generated a custom polyclonal antibody targeting the C-terminal region (Figure 1C) and performed western blot on a panel of mouse tissue lysates, revealing a ubiquitous 10-kDa protein enriched in cardiac and skeletal muscle, as well as adipose tissues (Figure 1D). Clustered regulary interspaced short palindromic repeats/spCas9 (CRISPR/Cas9) technology was used to produce knockout cells and mice by deleting the sORF from the homologous mouse locus (1500011K16Rik; Figure S1A). Notably, the 10-kDa protein was undetectable in knockout tissues (Figures S1B).
Figure 1. Human LINC00116 Encodes a Highly Conserved Muscle- and Heart-Enriched Microprotein.
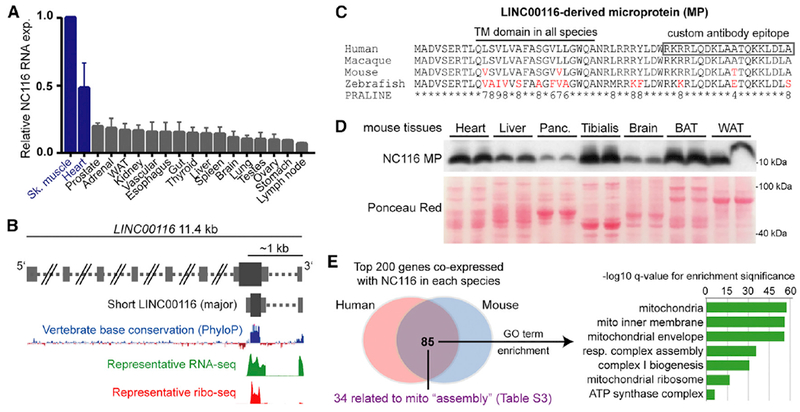
(A) Summarized LINC00116 RNA expression data from four independent human RNA-seq body-map datasets are plotted (mean ± SEM) relative to skeletal muscle, which had the highest LINC00116 expression levels in all four datasets.
(B) Schematic of the LINC00116 locus genomic architecture based on UCSC Genome Browser (hg19) annotations and expression data from GWIPS-viz ribosomal profiling database, the latter of which indicates the presence of a highly conserved sORF that is translated by ribosomes (ribo-seq).
(C) Multi-species alignment of the predicted LINC00116-derived microprotein (NC116-MP) performed using PRALINE algorithm after in silico translation of the conserved homologous sORFs in the indicated species. A conserved transmembrane (TM) domain is predicted in all species (PHOBIUS and TMHMM algorithms). The majority of non-perfectly conserved residues (red) represent conservative substitutions (higher PRALINE scores, range 0–10).
(D) A custom antibody was generated against the indicated C-terminal region, and western blot analysis on mouse tissue panels revealed a prominent 10-kDa band enriched in muscle and heart tissues. This band was not present in NC116-MP knockout mouse tissues (Figure S1).
(E) RNA co-expression analysis done by querying CoXpresDB database for LINC00116 and the mouse homolog, 1500011K16Rik indicated a clear overlap of co-expressed genes in both species. Gene ontology analyses revealed that the overlapping genes are strongly enriched for mitochondrial-related functions (Table S2). Among the 85 shared genes, 34 have reported functions related to mitochondrial complex and supercomplex assembly (Table S3).
To gain insight into the function of NC116 microprotein, we performed gene ontology enrichment analysis on genes that are highly co-expressed (i.e., positively correlated) with NC116/1500011K16Rik RNAs in human and mouse transcriptomic datasets (Obayashi et al., 2008). Among the top 200 co-expressed genes in both species (Table S1), 85 were shared, and these were strikingly enriched for mitochondrial genes corresponding to various protein machineries, including respiratory chain complexes and mitochondrial ribosomes (Figure 1E; Table S2); overall, 59 of these 85 genes encode proteins annotated as “mitochondrial part.” A closer evaluation of gene functions revealed that the shared co-expression network was specifically related to protein complexing and assembly, with >30 genes having reported roles in assembling respiratory chain complexes, mitochondrial ribosomes, and iron-sulfur clusters (Table S3). Notably, these include 9 of the 15 proteins found at inter-complex interaction sites within the respirasome supercomplex (I-III2-IV) (Letts et al., 2016). Together, these data indicated a strong likelihood that NC116 microprotein localizes within mitochondria, where it influences protein complex assembly, and our follow-up experiments (presented below) support this notion. As such, we named this microprotein mitoregulin (Mtln), keeping consistent with the “-regulin” theme for other lncRNA-derived microproteins (Anderson et al., 2016).
Mtln Microprotein Localizes to Inner Mitochondrial Membranes and Binds Cardiolipin
Localization of Mtln to mitochondria was assessed by microscopy and cell fractionation. In cultured cardiomyocytes, co-labeling revealed a clear overlap of Mtln localization with Mito-Tracker red (Figure 2A). Bioinformatic predictions indicated that Mtln lacks a classical mitochondrial-targeting sequence; however, Mtln has a high density of conserved, positively charged residues that likely promote mitochondrial import (Figure 1C), and fusing Mtln to the N terminus of GFP (Mtln-GFP) was sufficient to target GFP fluorescence to mitochondria (Figure 2B). Similarly, Mtln-FLAG-tagged protein also localized to mitochondria (Figure S2). In cell fractionation studies, Mtln was found to be enriched in mitochondrial pellets, along with other known mitochondrial proteins (Cox4 and Vdac1; Figure 2C). In addition, Mtln remained in the pellets upon treatment with increasing digitonin, which releases OMMs and associated proteins (e.g., Vdac1), supporting that Mtln predominantly localizes to IMMs, like Cox4 (Figure 2D). Further investigation using classical proteinase-K-based studies of mitochondrial protein topology corroborated localization to the IMM and revealed that the Mtln C terminus primarily projects into the intermembrane space (IMS), as indicated by a notable decrease in western blot detection of the C-terminal region in mitochondrial preparations treated with proteinase K in hypotonic buffer, which selectively disrupts OMMs; similar observations were made for cytochrome c (CytC; a known IMS protein; Figure 2E).
Figure 2. The LINC00116-Derived Microprotein Mitoregulin Localizes to Inner Mitochondrial Membranes and Binds Cardiolipin.
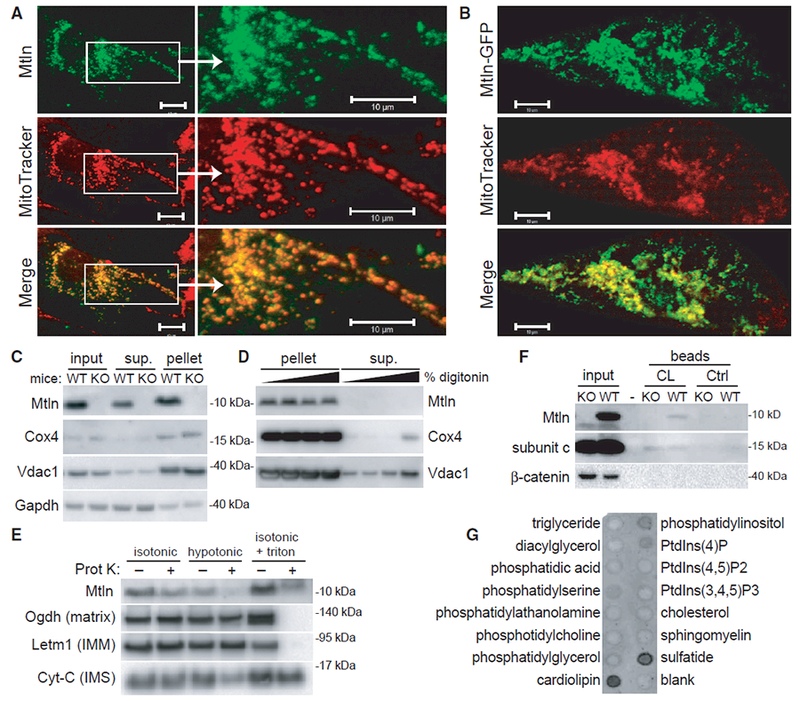
(A and B) Wild-type (A) and GFP-tagged (B) human Mtln were expressed in neonatal rat cardiomyocytes, and co-localization with MitoTracker red was evaluated. Representative photomicrographs are shown. Scale bars, 10 μm.
(C) Mitochondrial pellets were isolated from wild-type (WT) or Mtln-knockout (KO) C2C12 myoblast cells, and western blot was performed on various fractions.
(D) Mitochondrial pellets harvested from WT or Mtln-KO skeletal muscle tissues were treated with increasing digitonin concentrations to release OMMs, and pellet and supernatant fraction fractions were subjected to western blot analysis. Cox4 and Vdac1 are known IMM and OMM proteins, respectively. Gapdh is a cytosolic protein known to associate with mitochondria in some cases.
(E) Mitochondrial pellets harvested from WT skeletal muscle tissues were resuspended in isotonic, hypotonic, or isotonic plus triton buffers in the absence or presence of proteinase K and subjected to western blot analysis. Proteins with known localization to various mitochondrial compartments (e.g., matrix, IMM, and intermembrane space [IMS]) were evaluated as controls.
(F) Western blot analysis performed on WT and Mtln-KO cardiac tissue lysates subjected to pull-down assay using cardiolipin (CL)-coated or control beads. Subunit c, a known cardiolipin-binding protein, serves as the positive control.
(G) Lipid-strip binding assay performed using synthetic Mtln protein followed by anti-Mtln immunoblot.
Cardiolipin is a phospholipid found almost exclusively in IMMs, where it confers optimal mitochondrial functions by maintaining the structural integrity of the membrane and embedded protein complexes, including respiratory supercomplexes (Pfeiffer et al., 2003). Cardiolipin is known to bind to positively charged arginine and lysine residues on many mitochondrial proteins. Given that Mtln is a transmembrane protein rich in these residues, we investigated their potential interaction and found that cardiolipin-coated beads pulled down Mtln from wild-type mouse cardiac tissue lysates to a similar or greater degree compared to subunit c, an ATP synthase subunit with high affinity for cardiolipin (Figure 2F). A lipid-strip binding assay further supported that Mtln preferentially engages cardiolipin relative to other common cell membrane lipids (Figure 2G).
Mtln Overexpression Increases Mitochondrial Respiratory and Ca2+ Buffering Capacities while Decreasing ROS
To assess Mtln functions, we generated and tested doxycyclineinducible (“Dox-On”) Mtln overexpression vectors in cultured human HeLa cells. Mtln+Dox treatment elevated Mtln protein levels ~10-fold (p < 0.01) after 48 hr relative to control β-gal+Dox or Mtln-Dox (“no Dox”) but did not significantly influence relative levels of the five OXPHOS complexes per OXPHOS cocktail western blot or by Coomassie visualization after blue-native polyacrylamide gel electrophoresis (BN-PAGE; Figure 3A). In-gel complex I (CI) activity was also measured, given that co-expression data linked Mtln with CI assembly (Figure 1E; Table S2), but Mtln overexpression had no discernible effect on CI activity. Interestingly, overexpression of Mtln increased mitochondrial membrane potential (35%, p < 0.0001; tetramethylrhodamine, ethyl ester [TMRE] staining) with concomitant reductions in mitochondrial ROS (−60%, p < 0.0001; MitoSox staining) relative to controls (Figure 3B). Mtln+Dox cells also had increased basal and maximal mitochondrial respiration rates (70% and 35%, respectively, p < 0.05; Figure 3C) and decreased extracellular acidification rates (ECARs; glycolytic capacity down ~20%, p < 0.05; Figure S3A), supporting a Mtln-induced switch from glycolysis to OXPHOS. In addition, respiration appeared more tightly coupled in Mtln+Dox cells, as evidenced by increased ATP-linked respiration (70%, p < 0.001; Figure S3B). Notably, these Mtln-induced effects seem to occur independent of alterations in mitochondrial mass; i.e., no clear changes in (1) OXPHOS complex levels, (2) cellular mitochondrial densities observed in TMRE and MitoSox imaging studies, and (3) mitochondrial DNA content (Figure S4A). Cardiolipin levels were also unchanged (Figure S4C).
Figure 3. Mtln Overexpression Alters Mitochondrial Membrane Potential, ROS, Respiration, and Ca2+ Handling in Human HeLa Cells.
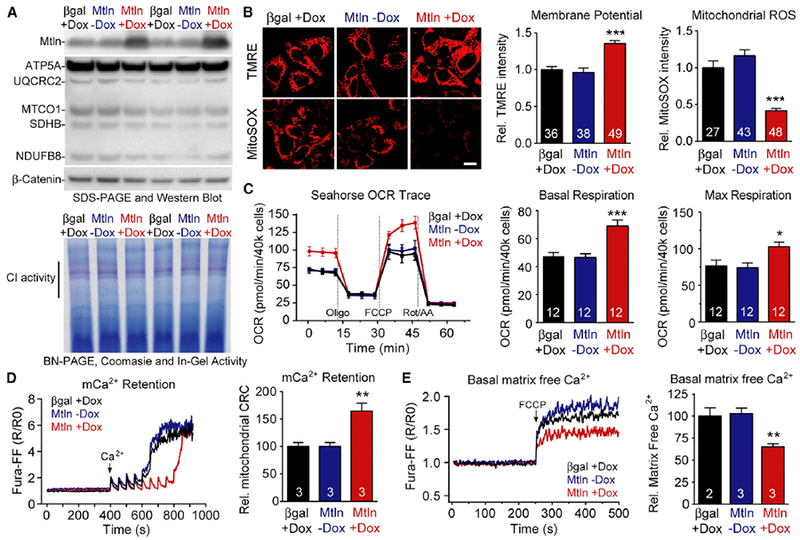
Doxycycline (DOX)-inducible expression vectors were used to overexpress Mtln and beta-galactosidase (βgal, control) in cultured human HeLa cells, and protein measures and mitochondria-related analyses performed 48 hr later. Mtln-Dox (i.e., no Dox) serves as additional control.
(A) Top, western blot shows clear Mtln overexpression in Mtln+Dox cells, relative to controls, but does not significantly alter respiratory complex levels by OXPHOS cocktail blot or BN-PAGE/Coomassie blue (bottom). Bottom, complex I (CI) in-gel activity assay.
(B) Representative photomicrographs depicting TMRE (mitochondrial membrane potential) and MitoSOX (ROS) probe intensities in treated cells. Scale bar, 20 μm. Quantified probe intensities are plotted at right.
(C) Standard mitochondrial respiration assays were performed in treated cells and oxygen consumption rates (OCR) plotted at right. Refer to Figure S3 for additional measures.
(D and E) Mitochondrial Ca2+ (mCa2+) dynamics were evaluated in treated cells; representative traces and quantified data are shown for Ca2+ retention capacity (CRC) assay (D) and measures of matrix-free Ca2+ (E). Graphs are plotted as mean ± SEM, sample n is indicated within each bar, and p values were determined by one-way ANOVA with Dunnett’s post hoc (*p < 0.05, **p < 0.01, and ***p < 0.001 compared to βgal+Dox).
Mitochondrial membrane potential and ATP generation is tightly coupled to mitochondrial Ca2+ (mCa2+) dynamics, which serve to activate mCa2+-dependent dehydrogenases in the TCA cycle and mediate mitochondrial permeability transition pore (mPTP) opening and cell death signaling. Thus, we evaluated mCa2+ dynamics and found that Mtln+Dox cells had increased mCa2+ retention capacity (65%; p < 0.01; Figure 3D), relative to controls, despite no differences in mCa2+ uptake kinetics or magnitude. Parallel experiments indicated that Mtln+Dox cells had significantly reduced matrix-free Ca2+ (35%, p < 0.01; Figure 3E), which along with decreased ROS levels could account for the increased mCa2+ retention capacity and delayed triggering of mPTP in Mtln+Dox cells.
Acute Mtln Inhibition Depresses Mitochondrial Membrane Potential and Increases Mitochondrial ROS
Given that gene overexpression studies may achieve supraphysiologic protein levels, we evaluated the effects of Mtln gene silencing in cultured HeLa cells. First, a panel of four small interfering RNAs (siRNAs) targeting human Mtln mRNA (siMtln1–4; Table S4) were tested for gene silencing efficiency; each suppressed Mtln protein expression in a dose-dependent manner within 48 hr post-transfection, with the most efficacious sequences (siMtln-1 and siMtln-3) achieving ~90% suppression at a higher dose (50 nM; Figure 4A) relative to negative control sequences. Subsequent imaging studies revealed that HeLa cells treated with siMtln-1 and siMtln-3 showed marked decreases in mitochondrial membrane potential (≥50%, p < 0.0001; TMRE staining) and concomitant increases in mitochondrial ROS (~2-fold, p < 0.0001; MitoSOX staining) relative to controls (Figure 4B). Notably, these results reciprocate our findings in overexpression studies (Figure 3), and similar to those studies, Mtln suppression did not significantly alter levels of respiratory chain complexes, as determined by OXPHOS cocktail western blot (Figure 4A), nor cardiolipin levels (Figure S4D).
Figure 4. Mtln Suppression Alters Mitochondrial Membrane Potential and ROS in Human HeLa Cells.
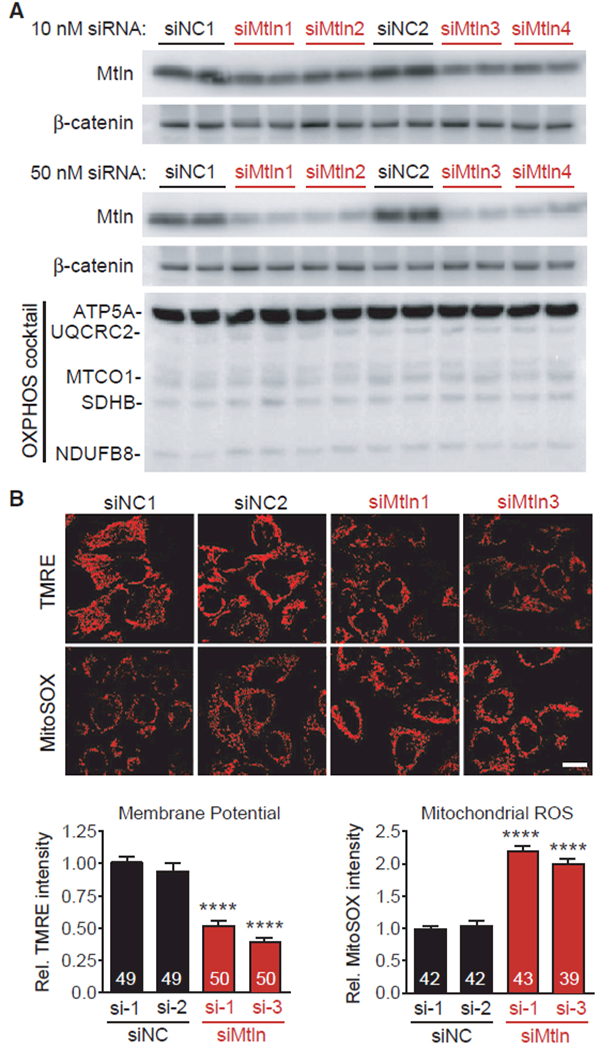
Mtln expression was inhibited in cultured human HeLa cells using siRNAs, and protein measures and mitochondrial imaging analyses were performed 48 hr later.
(A) Western blot shows clear reductions in Mtln expression in cells treated with Mtln-targeted siRNAs (siMtln1–4, each representing unique siRNA sequences), relative to two non-targeted negative control siRNAs (siNC1–2). 10 nM (top) and 50 nM (bottom) doses were tested. OXPHOS cocktail protein levels did not change in response to Mtln knockdown in any siMtln treatment group.
(B) Top, representative photomicrographs depicting TMRE (mitochondrial membrane potential) and MitoSOX (ROS) probe intensities in HeLa cells treated with 50 nM of the indicated siRNAs. Scale bar, 20 μm. Bottom, quantified probe intensities are plotted as mean ± SEM, sample n is indicated within each bar, and p values were determined by one-way ANOVA with Dunnett’s post hoc (****p < 0.0001 compared to βgal+Dox).
Mtln-KO Mice Exhibit Defects in Fatty Acid Oxidation and Mitochondrial Ca2+ Dynamics but Lack Overt Signs of Oxidative Stress
CRISPR/Cas9-derived Mtln-KO mice are viable and develop normally with no overt phenotypes later in life. Given that Mtln is enriched in skeletal muscle and cardiac tissues and overexpression altered mCa2+ dynamics in cells, we subjected Mtln-KO mice and wild-type (WT) littermates to exercise tolerance testing on treadmills using “sprint” and gradual acceleration (i.e., “endurance”) paradigms but found no genotype-based differences in performance (Figures S5A and S5B). We next evaluated mitochondrial respiratory activities in cardiac and skeletal muscle fibers prepared from fed mice or mice subjected to a 24-hr fast to activate FAO. In the fed state, Mtln-KO and WT muscle fibers showed comparable basal and maximal ADP-stimulated respiration rates in the presence of either carbohydrate-based (pyruvate, glutamate, succinate; Figure S5C) or fatty acid (palmitoyl-carnitine [PC]; Figure 5A) substrates. However, in fasted mice, Mtln-KO skeletal muscle (SKM) fibers showed significantly lower rates (~30%) of maximal fatty acid (PC) oxidation relative to WT controls (p = 0.02; Figure 5A), and left ventricular (LV) fibers showed a similar trend (20% decreased, p = 0.09). Also, mCa2+ uptake and retention assays performed on LV fibers indicated that Mtln-KO mitochondria have reduced mCa2+ retention capacity compared to WT (27%, p < 0.05, Figure 5B). Notably, mitochondrial DNA content and cardiolipin levels were not altered in Mtln-KO mice (Figures S4B and S4E). Overall, these data are consistent with and reciprocate our in vitro findings with Mtln overexpression.
Figure 5. Mtln-KO Mice Exhibit Alterations in Mitochondrial Metabolism, Ca2+ Retention, and Protein Complex Assemblies.
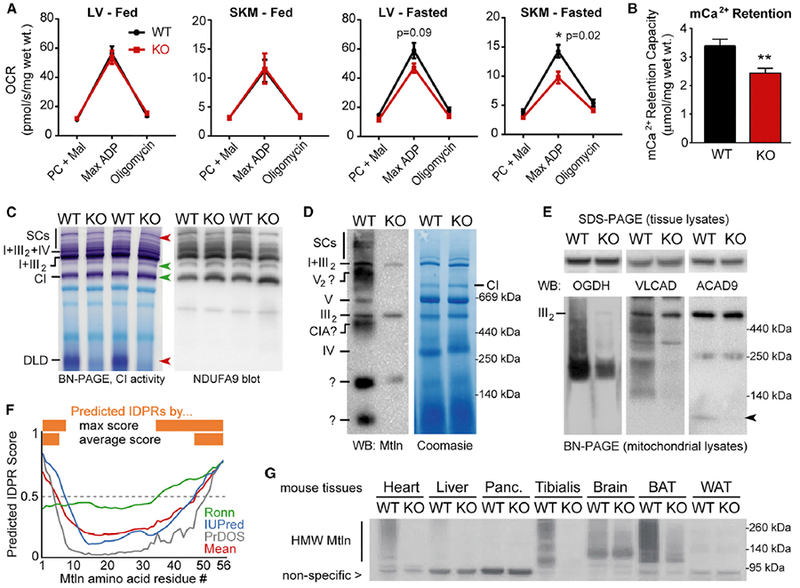
(A) Permeabilized muscle fibers (cardiac, left ventricular [LV], and skeletal muscle [SKM], gastrocnemius) were harvested from fed or fasted (24 hr) WT or Mtln-KO mice, and oxygen consumption rates (OCR) were measured during sequential addition of palmitoyl-carnitine/malate (PC + Mal), 1 mM ADP (Max ADP), and oligomycin; data are plotted as mean ± SEM. Fed: WT n = 7 (4 males, 3 females), KO n = 7 (3 males, 4 females); fasted: n = 4 females for both WT and KO.
(B) Mitochondrial Ca2+ retention capacities were measured in permeabilized LV fibers from fasted female mice (n = 4/genotype) and plotted as mean ± SEM; **p = 0.01.
(C) In-gel CI activity assay was performed on cardiac tissue mitochondrial lysates from fed WT female mice (n = 3–4/genotype, representative gels shown) (left). Red and green arrows denote bands with decreased or increased (respectively) CI activity in KO mice. Top red arrow points to a doublet band in WT hearts, with the top band virtually absent in KO mice. BN-PAGE and western blot for CI subunit NDUFA9 was performed on the same samples (right). See Figure S7B for SKM tissue data.
(D) Representative BN-PAGE western blot on cardiac tissue mitochondrial lysates shows co-migration of Mtln with various prominent mitochondrial complexes; see Figure S7D for SKM data. Some non-specific bands (e.g., in the KO lane) arise from the secondary antibody.
(E) SDS-PAGE (top, cardiac tissue lysates) and BN-PAGE (bottom, cardiac mitochondrial lysates) western blots for proteins involved in the TCA cycle (OGDH), CI assembly (ACAD9), and FAO (ACAD9 and VLCAD). Arrow indicates ACAD9 dimers. n = 3–4; representative blots are shown.
(F) IDPR prediction was run on the Mtln protein sequence (56 amino acids) using the three indicated independent algorithms and output scores plotted, along with the mean score (red). Scores above 0.5 indicate predicted IDPRs (orange).
(G) SDS-PAGE western blot for Mtln-containing high-molecular-weight (HMW) assemblies in WT and Mtln-KO mouse tissue panels. Nonspecific band provides loading control for comparing WT and KO lanes.
Given our in vitro data indicating that manipulation of Mtln expression alters mitochondrial ROS, we evaluated Mtln-KO mouse muscle and heart tissues for signs of oxidative stress. Levels of glutathione (both reduced [GSH] and oxidized [GSSG] forms) were not significantly different in WT and Mtln-KO mouse muscle whole-tissue lysates (Figure S6A), nor were aconitase activities in crudely isolated mitochondria, a surrogate measure of mitochondrial ROS, which is known to suppress aconitase activity (Figure S6B). Furthermore, western blot analyses indicted that expressions of key mitochondrial antioxidant enzymes (e.g., SOD2, GPX1, and GPX4) were similar in WT and Mtln-KO mouse tissues (Figure S6C).
Mtln Influences Mitochondrial Protein Complex Assembly and/or Stability
To gain insight into the mechanistic actions of Mtln, we performed OXPHOS cocktail western blot, BN-PAGE analyses, and in-gel activity studies to evaluate the effects of loss of Mtln on mitochondrial protein assemblies and to characterize Mtln distribution among them. Mtln-KO and WT cardiac and skeletal muscle tissue lysates showed no overt genotype-based differences in the major respiratory complex levels (OXPHOS cocktail blot; Figure S7A), though decreases in respiratory supercomplexes were evident in Mtln-KO hearts (Figure 5C) and skeletal muscles (Figure S7B). In-gel activity assays showed aberrations in CI distribution in Mtln-KO cardiac tissue samples, with a distinct loss of a high-molecular-weight supercomplex (SC; Figure 5C, left, top red arrow) and qualitative but consistent increases in free or disassembled CI-associated complexes in heart samples (green arrows; see Figure S8 for higher-magnification images). Western blotting for the CI subunit NDUFA9 indicated a correlation between CI activity and levels of the corresponding CI-containing complexes (Figure 5C, right). Mtln-KO skeletal muscle samples also showed reduced CI activities within both I+III2 supercomplexes and respirasomes (I+III2+IV) (Figure S7C). Of note, the previously characterized non-specific CI activity band, resulting from dihydrolipoamide dehydrogenase (DLD) activity (Yan and Forster, 2009), was overtly diminished in Mtln-KO samples. Subsequent BN-PAGE western blot revealed that Mtln co-migrates with several notable complexes, including the CI assembly module (CIA; Alston et al., 2016; Heide et al., 2012), complex V, and various supercomplexes, and appears in lower complexes of unknown identity (Figures 5D and S7D), thus supporting a broad distribution of Mtln throughout the IMM.
Consistent with the overt reduction in mitochondrial DLD activity, dimers of oxoglutarate dehydrogenase (OGDH), the first and rate-limiting step of the OGDHC, a multi-enzyme complex that includes DLD, were also severely decreased in mitochondria (216-kDa band; smearing may indicate possible membrane association). Notably, no expression changes were observed for these and other OGDHC subunits in whole-tissue lysates (Figures 5E and S9A).
To further investigate a potential Mtln-related impact on protein-protein interactions or membrane docking, we performed SDS-PAGE and BN-PAGE western blotting to evaluate levels and distributions of ACAD9 and VLCAD proteins, which are FAO pathway enzymes known to dimerize and tether with IMMs, where they function; interestingly, ACAD9 also plays a role in CI assembly (He et al., 2007; Nouws et al., 2010; Souri et al., 1998). Mtln-KO samples showed a clear loss of ACAD9 dimers, which appeared to co-migrate closely with a Mtln-positive band (Figures 5E and S9B); also, VLCAD distribution was significantly altered (i.e., lack of bands and smears, consistent with decreased membrane association). Of note, ACAD9 and VLCAD protein levels were not significantly different in whole-tissue lysates (Figures 5E and S9A). Together, these data, along with the DLD and OGDH findings, support that Mtln-KO mitochondria are unable to retain matrix localization of key FAO pathway and TCA cycle enzymes, perhaps resulting from decreased membrane associations and subsequent protein loss or leak during isolation of crude mitochondria from tissues.
To assess if Mtln has molecular chaperone-like characteristics to support protein-protein interactions and membrane docking, we analyzed Mtln amino acid sequence for the presence of intrinsically disordered protein regions (IDPRs), which are well known to support protein tethering and/or assembly and complex stabilizing functions (Uversky, 2015). Several independent algorithms predicted that Mtln N- and C-terminal domains harbor IDPRs (Figure 5F). Considering that IDPR-containing proteins are inherently sticky, Mtln may be found in high-molecular-weight (HMW) oligomeric complexes that remain stable under stringent denaturing conditions. To assess this, we performed SDS-PAGE western blot in WT and Mtln-KO mouse tissue panels and found predominant Mtln-containing HMW complexes (distinct bands and smears, not present in Mtln-KO samples) in heart and skeletal muscle (Figure 5G). These data, along with our other observations, support the notion that Mtln fine-tunes mitochondrial functions through broad interactions with an array of protein complexes, supercomplexes, and cardiolipin, possibly playing roles in protein-membrane recruitment, membrane dynamics, and complex assembly and stabilization.
DISCUSSION
Through exploration of gene expression and genome annotation databases, we discovered that the LINC00116 lncRNA gene encodes for a mitochondrial single-pass transmembrane protein that we named Mtln. Previous high-throughput ribosomal profiling studies have shown that LINC00116 harbors one of many transcriptome-wide sORFs that are engaged by actively translating ribosomes (Bazzini et al., 2014). Proteomics investigations have noted the existence of peptides mapping to this sORF (Catherman et al., 2013; Chen et al., 2015). One of these studies found Mtln peptides in the detergent insoluble fraction, which could explain (along with its tiny size) why this protein has not been identified in many unbiased proteomic analyses. This finding is consistent with our observation that Mtln has sticky IDPR domains and appears to form highly stable, SDS-resistant HMW complexes that may represent Mtln oligomeric structures and/or Mtln complexed with many other IDPR-containing mitochondrial proteins.
Our co-expression analyses revealed that Mtln is tightly linked to a distinct gene network dedicated to mitochondrial protein complex assembly. It is worth reiterating that this network includes several known respiratory chain complex assembly factors, and impressively, 6 of the top 10 most highly co-expressed genes encode for proteins found at inter-complex contact sites within the respirasome supercomplex (Table S3) (Letts et al., 2016). This is notable considering that only ~15 proteins have been identified as respirasome “contact-site” proteins. However, given that the current respirasome crystal structures lack unannotated transmembrane alpha-helices, it is unlikely that Mtln is part of the mature respirasome, unless it is lost during purification and/or crystallization steps.
In accord with co-expression data pointing to Mtln’s supportive role in protein complex assembly, our experimental data demonstrate that its physiologic function serves to enhance mitochondrial efficiency. Mtln overexpression in HeLa cells supercharged mitochondrial functions, as evidenced by increased membrane potential, respiration, and mCa2+ retention capacity, with a surprising decrease in mitochondrial ROS, all of which are consistent with improved respiratory chain complexing, super-complexing, and coupling (Lopez-Fabuel et al., 2016; Maranzana et al., 2013). Notably, this might account for the decreased ROS, which could subsequently desensitize mCa2+-induced triggering of mPTP, as was observed. Although Mtln overexpression did not cause overt changes in levels of OXPHOS machinery or respiratory supercomplexes, it is possible that subtle increases in supercomplexes could account for the improved respiratory capacity, as was previously observed with COX6B1 overexpression in cultured cells (Kim et al., 2015).
Our findings with Mtln overexpression in vitro were reciprocated, in part, in HeLa cells with Mtln knockdown, which showed decreased mitochondrial membrane potential and increased ROS, as well as in Mtln-KO mice, which exhibited decreased mCa2+ retention capacity and impaired FAO upon fasting, which is a well-established activator of FAO in muscle tissues and was more recently found to rewire respiratory supercomplex configurations in mice, likely in response to altered substrate usage (Lapuente-Brun et al., 2013). It is possible that Mtln-KO mice exhibit inefficient mitochondrial complex organization or reorganization to accommodate metabolic switches. Unlike our in vitro studies, Mtln-KO muscle tissues did show alterations in CI distribution and activity and levels of respiratory supercomplexes, along with changes in TCA cycle and FAO protein complex distributions. Increased levels of free CI and decreased supercomplexes, as observed in Mtln-KO mice, have been linked to a significant elevation in ROS (Lopez-Fabuel et al., 2016; Maranzana et al., 2013). However, our assessments of oxidative stress in the Mtln-KO mice yielded no significant findings. It is likely that the cells or mitochondria adapt to accommodate for mild deficiencies in respiratory activity and mCa2+ handling; future studies testing acute Mtln-knockdown or knockout in adult animals may reveal more profound physiological effects in mice.
Our BN-PAGE data suggest that Mtln elicits broad functions through interactions with an array of mitochondrial proteins. Thus far, we have been unsuccessful in our attempts to identify Mtln-interacting proteins by co-immunoprecipitation (co-IP) coupled with mass spectrometry; it is possible that during IP, our custom C-terminal-targeted antibody competes off key interactors bound to this region, which corresponds to the predominant extra-membrane portion of this small transmembrane protein. Alternatively, Mtln may interact, perhaps transiently, with many proteins, which could be “diluted” out in IP samples. Notably, our SDS-PAGE data also hint at the inherent sticky nature of this microprotein. Although not all sticky proteins are necessarily chaperones, given Mtln’s small size, lack of enzymatic domains, and lack of a clear single interacting complex, we speculate that Mtln, in part, serves as a mitochondrial molecular chaperone within the IMM; nevertheless, we cannot rule out other unforeseen functions that may be elucidated through future investigations.
Despite our observation that Mtln loss leads to deficiency in FAO, we believe that this occurs downstream of defects in respiratory chain activity (Nsiah-Sefaa and McKenzie, 2016) or tethering FAO and electron transport chain (ETC) systems (Wang et al., 2010), given that the co-expression data firmly integrate Mtln within a gene network related to respiratory complex and supercomplex assembly. Conversely, FAO pathway constituents (VLCAD, MCAD, and LCAD) are quite far removed from this co-expression module, and a dedicated co-expression analysis for VLCAD reveals a clear signature of known FAO pathway genes as expected. These findings reiterate the power of using gene co-expression analyses to help pinpoint the functions of newly identified mitochondrial proteins and/or known proteins with enigmatic roles in mitochondrial biology. Building and interrogating co-expression networks for known respiratory complex assembly factors could stimulate future searches for new related factors.
In summary, our findings highlight the need to continue exploring the vast non-coding RNA landscape for mitochondrial-related sORFs. In addition, follow-up studies are needed to (1) further define Mtln’s precise molecular functions, (2) determine how loss of Mtln alters the response to various metabolic stresses (e.g., high-fat diet, starvation, chronic exercise training, and cold exposure), and (3) begin to explore the translational relevance of Mtln expression changes in humans. For example, database searches indicate that LINC00116 is among the most significantly downregulated genes in aging human skeletal muscles (GEO: GSE362 and GSE674), and the GTEx portal annotates the existence of common genetic variants that strongly associate with LINC00116 expression in human tissues, including heart, SKM, and fat (GTEx Consortium, 2013). Although these variants have not yet been linked to human phenotypes in genetic association studies, targeted efforts may be required to evaluate their potential influence on human health and disease.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
CONTACT FOR REAGENT AND RESOURCE SHARING
- EXPERIMENTAL MODEL AND SUBJECT DETAILS
- Cultured cell lines
- Rodent studies
- METHOD DETAILS
- Analysis of human transcriptomic tissue expression profile data
- Bioinformatic assessment of LINC00116 sORF and the resulting Mtln microprotein
- Co-expression and gene ontology enrichment analyses
- Western blots
- Plasmids and viral constructs
- NRCM transfection, staining, and imaging
- Mitochondrial isolations
- Graded digitonin extraction of the outer mitochondrial membrane proteins
- Mitochondrial protein topology assessment with Proteinase K
- Cardiolipin binding assay
- Protein lipid strip binding assay
- Transduction of cells with adenovirus vectors
- Evaluation of mitochondrial calcium (mCa2+) retention capacity and matrix free-Ca2+ content
- Measurement of mitochondrial membrane potential (ΔΨm) and ROS in cells
- Oxygen consumption rate measures in cells
- Determination of mitochondrial and nuclear DNA contents
- Preparation of permeabilized cardiac and skeletal myofibers
- Measurement of mitochondrial O2 consumption (mO2) and Ca2+ retention capacity in permeabilized cardiac and skeletal myofibers
- Exercise tolerance testing
- Biochemical assays of oxidative stress
- Blue native gel electrophoresis (BNGE), blotting, Coomassie stain, and in-gel activity (IGA)
QUANTIFICATION AND STATISTICAL ANALYSES
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Mtln | custom Proteintech | N/A |
| Mouse CI monoclonal antibody (20E8C12) | ThermoFisher | Cat# A21348; RRID:AB_2535839 |
| Mouse VDAC1 monoclonal antibody (N152B/23) | NeuroMab | Cat# 75–204; RRID: AB_2214807 |
| Rabbit anti-subunit c of the mitochondrial ATP synthase Antibody | a gift from Dr. Elizabeth F. Neufeld | N/A |
| Total OXPHOS Rodent WB Antibody Cocktail | Abcam | Cat# ab110413, RRID:AB_2629281 |
| Rabbit polyclonal OGDH antibody | ABclonal | Cat# A6391 |
| Rabbit polyclonal ACAD9 antibody | AVIVA | Cat# OAAN02440 |
| Rabbit Anti-VLCAD Polyclonal Antibody | GeneTex | Cat# GTX114232S, RRID:AB_10728154 |
| Rabbit Anti-SOD2 Polyclonal Antibody | a gift from Dr. Frederick E. Domann | N/A |
| Rabbit Anti-Glutathione Peroxidase 1 Polyclonal Antibody | Abcam | Cat# ab22604, RRID:AB_2112120 |
| Rabbit monoclonal Glutathione Peroxidase 4 antibody | Abcam | Cat# ab125066, RRID:AB_10973901 |
| Rabbit anti-Beta-catenin Antibody | Sigma-Aldrich | Cat# PLA0230 |
| Peroxidase-AffiniPure Goat Anti-Mouse IgG (H+L) (min X Hu,Bov,Hrs,Rb,Sw Sr Prot) antibody | Jackson ImmunoResearch Labs | Cat# 115–035–146, RRID:AB_2307392 |
| Peroxidase-AffiniPure Goat Anti-Rabbit IgG (H+L) (min X Hu,Ms,Rat Sr Prot) antibody | Jackson ImmunoResearch Labs | Cat# 111–035–144, RRID:AB_2307391 |
| Mouse Anti-NDUFA9 Monoclonal Antibody, Clone 20C11 | Abcam | Cat# ab14713, RRID:AB_301431 |
| Rabbit Anti-Human LETM1 Polyclonal Antibody | GeneTex | Cat# GTX112455, RRID:AB_1950806 |
| Rabbit polyclonal CYCS (Cytochrome C) antibody | ABclonal | Cat# A0225 |
| Rabbit Anti-GAPDH Polyclonal antibody | Santa Cruz Biotechnology | Cat# sc-25778, RRID:AB_10167668 |
| Pyruvate Dehydrogenase (C54G1) Rabbit mAb antibody | Cell Signaling Technology | Cat# 3205S, RRID:AB_2162926 |
| Rabbit polyclonal DLD antibody | ABclonal | Cat# A13296 |
| Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 568 | Thermo Fisher Scientific | Cat# A-11011, RRID:AB_143157 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor Plus 488 | Thermo Fisher Scientific | Cat# A32723, RRID:AB_2633275 |
| Mouse Anti-FLAG Monoclonal Antibody, Clone M2 | Sigma-Aldrich | F3165 RRID:AB_259529 |
| Mouse Anti-UQCRC1 Monoclonal Antibody, Clone 16D10AD9AH5 | Abcam | Cat# ab110252 RRID: AB_10863633 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human Mtln (uniprot Q8NCU8–2) 56 amino acid protein, 96% purity by HPLC | Genescript | Custom order |
| MitoTracker® Red CMXRos | Invitrogen | M7512 |
| MitoSOX Red Mitochondrial Superoxide Indicator | Invitrogen | M36008 |
| Critical Commercial Assays | ||
| Worthington Neonatal Cardiomyocyte Isolation System | Worthington Biochemical Corporation | LK003300 |
| Cardiolipin Beads | Echelon Biosciences | P-BCLP |
| Control Beads | Echelon Biosciences | P-B000 |
| Lipid Strips | Echelon Biosciences | P-6002 |
| Glutathione Assay Kit | Cayman Chemicals | Item 703002 |
| EnzyChrom Aconitase Assay Kit | BioAssay Systems | EACO-100 |
| Cardiolipin Assay Kit, Fluorometric | BioVision | K944–100 |
| Experimental Models: Cell Lines | ||
| C2C12 myoblast | ATCC | CRL-1772 |
| HeLa | ATCC | CCL-2 |
| Neuro-2a (N2a) | ATCC | CCL-131 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6SJLF1/J | The Jackson Laboratory | JAX: 100012 |
| Mouse: C57BL/6J | JAX: 000664 | |
| Mouse: Mtln knockout mice backcrossed to C57BL/6J | This paper | N/A |
| Oligonucleotides | ||
| DNA oligonucleotides | Integrated DNA Technologies (IDT) | See Table S4 |
| Custom Dicer-substrate siRNAs targeting human Mtln | IDT | See Table S4 |
| Scrambled Dicer-substrate siRNA | IDT | IDT #1074346 |
| Negative Control Dicer-substrate siRNA | IDT | IDT #1074354 |
| Recombinant DNA | ||
| Multiplex Genome Engineering Using CRISPR/Cas Systems. | Addgene Plasmid #42229 | |
| Multiplex Genome Engineering Using CRISPR/Cas Systems. | Addgene Plasmid #42230 | |
| Ad5CMVcytoLacZ (Ad-ßgal) | University of Iowa Viral Vector Core | VVC-U of Iowa-3554 |
| Ad5TRENC116 (Ad-Mtln)) | University of Iowa Viral Vector Core | (Iowa-custom, Ad5TREnc116) |
| Ad5CMVeGFP (Ad-GFP) | University of Iowa Viral Vector Core | VVC-U of Iowa-4 |
| Software and Algorithms | ||
| coxpresdb ver.6 | NA | http://coxpresdb.jp/ |
| ToppFun gene ontology enrichment tool; ToppGene Suite | NA | https://toppgene.cchmc.org/ |
| RONN v3.2 | NA | https://www.strubi.ox.ac.uk/RONN |
| IUPred | NA | iupred.enzim.hu/ |
| PrDOS | NA | prdos.hgc.jp/cgi-bin/top.cgi |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ryan L. Boudreau (ryan-boudreau@uiowa.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cultured cell lines
Mouse C2C12 myoblast (ATCC, CRL-1772), human HeLa (ATCC, CCL-2), and mouse neuroblastoma N2a (ATCC, CCL-131) cell lines were maintained in DMEM with 100 U/ml penicillin, 0.1 mg/ml streptomycin and 10% FBS (DMEM10), in a cell culture incubator in standard conditions (37°C in 5% CO2). Cells were kept at low passage and routinely passaged prior to reaching 100% confluence.
Rodent studies
All animal studies were approved by the Institutional Animal Care and Use Committees (IACUC) at the University of Iowa, and all experiments conform to the appropriate regulatory standards. Rodents were housed in a controlled temperature environment on a 12 hr light/dark cycle, with food and water provided ad libitum, unless otherwise noted. Animal age and sex are noted within the mansucript where appropriate.
Neonatal rat cardiomyocytes
Neonatal rat cardiomyocytes (NRCMs) were isolated using the Worthington Neonatal Cardiomyocyte Isolation System (Worthington Biochemical Corporation, Cat. No LK003300). In brief, beating hearts were removed from 6 to 12 neonatal (2–3 day old) rat pups immediately after decapitation and immersed in ice cold calcium and magnesium free Hank’s Balanced Salt Solution (CMFHBSS). Non-cardiac and atrial tissue was separated from ventricular tissue and discarded. Ventricular pieces were washed three times in CMF-HBSS, minced into < 1 mm2 pieces, and processed through sequential treatments with trypsin (100 μg/mL, overnight, 4°C), Soybean trypsin inhibitor (182 μg/mL, 30 min, 37°C), and collagenase (1500 Units, reconstituted in Leibovitz L-15 media containing calcium and magnesium, 60 min, 37°C). Cardiac cells were dispersed via trituration through a 10 mL pipette, filtered through a cell strainer (70 μm pore size), centrifuged at 60 × G for 5 min to pellet cells, resuspended in pre-plating media (DMEM/F12 supplemented with 5% FBS, 10 mM HEPES, 1% ITS, and 15 μg/mL Gentamycin), transferred to a 10 cm tissue culture dish, and incubated for 60 min at 37°C with 5% CO2. After pre-plating, the non-adherent cell suspension containing cardiomyocytes was removed, centrifuged at 60 3 × for 5 min to pellet cells, resuspended in cardiomyocyte growth media (DMEM/F12 supplemented with 5% horse serum, 10 mM HEPES, 1% ITS, 15 μg/mL Gentamycin, and 100 μM BRDU), counted, plated on 0.2% gelatin coated tissue culture dishes at a density of 125,000 cardiomyocytes per cm2, and incubated at 37°C with 5% CO2.
Generation of LINC00116/Mtln knockout cells and mice
The CRISPR/Cas9 system was adapted using available plasmids and protocols (crispr.mit.edu/). CRISPR sgRNA expression plasmids were generated to target sequences flanking the mouse NC116 sORF homolog (1500011K16Rik gene), four upstream and four downstream. The 16 possible sgRNA up-down pairs were tested for activity in cultured mouse N2a cells (ATCC) by co-transfection of sgRNA plasmid pairs with a spCas9-expressing plasmid (pX260). On-target activity (i.e., cutting / sORF excision) was evaluated 48 h later by performing flanking PCR assay on genomic DNA (Figure S1; Table S4). The most efficient sgRNA pair was subsequently used to generate CRISPR/Cas9-based knockout (NC116-KO, also called Mtln-KO) mice via the University of Iowa Genome Editing Facility. CRISPR components for pronuclear injection were prepared using previously described methods (Yang et al., 2014). Briefly, spCas9 mRNA was in vitro transcribed after adding a T7 promoter to spCas9 coding region by PCR amplification of pX330 with T7-Cas9(F) and T7-Cas9(R) (Table S4). The two functional sgRNAs flanking NC116 were in vitro transcribed after generating templates with T7 promoters using primers T7-NC116(2) and T7-NC116(R) or primers T7-NC116(3) and T7-NC116(R). SpCas9 mRNA and sgRNAs were purified using a MEGAclear kit (Life Technologies). The injection mix consisted of 100 ng/μl spCas9 mRNA and 50 ng/μl each sgRNA. Zygotes from 4 week-old B6SJLF1/J mice (The Jackson Laboratory Stock: 100012) were prepared for pronuclear injection (Yang et al., 2014). Resulting offspring were genotyped to idenfity mice carrying the targeted NC116 / Mtln deletion (Figure S1; Table S4), and the Mtln-KO allele was then backcrossed to the C57BL/6J strain (The Jackson Laboratory Stock: 000664) for more than three generations prior to phenotyping.
METHOD DETAILS
Analysis of human transcriptomic tissue expression profile data
Four independent human tissue RNA profiling datasets were analyzed to determine LINC00116 RNA expression levels within each tissue, relative to SKM (set to 1) (Consortium and GTEx Consortium, 2013; Gibb et al., 2011; Uhlén et al., 2015; Wu et al., 2009). Within each dataset, relative expression data for similar tissues (e.g., different brain regions or segments of gut) were summarized by their mean. The mean relative expression values across datasets were then determined for tissues represented in at least three datasets, and mean ± SEM plotted in Figure 1A.
Bioinformatic assessment of LINC00116 sORF and the resulting Mtln microprotein
DNA sequences corresponding to the LINC00116 sORF and its homologs across various species were obtained from UCSC Genome Browser (http://genome.ucsc.edu/). Homologous sequences were identified using Lift-Over [or “View – In other genomes (convert)”] functions or by Blat search. DNA sequences were then input into the Translate tool (https://web.expasy.org), and resulting protein sequences derived from the putative sORF were aligned using Praline (https://www.ibi.vu.nl/programs/pralinewww/). Further evaluations of predicted protein structure and domains were performed using various online servers and tools: Phobius (http://phobius.sbc.su.se/), TMHMM (http://www.cbs.dtu.dk/services/TMHMM/), Ronn (https://www.strubi.ox.ac.uk/RONN), IUPred (iupred.enzim.hu/), and PrDOS (prdos.hgc.jp/cgi-bin/top.cgi), using default settings.
Co-expression and gene ontology enrichment analyses
Human LINC00116 and mouse 1500011K16Rik (NC116 homolog) gene symbols were input into the CoXpresDB database (coxpresdb.jp/), and the top 300 co-expressed genes for each species were downloaded. Gene symbols were matched across species using the NCBI HomoloGene database, and the top 200 genes, with identifiable shared gene symbols, for human and mouse were intersected (Table S1). The resultying 85 overlapping co-expressed genes were input into the ToppFun gene ontology enrichment tool (ToppGene Suite; https://toppgene.cchmc.org/), and the resulting significantly enriched GO terms were obtained and summarized in Table S2.
Western blots
Tissue lysates were generated by suspending fresh-frozen tissue pieces in lysis buffer (in mM, 150 NaCl, 10 NaF, 5 EGTA, 5 EDTA, 50 Tris-HCL pH 7.5 with 1.0% Triton X-100, 0.5% deoxycholate and 0.1% SDS) with freshly added 1X protease inhibitor (Roche), 1mM phenylmethylsulfonyl fluoride (PMSF), and 1mM sodium orthoavanadate, and shaking in a TissueLyser (QIAGEN). After 20 min on ice, lysates were clarified by centrifugation for 10 min 10,000 × G at 4°C. For adipose tissues, an upper layer of fat formed upon centrifugation, and the intranatant was collected and subjected to up to 3 additional clarification steps. Protein concentration was measured in clarified lysates using a BCA Protein Assay kit (ThermoFisher) and proteins were resolved by standard SDS-PAGE (Invitrogen NuPAGE system) before being transferred to a 0.2 μm PVDF membrane (Millipore) using NuPAGE transfer buffer (Invitrogen). The membrane was blocked with 2%–5% milk in 1X TBST (0.1% Tween 20), after which anti-Mtln antibody (1:2000; custom Proteintech, see Figure 1C), anti-Cox4 (1:3000, ThermoFisher, A21348), anti-VDAC1 (1:4000, NeuroMab, 75–204), anti-subunit c of the mitochondrial ATP synthase (1:3000, a gift from Dr. Elizabeth F. Neufeld), anti-OXPHOS cocktail (1:1000, Abcam, ab110413), anti-OGDH (1:2000, ABclonal, A6391), anti-ACAD9 (1:4000, AVIVA, OAAN02440), anti-VLCAD (1:2000, GeneTex, GTX114232), anti-SOD2 antibody (1:1000, a gift from Dr. Frederick E. Domann), anti-GPX1 antibody (1:1000, ab22604, Abcam), anti-GPX4 antibody (1:1000, ab125066, Abcam), anti-β-catenin (1:2000; Sigma-Aldrich, PLA0230), anti-DLD (ABlclonal A13296), or anti-PDHA1 (CST 3205S), diluted in blocking buffer was added and incubated overnight at 4°C with rocking. Membranes were washed with 1X TBST, incubated for 1 h with HRP-conjugated goat anti-mouse or anti-rabbit antibody (Jackson ImmunoResearch, 115–035–146 and 111–035–144) diluted 1:50,000 in blocking buffer, and then washed again. Chemiluminescence detection was performed using West Femto Maximum Sensitivity Substrate (ThermoFisher) followed by imaging on a BioRad Versadoc (Quantity One software used for acquisition and densitometry analyses).
Plasmids and viral constructs
Human and mouse NC116 sORFs (Mtln) were PCR amplified from genomic DNA using primers with added restriction enzyme sites and subsequently cloned into viral-based CMV or TRE (tetracycline/doxycyeline-inducible) expression plasmids (University of Iowa Viral Vector Core). Primers designed to add a C-terminal FLAG epitope were also used, as well as reverse primers that omit the stop codon, for downstream fusion to an eGFP cassette in custom CMV-based expression plasmids. Primer sequences are provided in Table S4. Replication incompetent adenoviral vectors including Ad-bgal (Iowa-3554, Ad5CMVcytoLacZ), Ad-GFP (Iowa-4,Ad5CMVeGFP), Ad-Mtln (Iowa-custom, Ad5TREnc116) were generated at the University of Iowa Viral Vector Core using the RapAdI system (Anderson et al., 2000).
NRCM transfection, staining, and imaging
NRCMs were transfected with various human Mtln expression plasmids (wild-type, C-terminal GFP-tagged, and N- or C-terminal FLAG tagged) using Lipofectamine 2000 (Invitrogen) or infected with Ad-Mtln (as described below). Transfection complexes were formed at a weight:volume ratio (wt:v) of 100–200 ng plasmid DNA to 1 μL Lipofectamine 2000 in 50 μL OPTIMEM, then diluted 4-fold in OPTIMEM and overlayed onto cells at 100 μL per cm2 after suctioning off the culture media. After 4 h incubation at 37°C with 5% CO2, the transfection media was replaced with cardiomyocyte growth media at a volume of 250 μL per cm2. The cells were incubated for 24–48 h at 37°C with 5% CO2 with daily media exchanges before visualization of Mtln localization by fluorescent microscopy and immunocytochemistry.
Mitochondria were labeled in live cells (non-transfected or 2 days post transfection or transduction, see below) by incubation with 100 nM MitoTrackerRed (Invitrogen) in pre-warmed media for 45 min at 37°C with 5% CO2. MitoTracker media was removed and cells washed 3X with culture media, prior to fixation with 4% paraformaldehyde in culture media for 10 min at 37°C. For immunofluorescent staining, fixed cells were washed with HBSS, permeabilized with 0.2% Triton X-100 for 15 min at RT, blocked in 2% BSA/HBSS for 1 h at RT, and incubated with primary antibodies diluted in 0.2% BSA/HBSS, overnight at 4°C with rocking. Primary antibody was rabbit anti-Mtln (1:500, custom Proteintech) or anti-FLAG mouse monoclonal antibody (clone M2, Sigma-Aldrich). Cells were washed in HBSS and stained with Alexa568-conjugated goat anti-rabbit or goat anti-mouse IgG at 1:2000 for 1 h at RT. After subsequent HBSS washes, image acquisition and anlaysis was done using a confocal laser scanning microscope (Zeiss LSM 510 meta) with the Zeiss ZEN software.
Mitochondrial isolations
For mitochondrial isolation from cultured cells, cells were harvested by trypsinization, washed in DMEM10, and crude mitochondria were isolated according to previously published methods (Clayton and Shadel, 2014), scaled down for 2 × 107 to 6 × 107 cells using a 2 mL Dounce homogenizer with tight fitting B pestle (Kontes), and with inclusion of 1x protease inhibitor cocktail in buffers. For C2C12 cells a sampling of pre-disrupted cells (input) and the supernatant above the crude mitochondrial pellet (sup) were collected for immunoblot analysis. Samples were analyzed on the day of isolation or stored at 80°C.
For mitochondrial isolation from tissues, tissues were dissected from mice under deep anesthesia, or immediately after euthanasia, and snap frozen in liquid nitrogen. Mitochondria were isolated by tissue disruption, homogenization, and differential centrifugation. Reagents, tubes and instruments were cold, tissue processing was performed on ice, and centrifugations were done at 4°C. Briefly, quadriceps, soleus, and gastrocnemius muscle from one mouse leg were minced together in isolation buffer for mitochondria-1 (IBM1) containing (in mM) 67 sucrose, 50 KCL, 10 EDTA, 50 Tris-HCL, and 0.2% fatty acid free BSA, pH 7.4 at 4°C. minced tissue was pelleted by 3 min centrifugation at 800 × G, digested for 30 min on ice in 1.5 mL 0.5% trypsin (Sigma T0303) in IBM1, transferred to an 8 mL Potter Elvehjem tissue grinder with PTFE Pestle (Kimble 886000–022) with 4.5 mL of IBM1 and freshly added HALT pro-tease inhibitor cocktail (Invitrogen) or Complete protease inhibitor cocktail (Roche), and homogenized manually with 6 strokes. After pelleting debris at 800 × G for 10 min the supernatant was transferred to a glass tube (Kimble HS) and centrifuged at 8000 × G for 10 min to obtain the mitochondrial pellet. The supernatant was poured off and the pellet suspended in IBM2 (in mM, 200 D-mannitol, 70 sucrose, 10 Tris-HCL, 5 EGTA, pH 7.4 at 4°C), transferred to a 2 mL eppendorf tube and washed twice in IBM2 by centrifugation for 10 min at 9000 × G. A sample of resuspended mitochondria was lysed in 1% Triton X-100 and protein concentration determined by DC (BioRad) or BCA (ThermoScientific) protein assay. Mitochondrial pellets were assayed on the same day or stored at −80. Cardiac mitochondria were isolated from the 1/3 to 1/2 of the mouse heart (posterior portion with atria removed). Mitochondrial isolation from heart was similar to that used for SKM except for modifications to buffers IBM1 (in mM, 200 sucrose, 10 Tris-HCL, 1 EGTA, 0.2% fatty acid free BSA, pH 7.4 at 4°C) and IBM2 (in mM, 200 D-mannitol, 75 sucrose, 10 Tris-HCL, 1 EGTA, pH 7.4 at 4°C), and trypsin digestion time reduced to 15 min.
Graded digitonin extraction of the outer mitochondrial membrane proteins
Crude mitochondria (isolated from SKM as described above and stored at −80°C) were suspended in IBM2 with protease inhibitors and divided into 4 equal parts (each approximately 150 μg mitochondrial protein in 100 μl) and incubated with increasing concentrations of digitonin (high purity digitonin, Millipore Cat #300410; at 0, 0.015, 0.05, or 0.15%) for 15 min on ice with intermittent vortexing, then centrifuged for 10 min 10,000 × G at 4°C to separate the supernatant containing extracted mitochondrial proteins, from pellets containing residual mitochondria and mitoplasts. Supernatants were transferred to fresh tubes and NuPAGE loading buffer (4X LDS and 10X reducing agent) was added to 1X. Pellets were suspended in 75 μL IBM2 with 1X NuPAGE loading buffer. Samples were heated to 70°C for 10 min, and 10 μL per well loaded onto precast NuPAGE 4%–12% Bis-Tris 15-well gels. SDS-PAGE and electro-transfer to 0.2 um PVDF membranes were performed using standard procedures with NuPAGE buffers (Invitrogen). After transfer, immunoblotting and chemiluminescent development were performed using standard procedures as described above. Primary antibodies (listed above) were anti-Vdac1 (OMM), anti-Cox4 (IMM) and anti-Mtln (all at 1:2000) and anti-GAPDH (1:3000).
Mitochondrial protein topology assessment with Proteinase K
Mitochondria were isolated from WT mouse SKM (combined quadriceps, gastrocnemius and tibialus) as described elsewhere in Methods, resuspended in isolation buffer and divided into 6 tubes (approximately 200 ug mitochondrial protein per tube), pelleted at 9000xg for 10 min then resuspended in 100 μl of either isotonic buffer (isolation buffer), hypotonic buffer (5 mM HEPES adjusted to pH 7.4 with KOH), or isotonic buffer containing 0.25% Triton X-100, with all steps performed on ice. After resuspension, each tube received either 4 μl water or 4 μl freshly prepared proteinase K (recombinant proteinase K, SKU pack 3115836001, Sigma-Aldrich) for a final concentration of 80 ug/ml. Tubes were incubated at 4°C for 30 min with gently mixing every 5 min. After the digest step, proteins were precipitated by addition of 100% trichloroacetic acid (TCA) to a final 10% volume and incubation on ice for 30 min. TCA precipitates were pelleted by centrifugation for 10 min at 14,000 x g at 4°C. Supernatants were discarded and pellets were washed twice by resuspension in 500 μl ice-cold acetone and centrifugation at 14,000 x g at 4°C. Pellets were allowed to air dry at room temperature in a fume hood for 45 min, then another 5 min at 95°C before addition of 2X NuPAGE loading buffer. Pellets were resuspended by pipetting, denatured at 95°C for 10 min, then 15 μl loaded per lane in 4%–12% bis-tris NuPage gels. SDS-PAGE was run in MES buffer and proteins were transferred to PVDF, membranes blocked, probed with primaries (overnight at 4°C) and developed as described elsewhere in Methods.
Membranes were immunoblotted with antibodies directed to antigens representative of different mitochondrial compartments. Oxoglutarate dehydrogenase (OGDH) is a soluble matrix protein. Letm1 is a single-pass IMM protein with the c-terminus oriented in the matrix (the antibody used recognizes c-terminus). Cytochrome c is a soluble protein in the intermembrane space (IMS). In this assay, hypotonic shock selectively disrupts the OMM allowing proteinase K access to the IMS, while the IMM remains intact.
Cardiolipin binding assay
Hearts from Mtln-KO mice or wild-type (WT) littermates were lysed in NP-40 lysis buffer [1% NP-40, 25 mM HEPES (pH 7.5), 50 mM NaCl, 10 mM NaF, 5 mM EDTA, 1 mM sodium orthoavanadate, 1 mM PMSF, proteinase and phosphatase inhibitor cocktails]. Insoluble material was removed by centrifugation at 10,000 × G for 10 min at 4°C. Protein concentration was measured in the cleared supernatant by BCA assay. Cardiolipin-bound agarose beads and control beads (Echelon) were incubated with 500 μg of mouse heart lysates for 2 h at 4°C, followed by 5 washes in 10 mM HEPES, 150 mM NaCl and 0.25% (v/v) NP-40. To elute proteins, an equal volume of 2X NuPAGE loading buffer (with LDS and reducing agent) was added to beads, incubated for 10 min at 70°C, and eluates were subjected to standard SDS-PAGE and western blotting.
Protein lipid strip binding assay
A protein-lipid overlay assay was performed using Membrane Lipid Strips (P-6002; Echelon Biosciences) according to the manufacturer’s recommendations. Briefly, The strips were first blocked in 1% (w/v) non-fat milk in TBST (50 mM Tris-HCl (pH 8.0), 150 mM NaCl, and 0.1% (v/v) Tween 20) for 1 h at room temperature in the dark, followed by 1 h incubation with synthetic NC116 protein (Genscript) at 0.5 μg/ml in blocking buffer at room temperature with gentle agitation. After washing the membranes three times for 10 min each with TBST, they were incubated for 2 hr with rabbit anti-NC116 polyclonal Ab (1:2000; custom from Proteintech). Membranes were washed as before and incubated for 1 h with anti-rabbit-HRP conjugated antibody (1:100,000; Jackson ImmunoResearch Laboratories) followed by three TBST washes and detection of lipid-bound protein by ECL.
Transduction of cells with adenovirus vectors
For Mtln overexpression experiments, HeLa cells were grown to approximately 60% confluency in DMEM10 at 37°C in 5% CO2, media was removed and Ad-βgal or Ad-Mtln diluted in OPTIMEM was added to cells at a multiplicity of infection of approximately 3–8 PFU per cell. After 4–5 h incubation, the vector solution was replaced with DMEM10 with or without doxycycline (+/−DOX, 0.5 μg/ml) to induce Mtln transgene expression. At 48 h to 72 h post-transduction, cells were immunostained or harvested for mitochondria isolation. Treatment groups include Ad-βgal (+DOX), Ad-Mtln (-DOX), and Ad-Mtln (+DOX).
Evaluation of mitochondrial calcium (mCa2+) retention capacity and matrix free-Ca2+ content
To evaluate mCa2+ retention capacity and content, HeLa cells from all groups including Ad-βgal (+DOX), Ad-Mtln (-DOX), and Ad-Mtln (+DOX), were transferred to an intracellular-like Ca2+ free buffer medium containing (in mM) 120 KCl, 10 NaCl, 1 KH2PO4, 20 HEPESTris and 3 μM thapsigargin to inhibit sarcoplasmic/endoplasmic reticulum calcium ATPases (SERCA) so that the movement of Ca2+ was only influenced by mitochondrial uptake, 80 μg/ml digitonin, protease inhibitors (Sigma EGTA-Free Cocktail), supplemented with 10 μM succinate and pH to 7.2. All solutions were cleared with Chelex 100 to remove trace Ca2+ (Sigma). For mCa2+ retention capacity: 3 × 106 digitonin-permeabilized HeLa cells were loaded with the ratiometric reporters FuraFF at concentration of 1 μM (Ca2+). At 20 s JC-1 (Enzo Life Sciences) was added to monitor mitochondrial membrane potential (ΔΨm). Fluorescent signals were monitored in a spectrofluorometer (Delta RAM, Photon Technology Int.) at 340 and 380 ex/510 em. After acquiring baseline recordings, at 400 s, a repetitive series of Ca2+ boluses (10 μM) were added at the indicated time points. At completion of the experiment the protonophore, 10 μM FCCP, was added to uncouple the ΔΨm and release matrix free-Ca2+. All experiments (3 replicates) were conducted at 37°C. All details have been previously reported (Luongo et al., 2017). For mCa2+ content, cells from all the groups were loaded with Fura2 and treated with digitonin and thapsigargin as previously reported (Luongo et al., 2017; Luongo et al., 2015). Upon reaching a steady-state recording, the protonophore, FCCP, was used to collapse ΔΨ and initiate the release of all matrix free Ca2+.
Measurement of mitochondrial membrane potential (ΔΨm) and ROS in cells
To assess mitochondrial membrane potential, HeLa cells from groups Ad-βgal (+DOX), Ad-Mtln (-DOX), and Ad-Mtln (+DOX) were stained with the ΔΨm indicator tetramethylrhodamine, methyl ester (TMRM; 100 nM) for 30 min at 37°C. Image acquisition was performed using a Carl Zeiss Axio Observer Z1 confocal microscope at 572/35 ex and 632/60 em. To measure mitochondrial superoxide production, cells were loaded with 5 μM MitoSOX Red for 30 min at 37°C and imaged at 545/30 ex and 585/40 em. Changes in fluorescence intensity was quantified using ImageJ as described (Luongo et al., 2017; Luongo et al., 2015).
Oxygen consumption rate measures in cells
HeLa cells infected with adenovirus including Ad-βgal (+DOX), Ad-Mtln (-DOX) and Ad-Mtln (+DOX) were subjected to oxygen consumption rate (OCR) measurements at 37°C in an XF96 extracellular flux analyzer (Seahorse Bioscience). Cells (3×104) were plated in XF media pH 7.4 supplemented with 25 mM glucose and 1 mM sodium pyruvate and sequentially exposed to oligomycin (2 μM), FCCP (2.5 μM), and rotenone plus antimycin A (1 μM each) as previously reported (Luongo et al., 2015).
Determination of mitochondrial and nuclear DNA contents
HeLa cells seeded in 24-well plates were infected with Ad virus as described above, and cells were treated with or without DOX for 48 h before DNA extraction in 100 μL QuickExtract DNA Extraction buffer (Epicenter Biosciences) per manufacturer instructions. Samples were diluted 1:4 and then serially diluted 1:5 Range (1–4, 1–20, 1–100, 1–500, 1–2500, 1–12500, 1–62500, 1–312500) before quantitative PCR analysis using primers specific for genomic DNA (LINC00116 locus) or mitochondrial DNA (see Table S4 for primer sequences), a Viia7 384-well plate QPCR machine and Power SYBR Green Master Mix (Thermo Fischer). We selected dilutions that showed a linear amplification between cycles 20 – 30, which corresponded to genomic DNA at dilutions a 1:20 – 1: 100, and mitochondria 1:12500 – 1:62500. Technical triplicates were performed to determine mean Ct per sample, and group means were calculated (n = 8 per group).
To determine mitochondrial DNA content in WT and Mtln-KO mouse muscles, gastrocnemius muscles were harvested from n = 7 mice per genotype, snap frozen in liquid nitrogen, and crushed into powder. DNA was extracted from approximately 5–10 mg of tissue powder after digestion with Proteinase K (0.2 mg/mL) overnight at 56°C. Sample were subsequently heated for 45 mins at 85°C, centrifuged to remove debris and then supernatants were removed for further DNA isolation by standard isopropanol precipitation. DNA was resuspended in TE buffer and concentrations determined via Nanodrop spectrophotometry. DNA samples were diluted to a standard concentration before serially diluting each 1:5 over range (1–5, 1–25, 1–125, 1–625, 1–3125, 1–15625, 1–78125) before QPCR analysis using primers specific for genomic DNA (TCRD locus) or mitochondrial DNA (Table S4). We selected dilutions that showed a linear amplification between cycles 20 – 30, which corresponded to genomic DNA at dilutions a 1:5 – 1: 125, and mitochondria 1:3125 – 1:78125. Technical triplicates were performed to determine mean Ct per sample, and group means were calculated (n = 7 per genotype).
Preparation of permeabilized cardiac and skeletal myofibers
This technique has been described by our group (Anderson et al., 2007) and others (Saks et al., 1998) in detail. Following dissection, muscle samples were placed in ice-cold (4°C) Buffer X containing (in mM) 7.23 K2EGTA, 2.77 CaK2EGTA, 20 Imidazole, 20 Taurine, 5.7 ATP, 14.3 Phosphocreatine, 6.56 MgCl2-6H2O and 50 MES (pH 7.1, 295 mOsm). Under a dissecting microscope, fat and connective tissue were removed from muscle samples and then small bundles of fibers were prepared (~1–3.5 mg wet weight/fiber bundle). Fiber bundles were then treated with 50 μg/ml saponin for 30 min as previously described (Anderson and Neufer, 2006). Following permeabilization, myofiber bundles (PmFBs) were washed in ice-cold Buffer Z containing (in mM) 110 K-MES, 35 KCl, 1 EGTA, 5 K2HPO2, 3 MgCl2-6H2O, and 5 mg/ml BSA (pH 7.4, 295 mOsm) and remained in Buffer Z on a rotator at 4°C until analysis (< 4 hr). We have observed that PmFBs undergo a Ca2+-independent contraction that is temperature sensitive and can occur even at 4°C (Perry et al., 2011), therefore, 20 μM Blebbistatin was added to the wash buffer and to the respiration medium during experiments, to prevent contraction. Chemicals and reagents used for OCR and calcium measures (see below) in myofibers were all obtained from Sigma-Aldrich, except for Amplex Red and Calcium Green 5-N, which were obtained from Invitrogen.
Measurement of mitochondrial O2 consumption (mO2) and Ca2+ retention capacity in permeabilized cardiac and skeletal myofibers
All mitochondrial measurements were performed at 37°C and contained 20 μM Blebbistatin in the assay buffer to prevent contraction during the course of the experiments. The Oroboros O2K Oxygraph system (Oroboros Instruments, Innsbruck, Austria) was used for O2 consumption (JO2) measurements. Ca2+ measurements were performed in a spectrofluorometer (Photon Technology Instruments, Birmingham, NJ), equipped with a thermal-jacketed cuvette chamber. The JO2 experiments were performed in Buffer Z + 0.5 mg/ml BSA, with the exception of Ca2+ retention experiments, which were performed in Buffer Y, containing (in mM) 250 Sucrose, 10 Tris-HCl, 20 Tris base, 10 KH2PO4, 2 MgCl2 6H2O, 0.5 mg/ml BSA). During the course of the JO2 experiments, substrates, nucleotides, and respiratory inhibitors were provided as indicated in the Figures and legends. For mitochondrial Ca2+ retention measurements, Buffer Z (or Y for SKM fibers) contained 1 μM Calcium Green 5-N (Invitrogen) and 5 mM pyruvate, 2 mM malate, and 5 mM succinate. At start of Ca2+ retention experiments, 1 μM Thapsigargin was added to inhibit endogenous SERCA in the myofibers, and 50 μM EGTA added to chelate residual Ca2+ and to establish Fmin. Pulses of 120 nmols Ca2+ (CaCl2) were added at fixed time intervals and Ca2+ uptake followed until mPTP opening as in (Anderson et al., 2011). At the end of experiment, 5 mM CaCl2 was added to saturate the probe and establish Fmax. Changes in free Ca2+ in cuvette during mCa2+ uptake were then calculated using the known Kd for Calcium Green 5-N and the equations established by Tsien’s group for calculating free ion concentrations using ion-sensitive fluorophores (Tsien, 1989). Prior to or following each experiment, fiber bundles were blot dried on wax paper and weighed on a micro-balance. Data are expressed as nmol min−1·mg dry wt.−1 (JO2) or nmol mg dry wt.−1 (Ca2+ retention).
Exercise tolerance testing
Exercise tolerance testing was conducted using a multilane motor-driven treadmill (Columbus Instruments, Model Exer 3/6 Treadmill) equipped with a wire grid at the end the tread belt that delivers a mild electric shock when mice cease running or cannot maintain the speed of the belt. The shock grid was set to deliver a shock (0.2 mA at 2 Hz) to motivate running. Time to exercise intolerance (i.e., failure to run) is the experimental endpoint defined as the mouse remaining on the shock grid for 10 s despite shock motivation. Mice were watched continuously during testing and were removed from the test at the time of failure. Wild-type and Mtln-KO mice were subjected to two different protocols designed to measure “sprint” or “endurance” capacity.
Exercise tolerance on the sprint protocol was performed as previously described (Kwong et al., 2015). Mice were not acclimatized to the treadmill prior to the day of the test. On the test day, mice were placed on the stationary treadmill for five min, then the treadmill was started at a speed of 10 m/min for one min, and then increased to a speed of 15 m/min for one min. The clock is started after this 7 min acclimation/warm-up period and the treadmill speed is increased to 20 m/min for a maximum run time of 20 min.
For exercise tolerance on the gradual acceleration (i.e., “endurance”) protocol, mice were acclimatized to the treadmill during a two day training period and then the test was done on day 3. On training day one, mice were placed on the treadmill at a starting speed of 3 m per min (m/min) and an incline of 15°. The treadmill speed was increased to 5 m/min after 5 min, increased to 7 m/min after 10 min, and stopped after 15 min. On training day two, mice were placed on the treadmill at a starting speed of 3 m per min (m/min) and an incline of 15°. The treadmill speed was increased to 5 m/min after 5 min, increased to 7 m/min after 10 min, increased to 10 m/min after 15 min, and stopped after 18 min. For “endurance” testing on day three, mice were placed on the treadmill at a starting speed of 5.5 m per min (m/min) and an incline of 0°. At 3 min intervals either the speed or incline was increased stepwise. Incline started at 0° and was increased to 5° after 3 min, 10° after 6 min, and 15° after 12 min for the duration. Speed started at 5.5 m/min and was increased to 7 m/min after 9 min, 9 m/min after 15 min, and increased an addition 1 m/min up to 22 m/min at every 3 min interval for duration.
Biochemical assays of oxidative stress
Biochemical assays were performed in isolated mitochondria or whole tissue lysates. The GSH/GSSG levels were measured on whole-tissue lysates with a commercially available kit (Cayman Chemical). The aconitase enzyme activity was measured in isolated mitochondria with an Aconitase Assay Kit (BioAssay Systems), and Cardiolipin level was measured with a Cardiolipin Assay Kit (BioVision) according to manufacturer’s instructions.
Blue native gel electrophoresis (BNGE), blotting, Coomassie stain, and in-gel activity (IGA)
BNGE procedures were derived from published methods (Diaz et al., 2009; Jha et al., 2016). Mitochondrial pellets were resuspended in aminocaproic acid buffer (ACAB, 0.75 M 6-aminocaproic acid, 50 mM bis-tris, pH 7.0) with freshly added protease inhibitors. Digitonin (high purity, Millipore) was added from a 10% stock in water to give a 4 g:g ratio of digitonin to mitochondrial protein and samples were solubilized on ice for 20 min, then centrifuged for 30 min at 20,000 × G and 4°C. Supernatants were transferred to fresh tubes and coomasie brilliant blue G (CBBG, Sigma B0770) was added from a 5% stock to give a 4 g:g digitonin:CBBG ratio. Glycerol was added to a final 5% volume, and 10–20 μL of sample containing 10–70 μg of protein were loaded into wells of precast NativePAGE Novex® 3%–12% Bis-Tris 10-well Gels (Invitrogen). High molecular weight (HMW) native markers (GE Healthcare) reconstituted in 100 μL ACAB plus protease inhibitors and 10 mL 5% CBBG, were loaded at 10–15 μL per lane. BNGE was run in an Invitrogen Novex mini-Cell tank in a 4°C cold room. Cathode buffer (15 mM bis-tris, 50 mM tricine,) and anode buffer (50 mM bis-tris) were pH 7.0 at 4°C. CBBG was added to the cathode buffer to a final 0.02% (dark blue) or 0.0005% (light blue). Electrophoresis was initiated at constant 150V with dark blue cathode buffer, and after approximatelys 50 min the dark blue was replaced with light blue cathode buffer and electrophoresis resumed at 250V for a maximum of 3 h total running time (until the dye front began to run into the anode buffer). Gels were transferred to 0.2 μm PVDF membrane using the Invitrogen mini Blot Module and NuPAGE transfer buffer at 4°C at 20V for 2–5 h. After transfer, membranes were fixed for 10 min in 8% acetic acid, rinsed in water and air-dried. Membranes were re-wet in methanol and washed in several changes of methanol to remove most of the CBBG, equilibrated in water, blocked with 5% milk (skim milk powder) in PBS-0.05% tween (PBST), then incubated with primary antibody diluted in PBST with 2.5% milk overnight at 4°C on a rocker. Primary antibodies were anti-Mtln (1/2000–1/3000, custom antibody) anti-NDUFA9 (1/5000 Abcam ab14713), anti-UQCRC1 (1/5000 Abcam ab110252), anti-OGDH (1/2000), anti-DLD (1/2000), anti-ACAD9 (1/4000). Blots were washed in PBST and incubated with HRP-conjugated secondary antibody from goat (1:50,000 to 1:100,000), washed and developed using ECL-prime (GE Health-care) or SuperSignal Femto (ThermoScientific) western blot developing reagent, and chemiluminescent images captured on a VersaDoc Imaging system with Quantity One software (Bio-Rad). For some experiments, the PVDF was incubated for 15 min in 2%–4% SDS in PBST with or without 4% beta-mercaptoethanol before blocking, to enhance primary antibody binding.
For Coomassie staining after BNGE, gels were immersed in stain (0.5% CBBG in 50% methanol, 10% acetic acid) on a rocker for 5 min, rinsed several times in MilliQ water, destained in 40% methanol / 10% acetic acid with rocking for 3 exchanges of destain every 15 min. Destaining was continued overnight in MilliQ water.
For IGA staining, BNGE conditions were similar to that described above except anode and cathode buffers were ice-cold and the gel box was packed in ice during the run, the starting cathode buffer contained 0.002% CBBG and was replaced after 50 min with cathode buffer without CBBG. Oxphos CI IGA staining was performed immediately after BNGE, as described (Jha et al., 2016). Briefly, the gel was incubated in freshly prepared CI substrate solution (2 mM Tris-HCl pH 7.4, 0.1 mg/ml NADH (Sigma-Aldrich), 2.5 mg/ml Nitrotetrazolium Blue chloride (NTB, Sigma-Aldrich)) for 20–30 min on a rocker at room temperature. Appearance of violet bands indicated CI activity. Reaction was stopped by incubation in 10% of acetic acid for 10 min and the gel was then washed in water.
Scanned images were obtained for PVDF after BNGE (before blotting), Coomassie stained gels, and IGA stained gels on an Epson Perfection V550 Photo scanner.
QUANTIFICATION AND STATISTICAL ANALYSES
Statistical analyses were performed using Prism GraphPad software, with guidance from the University of Iowa Department of Biostatistics (Dr. Patrick Breheny). GraphPad was used to generate plots and to perform two-tailed t tests or one-way ANOVAs with Dunnett’s post hoc to obtain the reported p values. The specific statistical tests for each experiment are indicated throughout the manuscript; for each, all data met the test assumptions and groups showed similar variance. Results were considered to be significant if p values were ≤ 0.05.
KEY RESOURCES TABLE
Supplementary Material
Highlights.
LINC00116 encodes a single-pass transmembrane protein known as mitoregulin (Mtln)
Mtln appears to be a sticky inner mitochondrial membrane protein that binds cardiolipin
Mtln levels influence mitochondrial respiration, ROS, and Ca2+ retention capacity
Mtln-KO mice exhibit deficiencies in FAO, mCa2+ retention, and supercomplexes
In Brief.
Stein et al. show that the long non-coding RNA LINC00116 encodes a highly conserved single-pass transmembrane protein named mitoregulin (Mtln). Studies in cells and mice demonstrate that Mtln localizes to inner mitochondrial membranes, where it interacts with several complexes to influence mitochondrial membrane potential, respiration, Ca2+ retention capacity, ROS, and supercomplex levels.
ACKNOWLEDGMENTS
This work was supported by the American Heart Association (grant 14SDG18590008 to R.L.B.), the Roy J. Carver Trust (University of Iowa to R.L.B.), a Fraternal Order of Eagles Diabetes Research Center Pilot Grant (University of Iowa to R.L.B.), and the NIH NHLBI and NIA (grants HL136954, HL123966, and DA037830 to J.W.E. and grants HL122863 and AG057006 to E.J.A.). J.M.M. was supported by the Abboud Cardiovascular Research Center NIH T32 (grant HL007121). We also acknowledge members of the laboratories of Dale E. Abel, Eric Taylor, Leonid Zingman, and Leonid Sazanov for technical assistance and/or relevant scientific discussions, as well as University of Iowa core facilities that made significant contributions to this work, including the Genome Editing Facility, Viral Vector Core, Genomics Division, and Metabolic Phenotyping Core.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes nine figures and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.002.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Abdulhag UN, Soiferman D, Schueler-Furman O, Miller C, Shaag A, Elpeleg O, Edvardson S, and Saada A (2015). Mitochondrial complex IV deficiency, caused by mutated COX6B1, is associated with encephalomyopathy, hydrocephalus and cardiomyopathy. Eur. J. Hum. Genet 23, 159–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alston CL, Compton AG, Formosa LE, Strecker V, Oláhová M, Haack TB, Smet J, Stouffs K, Diakumis P, Ciara E, et al. (2016). Biallelic mutations in TMEM126B cause severe complex I deficiency with a variable clinical phenotype. Am. J. Hum. Genet 99, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunts A, Brown A, Toots J, Scheres SHW, and Ramakrishnan V (2015). Ribosome. The structure of the human mitochondrial ribosome. Science 348, 95–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson EJ, and Neufer PD (2006). Type II skeletal myofibers possess unique properties that potentiate mitochondrial H(2)O(2) generation. Am. J. Physiol. Cell Physiol 290, C844–C851. [DOI] [PubMed] [Google Scholar]
- Anderson RD, Haskell RE, Xia H, Roessler BJ, and Davidson BL (2000). A simple method for the rapid generation of recombinant adenovirus vectors. Gene Ther. 7, 1034–1038. [DOI] [PubMed] [Google Scholar]
- Anderson EJ, Yamazaki H, and Neufer PD (2007). Induction of endogenous uncoupling protein 3 suppresses mitochondrial oxidant emission during fatty acid-supported respiration. J. Biol. Chem 282, 31257–31266. [DOI] [PubMed] [Google Scholar]
- Anderson EJ, Rodriguez E, Anderson CA, Thayne K, Chitwood WR, and Kypson AP (2011). Increased propensity for cell death in diabetic human heart is mediated by mitochondrial-dependent pathways. Am. J. Physiol. Heart Circ. Physiol 300, H118–H124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DM, Makarewich CA, Anderson KM, Shelton JM, Bezprozvannaya S, Bassel-Duby R, and Olson EN (2016). Widespread control of calcium signaling by a family of SERCA-inhibiting micropeptides. Sci. Signal 9, ra119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, Obermayer B, Fleming ES, Vejnar CE, Lee MT, Rajewsky N, Walther TC, and Giraldez AJ (2014). Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 33, 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, and Mootha VK (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 44 (D1), D1251–D1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catherman AD, Li M, Tran JC, Durbin KR, Compton PD, Early BP, Thomas PM, and Kelleher NL (2013). Top down proteomics of human membrane proteins from enriched mitochondrial fractions. Anal. Chem 85, 1880–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Li Y, Zhong J, Zhang J, Chen Z, Yang L, Cao X, He QY, Zhang G, and Wang T (2015). Identification of missing proteins defined by chromosome-centric proteome project in the cytoplasmic detergent-insoluble proteins. J. Proteome Res 14, 3693–3709. [DOI] [PubMed] [Google Scholar]
- Clayton DA, and Shadel GS (2014). Isolation of mitochondria from tissue culture cells. Cold Spring Harb. Protoc 2014, pdb.prot080002. [DOI] [PubMed] [Google Scholar]
- Cogliati S, Calvo E, Loureiro M, Guaras AM, Nieto-Arellano R, Garcia-Poyatos C, Ezkurdia I, Mercader N, Vázquez J, and Enriquez JA (2016). Mechanism of super-assembly of respiratory complexes III and IV. Nature 539, 579–582. [DOI] [PubMed] [Google Scholar]
- D’Lima NG, Ma J, Winkler L, Chu Q, Loh KH, Corpuz EO, Budnik BA, Lykke-Andersen J, Saghatelian A, and Slavoff SA (2017). A human microprotein that interacts with the mRNA decapping complex. Nat. Chem. Biol 13, 174–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz F, Barrientos A, and Fontanesi F (2009). Evaluation of the mitochondrial respiratory chain and oxidative phosphorylation system using blue native gel electrophoresis. Curr. Protoc. Hum. Genet Chapter 19, Unit19.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Vizarra E, Bugiani M, Goffrini P, Carrara F, Farina L, Procopio E, Donati A, Uziel G, Ferrero I, and Zeviani M (2007). Impaired complex III assembly associated with BCS1L gene mutations in isolated mitochondrial encephalopathy. Hum. Mol. Genet 16, 1241–1252. [DOI] [PubMed] [Google Scholar]
- Gibb EA, Vucic EA, Enfield KS, Stewart GL, Lonergan KM, Kennett JY, Becker-Santos DD, MacAulay CE, Lam S, Brown CJ, and Lam WL (2011). Human cancer long non-coding RNA transcriptomes. PLoS ONE 6, e25915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GTEx Consortium (2013). The Genotype-Tissue Expression (GTEx) project. Nat. Genet 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, and Nijtmans L (2017). The assembly pathway of mitochondrial respiratory chain complex I. Cell Metab. 25, 128–139. [DOI] [PubMed] [Google Scholar]
- He M, Rutledge SL, Kelly DR, Palmer CA, Murdoch G, Majumder N, Nicholls RD, Pei Z, Watkins PA, and Vockley J (2007). A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am. J. Hum. Genet 81, 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heide H, Bleier L, Steger M, Ackermann J, Dröse S, Schwamb B, Zörnig M, Reichert AS, Koch I, Wittig I, and Brandt U (2012). Complexome profiling identifies TMEM126B as a component of the mitochondrial complex I assembly complex. Cell Metab. 16, 538–549. [DOI] [PubMed] [Google Scholar]
- Jha P, Wang X, and Auwerx J (2016). Analysis of mitochondrial respiratory chain supercomplexes using blue native polyacrylamide gel electrophoresis (BN-PAGE). Curr. Protoc. Mouse Biol 6, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SE, Mori R, Komatsu T, Chiba T, Hayashi H, Park S, Sugawa MD, Dencher NA, and Shimokawa I (2015). Upregulation of cyto-chrome c oxidase subunit 6b1 (Cox6b1) and formation of mitochondrial super-complexes: implication of Cox6b1 in the effect of calorie restriction. Age (Dordr.) 37, 9787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong JQ, Lu X, Correll RN, Schwanekamp JA, Vagnozzi RJ, Sargent MA, York AJ, Zhang J, Bers DM, and Molkentin JD (2015). The mitochondrial calcium uniporter selectively matches metabolic output to acute contractile stress in the heart. Cell Rep. 12, 15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapuente-Brun E, Moreno-Loshuertos R, Acín-Pérez R, Latorre-Pellicer A, Colás C, Balsa E, Perales-Clemente E, Quirós PM, Calvo E, Rodríguez-Hernández MA, et al. (2013). Supercomplex assembly determines electron flux in the mitochondrial electron transport chain. Science 340, 1567–1570. [DOI] [PubMed] [Google Scholar]
- Letts JA, Fiedorczuk K, and Sazanov LA (2016). The architecture of respiratory supercomplexes. Nature 537, 644–648. [DOI] [PubMed] [Google Scholar]
- Long J, Badal SS, Ye Z, Wang Y, Ayanga BA, Galvan DL, Green NH, Chang BH, Overbeek PA, and Danesh FR (2016). Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Invest 126, 4205–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Fabuel I, Le Douce J, Logan A, James AM, Bonvento G, Murphy MP, Almeida A, and Bolaños JP (2016). Complex I assembly into super-complexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 113, 13063–13068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Yuan A, Zhang X, Gross P, Song J, Shanmughapriya S, Gao E, Jain M, Houser SR, et al. (2015). The mitochondrial calcium uniporter matches energetic supply with cardiac workload during stress and modulates permeability transition. Cell Rep. 12, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luongo TS, Lambert JP, Gross P, Nwokedi M, Lombardi AA, Shanmughapriya S, Carpenter AC, Kolmetzky D, Gao E, van Berlo JH, et al. (2017). The mitochondrial Na+/Ca2+ exchanger is essential for Ca2+ homeostasis and viability. Nature 545, 93–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maranzana E, Barbero G, Falasca AI, Lenaz G, and Genova ML (2013). Mitochondrial respiratory supercomplex association limits production of reactive oxygen species from complex I. Antioxid. Redox Signal. 19, 1469–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Pasut A, Matsumoto M, Yamashita R, Fung J, Monte-leone E, Saghatelian A, Nakayama KI, Clohessy JG, and Pandolfi PP (2017). mTORC1 and muscle regeneration are regulated by the LINC00961-encoded SPAR polypeptide. Nature 541, 228–232. [DOI] [PubMed] [Google Scholar]
- Michel AM, Fox G, M Kiran A, De Bo C, O’Connor PB, Heaphy SM, Mullan JP, Donohue CA, Higgins DG, and Baranov PV (2014). GWIPS-viz: development of a ribo-seq genome browser. Nucleic Acids Res. 42, D859–D864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nouws J, Nijtmans L, Houten SM, van den Brand M, Huynen M, Venselaar H, Hoefs S, Gloerich J, Kronick J, Hutchin T, et al. (2010). Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 12, 283–294. [DOI] [PubMed] [Google Scholar]
- Nsiah-Sefaa A, and McKenzie M (2016). Combined defects in oxidative phosphorylation and fatty acid b-oxidation in mitochondrial disease. Biosci. Rep 36, e00313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi T, Hayashi S, Shibaoka M, Saeki M, Ohta H, and Kinoshita K (2008). COXPRESdb: a database of coexpressed gene networks in mammals. Nucleic Acids Res. 36, D77–D82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogilvie I, Kennaway NG, and Shoubridge EA (2005). A molecular chaperone for mitochondrial complex I assembly is mutated in a progressive encephalopathy. J. Clin. Invest 115, 2784–2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostojić J, Panozzo C, Lasserre JP, Nouet C, Courtin F, Blancard C, di Rago JP, and Dujardin G (2013). The energetic state of mitochondria modulates complex III biogenesis through the ATP-dependent activity of Bcs1. Cell Metab. 18, 567–577. [DOI] [PubMed] [Google Scholar]
- Perry CG, Kane DA, Lin CT, Kozy R, Cathey BL, Lark DS, Kane CL, Brophy PM, Gavin TP, Anderson EJ, and Neufer PD (2011). Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem. J 437, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer K, Gohil V, Stuart RA, Hunte C, Brandt U, Greenberg ML, and Schägger H (2003). Cardiolipin stabilizes respiratory chain supercomplexes. J. Biol. Chem 278, 52873–52880. [DOI] [PubMed] [Google Scholar]
- Saada A, Vogel RO, Hoefs SJ, van den Brand MA, Wessels HJ, Willems PH, Venselaar H, Shaag A, Barghuti F, Reish O, et al. (2009). Mutations in NDUFAF3 (C3ORF60), encoding an NDUFAF4 (C6ORF66)-interacting complex I assembly protein, cause fatal neonatal mitochondrial disease. Am. J. Hum. Genet 84, 718–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saks VA, Veksler VI, Kuznetsov AV, Kay L, Sikk P, Tiivel T, Tranqui L, Olivares J, Winkler K, Wiedemann F, and Kunz WS (1998). Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol. Cell. Biochem 184, 81–100. [PubMed] [Google Scholar]
- Souri M, Aoyama T, Hoganson G, and Hashimoto T (1998). Very-long-chain acyl-CoA dehydrogenase subunit assembles to the dimer form on mitochondrial inner membrane. FEBS Lett. 426, 187–190. [DOI] [PubMed] [Google Scholar]
- Stroud DA, Surgenor EE, Formosa LE, Reljic B, Frazier AE, Dibley MG, Osellame LD, Stait T, Beilharz TH, Thorburn DR, et al. (2016). Accessory subunits are integral for assembly and function of human mitochondrial complex I. Nature 538, 123–126. [DOI] [PubMed] [Google Scholar]
- Sugiana C, Pagliarini DJ, McKenzie M, Kirby DM, Salemi R, Abu-Amero KK, Dahl HH, Hutchison WM, Vascotto KA, Smith SM, et al. (2008). Mutation of C20orf7 disrupts complex I assembly and causes lethal neonatal mitochondrial disease. Am. J. Hum. Genet 83, 468–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsien RY (1989). Fluorescent indicators of ion concentrations. Methods Cell Biol. 30, 127–156. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Uversky VN (2015). The multifaceted roles of intrinsic disorder in protein complexes. FEBS Lett. 589 (19 Pt A), 2498–2506. [DOI] [PubMed] [Google Scholar]
- Wang Y, Mohsen AW, Mihalik SJ, Goetzman ES, and Vockley J (2010). Evidence for physical association of mitochondrial fatty acid oxidation and oxidative phosphorylation complexes. J. Biol. Chem 285, 29834–29841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C, Orozco C, Boyer J, Leglise M, Goodale J, Batalov S, Hodge CL, Haase J, Janes J, Huss JW 3rd, and Su AI (2009). BioGPS: an extensible and customizable portal for querying and organizing gene annotation resources. Genome Biol. 10, R130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan LJ, and Forster MJ (2009). Resolving mitochondrial protein complexes using nongradient blue native polyacrylamide gel electrophoresis. Anal. Biochem 389, 143–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, and Jaenisch R (2014). Generating genetically modified mice using CRISPR/Cas-mediated genome engineering. Nat. Protoc 9, 1956–1968. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.