

Summary
Bacterial community acquisition in the infant gut impacts immune education and disease susceptibility. We compared bacterial strains across and within families in a prospective birth cohort of 44 infants and their mothers, sampled longitudinally in the first months of each child’s life. We identified mother-to-child bacterial transmission events and describe the incidence of family-specific antibiotic resistance genes. We observed two inheritance patterns across multiple species, where often the mother’s dominant strain is transmitted to the child, but occasionally her secondary strains colonize the infant gut. In families where the secondary strain of B. uniformis was inherited, a starch utilization gene cluster that was absent in the mother’s dominant strain was identified in the child, suggesting the selective advantage of a mother’s secondary strain in the infant gut. Our findings reveal mother-to-child bacterial transmission events at high-resolution and give insights into early colonization of the infant gut.
eToC blurb
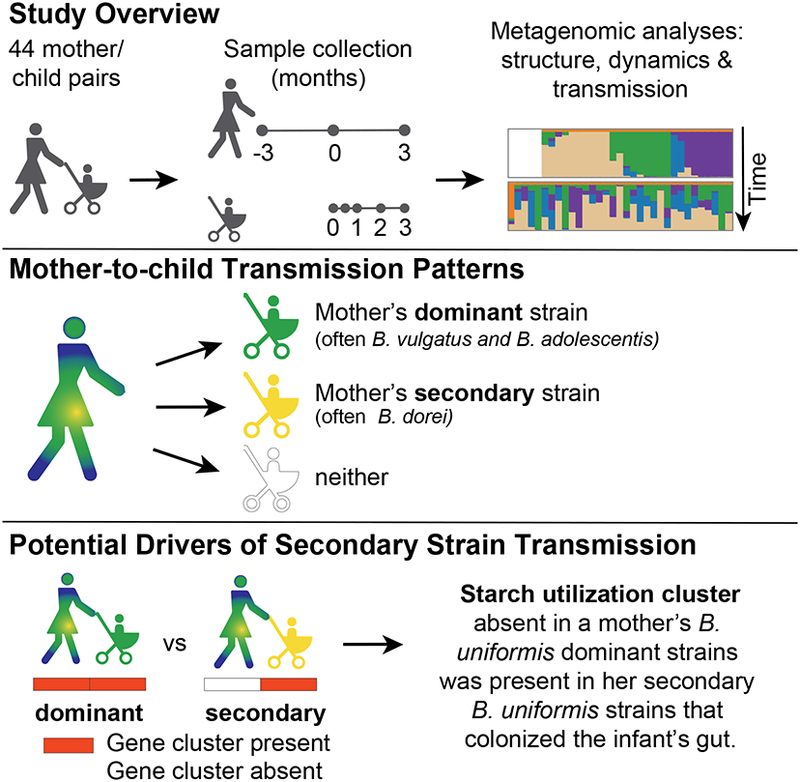
Using longitudinal metagenomic sequencing from 44 mother/child pairs, Yassour et al. characterized mother-to-child strain transmission patterns. While mothers’ dominant strains were often inherited, nondominant secondary strain transmissions were also observed. Microbial functional analysis reveal inherited maternal secondary strains may have a selective advantage to colonize infant guts.
Introduction
The infant gut microbial community plays an important role in human development, including the maturation of the immune system (Smith et al., 2013), nutrient utilization and modification (Nicholson et al., 2012; Sela et al., 2008), and the prevention of pathogen colonization (Sela and Mills, 2010). This dynamic community is strongly affected by delivery mode (Biasucci et al., 2010; Dominguez-Bello et al., 2010; Jakobsson et al., 2014; Penders et al., 2006; Yassour et al., 2016) and feeding (Bäckhed et al., 2015), and stabilizes by age 3, when it starts to resembles an adult gut community (Yatsunenko et al., 2012). Although considerable progress has been made in characterizing the taxonomic profiles of these early life gut communities (Bäckhed et al., 2015; Dominguez-Bello et al., 2010; Koenig et al., 2011; Yassour et al., 2016), questions concerning its origin and the extent of maternal transmission contributions remain largely unanswered (Perez-Muñoz et al., 2017). Here, longitudinal sampling from both mother and child, coupled with deep metagenomic sequencing, enables us to use strain-level analysis to bridge this knowledge gap.
Children are exposed to diverse environmental sources of bacteria from the day they are born, yet which of these bacteria will end up colonizing their gut is still only partially understood. Multiple factors can influence this selection process, including the microbial composition of potential maternal sources (e.g., vaginal/gut communities); the state of immune activation in the host (Cahenzli et al., 2013; Olszak et al., 2012); and the carbon sources found in the unique environment of the infant gut (Marcobal et al., 2011; Sela et al., 2008). Importantly, while the mother’s own gut bacteria provide a likely source of continual exposure, we know little about vertical inheritance events in determining early childhood gut composition. Such postnatal vertical transmission of the mother’s gut bacteria may provide early colonizers that can reside in the child’s gut for many years (Faith et al., 2013).
Strain-level variations among isolated bacteria have been studied for over a century using culture-based methods, but metagenomic sequencing and new computational approaches have only recently become available as important new tools to study strain variation in culture-free environmental samples and complex mixtures of bacteria (Luo et al., 2015; Truong et al., 2017). Different strains of the same species often share 80–90% of their genes, referred to as the species core genome. However, the non-core genome confers important functional differences, such as antibiotic resistance and carbon source utilization, which may play an important role in the selection process in various environmental niches. The ability to detect such strain differences in early life and infer their functional consequences remains a challenge for the field.
Here, we present a longitudinal study of paired mother-child stool samples from 44 Finnish families. We describe the structure and dynamics of the gut microbial communities in the first 3 months of life using whole-genome metagenomic sequencing. By examining mother and child stool at multiple time points, we assessed changes in individuals as well as the relatedness of taxonomic profiles within families. Additionally, we developed a novel strain-level approach to characterize mother-to-child bacterial transmission patterns. We found that, while in most cases the mother’s dominant strain was transmitted to her child, sometimes her secondary strains preferentially colonized the infant gut. Furthermore, in those cases of secondary strain transmission we sought to identify the functional genes that were missing in the mother’s dominant strain. For example, we identified a glycan utilization system in Bacteroides uniformis infant strains that may specifically facilitate the metabolism of mother’s milk. The lack of such niche-specific genes may partly explain the inheritance of mother’s’ secondary strains.
Results
A paired longitudinal study tracking mothers and infants in the first 3 months of life
To elucidate the nature of maternal-child microbiota transmission in early life we designed a paired longitudinal study of mother and child stools samples, starting from birth. We collected longitudinal stool samples and clinical metadata variables from a cohort of 44 Finnish families (7/44 children delivered by c-section, Table S1). For 33 of those families, we collected five child stool samples: at birth (meconium), two weeks, and one, two and three months of age, together with three maternal stool samples: gestational week 27, delivery, and three months postdelivery. The remaining 11 families have only three samples: mother gestational week 27, delivery and child meconium samples. To analyze the species and subspecies composition of the microbial communities in this cohort, we performed whole-genome metagenomic sequencing and constructed species-level composition profiles for all 287 samples (see Methods). Here, the metagenomic sequencing approach allowed us to investigate microbiome samples at subspecies resolution and further characterize these communities to identify the strain composition and to examine postnatal vertical inheritance patterns between mother and child.
Distinct microbial features of the maternal and infant gut communities
We characterized the relative abundance patterns of microbial species found in mother and child stool samples. As expected, each individual showed unique microbial composition and the adult and infant communities were clearly distinctive, even at a coarse phylum-level (Figure 1A, Figure S1). Child communities were dominated by species from the Actinobacteria and Proteobacteria phyla (Figure 1A), whereas mothers had a higher fraction of Bacteroidetes and Firmicutes (Figure 1B), as expected for the healthy western adult gut (Yatsunenko et al., 2012).
Figure 1: Gut microbiome trajectories of mother and child in the first 3 months of life.
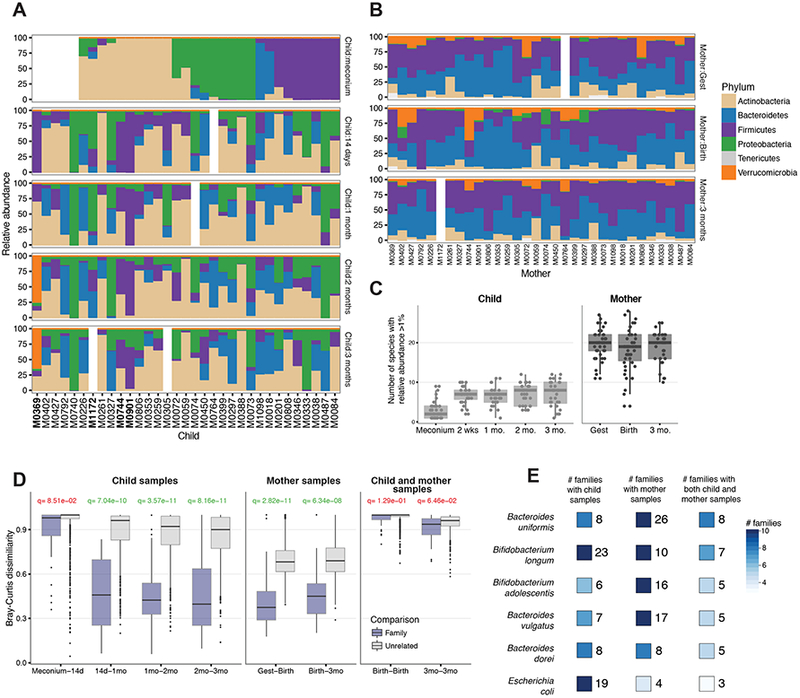
(A–B) Relative phylum-level relative abundance profiles for the child (A) and mother (B) samples. Children born by C-section are highlighted in bold. (C) Microbial community complexity over time as estimated by the number of species occurring in a given sample above 1% relative abundance. (D) Stability of species-level composition profiles over time as measured by Bray-Curtis dissimilarity of individual children (left), mothers (center), and child-mother paired samples collected at birth and 3 months (right). The FDR-corrected q-value is shown for each comparison (calculated with a t-test) and colored green if q<0.05. (E) Species observed across multiple families, enumerating how many families have these species present in children (left), mothers (center), or both (right), with relevant abundance of >=5%.
The child meconium communities had very low complexity on the phylum-level (often dominated by a single phylum). We confirmed this observation on the species-level profiles. We measured the complexity of each community by calculating the number of species above 1% relative abundance, and found that child stool samples had lower overall microbial complexity, with the meconium sample containing very few unique species (Figure 1C; this trend was also evident using Shannon diversity as the complexity measure, Figure S2). Maternal samples contained 20 or more species above 1% relative abundance, while child samples typically contained 10 or fewer (Figure 1C). Furthermore, very few species (typically 3–7) were found in the meconium communities. We confirmed that the low complexity was not a side effect of the lower number of reads in the meconium samples by repeating this analysis with downsampling of all samples to have the same number of reads (see Methods and Figure S2).
To compare the maternal and child microbial communities, we examined the development and variability of microbial communities within and across families. We compared the stability of early child gut microbiota by testing the species-level dissimilarity between adjacent time points for each individual over time, and within families (using Bray-Curtis dissimilarity, see Methods). The maternal microbial communities displayed stability with significant overlap in the species-level composition between time points relative to random subject pairings (p=2.82e-11, 6.34e-08, calculated with t-test, Figure 1D). The child microbial communities also exhibit significantly similar features over time, with the exception of the first 2 weeks of life, where the community is rapidly changing (p=8.51e-02, 7.04e-10, 3.57e-11, 8.16e-11, Figure 1D). However, we found no significant similarity by this metric between the microbial communities of a mother and her child (p=8.16e-11, 6.34e-08, Figure 1D). These comparisons were not significantly different when segregated by delivery mode, but this study is underpowered to study the effect of delivery-mode on bacterial transmission as only few children were born by C-section (see Discussion). Despite the lack of similarity at the level of the entire community structure, we examined whether there were shared species in the mother and her offspring to explore possible mother-to-child bacterial transmission. Indeed, we found six common species in the maternal and infant communities with 5% relative abundance or greater both mother and child: three Bacteroides species (B. uniformis, B. vulgatus, and B. dorei), two Bifidobacterium species (B. adolescentis and B. longum), and Escherichia coli (Figure 1E). Each of these species was observed in at least 3 mother-child pairs in the cohort, enabling higher-resolution strain characterization and matching maternal and child strains.
Strain-level variation suggests two patterns of mother-to-child bacterial transmission
For the purposes of this study, we defined the ‘dominant strain’ within a microbial species in a sample to be the single most common haplotype detectable by metagenomic sequencing (see Methods). In particular, this captures only the single ‘dominant strain’ per species, which typically comprises of at least 70% relative abundance of that species (Luo et al., 2015; Truong et al., 2017; Yassour et al., 2016). In the underlying microbial community, this dominant strain may not represent a perfectly clonal population, nor the only genetic diversity within a species, but we use the language here to describe the variants detectable metagenomically. In this study, we denote all the non-dominant strains as the collection of ‘secondary strains’. At any given genomic position, the dominant strain has the highest read count (by definition), and we denote the nucleotide found in that position as the ‘major nucleotide’. Conversely, the secondary strains contribute to the second most frequent nucleotide at each position, which is denoted as the ‘minor nucleotide’ (see Methods); we ignored other, less frequent nucleotides. To characterize each sample’s population and identify its dominant strain, we examined patterns of SNVs in genomic regions that are unique to each species within and across families (‘species-specific marker genes’; see Methods). Observing highly-similar nucleotide variation patterns in two individuals suggests they share the same strain, and thereby a potential strain inheritance from mother to child.
In order to assess whether a child shared the dominant or a secondary strain observed in the mother, we enumerated all positions where the child’s major nucleotide matched the major nucleotide observed in the mother (Figure 2A). Observing multiple positions with identical nucleotides within a family but not across families, suggests a transmitted dominant strain between a mother and her child. When we compared the major-nucleotide match rates (on species-specific marker genes) we found that unrelated child-mother pairs commonly had 98.5–99.0% identity, whereas true child-mother pairs often had at least 99.8% identity. To quantify within vs. across families match rates, we assigned z-scores for the match percentages (see Methods). For example, when examining the strains of Bacteroides uniformis, we found evidence that in some families the dominant strain was transmitted between mother and child (e.g., M0074 has a z-score of 4.27, Figure 2B), while other families do not exhibit shared dominant strains (e.g. M0297 and M0399 with z-scores of 0.43 and −0.43, respectively, Figure 2B). We denote these z-scores as the ‘dominant strain z-scores’. Next, we studied whether instead of the mother’s dominant strain, perhaps one of her secondary strains was transmitted to the child. Transmission of the mother’s secondary strains could be the result of important functional differences, as such strains could harbor selective advantages in the unique environment of the infant gut.
Figure 2: Patterns of dominant and secondary strains shared in mother-child pairs.
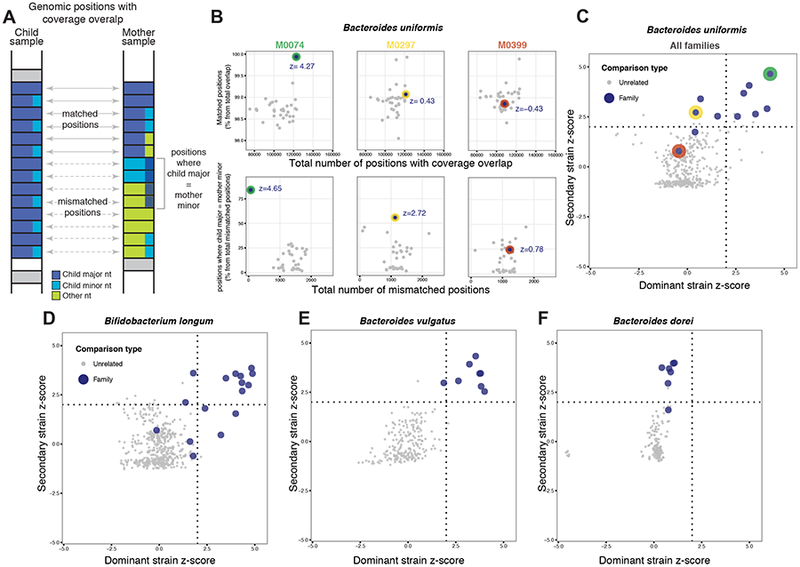
(A) Schematic view of matched and mismatched nucleotides counts based on reads aligning to species-specific marker genes. At each position of shared coverage, counts of major nucleotide matched positions are enumerated relative to those positions where the child’s major nucleotide matches the mother’s minor nucleotide. (B) Data from three mother-child pairs. Relative counts of major (top) and minor (bottom) nucleotide match events are shown together with their z-scores. (C–F) Plots depicting the distribution of dominant- and secondary-strain z-scores for individual species. Families occuring in the upper right, upper left, or bottom left quadrants show evidence of maternal transmission of dominant strain, secondary strain, or none, respectively, for that species (see Methods). Red, green, and yellow dots represent families shown in panel B.
In order to identify secondary strain transmissions, we also inspected patterns of minor nucleotide identity in species-specific marker genes, within- and across- families. Specifically, we focused on genomic positions where the major nucleotide in the mother and child do not match. For those, we asked whether the child’s major nucleotide perhaps matched the mother’s minor nucleotide (Figure 2A, see Methods). As with the dominant strain, we transformed these measurements to z-score, and a high z-score would suggest transmission of a secondary strain from mother to child (Figure 2B). We denote these z-scores as the ‘secondary strain z-scores’. For example, for the families stated above with low ‘dominant strain z-scores’ for B. uniformis, M0297 and M0399 have a ‘secondary strain z-score’ of 2.72 and 0.78, respectively, suggesting a secondary strain transmission only in the M0297 family. Interestingly, the M0074 family has high dominant and secondary strain z-scores (4.27 and 4.65, respectively), suggesting the mother transmitted both her dominant and secondary strains to her child.
To examine whether different species had unique transmission patterns, we compared these dominant and secondary z-scores across various species (Figure 2C–F, Figure S3). Overall, when combining the z-scores from all families, we found that B. vulgatus and B. adolescentis showed dominant strain transmission from mother to child, whereas B. dorei indicated mostly secondary strain transmission (Figure 2C–F, Figure S3). Interestingly, for B. longum, B. uniformis, and E. coli the transmission patterns vary by family (Figure 2C–F, Figure S3). In B. uniformis, for example, some families exhibit transmission of the dominant strain (e.g., M0074), or secondary strain (e.g., M0297), or none (e.g., M0399; Figure 2C).
Family-specific antibiotic resistance factors are found in the microbiome of mother-child pairs
In addition to characterizing the shared strains found in mother-child pairs, we set out to further characterize vertical inheritance by examining transmission patterns of individual genes. Antibiotic resistance (AR) genes serve as good examples for this question, as they are both well annotated (McArthur et al., 2013) and there is a great interest in studying their inheritance patterns. Toward this end, we searched for the presence of known AR genes in our metagenomic sequences, using the Comprehensive Antibiotic Resistance Database (McArthur et al., 2013) (see Methods). Unlike the strain profiles, where we focused on single nucleotide variation to study transmission patterns, individual genes do not necessarily have sufficient coverage to examine their nucleotide-level variation. To examine the transmission patterns of the AR genes, we focused on genes whose presence alone confers resistance (rather than commonly found genes in which resistance is caused by single-nucleotide mutations).
First, we profiled the abundance of AR genes in our samples and found AR genes to be more prevalent in child samples compared to mother samples, consistent with our own (Yassour et al., 2016) and others’ (Moore et al., 2015) previous findings (Figure S4). Maternal and child AR profiles differed both in their prevalence and in the identity of detected AR genes; many AR genes were mutually exclusive and occurred only in either maternal or in child samples (Figure S4). We next sought to identify potential transmission events of AR genes, as defined by matching patterns of presence/absence profiles in mothers and children (since, as noted above, we do not have sufficient coverage to examine SNVs for each AR gene). We hypothesized that AR inheritance would be observed more frequently in samples from the same family compared to samples from unrelated individuals. Because the most common AR genes are present in too many samples to reliably distinguish transmission events from random co-occurrence, we focused on the less common AR genes. These comprised 249 AR genes which appear in up to 50 samples in our cohort. For each of these genes, we asked whether it tends to co-occur within families or is randomly distributed across individuals; for each pair of gene and family, we calculated a p-value (using the hypergeometric distribution, see Methods). Of these AR genes, 59 had at least one significant family transmission (q<=0.05; Figure 3A, Table S1).
Figure 3: Antibiotic resistance gene profiles show family-specific patterns.
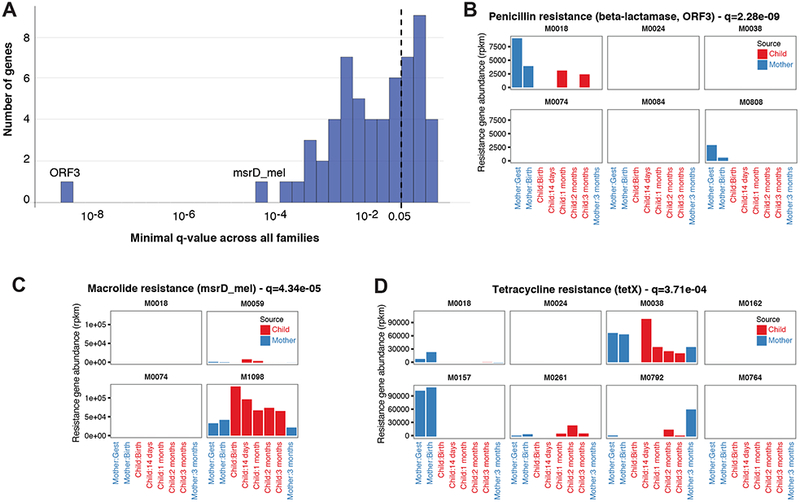
(A) Enrichment pattern of AR genes in at least one family relative to background. (B–D) These genes were identified as highly-associated with only one or two families in the cohort: (B) Penicillin resistance gene (ORF3), and (C) Macrolide resistance gene (msrD). (D) Tetracycline resistance gene (tetX) shows specific mother-only or mother-and-child family patterns.
The inherited AR genes were not limited to a specific type of resistance. For example, the genes with the lowest q-value were ORF3, which confers penicillin resistance and was found in the child samples of a single family (Figure 3B), and Msr3_mel, which confers macrolide resistance and found in the child samples of only two families (Figure 3C). The tetX gene, which confers tetracycline resistance, was found in samples from several families, sometimes in both mother and child samples, sometimes only in mother samples (Figure 3D). The inheritance profiles of AR genes are especially interesting, as they could be encoded either in the genome or on mobile elements. According to current annotations (McArthur et al., 2013), the Msr3_mel gene seems to be chromosomally encoded, while the ORF3 and tetX genes appear to be encoded on mobile elements; regardless of their genomic positions, we were able to detect their inheritance.
Gene abundance differences are identified as potential drivers in secondary strain transmission events
Finally, in addition to describing putative transmission events of strains and/or genes, we sought to identify gene presence/absence signatures that may explain the different inheritance patterns. Specifically, we wondered why a mother’s secondary strain transmitted in some families but not others. We hypothesized that the dominant strains in some mothers lacked specific genes that are advantageous in the infant gut. An ideal species in which to test this hypothesis would be one that has different inheritance patterns across families, such as Bacteroides uniformis. We therefore searched for B. uniformis genes that were present in the mother and child samples of families who shared a dominant strain, but only in the child sample of families in which the mother’s secondary strain was transmitted (see Methods, Figure 4A). We examined 7,410 genes found in the pangenome of B. uniformis and identified nine genes that vary by mode of transmission (for at least the majority of families) and may confer a selective advantage to B. uniformis in the infant gut, but may not always be present in the mother’s dominant strain (Figure 4B, see Methods). We confirmed that these genes are present at detectable levels, and that in both families this is indeed a minor strain (M0297 and M0353, as detected by qPCR in the mother samples; Figure 4B). By contrast, in the three families that did not transmit the genes, this strain is not present in the mother samples (M0201, M0305 and M1172; Figure 4B).
Figure 4: B. uniformis starch utilization system.
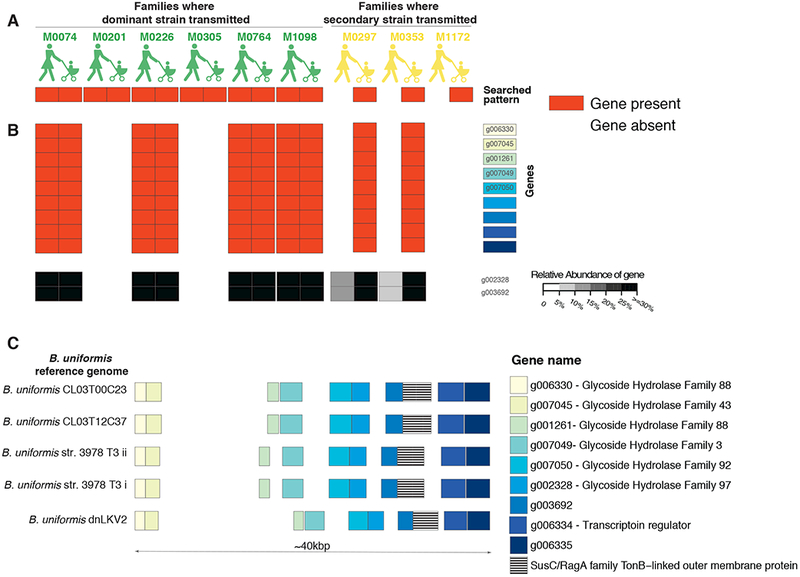
(A) A schematic showing the gene presence/absence (red/gray) patterns we inspected to identify functions that potentially drive the secondary strain inheritance. (B) Presence/absence profiles of nine B. uniformis genes that differ based on their inheritance patterns in a majority of mothers (red/white), together with qPCR results for two of the nine genes (grayscale). (C) The genomic location of the nine differential genes (yellow to blue) in five B. uniformis reference genomes, together with the SusC gene found in that location (stripes). White boxes are other genes.
Importantly, these nine genes were located in a single genomic region in five B. uniformis reference genomes (out of seven available reference genomes) (Figure 4C). This genomic region appears to be a starch utilization system (Sus), commonly found in Bacteroidetes species, that enables them to process complex glycans by a cell envelope-associated multiprotein system (Martens et al., 2009). Glycan metabolism is especially interesting in the context of the infant gut, as one of the major differences between the infant and the adult gut is the utilization of unique glycans found in mothers’ breast milk (human milk oligosaccharides), suggesting the potential selective advantage conferred by the existence of this specific Sus module in the infant gut. Interestingly, although there are 77 different SusC modules in pangenome of B. uniformis (17 of which are found in the ATCC 8492 type strain, as annotated by uniref90, Methods), we found only a single susC module which varies by mode of transmission, suggesting the specificity of this individual Sus module.
Discussion
We followed 44 mothers in the months before and after the birth of their child, and collected multiple stool samples from both mothers and children. The longitudinal nature of our cohort, coupled with deep metagenomic sequencing allowed us to characterize the microbial composition and dynamics of these communities. In addition, we developed a single nucleotide variation (SNV) profiling approach to compare the shared maternal and infant species, by examining not only the mother’s dominant strains but also her less abundant strains to study the various mother-to-child bacterial transmission patterns. Finally, we focused on the maternal transmission of Bacteroides uniformis and found functional differences in the mother’s strains that may drive its various transmission patterns.
The infant gut microbial communities were often dominated by the Actinobacteria, Bacteroidetes and Proteobacteria phyla, commonly with species from the Bifidobacterium, Bacteroides and Escherichia genera, respectively. We found limited species-level complexity in the meconium samples collected within 24 hours of birth, in concordance with previous infant studies (Bäckhed et al., 2015; Chu et al., 2017). While the maternal and child microbial communities were often statistically-significantly stable, they did not resemble one another, as there were striking differences between infant and typical adult gut microbial community compositions. Despite these gross differences in composition, we and others (Asnicar et al., 2017; Nayfach et al., 2016) were still able to identify multiple species that were commonly found in both maternal and child samples of the same family, enabling the study of maternal bacterial inheritance.
Quantifying the extent to which mothers directly shape the microbial composition of the gut in early childhood has remained a difficult challenge in the field of infant microbial research. Several studies have characterized vertical inheritance profiles from mother to child (Asnicar et al., 2017; Milani et al., 2015; Nayfach et al., 2016), however, most of them examined only a handful of mother-child pairs, and all of them focused only on the mother’s dominant strain. Here, we used single-nucleotide variation to examine the mother’s dominant and secondary strains and identify transmissions of either one to the child’s gut community. We found evidence of vertical transmission events in a number of species shared in both mother and child samples including Bifidobacterium longum, Bacteroides vulgatus, Bacteroides dorei, Bacteroides uniformis and Bifidobacterium adolescentis, with secondary strain inheritance occurring most frequently in B. dorei and B. uniformis. In addition to looking at SNVs, we identified antibiotic resistance genes that were present in both mother and child samples from the same family, but rarely in others.
To characterize the functional significance of dominant- vs. secondary-strain inheritance events, we compared the functional potential of the mother and child strains depending on their mode of inheritance. We examined families with evidence of secondary strain transmission and studied the functional differences between the child and mother strains. We focused on genes that were present in the child regardless of the inheritance pattern, but were present in the mother only in cases when her dominant strain was transmitted. We found that when the child was colonized with a mother’s secondary strain of B. uniformis, her dominant strain often lacked a specific starch utilization system (Sus). B. uniformis has dozens of such Sus modules, yet only a single one was different between the mother and child in these cases. We hypothesize that this Sus module may have a selective advantage in breaking down the glycans from the mother’s milk found in the infant gut, hence the mother’s secondary strain which harbors this module would preferentially colonize the infant gut.
In this context, our study is unique in two aspects. First, unlike previous studies which examined only the dominant strains in each sample, we were able to identify transmission events of mother’s secondary strain to the child. Second, we used the metagenomic not only to characterize the strain SNV profiles but also to examine the functional differences that may drive these different inheritance patterns. One question illuminated by this research is why do we observe two inheritance patterns? What external variables would influence which strain infants will inherit from their mother? Interesting variables in this context include, for example, mode of delivery and breastfeeding. While we have gathered this information for all subjects in our cohort (Table S1), this study is underpowered to answer this question, as it is heavily biased toward vaginally-delivered, breastfed infants. Large, well-balanced, cohorts will be needed to study the effect of these and other variables on microbial acquisition patterns.
The canonical challenge of bacterial transmission studies is to resolve the directionality of strain inheritance events. While it is possible for an infant to acquire a strain and pass it to the mother, this is unlikely due to the adults more diverse and stable gut community. Similarly, a shared strain might be acquired independently from a shared environmental source. However, given the more established nature of most adult gut microbiomes (Yatsunenko et al., 2012), these scenarios seem likely to represent the minority of transmission events. Here, to best address these issues for this cohort and experimental design, we analyzed strain transmissions for which the mother sample predates the child sample. Follow-up studies with denser samples around birth, with additional samples from potential environmental sources and even deeper sequencing will shed more light on this process. Each study examining mother-to-child bacterial transmission brings us closer to understanding the postnatal vertical inheritance events and their contribution to the establishment of the infant gut microbial community.
STAR Methods
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Ramnik J. Xavier (xavier@molbio.mgh.harvard.edu).
Experimental Model and Subject Details
Study cohort
44 otherwise healthy pregnant women were recruited from January 28, 2013 to February 26, 2015. Families were contacted at the fetal ultrasonography visit, which is arranged for all pregnant women in Finland around gestational week 20. Written informed consent was signed by the parents at the beginning of the third trimester to analyze the offspring’s HLA genotype. Patient consent was overseen by the Ethical Committee of the Joint Municipal Authority of the Pirkanmaa Hospital District. Inclusion criteria for the study were informed consent signed by the parents and an eligible HLA genotype of the newborn conferring increased risk for type 1 diabetes (T1D), as this cohort is a subset of another larger T1D-centered cohort. Eligible genotypes, determined as previously described (Ilonen et al., 2016) were the high-risk genotype combining (DR3)-DQA1*05-DQB1*02 and DRB1*0401/2/4/5-DQA1*03-DQB1*03:02 haplotypes or the moderate-risk genotypes defined as homozygosity for either one, DRB1*04:01/2/5-DQA1 *03-DQB1 *03:02 with a neutral haplotype, and the (DR3)-DQA1*05-DQB1*02/DRB1*09-DQA1*03-DQB1*03:03 genotype. Haplotypes defined as neutral in this context were: (DR1/10)-DQB1*05:01, (DR8)-DQB1*04, (DR7)-DQA1*02:01-DQB1*02, (DR9)-DQA1*03-DQB1*03:03 and (DR13)-DQB1*06:03. DR molecules marked in parentheses are deduced based on the strong linkage disequilibrium. The average age of mothers was 31.9 years. There were 23 female and 21 male infants enrolled in the study. The gender of children was not found to be significantly associated with the number of microbial species present in the child stool samples across the five time points tested (Wilcoxon rank-sum test, p=0.651). Similarly, t-tests were used to compare the Bray-Curtis dissimilarity of child samples within and between genders and no significant differences were found at any of the five time points tested (tests were FDR corrected with a threshold of q<0.05). Subject metadata is provided in Table S1.
Exclusion criteria were (i) an older sibling participating in the study; (ii) multiple gestation; (iii) The parents unwilling or unable to feed the infant with cow’s milk based products; (iv) the gestational age at birth less than 35 weeks; (v) technical challenges to take part in the study (e.g. the family had no access to the study center or telephone); (vi) no HLA sample was obtained before the age of 8 days.
Microbial strains
The following Bacteroides uniformis strains were used in experimental: CL03T12C37, CL03T00C23, ATCC 8492. Cultures were grown anaerobically at 37C for 48 hours on Brain Heart Infusion Broth (BD, Franklin Lakes, NJ) supplemented with 1% Vitamin K-Hemin solution (BD, Franklin Lakes, NJ). The Coy vinyl anaerobic chamber was maintained at >2.5% H2 by flushing with gas mix containing 5% H2, 5% CO2 and 90% N2.
Method Details
Sample collection and DNA extractions
The mothers collected their stool samples at home or in the delivery hospital. The meconium sample was collected in the delivery hospital and the other infant stool samples by the mothers at home. The samples collected at home were stored in the households freezer (−20°C) until the next visit to the study center. The samples were then shipped on dry ice to the EDIA Core Laboratory in Helsinki, where the samples were stored at −80°C until shipping to the University of Tampere for DNA extraction. DNA extractions from stool were carried out using the vacuum protocol of PowerSoil DNA Isolation Kit.
Metagenome library construction
Metagenomic DNA samples were quantified by Quant-iT PicoGreen dsDNA Assay (Life Technologies) and normalized to a concentration of 50 pg μL−1. Illumina sequencing libraries were prepared from 100–250 pg DNA using the Nextera XT DNA Library Preparation kit (Illumina) according to the manufacturer’s recommended protocol, with reaction volumes scaled accordingly. Batches of 24, 48, or 96 libraries were pooled by transferring equal volumes of each library using a Labcyte Echo 550 liquid handler. Insert sizes and concentrations for each pooled library were determined using an Agilent Bioanalyzer DNA 1000 kit (Agilent Technologies).
Analysis of whole-genome shotgun (WGS) sequencing
WGS libraries were sequenced on the Illumina HiSeq 2500 platform, targeting ~2.5 Gb of sequence per sample with 101 bp paired-end reads (number of reads per sample is mentioned in Table S1). Reads were quality controlled by trimming low-quality bases, removing reads shorter than 60 nucleotides. We identified and filtered out potential human contamination using the KneadData Tool, v0.5.1 with the hg19 human reference genome. Quality controlled samples were profiled taxonomically using MetaPhlAn 2.0 (Segata et al., 2012; Truong et al., 2015), following Bowtie 2–2.1.0 (Langmead and Salzberg, 2012) alignment to the MetaPhlAn 2.0 unique marker database. To calculate the abundance of genes from multiple pan-genomes, we applied PhanPhlan (v1.2.2) (Scholz et al., 2016) to the all metagenomic samples after filtering out reads mapping to human reference.
Sample richness and stability
To assess alpha-diversity for each sample, we counted the number of species reported in the MetaPhlAn output appearing at greater than 1% relative abundance. To confirm that these findings were robust to the variation in read counts seen across sequenced samples, we repeated measures of community diversity after downsampling the number of reads. To asses the alpha-diversity findings with comparable read counts across samples, 3 million read pairs were chosen at random for each sample and the downsampled files were re-run through the Metaphlan (samples that had less than 3M reads were not included in this analysis). Shannon diversity scores were calculated before and after downsampling and species diversity estimates were confirmed (Figure S2).
We evaluated community stability by comparing species-level composition profiles of related samples measured at consecutive time points. Bray-Curtis distance was first measured for each subject by comparing consecutive time points. The distribution of mother and child samples was evaluated separately for each comparison and unrelated samples were also compared to serve as a background distribution. Next, Bray-Curtis distance was measured comparing mother and child samples belonging to the same individual at birth and at 3 months. Here, these distributions were also compared relative to unrelated mother-child pairs.
Species-specific strain analysis
For each sample, the whole genome sequences that mapped to species marker genes were identified with Metaphlan as described above. The ‘mpileup’ feature of Samtools (v1.3.1) was used to calculate coverage and describe sites of variation along each marker gene for the subset of species examined (Figure 1E). The mpileup results for the marker genes corresponding to each species of interest were concatenated across all samples for downstream analysis. To identify positions of polymorphism, sites of single-nucleotide variation in a given sample were tested for significance using a binomial test assuming 1% sequencing error. Coverage overlap events were assessed to find where in mother-child pairs each sample contained reads mapping to the same segment(s) of marker genes. Within a given family, a mother sample was compared to child sample when the pair was found to have coverage overlap along at least one species-specific marker gene. Mother-child comparisons were also performed among samples belonging to different families.
After identifying coverage overlap sites in mother-child pairs, each site was evaluated for a number of match and polymorphic outcomes possible for a given pair (Figure 2A). Briefly, the most abundant nucleotide observed at a given position was assigned to be either a match in both samples or a “dominant”-base mismatch. Similarly, the second most abundant nucleotide or “secondary” base at a given position was evaluated for match or mismatch between the samples of a given pair. Secondary base match and mismatch events were only considered at those sites deemed polymorphic in at least one sample of the pair. Samples with fewer than 1000 reads mapping to species specific marker genes were excluded from downstream analysis. We focused on the mother-child pairs for which mother sample predates child sample and then selected a most representative sample pair based on the samples that had the largest coverage overlap across the marker genes of a given species.
First, the number of “major-major” nucleotide match events were computed relative to the number of overlap sites per pair of samples. Second, “MotherMinor-ChildMajor” match events were calculated as a ratio compared to the total number of major-major mismatch events. The relative frequency of match and mismatch events at the dominant or secondary bases were evaluated by conversion to z-scores. These scores were taken together to infer modes inheritance of major and minor strains. Mother-child pairs that scored above z=2 in the “dominant-dominant” and “MotherMinor-ChildMajor” base matches were inferred to have shared dominant and secondary strains. Those pairs that had “MotherMinor-ChildMajor” scores above z=2, but had “dominant-dominant” scores z<2 were inferred to have shared secondary strains but not a shared dominant strain. Those family pairs with scores below z=2 on both axis were inferred to not share major or minor strains.
Measuring antibiotic resistance genes
The abundance of antibiotic resistance genes were quantified using an existing approach we have developed (Yassour et al., 2016) to map metagenomic reads to a database of protein sequences curated from the Comprehensive Antibiotic Resistance Database (McArthur et al., 2013). To detect and quantify the abundance of the antibiotic resistance (AR) genes in our WGS data, we used shortBRED (Kaminski et al., 2015). Briefly, given a set of AR protein sequences, shortBRED clusters them into similar families based on their sequence, extracts a set of distinctive strings (“markers”) per family, and then searches for these markers in metagenomic data. We did not take into consideration genes that are normally present in the core genome of the species and in which point mutations can give rise to antibiotic resistance, as we need very high read coverage to clearly identify these mutations. Instead, we focused on genes whose presence is sufficient to confer resistance. Specifically, we used the sequences of 3,060 proteins from The Comprehensive Antibiotic Resistance Database version 1.0.0 (McArthur et al., 2013).
Functional differences in B. uniformis based on transmission patterns
Using the panphlan profiles, which were calculated above, we looked for genes that had the following pattern. For families who had the dominant strain transmitted, the genes are present in both maternal and child samples. However, for the families who had the secondary strain transmitted, the gene was present only in the child and not in the mother. This analysis examined 7,410 genes found in the pangenome of B. uniformis, and revealed only nine genes that matched this pattern for most families. Next, we wanted to assign a function to each of these genes, when possible. We used the panphlan mapping to assign a UniRef90 id to each gene, and then a Pfam family. Finally, we mapped the Pfam families to Glycoside Hydrolase (GH) families using the Carbohydrate Active Enzymes (CAZy) database (http://www.cazy.org/) (Lombard et al., 2014). Gene annotations for uniref 90 IDs were extracted from the Humann2 utility mapping (v0.10.0) and multiple B. uniformis strains were inspected for SusC genes using PanPhalAn (v1.2.2).
qPCR detection of B. uniformis cluster
Primers were designed for the detection of B. uniformis genes within the cluster for strain-level identification, (WP_005832980_317F 5’TCACGAACCGCATTGAT ACA3’, WP_005832980_426R 5’CGTGACATCGTGTTGTTTCG 3’ and WP_005832976_231 F 5’ CCTGTTCGCGAGCATCATTA 3’, WP_005832976_340R 5’ TGATACGCAGGTGCTTCAAG 3’), all B. uniformis for species-level identification (NZ_DS362232.1_1476F 5’ CGGAAGTGTGCATGAACATG 3’, NZ_DS362232.1_1585R 5’ AGTCAACCTCTTTGTCACCG 3’) and overall 16S (Atarashi16S_F 5’ GGTGAATACGTTCCCGG 3’, Atarashi16S_R 5’ TACGGCTACCTTGTTACGACTT 3’ (Ivanov et al., 2009). Standard curves were generated with qPCR to determine priming efficiencies for each primer pair (range 87.4–108.1%). qPCR was performed on extracted DNA (reaction volumes: 2uL 2.5ng/uL DNA, 0.75uL forward primer, 0.75uL reverse primer, 1.5uL Ambion Nuclease-free water, 5uL Power SYBR Green PCR Master Mix, conditions: 1. 95C for 10 minutes, 2. 95C for 15 seconds, 3. 60C for 30 seconds and 4. Repeat 2 and 3 39 times) in a BioRad CFX96 Real-Time System. The samples that had this gene cluster in their dominant strain showed three-four fold higher values than those predicted to have this cluster in their secondary strain.
Quantification and Statistical Analysis
Microbiome stability and gender analysis
We compared the stability of early child gut microbiota by t-test using the species-level dissimilarity between adjacent time points for each individual over time, and within families (using Bray-Curtis dissimilarity). A t-test was also used to evaluate the dissimilarity of species-level metagenomic profiles of child samples belonging to the same gender as compared to the distribution of child samples belonging to different genders at each time point. Both of these dissimilarity tests were FDR corrected with a threshold of q<0.05.
Finding family-specific antibiotic resistance genes
To quantify the occurrence of family-specific events, we calculated the Hypergeometric distribution of gene-family occurrences. This tested the null hypothesis that a given gene of interest is evenly distributed across the samples of all families. We rejected this null hypothesis in cases where a gene of interest was found within samples of the same family at a much higher rate than its background frequency in the cohort. To look across many antibiotic resistance factors to identify such events in families, we focused only on those genes that appear at low or moderate frequency in our cohort (50 or fewer samples in the cohort with measurable abundance of the gene). For each family-gene pair, a p-value was assigned using the hypergeometric distribution to quantify how specific a given gene was to a family. The results across all comparisons were false discovery rate (FDR) corrected at q<0.05.
Data And Software Availability
Metagenomic sequencing data for all subjects have been deposited to the SRA database under BioProject ID PRJNA475246. All sequencing reads that map to the human reference genome (hg19) have been removed from the sequencing files.
Supplementary Material
Listing all samples in this study with number of metagenomic reads obtained for each sample. Additional information is provided per family for delivery mode (C-section or vaginal), early feeding (total days of breastfeeding, and first introduction of formula), maternal and child antibiotic and probiotic use.
Corrected p-values for antibiotic resistance genes from the Comprehensive Antibiotic Resistance Database (McArthur et al., 2013), scored for their family specificity using the hypergeometric test. Only the smallest q-value associated with a single family is listed per gene. Gene names and full annotations from the database are listed the rightmost columns. Entries are ordered according to significance.
Highlights.
Gut bacterial transmission patterns assessed longitudinally in 44 mother-infant pairs
Metagenomic sequencing reveals transmission patterns beyond dominant strains
Mother’s minor strain sometimes colonizes infant, likely driven by functional selection
Some antibiotic resistance genes co-occur in families, suggesting their inheritance
Acknowledgements
We thank Tiffany Poon, James Bochicchio and Scott Steelman (Broad Institute) for help in sequence production and sample management, and Theresa Reimels for text and graphical support. The study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK), National Institutes of Health (grant number 1DP3DK094338–01), the Academy of Finland Centre of Excellence in Molecular Systems Immunology and Physiology Research 2012–17 grant 250114), Juvenile Diabetes Research Foundation (grant 2-SRA-2016–247-S-B), Sigrid Juselius Foundation and the Medical Research Funds, Tampere and Helsinki University Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Authors contribution
M.Y. and L.H performed the data analysis. M.Y, L.H., H.V., E.S.L., C.H. and R.J.X. assembled and wrote the paper. E.J., H.S., J.S., S.O., H.H., J.I., and M.K. designed the cohort study and collected clinical samples. S.M.V. collected infant nutritional data. T.A. and S.T. performed the experiments. P.F., E.P, A.T., F.A. and N.S. provided valuable feedback, M.Y., L.H., C.H., H.V., and E.S.L. led method and research development. M.K., and R.J.X. acquired funding, C.H., E.S.L., M.K., and R.J.X. served as principal investigators.
Declaration of Interests
The authors declare no competing interests
References
- Asnicar F, Manara S, Zolfo M, Truong DT, Scholz M, Armanini F, Ferretti P, Gorfer V, Pedrotti A, Tett A, et al. (2017). Studying Vertical Microbiome Transmission from Mothers to Infants by Strain-Level Metagenomic Profiling. mSystems 2. [DOI] [PMC free article] [PubMed]
- Bäckhed F, Roswall J, Peng Y, Feng Q, Jia H, Kovatcheva-Datchary P, Li Y, Xia Y, Xie H, Zhong H, et al. (2015). Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 17, 690–703. [DOI] [PubMed] [Google Scholar]
- Biasucci G, Rubini M, Riboni S, Morelli L, Bessi E, and Retetangos C (2010). Mode of delivery affects the bacterial community in the newborn gut. Early Hum. Dev 86 Suppl 1, 13–15. [DOI] [PubMed] [Google Scholar]
- Cahenzli J, Köller Y, Wyss M, Geuking MB, and McCoy KD (2013). Intestinal microbial diversity during early-life colonization shapes long-term IgE levels. Cell Host Microbe 14, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu DM, Ma J, Prince AL, Antony KM, Seferovic MD, and Aagaard KM (2017). Maturation of the infant microbiome community structure and function across multiple body sites and in relation to mode of delivery. Nat. Med 23, 314–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez-Bello MG, Costello EK, Contreras M, Magris M, Hidalgo G, Fierer N, and Knight R (2010). Delivery mode shapes the acquisition and structure of the initial microbiota across multiple body habitats in newborns. Proc. Natl. Acad. Sci. U. S. A 107, 11971–11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith JJ, Guruge JL, Charbonneau M, Subramanian S, Seedorf H, Goodman AL, Clemente JC, Knight R, Heath AC, Leibel RL, et al. (2013). The long-term stability of the human gut microbiota. Science 341, 1237439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilonen J, Kiviniemi M, Lempainen J, Simell O, Toppari J, Veijola R, Knip M, and Finnish Pediatric Diabetes Register (2016). Genetic susceptibility to type 1 diabetes in childhood - estimation of HLA class II associated disease risk and class II effect in various phases of islet autoimmunity. Pediatr. Diabetes 17 Suppl 22,8–16. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobsson HE, Abrahamsson TR, Jenmalm MC, Harris K, Quince C, Jernberg C, Björkstén B, Engstrand L, and Andersson AF (2014). Decreased gut microbiota diversity, delayed Bacteroidetes colonisation and reduced Th1 responses in infants delivered by caesarean section. Gut 63, 559–566. [DOI] [PubMed] [Google Scholar]
- Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G, and Huttenhower C (2015). High-Specificity Targeted Functional Profiling in Microbial Communities with ShortBRED. pLoS Comput. Biol 11, e1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koenig JE, Spor A, Scalfone N, Fricker AD, Stombaugh J, Knight R, Angenent LT, and Ley RE (2011). Succession of microbial consortia in the developing infant gut microbiome. Proc. Natl. Acad. Sci. U. S. A 108 Suppl 1, 4578–4585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard V, Golaconda Ramulu H, Drula E, Coutinho PM, and Henrissat B (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo C, Knight R, Siljander H, Knip M, Xavier RJ, and Gevers D (2015). ConStrains identifies microbial strains in metagenomic datasets. Nat. Biotechnol 33, 1045–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcobal A, Barboza M, Sonnenburg ED, Pudlo N, Martens EC, Desai P, Lebrilla B, Weimer BC, Mills DA, German JB, et al. (2011). Bacteroides in the infant gut consume milk oligosaccharides via mucus-utilization pathways. Cell Host Microbe 10, 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Koropatkin NM, Smith TJ, and Gordon JI (2009). Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J. Biol. Chem 284, 24673–24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, Baylay AJ, Bhullar K, Canova MJ, De Pascale G, Ejim L, et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57,3348–3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani C, Mancabelli L, Lugli GA, Duranti S, Turroni F, Ferrario C, Mangifesta M, Viappiani A, Ferretti P, Gorfer V, et al. (2015). Exploring Vertical Transmission of Bifidobacteria from Mother to Child. Appl. Environ. Microbiol 81,7078–7087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AM, Ahmadi S, Patel S, Gibson MK, Wang B, Ndao MI, Deych E, Shannon W, Tarr PI, Warner BB, et al. (2015). Gut resistome development in healthy twin pairs in the first year of life. Microbiome 3, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Rodriguez-Mueller B, Garud N, and Pollard KS (2016). An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res. 26, 1612–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, and Pettersson S (2012). Host-gut microbiota metabolic interactions. Science 336, 1262–1267. [DOI] [PubMed] [Google Scholar]
- Olszak T, An D, Zeissig S, Vera MP, Richter J, Franke A, Glickman JN, Siebert R, Baron RM, Kasper DL, et al. (2012). Microbial exposure during early life has persistent effects on natural killer T cell function. Science 336, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penders J, Thijs C, Vink C, Stelma FF, Snijders B, Kummeling I, van den Brandt PA, and Stobberingh EE (2006). Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521. [DOI] [PubMed] [Google Scholar]
- Perez-Muñoz ME, Arrieta M-C, Ramer-Tait AE, and Walter J (2017). A critical assessment of the “sterile womb” and “in utero colonization” hypotheses: implications for research on the pioneer infant microbiome. Microbiome 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz M, Ward DV, Pasolli E, Tolio T, Zolfo M, Asnicar F, Truong DT, Tett A, Morrow AL, and Segata N (2016). Strain-level microbial epidemiology and population genomics from shotgun metagenomics. Nat. Methods 13,435–438. [DOI] [PubMed] [Google Scholar]
- Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, and Huttenhower C (2012). Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 9,811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela DA, and Mills DA (2010). Nursing our microbiota: molecular linkages between bifidobacteria and milk oligosaccharides. Trends Microbiol. 18, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sela DA, Chapman J, Adeuya A, Kim JH, Chen F, Whitehead TR, Lapidus A, Rokhsar DS, Lebrilla CB, German JB, et al. (2008). The genome sequence of Bifidobacterium longum subsp. infantis reveals adaptations for milk utilization within the infant microbiome. Proc. Natl. Acad. Sci. U. S. A 105, 18964–18969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, and Garrett WS (2013). The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science 341, 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, and Segata N (2015). MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods 12,902–903. [DOI] [PubMed] [Google Scholar]
- Truong DT, Tett A, Pasolli E, Huttenhower C, and Segata N (2017). Microbial strain-level population structure and genetic diversity from metagenomes. Genome Res. 27, 626–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yassour M, Vatanen T, Siljander H, Hämäläinen A-M, Härkönen T, Ryhänen SJ, Franzosa EA, Vlamakis H, Huttenhower C, Gevers D, et al. (2016). Natural history of the infant gut microbiome and impact of antibiotic treatment on bacterial strain diversity and stability. Sci. Transl. Med 8, 343ra81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Listing all samples in this study with number of metagenomic reads obtained for each sample. Additional information is provided per family for delivery mode (C-section or vaginal), early feeding (total days of breastfeeding, and first introduction of formula), maternal and child antibiotic and probiotic use.
Corrected p-values for antibiotic resistance genes from the Comprehensive Antibiotic Resistance Database (McArthur et al., 2013), scored for their family specificity using the hypergeometric test. Only the smallest q-value associated with a single family is listed per gene. Gene names and full annotations from the database are listed the rightmost columns. Entries are ordered according to significance.