

Abstract
Access to streamlined computational resources remains a significant bottleneck for new users of cryo-electron microscopy (cryo-EM). To address this, we have developed tools that will submit cryo-EM analysis routines and atomic model building jobs directly to Amazon Web Services (AWS) from a local computer or laptop. These new software tools (“cryoem-cloud tools”) have incorporated optimal data movement, security, and cost-saving strategies, giving novice users access to complex cryo-EM data processing pipelines. Integrating these tools into the RELION processing pipeline and graphical user interface we determined a 2.2 Å structure of ß-galactosidase in ~55 hours on AWS. We implemented a similar strategy to submit Rosetta atomic model building and refinement to AWS. These software tools dramatically reduce the barrier for entry of new users to cloud computing for cryo-EM and are freely available at cryoem-tools.cloud.
INTRODUCTION
Cryo-electron microscopy (cryo-EM) is a structural biology technique that has undergone rapid growth over the past few years (Nogales 2016). Technical developments in direct electron detection and electron optics in conjunction with improvements in image analysis (S. H. Scheres 2014; Punjani et al. 2017) have led to the widespread adoption of cryo-EM as a structural biology technique. Furthermore, the advent of GPU-accelerated cryo-EM structure determination (Punjani et al. 2017; Kimanius et al. 2016) has helped to reduce the overall cost for computing hardware for a single user. While these improvements have helped to spread cryo-EM, it becomes difficult to scale the required hardware to accommodate large cryo-EM facilities that have a large number of users. These facilities have to balance cost with availability of resources: idle computing infrastructure is wasted capital whereas queuing times for compute resources waste personnel salaries. The challenge is how to create a computing facility that is cost-effective while also delivering compute resources on-demand without wait times.
Previous work has shown that Amazon Web Services (AWS), the world’s largest cloud computing provider, is a cost-effective resource for cryo-EM structure determination (Cuenca- Alba et al. 2017; Cianfrocco and Leschziner 2015). Since this original publication, AWS released GPU-accelerated virtual machines (‘VMs’) (named ‘p2’, ‘p3, and ‘g3’) with 1, 8, or 16 NVIDIA K80 GPUs on p2, 1, 8, or 16 NVIDIA V100 GPUs on p3, 1, 2, or 4 NVIDIA M60 GPUs on g3, while also reducing prices for data storage on the block storage service (‘S3’) and archival storage (‘Glacier’).
Despite its power, previous implementations for cryo-EM required users to manually deploy AWS resources. To streamline the process, we have developed software tools that allow for the remote management of AWS resources from the local computer of a user. These tools were then combined with the standard suite of cryo-EM software tools MOTIONCOR (Li et al. 2013), MOTIONCOR2 (Zheng et al. 2017), UNBLUR (Grant and Grigorieff 2015), GCTF (Zhang 2016), CTFFIND4 (Rohou and Grigorieff 2015), RELION (S. H. W. Scheres 2012) and Rosetta (Wang et al. 2016, 2015), allowing users to submit jobs directly to AWS from their local project directory while syncing results back in real time. In contrast to our previous implementation, we are now using ‘on-demand’ VMs from AWS, which eliminates the risk of users being ‘kicked-off due to price changes. Finally, by combining the full RELION pipeline (and associated software) with atomic model building and refinement with Rosetta (Wang et al. 2016, 2015) with AWS, cryoem-cloud-tools provides users with all aspects of cryo-EM structure determination in a single pipeline - from micrograph motion correction to atomic model refinement.
APPROACH
We realized that manual workflows for managing AWS resources was cumbersome, requiring the use of complex commands. To streamline this process, we wrote software tools that leverage the capabilities of command-line tools provided by AWS. Then, we incorporated these commands directly into the RELION GUI to allow users to submit RELION jobs directly to AWS (Figure 1).
Figure 1 -. AWS architecture for ‘advanced’ cryo-EM data processing with RELION.
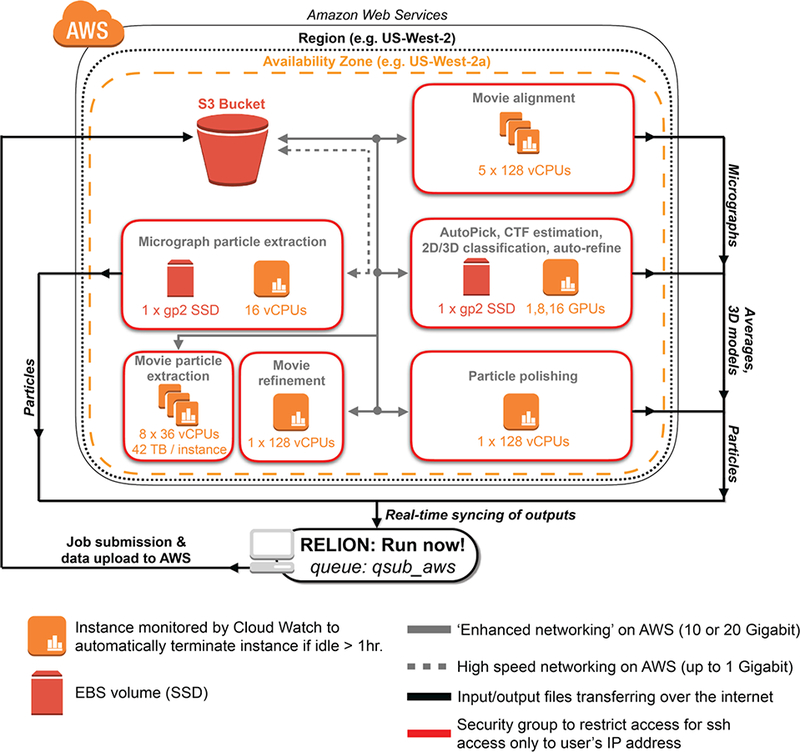
Shown is a schematic of AWS resources deployed by cryoem-cloud-tools through the program ‘qsub_aws’. For all job types shown, the software places VMs within security groups that restrict access to the IP address of the end-user. Within a security group, the software determines the appropriate VM and storage choices, using S3 as a distribution point between local and AWS resources.
The overall approach takes advantage of the cluster submission feature of RELION by providing users with a new submission command (‘qsub_aws’) to do the following: 1) identify the type of RELION job, 2) upload data to AWS block storage (S3), 3) start VM(s) required for the task, 4) download data from S3 to VM, 5) Run RELION commands, 6) Sync output results back to the local machine in real time, and 7) Turn off machines when finished (or if an error is detected). Given this workflow, for large upload tasks (such as uploading movies), there is an initial data movement step onto S3 prior to any calculations being performed. We are maintaining the latest stable version of RELION, which will inform users of the RELION version and detect any discrepancies between local and cloud versions of RELION. As shown in Figure 1, we implemented job type-dependent data processing strategies for RELION analysis routines. This means that GPU-accelerated steps (Auto Pick, CTF estimation, 2D/3D classification, auto-refine) are run on VMs with GPUs (p2 VMs), whereas CPU-based steps are run on VMs with 16 or 128 virtual CPUs (vCPUs) (See (Cianfrocco and Leschziner 2015) for a detailed discussion of vCPUs vs. CPUs).
In building this software, we are providing users with workflows that have been optimized for data transfer and computing. For instance, all data is first uploaded into AWS’s S3 ‘buckets’. This allows for fast uploads (up to 300 MB/sec) and also for cost-effective storage of data in between analysis routines. Storing data on S3 between RELION runs removes the latency that results from re-uploading the same data multiple times. Next, we implemented data storage policies that allow for high input/output tasks and large dataset sizes, which included 42 terabyte drives for movie particle extraction on d2 VMs. Finally, for computational tasks that can be distributed (Movie alignment and Movie particle extraction), we boot up and manage multiple VMs in parallel to finish analysis routines quickly.
RESULTS & DISCUSSION
To assess the performance of our approach, we compared processing times for the determination of a 2.2 Å ß-galactosidase structure (Bartesaghi et al. 2015) that was recently solved using a stand-alone GPU workstation (Kimanius et al. 2016). While the comparison is testing very different computing environments, we chose it because many new cryo-EM users are purchasing stand-alone GPU workstations and we wanted to compare performance relative to AWS. The following discussions of AWS assume that the user has setup an AWS account and followed installation instructions for using ‘cryoem-cloud-tools’, including setting up security access keys on local computing resources.
Below we lay out two computing scenarios: ‘advanced’ and ‘common’. The advanced computing scenario (Figure 1) describes all steps in the RELION processing pipeline, which include movie alignment and particle polishing, as previously described. The common approach involves (Figure 3) using particles that have already been extracted from motion-corrected micrographs, and thus does not require S3 storage or particle polishing steps. We will first describe the advanced pipeline and then the common pipeline.
Figure 3 -. AWS architecture for ‘common’ cryo-EM data processing with RELION.
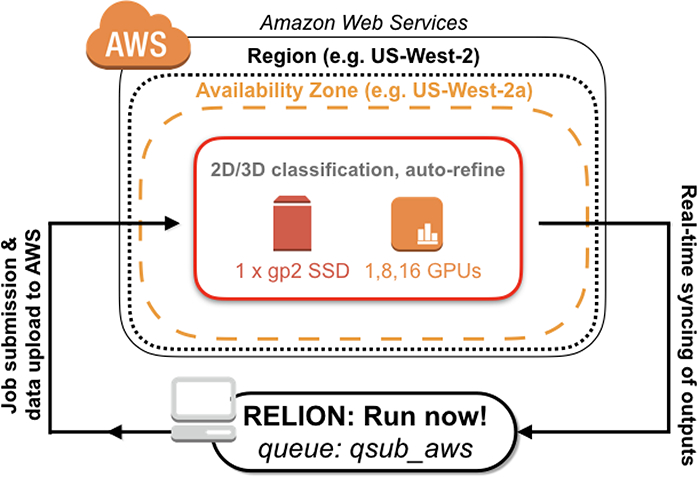
Shown is a schematic of AWS resources deployed by cryoem-cloud-tools through the program ‘qsub_aws’. For this common pipeline, there is no data storage on S3 and RELION jobs are run directly on p2 instances.
Using our integrated AWS software tools in the RELION GUI and launching all RELION analysis commands remotely for the advanced pipeline, we were able to determine a 2.2 Å structure in 54.5 hours on AWS (Figure 2A & 2B, Figure 2 - Supplement 1), which is 2X faster than a standalone GPU workstation (Figure 2C). These processing times also included the time required for movement of data into and between resources on AWS, thus reflecting the full processing times experienced by a user (For full list of data transfer times, see Figure 2 - supplement 1). For GPU-accelerated RELION processing steps, VMs with 8 GPUs (p2.8xlarge) performed equally well or slightly faster than a 4 GPU workstation (Figure 2D). This likely results from faster GPUs in the workstation (NVIDIA GTX1070: 1683 MHz clock speed) compared to those on AWS (NVIDIA K80: 875 MHz clock speed). Expectedly, the largest time savings were seen in steps that could be distributed across multiple VMs (Movie alignment and Movie particle extraction) (Figure 2D). For these processes, we were able to select VMs that were appropriate for the process - CPU machines for movie alignment (x1.32xlarge), large storage arrays for movie particle extraction (d2.8xlarge), and high vCPU numbers for movie refinement and polishing (x1.32xlarge: 128 vCPUs). As an alternative to cryoem-cloud-tools, a common processing pipeline can also be used. The common pipeline assumes particles that have already been extracted from motion-corrected micrographs (e.g. using MotionCor2) (Figure 3).
Figure 2 -. Performance of AWS vs. local GPU workstation.
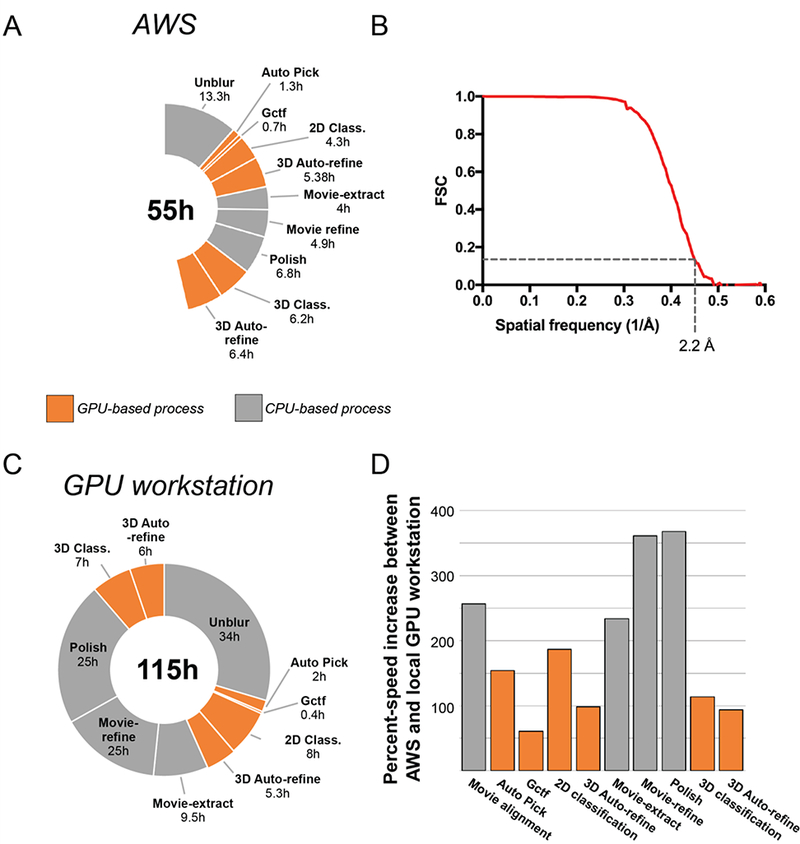
Processing times (A) and FSC curve (B) for the determination of a 2.2 Å ß-galactosidase structure on AWS. (C) Processing times from the determination of 2.2 Å ß-galactosidase structure on GPU workstation (Kimanius et al. 2016). (D) Comparison of percent speed-up increases between AWS and a GPU workstation.
In order to build the atomic model for ß-galactosidase into this density, we used the molecular modelling program Rosetta (Wang et al. 2016, 2015). As modelling software, Rosetta needs CPU computing clusters because its sampling of hundreds of atomic models relative to the cryo-EM density requires a dedicated CPU for each model. Therefore, we incorporated Rosetta tools for model building and refinement into our AWS-based pipeline, allowing users to submit a Rosetta refinement to AWS from their local computer or laptop (Figure 4A). By distributing the Rosetta refinement over multiple VMs on AWS, each with 36 vCPUs (c4.8xlarge), we were able to generate 200 models using RosettaCM and Rosetta FastRelax 6.1 hours on AWS, a speedup of about 7X over a single workstation with 16 processors (41.8 hours) (Figure 4B). The resulting model showed good agreement with the density, with a r.m.s.d for the top 10 Rosetta models of < 0.5 Å. (Figure 4C & Figure 4D, Figure 4 - Supplement 1).
Figure 4 -. Rosetta atomic model refinement in the cloud.
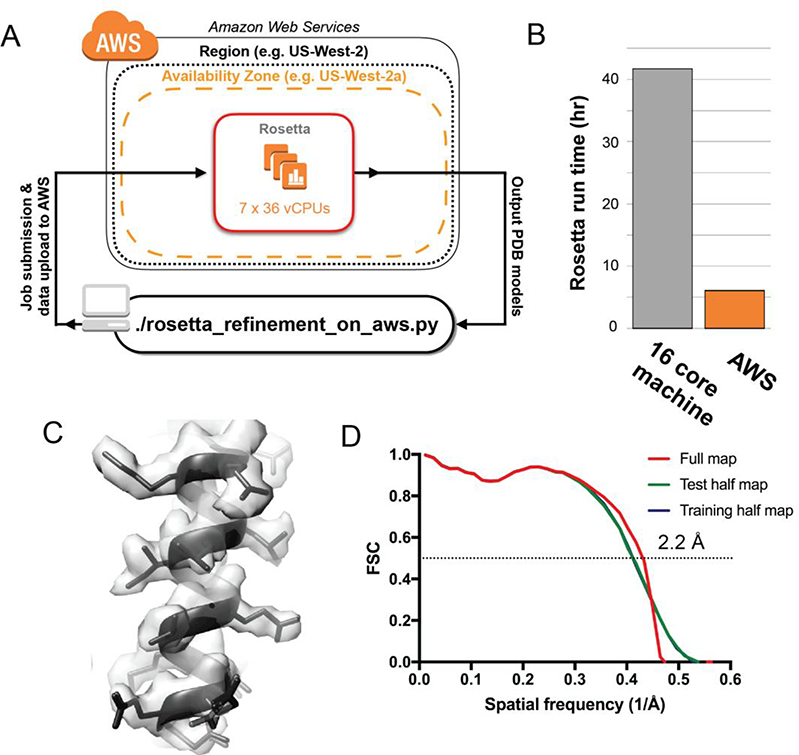
(A) WS architecture for running Rosetta model refinement across multiple VMs. (B) Run time comparisons between a local workstation (16 cores) and AWS (252 vCPUs). (C) Representative region of the cryo-EM map with the top five atomic models built by Rosetta FastRelax (D) FSC curves between the best atomic model from FastRelax and the cryo-EM map of ß- galactosidase. The resolution corresponding to the FSC value of 0.5 for the full map is shown.
The cost for determining a 2.2 Å structure using the advanced computing pipeline with RELION and building an atomic model with Rosetta, both using AWS, was $1,468 USD (Figure 2 - Supplement 1). This cost represents both storage and computing on AWS, with the top three expenditures (71% of the total) coming from 30 terabytes of data storage on AWS S3 ($690.00), Movie particle extraction ($179.73), and Movie alignment ($146.72) (Figure 2 - Supplement 1). The common pipeline (including Rosetta modeling) was ~6X cheaper than the advanced pipeline, with an overall price of $247 (Figure 3 - Supplement 1). As a typical user likely already has extracted, motion-corrected particles, this common pipeline cost represents a realistic price for users of cryoem-cloud-tools.
This approach for cryo-EM data analysis has the potential to benefit many different types of cryo-EM users. Since this software package integrates directly into a user interface, individual users will have the option to perform multiple analysis routines from a single workstation by pushing additional jobs to AWS instead of waiting to run them sequentially on a local GPU workstation. For research teams, this software provides ‘burstable’ processing power, ensuring that data processing does not become rate-limiting ahead of grant and manuscript deadlines. Finally, this software can have a significant impact on cryo-EM facilities with a large user base. Given the scale of AWS, a cryo-EM facility could not only provide many users with access to microscopes but also allow those users to push cryo-EM jobs to AWS without having to accommodate their computing needs locally.
METHODS
Integrating cryoem-cloud-tools into the RELION GUI
The overall strategy for users accessing cryoem-cloud-tools from the RELION GUI used the cluster submission of RELION. When users submit jobs to a cluster, they indicate the submission command directly into the RELION GUI (e.g. ‘qsub’). Within this framework, we built cryoem-cloud-tools to be specified directly from the GUI using a python program named ‘qsub_aws.’ This program will automatically determine the type of RELION command that needs to be run and determine the AWS resources required to execute the task. This approach does not require users to compile RELION using cryoem-cloud-tools; instead cryoem-cloud-tools is a software extension for RELION to submit jobs to AWS.
ß-galactosidase image processing
To replicate the published work on ß-galactosidase (Kimanius et al. 2016; Bartesaghi et al. 2015), we used an almost identical processing strategy using RELION v2.1 compiled with CUDA v7.5. A summary of processing times, VM types, and costs can be found in Figure 2 -Supplement 1. All VMs were ‘on-demand’, which means that we paid full price and did not risk being ‘kicked off by being outbid due to spot price markets. We uploaded 1536 7676 × 7420 pixels super-resolution movies of ß-galactosidase (EMPIAR 10061) (Bartesaghi et al. 2015) to AWS and aligned them using Unblur v1.0.2 (Grant and Grigorieff 2015) on 5 x x1.32xlarge instances. From our data servers at UCSD that have a 10 Gb connection to AWS, we were able to achieve ~350 MB/sec upload speeds to S3 using multi-file uploads with ‘rclone’. Note that data upload times reflect data movement into S3, which is a separate process from data processing. For users with 1 Gb connections to AWS, we expect speeds of 35 MB/sec, which would increase upload time by 10X, requiring 110 hours for movie upload (4.5 days). Gctf v0.50 (Zhang 2016) was used to estimate the CTF of the aligned micrographs on a single p2.8xlarge VM (8 GPUs). Then, 138,901 particles were picked using GPU-accelerated AutoPick on a single p2.8xlarge VM and extracted at a pixel size of 1.274 Å (binned by 4 from the original data) in a box size of 192 × 192 pixels on a single m4.4xlarge VM (16 vCPUs). This stack of particles was subjected to 2D classification into 200 classes over 25 iterations on a p2.16xlarge VM (16 GPUs). Selection of the best class averages resulted in a stack of 119,443 particles that were then re-extracted at a pixel size of 0.637 Å in a box size of 384 × 384 pixels on a m4.4xlarge VM. These particles were refined with PDB 3I3E (Dugdale et al. 2010) as the initial model using auto-refine to a resolution of 3.5 Å (unmasked) on a single p2.8xlarge VM. These refined coordinates were used for Movie particle extraction on 8 x d2.8xlarge VMs (36 vCPUs and 48 Terabytes on each VM) and Movie refinement on a single x1.32xlarge VM (128 vCPUs) with a running average of 7 movie frames and a standard deviation of 2 pixels on particle translations. These particles were subjected to Polishing on a single x1.32xlarge VM, yielding an unmasked resolution of 3.3 Å, after which they were used for 3D classification into 8 classes over 25 iterations using an angular step of 7.5 degrees on a single p2.8xlarge VM. From the 4 best classes, 106,237 particles were used for 3D auto-refine on a single p2.8xlarge instance to obtain a final, post-processed structure at 2.2 Å, as previously reported(Kimanius et al. 2016; Bartesaghi et al. 2015). During the course of this work, we moved data using ‘rclone’ for data synchronization, uploading 12 TB of movie files and downloading approximately 400 GB of data, which would incur $36 in data egress charges ($0.09 USD/GB).
Atomic model building with Rosetta on AWS
We extended cryoem-cloud-tools to allow users to build atomic models into cryo-EM maps using Rosetta, specifically RosettaCM and Rosetta’s FastRelax protocols. We ran these protocols on c4.8xlarge instances with a single solution requested per vCPU. Using this method we generated atomic models for the 2.2 Å ß-galactosidase map determined on AWS. We used atomic coordinates of 1JZ7 chain A as the starting model for the asymmetric unit of the ß-galactosidase map and generated the initial aligned reference structure using rosetta_refinement_on_aws.py routine from cryoem-cloud-tools. Following this step, we generated the symmetry definition file for Rosetta describing the D2 symmetry of ß- galactosidase in the context of 1JZ7 using the script rosetta_prepare_symmfile.py. All these initial steps were carried out on t2.micro instances. We used the initial reference structure and the symmetry definition file as input and used RosettaCM to generate 200 output models. RosettaCM was run using using rosetta_refinement_on_aws.py routine running on 10 x c4.8xlarge instances with 20 models per instance. The best model in terms of Rosetta energy (including fit-to-density energy) was used as an input for a final refinement with Rosetta’s FastRelax. We generated 8 models from FastRelax using one of the two half maps generated during refinement (training half map) low-pass filtered to a resolution of 2.24 Å and sharpened with a B-factor of −49.52. To estimate overfitting, FSCWOrk (FSC curve between the refined model and the training half map) and and FSCfree (FSC curve between the refined model and the other half map generated during refinement, the test half map) were compared and the the spatial frequency at which the FSC value was 0.5 was 1/2.4 Å−1 in both cases (Figure 4D green and blue curves). The nearly identical FSC curves obtained with the two half maps indicate that there was no over-refinement of the model. The FSC curve between the refined model and the final cryo-EM map (obtained by combining data from both the half maps) showed that FSC value was 0.5 at a spatial frequency of 1/2.2 Å−1 (Figure 4D red curve). The FSC curves were calculated in Rosetta and the plots were made using GraphPad Prism (GraphPad software). The best model in terms of Rosetta energy and model geometry (as determined by MolProbity) was selected as the final atomic model for the ß-galactosidase map.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank all of the members of the Leschziner lab at UC San Diego for helping to test and debug commands to run on AWS. M.A.C. was an HHMI Fellow of the Damon Runyon Cancer Research Foundation. A.E.L. and F. D. acknowledge support from the National Institutes of Health (R01GM107214 to A.E.L and R01GM123089 to F.D.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DATA ACCESSIBILITY
Information and tutorials related to using the software presented here are available at cryoem-tools.cloud. All software is freely available at Github: https://github.com/cianfrocco-lab/cryoem-cloud-tools. The ß-galactosidase cryo-EM structure can be accessed at EMD XXXX and PDB XXXX.
REFERENCES
- Bartesaghi Alberto, Merk Alan, Banerjee Soojay, Matthies Doreen, Wu Xiongwu, Milne Jacqueline L. S., and Subramaniam Sriram. 2015. “2.2 Å Resolution Cryo-EM Structure of ß- Galactosidase in Complex with a Cell-Permeant Inhibitor.” Science 348 (6239): 1147–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cianfrocco Michael A., and Leschziner Andres E.. 2015. “Low Cost, High Performance Processing of Single Particle Cryo-Electron Microscopy Data in the Cloud.” eLife 4 (May). 10.7554/eLife.06664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuenca-Alba Jesús, Del Cano Laura, Blanco Josué Gómez, de la Rosa Trevín José Miguel, Mingo Pablo Conesa, Marabini Roberto, Sorzano Carlos Oscar S, and Carazo Jose Maria. 2017. “ScipionCloud: An Integrative and Interactive Gateway for Large Scale Cryo Electron Microscopy Image Processing on Commercial and Academic Clouds.” Journal of Structural Biology 200 (1): 20–27. [DOI] [PubMed] [Google Scholar]
- Dugdale Megan L, Dymianiw Dayna L., Minhas Bhawanjot K., D’Angelo Igor, and Huber Reuben E.. 2010. “Role of Met-542 as a Guide for the Conformational Changes of Phe-601 That Occur during the Reaction of ß-Galactosidase (Escherichia Coli).” Biochemistry and Cell Biology = Biochimie et Biologie Cellulaire 88 (5): 861–69. [DOI] [PubMed] [Google Scholar]
- Grant Timothy, and Grigorieff Nikolaus. 2015. “Measuring the Optimal Exposure for Single Particle Cryo-EM Using a 2.6 Å Reconstruction of Rotavirus VP6.” eLife 4 (May): e06980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimanius Dari, Forsberg Björn O., Scheres Sjors Hw, and Lindahl Erik. 2016. “Accelerated Cryo-EM Structure Determination with Parallelisation Using GPUs in RELION-2.” eLife 5 (November). 10.7554/eLife.18722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Xueming, Mooney Paul, Zheng Shawn, Booth Christopher R., Braunfeld Michael B., Gubbens Sander, Agard David A., and Cheng Yifan. 2013. “Electron Counting and Beam-Induced Motion Correction Enable near-Atomic-Resolution Single-Particle Cryo-EM.” Nature Methods 10 (6): 584–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales Eva. 2016. “The Development of Cryo-EM into a Mainstream Structural Biology Technique.” Nature Methods 13 (1): 24–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Punjani Ali, Rubinstein John L., Fleet David J., and Brubaker Marcus A.. 2017. “cryoSPARC: Algorithms for Rapid Unsupervised Cryo-EM Structure Determination.” Nature Methods 14 (3): 290–96. [DOI] [PubMed] [Google Scholar]
- Rohou Alexis, and Grigorieff Nikolaus. 2015. “CTFFIND4: Fast and Accurate Defocus Estimation from Electron Micrographs.” Journal of Structural Biology 192 (2): 216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinstein John L., and Brubaker Marcus A.. 2015. “Alignment of Cryo-EM Movies of Individual Particles by Optimization of Image Translations.” Journal of Structural Biology 192 (2): 188–95. [DOI] [PubMed] [Google Scholar]
- Scheres Sjors H. W. 2012. “RELION: Implementation of a Bayesian Approach to Cryo-EM Structure Determination.” Journal of Structural Biology 180 (3): 519–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheres Sjors Hw. 2014. “Beam-Induced Motion Correction for Sub-Megadalton Cryo-EM Particles.” eLife 3 (August): e03665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Ray Yu-Ruei, Kudryashev Mikhail, Li Xueming, Egelman Edward H., Basler Marek, Cheng Yifan, Baker David, and DiMaio Frank. 2015. “De Novo Protein Structure Determination from near-Atomic-Resolution Cryo-EM Maps.” Nature Methods 12 (4): 335–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Ray Yu-Ruei, Song Yifan, Barad Benjamin A., Cheng Yifan, Fraser James S., and DiMaio Frank. 2016. “Automated Structure Refinement of Macromolecular Assemblies from Cryo-EM Maps Using Rosetta.” eLife 5 (September). 10.7554/eLife.17219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Kai. 2016. “Gctf: Real-Time CTF Determination and Correction.” Journal of Structural Biology 193 (1): 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Shawn Q., Palovcak Eugene, Armache Jean-Paul, Verba Kliment A., Cheng Yifan, and Agard David A.. 2017. “MotionCor2: Anisotropic Correction of Beam-Induced Motion for Improved Cryo-Electron Microscopy.” Nature Methods 14 (4): 331–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.