

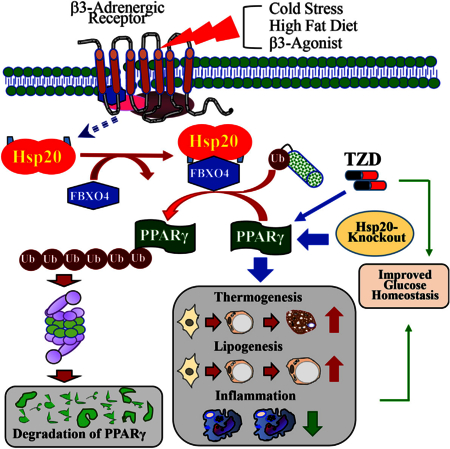
SUMMARY
Exposure to cold temperature is well known to upregulate heat shock protein (Hsp) expression and recruit and/or activate brown adipose tissue and beige adipocytes in humans and animals. However, whether and how Hsps regulate adipocyte function for energy homeostatic responses is poorly understood. Here, we demonstrate a critical role of Hsp20 as a negative regulator of adipocyte function. Deletion of Hsp20 enhances non-shivering thermogenesis and suppresses inflammatory responses, leading to improvement of glucose and lipid metabolism under both chow diet and high-fat diet conditions. Mechanistically, Hsp20 controls adipocyte function by interacting with the subunit of the ubiquitin ligase complex, F-box only protein 4 (FBXO4), and regulating the ubiquitin-dependent degradation of peroxisome proliferation activated receptor gamma (PPARγ). Indeed, Hsp20 deficiency mimics and enhances the pharmacological effects of the PPARγ agonist rosiglitazone. Together, our findings suggest a role of Hsp20 in mediating adipocyte function by linking β-adrenergic signaling to PPARγ activity.
Graphical Abstract
In Brief
Peng et al. identify a dual function for heat shock protein 20 (Hsp20) in regulating white adipocyte maturation and function through the FBXO4-PPARγ axis. The absence of Hsp20 improves the metabolic profile even under obesity conditions.
INTRODUCTION
Adipose tissue is a remarkably complex organ with profound effects on energy balance and glucose homeostasis (Rosen and Spiegelman, 2014). In particular, white adipose tissue (WAT) can functionally adapt and expand to store excess calories following chronic calorie overconsumption, whereas brown adipose tissue (BAT) and beige adipocytes within WAT are characterized by their thermogenic function in rodents (Kajimura et al., 2015) and humans (Cypess et al., 2009), mainly mediated by the action of uncoupling protein 1 (UCP1) following adrenergic stimulation by the sympathetic nervous system (Fedorenko et al., 2012), and have the capacity to burn glucose and stored fat to dissipate energy as heat.
“Lipotoxicity” arises when chronic overconsumption of calories surpasses the capability of WAT and BAT and/or beige adipocytes to adequately store or dissipate the excess fuel, respectively. This involves ectopic accumulation of lipids in other tissues, inflammation and whole-body insulin resistance, and dyslipidemia (Peirce et al., 2014). Conceptually, lipotoxicity can be ameliorated by increasing the capacity of WAT to store fat, which results in reduced inflammation and improved glycemic and lipid parameters (Cao, 2013). Alternatively, the excess calories could also be eliminated by activating BAT and/or beige adipocytes (Bartelt et al., 2011). Thus, mechanisms that simultaneously control both processes would be optimal targets for treating metabolic disease.
As a master regulator of adipocyte development, peroxisome proliferation activated receptor gamma (PPARγ) is best known for its role in regulating adipogenic and lipogenic pathways in white adipocytes (Tontonoz et al., 1994), but it is also necessary for the differentiation and control of the thermogenic program in brown or beige adipocytes (Kajimura et al., 2009). Thiazolidinediones (TZDs) exert their insulin-sensitizing effects and adipogenic as well as thermogenic functions via regulating PPARγ’s posttranslational modifications, including phosphorylation and acetylation (Hu et al., 1996; Qiang et al., 2012). Likewise, FGF21 increases PPARγ activity in adipocytes by preventing PPARγ sumoylation at Lys107 (Dutchak et al., 2012). Additionally, recent evidence has demonstrated that PPARγ can also be covalently attached to ubiquitin proteins (i.e., Siah2, Mkrn1, Fbxo9, and Trim23) and degraded in a proteasome-dependent manner (Kilroy et al., 2012; Kim et al., 2014; Lee et al., 2016; Watanabe et al., 2015), which represents another level of control. However, the in vivo relevance of the change in absolute magnitude of PPARγ because of its ubiquitination remains obscure.
Heat shock proteins (Hsps) are a family that exerts chaperone functions by protecting cellular proteins from denaturation and aggregation in response to a variety of stimuli (Craig et al., 1993). Interestingly, among Hsp family members, Hsp20 is the most upregulated protein during differentiation of human adipose-derived stem cells into mature adipocytes (DeLany et al., 2005); however, the exact role of Hsp20 in adipocytes remains unknown. By combining in vivo and in vitro gain- and loss-of-function studies, we demonstrate in this study that Hsp20 is a critical repressor of adrenergic signaling on adipocyte function by promoting FBXO4-dependent ubiquitination of PPARγ. Our results support the hypothesis that inhibition of Hsp20 activity is a potential target of therapies seeking to enhance metabolic control by improving adipose tissue function.
RESULTS
Genetic Disruption of Hsp20 Increases Whole-Body Energy Expenditure
Using qRT-PCR analysis, Hsp20 was shown to be highly expressed in the heart, testis, and BAT compared with other tissues, including inguinal WAT (iWAT) and epididymal WAT (eWAT) (Figure 1A). Given the critical role of BAT in generating heat, we investigated the effect of cold exposure (48 hr at 4°C) on Hsp20 levels in wild-type (WT) mice. Cold exposure significantly increased both the mRNA and protein levels of Hsp20 in BAT but also in iWAT (Figures 1B and S1A), suggesting a role for Hsp20 in adaptive thermogenesis. We thus generated animals lacking Hsp20 (referred to as knockout [KO] in this paper) (see molecular characterization in Figures 1C and S1B-S1E) and performed histological analysis of BAT, eWAT, and iWAT collected at room temperature (23°C) by H&E staining as well as by Ucp1 immunoreactivity. Intriguingly, BAT and eWAT of KO mice appeared histologically similar to those from WT controls (Figures 1D and S1F). However, iWAT from KO mice contained more multilocular, Ucp1-positive, beige adipocytes (Figure 1D). Immunoblot of Ucp-1 protein confirmed the histological evidence of increased Ucp-1 expression in the iWAT of KO mice (Figures 1E and 1F). We also quantified mRNA expression of thermogenic genes by qRT-PCR in various adipose depots of WT and KO mice housed at 23°C. Consistent with histological inspection and protein expression analysis, Ucp1 mRNA expression in BAT was similar in KO and WT mice (Figure 1G). Indeed, BAT of KO mice exhibited significantly reduced mRNA levels of Prdm16, Dio2, and Acox1 mRNA levels compared with WT controls, although Pparα levels were significantly increased in BAT of KO mice (Figure 1G). In contrast, Ucp1 mRNA levels exhibited a 15-fold increase in the iWAT of KO mice compared with WT controls (Figure 1H), an increase that is consistent with the differences in Ucp1 protein levels (Figures 1E and 1F). The expression of other genes involved in the control of thermogenesis (Cidea, Cox8b, and Dio2), fatty acid oxidation (Pparα, Mcad, and Cpt1a), and mitochondrial metabolism (Cox7a1 and Cytc) was also increased in iWAT of KO mice (Figure 1H). Interestingly, mRNA levels of Cidea, Elovl3, Pparα, Mcad, and Cytc were also significantly higher in eWAT of KO mice, although a trend toward increased Ucp1 mRNA did not reach statistical significance (Figures S1G and S1H).
Figure 1. Genetic Disruption of Hsp20 Increases Whole-Body Energy Expenditure.
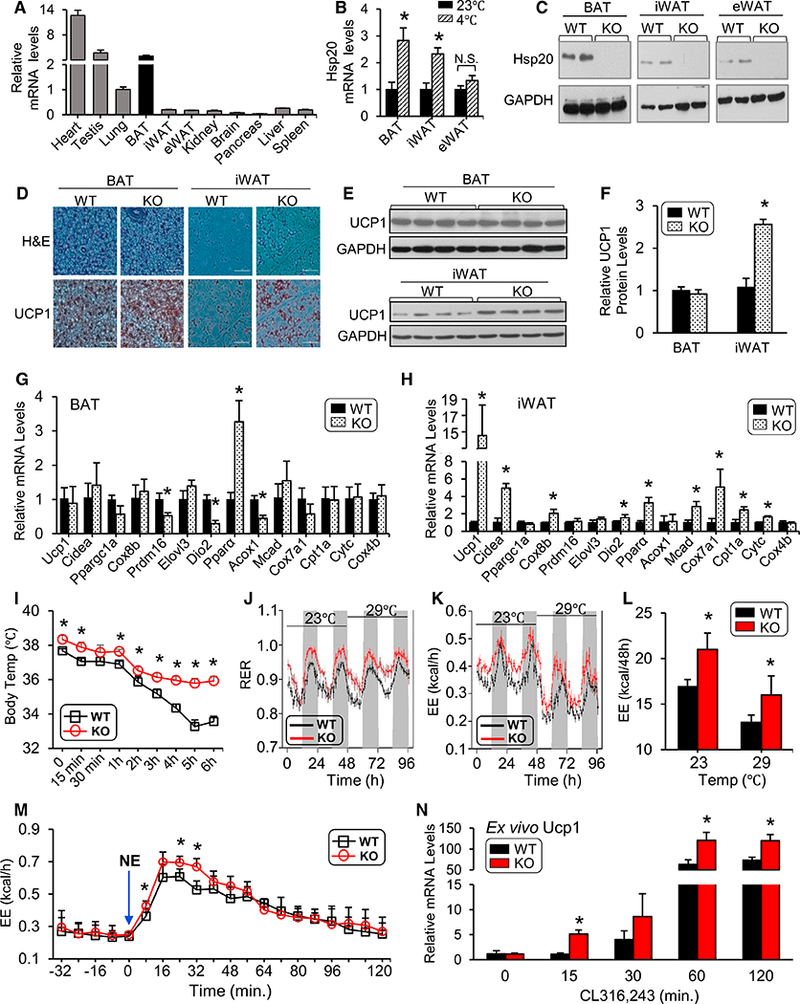
(A) Hsp20 mRNA expression in different tissues was measured by qRT-PCR.
(B) mRNA levels of Hsp20 were determined using RT-PCR in the BAT, iWAT, and eWAT of mice housed at 23°C or 4°C for 48 hr (n = 5 per group).
(C) Western blot analysis of Hsp20 in various tissues of WT and KO mice.
(D) H&E (top) and UCP1-IHC staining (bottom) of the BAT and iWAT of WT and KO mice housed at room temperature (RT) (scale bars, 100 um; n = 6 per genotype).
(E and F) Immunoblot analysis (E) of UCP1 in the BAT and iWAT of WT and KO mice housed at RT and quantification (F) (n = 8 per genotype).
(G and H) mRNA levels of thermogenic, mitochondrial, and fatty acid oxidation genes in the iWAT (G) and BAT (H) of WT and KO mice.
(I) Core body temperature of WT and KO mice during a 6-hr cold challenge (n = 5 per genotype).
(J) Respiratory exchange ratio (RER) in WT and KO mice at different environmental temperatures (n = 8 per genotype).
(K and L) Energy expenditure (EE) (K) and quantification (L) of WT and KO mice from (J).
(M) Energy expenditure in WT and KO mice when injected intraperitoneally (i.p.) with norepinephrine (NE).
(N) SVCs isolated from iWAT of WT and KO mice were differentiated ex vivo over an 8-day period. qRT-PCR was performed on day 8, when adipocytes were treated with CL316,243 for the indicated lengths of time.
*p < 0.05. Data are represented as the mean ± SEM. See also Figure S1.
We then studied whether the increased thermogenic molecular signature in iWAT of KO mice resulted in increased thermogenesis in vivo. First, we challenged KO and WT controls with acute cold exposure. Consistent with the changes in thermogenic gene expression, KO mice were able to maintain thermal homeostasis during acute cold stress; in contrast, the body temperature of WT mice dropped significantly (Figure 1I). We next assessed home cage energy expenditure in mice via indirect calorimetry while the mice were housed at standard room temperature (23°C) or at thermoneutrality (29°C) for 48 hr. The KO mice exhibited a significant increase in respiratory exchange ratio (RER) regardless of the housing temperature (Figure 1J), suggesting that Hsp20 deletion stimulated a substantial shift from lipid to carbohydrate fuels. The increase in external temperature led to a similar decrease in energy expenditure (EE) in KO and WT mice, suggesting that KO mice retain intact neural control of adaptive thermogenesis (Figures 1K and 1L). However, energy expenditure was significantly increased in KO mice compared with body weight-matched WT controls, regardless of the housing temperature (Figures 1K and 1L), and a similar trend was also noted in oxygen consumption (VO2) and carbon dioxide production (VCO2) (data not shown). Locomotor activity did not differ between the two genotypes (data not shown). We reasoned that the persistence of increased energy expenditure in KO mice, despite normal dynamic adjustment to external temperatures, was consistent with increased intrinsic adaptive thermogenesis in adipose tissue. To test this hypothesis, we challenged KO and WT mice, which were maintained at thermoneutrality for 18 hr, with the β-adrenergic receptor agonist norepinephrine (NE). KO mice exhibited a significantly higher increase in energy expenditure than WT controls following NE injection (Figure 1M), which is consistent with increased Ucp-1-dependent thermogenesis (Feldmann et al., 2009). These results suggest a role for Hsp20 in inhibiting Ucp-1-dependent thermogenesis in adipocytes. However, it has been reported that Hsp20 can be found in exosomes released into the circulation, raising the possibility of cross-talk between organs (Wang et al., 2016). To investigate whether Hsp20 regulates the thermogenic program in a cell-autonomous manner, we isolated stromal vascular cells (SVCs) from iWAT of WT and KO mice. When differentiated into mature adipocytes ex vitro, we treated them with the β3 adrenoceptor agonist CL316,243 and monitored the changes in Ucp-1 gene expression. Adipocytes from KO mice exhibited a significant increase in Ucp-1 expression (at 15, 60, and 120 min) compared with those from WT controls (Figure 1N), suggesting that cell-autonomous mechanisms regulated by Hsp20 contribute to the ‘‘browning’’ of iWAT (Figure 1D) and the increased energy expenditure (Figure 1J) exhibited by the KO mice.
KO of Hsp20 Leads to Increased Fat Mass but Improved Glucose Metabolism
Despite increased thermogenic capacity, KO mice maintained on a chow diet (CD) exhibited increased body weight compared with WT controls (Figures 2A and 2B), which was accompanied by a proportional (15%) increase in food intake (Figure 2C). The increased weight gain was due to increased weight of both visceral and subcutaneous fat depots (Figures 2D and 2E). Consistently, nuclear magnetic resonance (NMR) analysis of body composition revealed a 68% and a 15% increase in fat and lean mass, respectively, in KO mice compared with WT controls (Figure 2F). Histological analysis of iWAT and eWAT from 12-week-old mice showed that the average adipocyte sizes in KO mice were 2- to 4-fold larger than those in WT mice (Figures 2G–2I). Consistent with the increased adiposity, serum leptin levels were significantly higher in KO mice (Figure 2J). These results suggest that increased food intake leads to weight gain beyond the effect of higher thermogenic capacity in Hsp20-deficient mice at room temperature.
Figure 2. KO of Hsp20 Leads to Increased Fat Mass but Improved Glucose Metabolism.
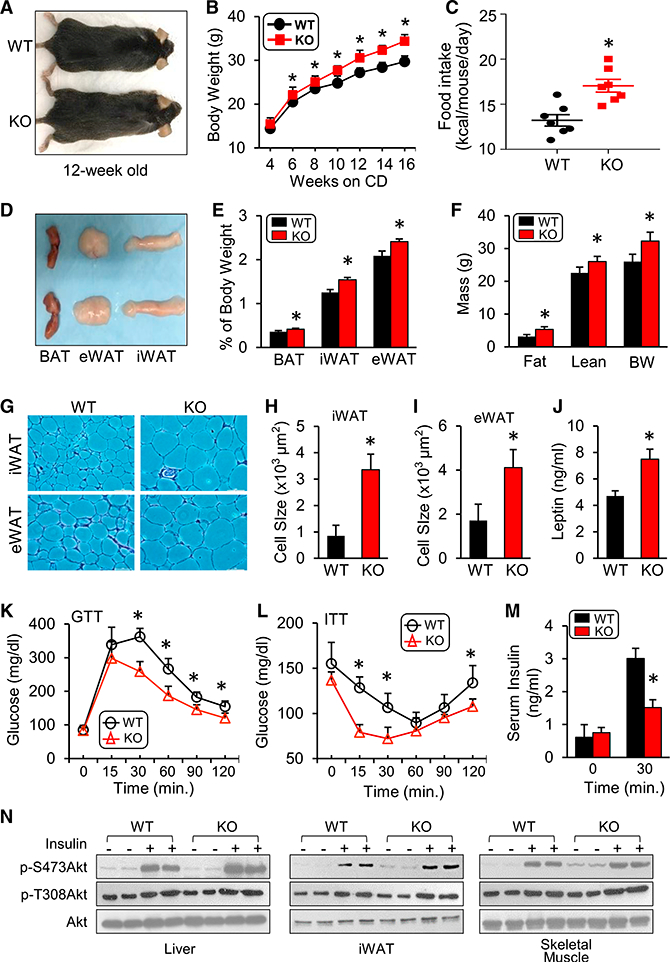
(A) Gross morphology of KO mice (12 weeks old) and 3 fat pads on a chow diet (CD)
(B) Body weight of WT and KO mice on a CD (n = 9–10 per group).
(C) Food intake in WT and KO mice on a CD (n = 9–10 per group).
(D and E) Gross morphology (D) and weight of different fat pads to body weight ratio (E) (n = 4 per genotype).
(F) Body composition of WT and KO mice (n = 8 per genotype).
(G-I) H&E staining (G) and cell size quantification of iWAT (H) and eWAT (I) adipocytes (n = 6 per genotype; scale bars, 100 μm).
(J) Serum leptin levels in mice after 6-hr fasting (n = 3 per genotype).
(K and L) IPGTT (1.5 g/kg) (K) and IPITT (1 U/kg) (L) analyses were performed in WT and KO mice on a CD (n = 5 per genotype).
(M) Fasting basal and glucose-stimulated (1.5g/kg) insulin levels (n = 3–5 per genotype).
(N) Phosphorylation of AKT (Ser-473 and Thr-308) was measured in the iWAT, liver, and skeletal muscle after insulin stimulation (n = 5 per genotype).
*p < 0.05. Data are represented as the mean ± SEM. See also Figure S1.
Because body weight and thermogenic capacity are tightly linked to regulation of systemic glucose metabolism (Peirce et al., 2014), we next conducted glucose and insulin tolerance tests at 12 weeks of age, when the body weight is significantly higher in KO mice. Despite the higher body weight, Hsp20-deficient mice exhibited a lower blood glucose level during an intraperitoneal glucose tolerance test (IPGTT) compared with their WT controls (Figure 2K). Furthermore, KO mice displayed a significant reduction in blood glucose during an insulin tolerance test compared with WT controls (Figure 2L) and reduced glucose-stimulated insulin levels (Figure 2M), suggesting increased whole-body insulin sensitivity in KO mice. We also directly investigated the action of insulin in metabolically active tissues, including the liver, iWAT, and skeletal muscle, by measuring Akt phosphorylation at Ser-473 (S473) and Thr-308 (T308) upon insulin stimulation. Insulin-stimulated Akt phosphorylation was significantly increased in all three tissues of KO mice compared with the insulin-treated WT controls (Figure 2N). Altogether, these findings indicate that loss of Hsp20 in vivo results in increased weight gain but enhanced insulin sensitivity and systemic glucose metabolism.
Gene Enrichment Analyses from iWAT Reflect a Comparable Role of Hsp20 Deletion in Lipid Metabolism and Inflammation with Cold Treatment
The unique characteristics displayed above in Hsp20-deficient mice provoked us to dissect the molecular mechanism by which Hsp20 regulates energy metabolism, especially in adipose tissues. Toward this end, we performed RNA sequencing (RNA-seq) analyses of the gene expression profile of iWAT in both WT and KO mice under conditions of room temperature and cold exposure (4°C) for 48 hr (Figure 3A). Surprisingly, comparisons of gene expression patterns in iWATs revealed that Hsp20 deficiency highly mimicked the gene signature of the cold-treated WT (Figure 3A). Specifically, cold exposure drastically downregulated the expression of pro-inflammatory genes (i.e., Ccl2, Cxcl2, Cxcl10, IL-1β, IL1RL1, and Vcaml) and upregulated the expression of lipogenesis genes (i.e., Acly, Acaca, Dgat2, Fasn, Me1, and Scd1) and thermogenesis genes (i.e., Elovl3, Dio2, Pparα, Ucp1, Cidea, Cox8b, Cox7a, Acadam, and Cytc) in iWAT, which were similar to alterations in Hsp20-deficient iWAT without cold exposure (Figures 3B and 3C). Subsequent gene network analysis identified that a total of 278 genes were upregulated to a similar degree between KO- and cold-treated iWATs, whereas there were 204 genes similarly downregulated in both KO- and cold-treated iWATs (Figures 3D and S2A). Gene ontology (GO) enrichment analysis showed that the PPAR pathway was greatly activated in both KO- and cold-treated iWAT compared with WT controls (Figures 3E and 3F). Conversely, the signaling cascades involved in diabetes mellitus, nuclear factor kB (NF-kB), mitogen-activated protein kinase (MAPK), cytokine-cytokine, and chemokine, were significantly attenuated (Figures 3E and 3F). These results suggest that ablation of Hsp20 shares common mechanistic underpinnings with cold-treated lean WT mice, which leads to enhanced thermogenic capacity and activation of the PPARγ pathway, known to enhance adiposity, improve insulin sensitivity, and exert anti-inflammatory effects (Ahmadian et al., 2013). The unique expression pattern displayed in KO iWAT may explain, at least in part, the increased fat mass with higher thermogenic capacity and improved insulin sensitivity observed in Hsp20-deficient mice.
Figure 3. Gene Enrichment Analyses from iWAT Reflect a Comparable Role of Hsp20 Deletion in Lipid Metabolism and Inflammation with Cold Treatment.
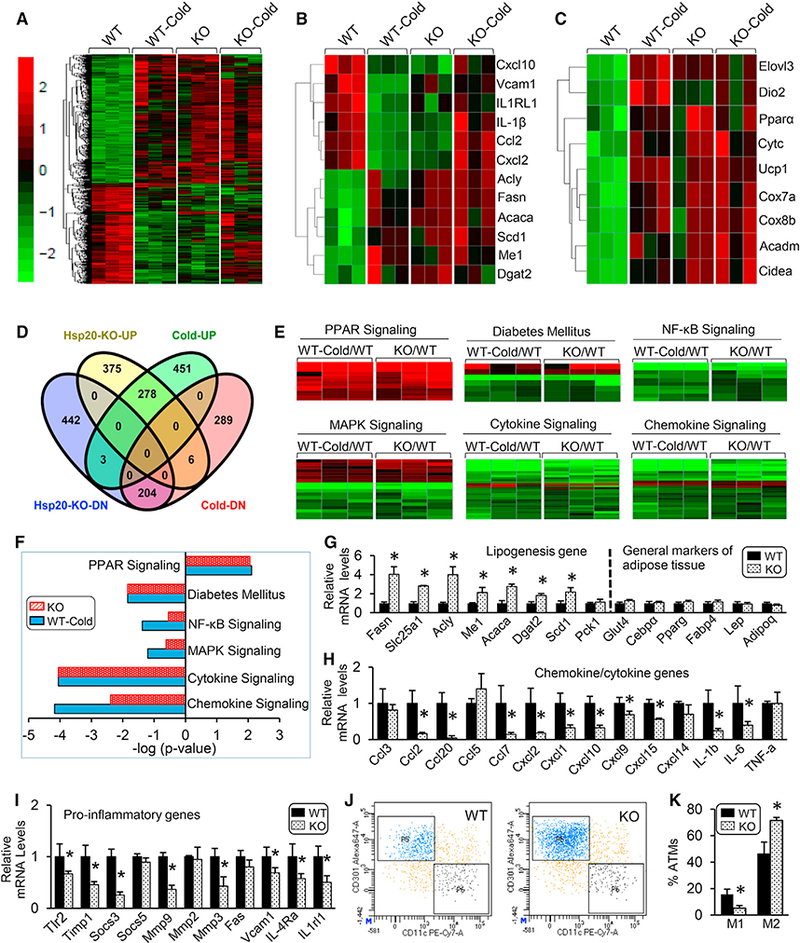
(A) Normalized heatmap comparison of gene expression alterations in iWAT isolated from WT or KO mice housed at RT or cold using RNA-seq (n = 3 per genotype).
(B and C) Normalized heatmap comparison of genes involved in inflammation, lipogenesis (B), and thermogenesis (C) in iWAT isolated from WT or KO mice housed at RT or cold using RNA-seq.
(D) Venn diagram comparing differentially expressed genes following cold treatment with those from KO mice.
(E) Normalized heatmap comparison of gene expression alterations in iWAT isolated from WT mice housed at cold (WT-Cold) versus at RT (WT) and KO mice versus WT mice housed at RT using RNA-seq.
(F) Kegg pathway analysis of significantly activated or inhibited genes from (A).
(G-I) mRNA expression of lipogenic genes (G), chemokines and cytokines (H), and genes involved in pro-inflammatory pathways (I) in iWAT ofWT and KO mice.
(J and K) Flow cytometry analysis (J) and its quantification (K) for M1 and M2 macrophages in iWAT ofWT and KO mice (ATM, adipose tissue macrophage) (n = 5 per genotype).
*p < 0.05. Data are represented as the mean ± SEM. See also Figures S2 and S3.
Consistent with the RNA-seq data, qRT-PCR further validated a significant increase in the expression of the Fasn, Slc25a1, Acly, Me1, Acaca, Dgat2, and Scd1 genes, which are involved in the control of lipogenesis in iWAT of KO mice, whereas general markers for adipose tissue demonstrated no difference between the two genotypes (Figure 3G). To determine the effects of altered lipogenesis on lipid composition in KO mice, a lipidomics analysis was performed in iWAT and liver. iWAT of KO mice exhibited a significant enrichment of C16:1n7 palmitoleate (Figure S3A), enrichment consistent with the increased Scd1 expression and Scd1 desaturation index (C16:1/C16:0) in iWAT (Figure S3A). Palmitoleate levels in the livers of KO mice were also significantly elevated, albeit to a lesser extent (Figure S3B). In contrast to the activation of lipogenesis, additional qRT-PCR analyses validated that loss of Hsp20 had a profound inhibitory effect on a whole array of chemokines, such as Ccl2, Ccl7, Cxcl1, Cxcl2, and Cxcl10, and cytokines, such as IL-1β and IL-6 (Figure 3H). A similar significant reduction was also observed in the expression of genes important for inflammatory processes, such as Tlr2, Timp1, Socs3, Mmp3, Mmp9, and Vcam1 (Figure 3I). In line with the changes in inflammatory markers, flow cytometry analysis revealed that the number of classically activated macrophages (M1 macrophages) was significantly decreased, whereas the number of alternatively activated macrophages (M2 macrophages) was significantly elevated in the iWAT of KO mice compared with WT mice (Figures 3J and 3K). Likewise, the mRNA levels of inducible nitric oxide synthase (iNOS) (an M1 macrophage marker) were significantly decreased in iWAT, eWAT, and BAT, although the M2 macrophage marker Arg1 was decreased only in iWAT (Figure S3C). Taken together, these results indicate that Hsp20-KO-mediated functions in iWAT are highly associated with the activation of lipogenesis and suppression of inflammatory responses, both of which affect insulin sensitivity and systemic glucose metabolism.
Inflammation and Insulin Resistance Associated with Increased Obesity Are Ameliorated because of Hsp20 Deficiency
Given that ablation of Hsp20 improves systemic glucose metabolism, we next asked whether nutrient challenge might accentuate Hsp20’s role in metabolic regulation. To address this question, we employed a high-fat diet (HFD)-induced obesity model that provokes insulin resistance, glucose intolerance, and hepatic steatosis. Interestingly, we observed that HFD feeding of WT mice elevated Hsp20 protein levels in all three fat depots (eWAT, iWAT, and BAT) but not in other metabolically relevant tissues, such as muscle or liver (Figures S4A and S4B). During the 16-week period of HFD feeding, KO mice exhibited a significant increase in body weight (Figures 4A and 4B), accompanied by a significant increase of food intake (Figure 4C). MRI analysis revealed a nearly 30% increase in whole-body fat content, whereas there was no difference in lean body mass (Figure 4D). The increase of fat mass was in part due to a 60% increase in iWAT depot weight and a 20% increase in eWAT depot in KO mice compared with their WT controls (Figure 4E). Analysis of energy expenditure by indirect calorimetry after 11 weeks of HFD feeding revealed a significant increase in energy expenditure in KO mice compared with WT mice (Figures 4F and 4G), whereas the RER remained unchanged (Figure 4H). The increased energy expenditure was consistent with a significant increase in oxygen consumption and carbon dioxide production (Figures S4C-S4F) and was not the result of increased locomotor activity, which was, in fact, significantly reduced in the KO mice (Figure S4G).
Figure 4. Inflammation and Insulin Resistance Associated with a HFD Are Ameliorated Because of Hsp20 Deficiency.
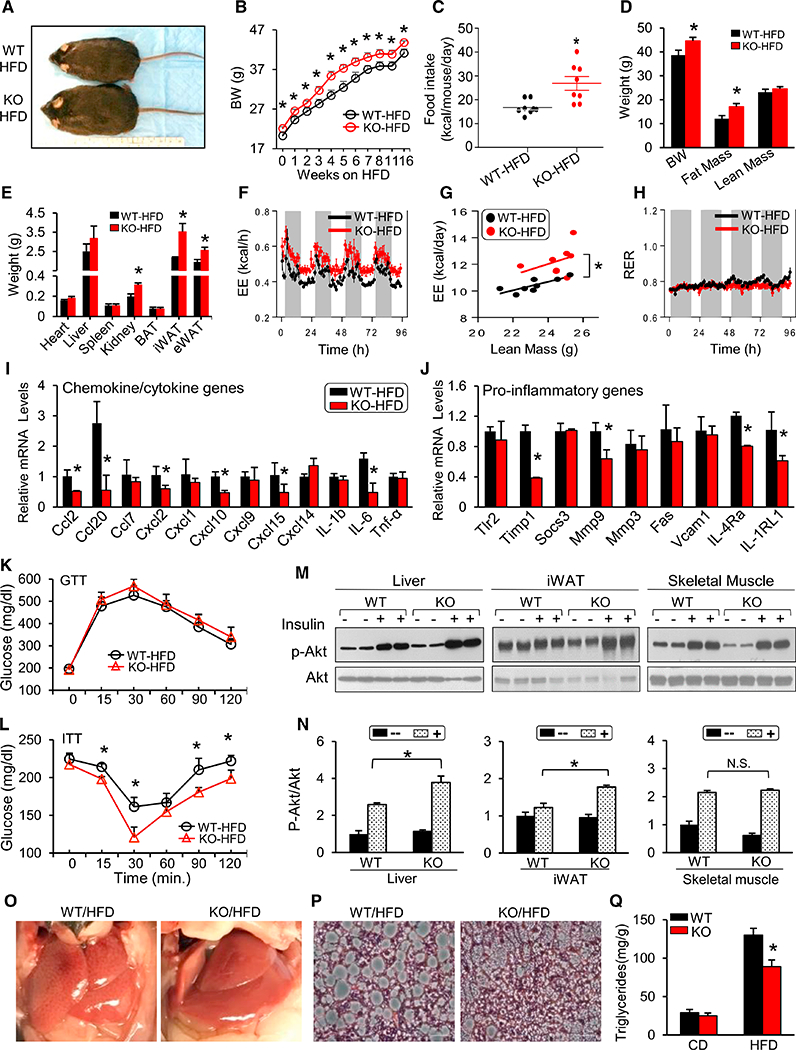
(A) Gross morphology of WT and KO mice after 16-week HFD feeding.
(B) Body weight of WT and KO mice on an HFD (n = 7–8 per group).
(C) Food intake of WT and KO mice fed with HFD (n = 8 per genotype).
(D and E) Body composition (D) and weight of various tissues (E) in WT and KO mice.
(F) Energy expenditure after 12 weeks of HFD feeding (n = 8 per genotype).
(G) Linear regression analysis of energy expenditure against lean body mass.
(H) RER in WT and KO mice from (F).
(I and J) mRNA levels of chemokines and cytokines (I) and genes involved in pro-inflammatory pathways (J) in iWAT ofWT and KO mice fed an HFD for16weeks.
(K and L) IPGTT (K) and IPITT (L) analyses were performed in WT and KO mice after 16 weeks of HFD feeding (n = 5 per genotype).
(M and N) Phosphorylation of AKT (Ser-473) in iWAT, liver, and skeletal muscle (M) after insulin stimulation and quantification (N) (n = 5 per genotype).
(O and P) Gross morphology (O) and representative liver sections (H&E) (P) from WT and KO mice after 16-week HFD feeding (n = 5).
(Q) Hepatic triglyceride content after 16 hr fasting.
*p < 0.05. Data are represented as the mean ± SEM. See also Figure S4.
Because diet-induced obesity (DIO) is associated with the development of inflammation, we then assessed the expression of chemokine and proinflammatory genes by qRT-PCR. Consistent with the findings in their CD counterparts (Figures 3H and 3I), some chemokines (i.e., Ccl2, Cxcl2, Cxcl10, Cxcl15, and IL-6) and pro-inflammatory markers (i.e., Timpl, Mmp9, IL4ra, and IL1rl1) were significantly reduced in DIO-KO mice (Figures 4I and 4J). We next assessed glucose homeostasis in HFD-fed WT and KO mice. An IPGTT revealed similar glucose tolerance (Figure 4K) and similar insulin levels (Figure S4H) in DIO-WT and DIO-KO animals. Given that KO mice are more obese, this similarity suggested relatively improved whole-body glucose control. Indeed, blood glucose levels in DIO-KO mice remained significantly lower than in WT controls at several time points (15, 30, 90, and 120 min) throughout the intraperitoneal insulin tolerance test (IPITT), suggesting elevated insulin sensitivity (Figure 4L). Importantly, this was confirmed by significantly increased insulin-stimulated Akt phosphorylation at Ser-473 in the liver and iWAT of KO mice compared with WT mice (Figures 4M and 4N). In addition, fasting serum nonesterified fatty acid (NEFA) levels were significantly reduced by 26% in KO mice (Figure S4I), in line with their increased insulin sensitivity. Serum triglycerides, but not cholesterol (Figures S4J and S4K), were increased in ad libitum-fed DIO-KO mice. Interestingly, DIO-KO mice showed less liver steatosis and decreased hepatic triglyceride content (Figures 4O–4Q) as well as significantly reduced expression of genes involved in lipogenesis (i.e., Fasn and Scd1) and gluconeogenesis (i.e., Pepck and G6pase; Figures S4L and S4M), features also consistent with improved hepatic insulin signaling. Furthermore, the expression of Mcad was significantly increased, suggesting increased fatty acid oxidation in DIO-KO mice (Figure S4M). Collectively, these data indicate that DIO-induced insulin resistance, meta-inflammation, and liver steatosis were substantially alleviated in KO mice compared with WT mice.
Hsp20 Regulates PPARγ Protein Ubiquitination and Stability in Isolated Adipocytes
The concurrence of increased fat mass with increased thermogenic capacity, reduced inflammation, and improved insulin sensitivity in KO-iWAT could be explained by an increase in PPARγ’s activity as a result of reduced Hsp20 activity. Indeed, our RNA-seq analysis had shown that the PPAR signaling pathway was activated in KO-iWAT. To further clarify this issue and exclude the in vivo effects of neurohumoral and inflammatory factors, we first examined the differentiation of SVCs isolated from iWAT of WT and KO mice into adipocytes. Consistent with the in vivo findings, cells derived from KO mice exhibited more lipid droplets than those from WT controls (Figure S5A). Expression analysis of lipogenic genes revealed that PPARγ, Cebpa, Fabp4, Fasn, Me1, and Slc25a1 levels were significantly increased in KO adipocytes after day 6 post-differentiation (Figure S5B). The expression of thermogenic genes was minimally (i.e., Prdm16) or unchanged (Ucp1) between two genotypes (Figure S5B). These results suggest that Hsp20 restrains adipocyte differentiation in a cell-autonomous manner.
The ex vivo analysis of PPARγ total protein levels revealed a significant increase (5-fold) in KO adipocytes (Figure S6A). Despite this increase, the levels of phospho-PPARγ (both S112 and S273) were similar to those of WT controls (Figure S6A), which resulted in a significant reduction in the relative levels of each of the phosphorylated forms in KO adipocytes compared with WT adipocytes (Figure S6A). Total PPARγ protein was also significantly increased in iWAT tissue samples from KO mice (Figure S6B), with a significant reduction in the relative levels of phospho-PPARγ (S112) and no change in phospho-PPARγ (S273) in iWAT (Figure S6B). This relative decrease in PPARγ phosphorylation did not correlate with changes in phospho-extracellular signal-related kinase (ERK), phospho-MAPK, CDK5, or CDK9, which were comparable between WT and KO adipocytes (Figures S6A-S6D). Taken together, the ex vivo and in vivo data suggest that disruption of Hsp20 may enable an increase in PPARγ transcriptional activity by increasing PPARγ protein levels.
The discrepancy between the increased PPARγ gene (1.4-fold; Figure S5B) and protein (5-fold; Figure S6A) expression in KO adipocytes led us to assess the role of Hsp20 in the control of PPARγ degradation. Given that a physiological increase in adrenergic signaling (i.e., cold exposure) increases Hsp20 in iWAT (Figure S1A), we treated adipocytes differentiated from iWAT-derived SVCs with the β3 adrenoceptor agonist CL316,243 to indirectly promote (WT) or not (KO) Hsp20 gain of function, and then we measured PPARγ protein levels. Consistent with previous reports (Lindgren et al., 2004), adrenergic stimulation reduced PPARγ levels, but this reduction was significantly prevented in KO adipocytes (Figure 5A). These data link the control of PPARγ protein levels by adrenergic signaling to Hsp20 activity. Consistently, inhibition of protein translation with cycloheximide (CHX) revealed a significantly extended half-life of PPARγ protein levels in KO-derived adipocytes compared with the WT control (Figure 5B). Altogether, ubiquitin-proteasome-dependent degradation is one of the mechanisms that control PPARγ protein levels, and our in vitro analysis revealed that PPARγ polyubiquitination was diminished upon Hsp20 ablation (Figure 5C).
Figure 5. Hsp20 Regulates PPARγ Protein Ubiquitination and Stability in Isolated Adipocytes.
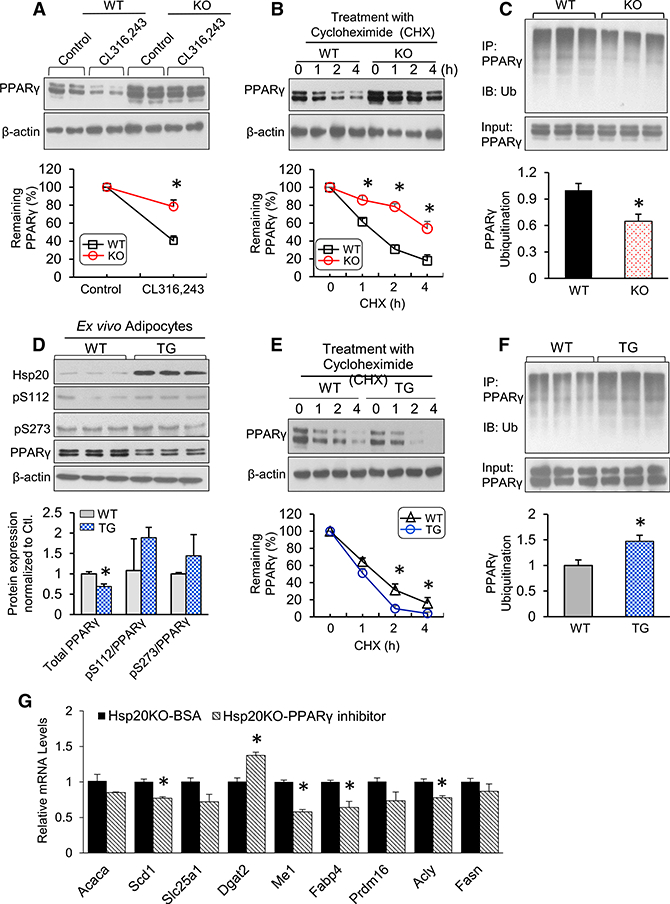
(A) Protein levels of PPARγ in differentiated WT or KO adipocytes upon CL316,243 treatment. The expression level of PPARγ in control cells was set to 100%.
(B) The differentiated cells were treated with cycloheximide (CHX) (5 μM) for 0, 1, 2, or 4 hr. Immunoblot analysis results of PPARγ are reported as a percentage of the value at 0 hr.
(C) The differentiated cells were treated with MG132 (10 μM) for 4 hr, and protein extracts were immunoprecipitated with anti-PPARγ, followed by immunoblotting with anti-ubiquitin (Ub).
(D) Immunoblot analysis and quantification of total PPARγ and phospho-PPARγ at Ser-112 and Ser-273 on day 8 of SVC differentiation.
(E) The differentiated cells were treated with CHX (5 μM) for 0, 1, 2, or 4 hr. Immunoblot analysis of PPARγ was conducted, and quantification results are indicated as a percentage of the value at 0 hr.
(F) The differentiated cells were treated with MG132 (10 μM) for 4 hr, and protein extracts were immunoprecipitated with anti-PPARγ, followed by immunoblotting with anti-Ub.
(G) mRNA expression of lipogenic genes in Hsp20-KO adipocytes treated with PPARγ (5 μM, 8 hr).
*p < 0.05. Data are represented as the mean ± SEM. See also Figures S5 and S6.
To further investigate the extent to which gain of Hsp20 activity in adipose tissue regulates adipocyte function, we generated transgenic (TG) mice specifically overexpressing Hsp20 in adipose tissue by using adiponectin promoter cassette (data not shown). These transgenic mice bred normally, and they were morphologically and behaviorally similar to WT controls. Interestingly, the food intake and body weight gain of TG mice were similar to their WT controls when fed with a CD or HFD (data not shown), suggesting that physiological levels of Hsp20 are sufficient to mediate its effects on whole-body energy balance. Assessment of Hsp20 levels by immunoblotting demonstrated a 3- to 10-fold increase in Hsp20 protein levels in different fat pads (data not shown). We then isolated SVCs from iWAT of WT and TG mice for an 8-day differentiation process into mature adipocytes. We found no significant difference in lipid accumulation between both types of adipocytes (data not shown). Transgenically derived adipocytes exhibited a dramatic increase in Hsp20 levels compared with WT controls (Figure 5D). Interestingly, and opposing the findings seen in KO, total PPARγ protein levels were significantly reduced by 20% compared with WT adipocytes, although the ratio of phospho-PPARγ at S112 or S273 did not differ between groups (Figure 5D). Furthermore, the half-life of endogenous PPARγ was significantly reduced in adipocytes derived from Hsp20-TG mice (Figure 5E). This reduction was consistent with the significant increase in PPARγ polyubiquination exhibited by TG adipocytes (Figure 5F). To further investigate whether Hsp20 regulates adipocyte function through PPARγ, we measured genes involved in lipogenesis from KO adipocytes after treating the cells with the PPARγ inhibitor. The results showed a significant reduction in genes, including Scd1, Me1, Fabp4, and Acly, which indicated that Hsp20-KO-induced lipogenesis was PPARγ-dependent (Figure 5G). Altogether, these loss- and gain-of-function studies further demonstrate a role of Hsp20 in the control of adipocyte function by regulating ubiquitination-dependent PPAR γ2 degradation.
FBXO4 Is Required for Hsp20-Mediated Control of PPARγ Stability
To dissect the mechanisms underlying the Hsp20-regulated ubiquitin-dependent degradation of PPARγ, we next measured the protein levels of major E3 ligase family members (i.e., SIAH2, FBXO4, TRIM23, and MKRN1) known to be expressed in iWATs (Kilroy et al., 2012; Kim et al., 2014; Watanabe et al., 2015). Our analyses indicated that FBXO4 was the only E3 ubiquitin ligase exhibiting a significant change (20% decrease in KO versus WT [Figures S7A and S7B] and 50% increase in TG versus WT [Figures S7C and S7D]) among several candidates in iWATs. Using ex vivo differentiated adipocytes from SVCs, we further confirmed a 1.8-fold induction of FBXO4 protein upon differentiation with concurrent upregulation of Hsp20, whereas other endogenous Hsps (i.e., Hsp90 and Hsp70) were not altered (Figure S7E). These results imply that Hsp20 may control PPARγ ubiquitination through FBXO4. Indeed, our biochemical analyses identified that endogenous Hsp20 was co-immunoprecipitated with FBXO4 in ex vivo-differentiated WT adipocytes (Figure 6A). Notably, PPARγ was co-precipitated with FBXO4 in WT adipocytes, and the strength of interaction was substantially reduced or increased in samples obtained from KO or TG adipocytes, respectively (Figure 6B). Together, our findings suggest that Hsp20 interacts with FBXO4 and, likely through its chaperone-like activity, enhances the ability of FBXO4 to bind and ultimately promote the proteasomal degradation of PPARγ. To directly demonstrate that FBXO4 is necessary for Hsp20 to regulate PPARγ levels, we infected 3T3-L1 preadipocytes with an adenovirus vector carrying either Hsp20 antisense RNA (Hsp20-KO), cDNA (adenovirus [Ad]-Hsp20), or β-galactosidase (β-gal) as a control (Ad-β-gal) and with an adeno-associated virus (AAV) expressing short hairpin RNA (shRNA) for FBXO4 (shFBXO4) or a non-targeting control shRNA (shControl). Ad-Hsp20 and Ad-Hsp20+shFBXO4 cells displayed reduced and increased differentiation, respectively (Figure 6C). In line with our hypothesis, treatment of shFBXO4 alone showed results similar to the Hsp20-KO group; protein levels of PPARγ increased after either FBXO4 or Hsp20 was downregulated (Figures 6D and 6E). Consistent with the results seen in iWAT of TG mice (Figure S7C), Ad-Hsp20 significantly increased FBXO4 while significantly decreasing PPARγ levels (Figures 6D and 6E). Importantly, downregulation of FBXO4 (shFBXO4 cells) was able to prevent such a reduction (Figures 6D and 6E). These changes in PPARγ levels were predictably consistent with an increase and decrease in PPARγ polyubiquitination in Ad-Hsp20- and Ad-Hsp20+shFBXO4-transfected cells, respectively (Figures 6F and 6G). qRT-PCR analysis indicated that the expression of the lipogenic markers Fabp4 and Fasn was unaffected in Ad-Hsp20 cells, but it was significantly increased following FBXO4 depletion (Figure 6H). We also investigated whether the Hsp20-FBXO4 interaction played a role in control of the thermogenic program by measuring Ucp1 gene expression upon CL316,243 treatment. As expected, CL316,243 promoted a robust increase in Ucp1 expression in the control cells (Figure 6I). This increase was significantly attenuated and enhanced in Ad-Hsp20- and Ad-Hsp20+shFBXO4-transfected cells, respectively (Figure 6I). Altogether, these data demonstrate that the cell-autonomous control of adipocyte function by Hsp20 requires FBXO4-dependent regulation of PPARγ stability.
Figure 6. FBXO4 Is Required for Hsp20-Mediated Control of PPARγ Stability.
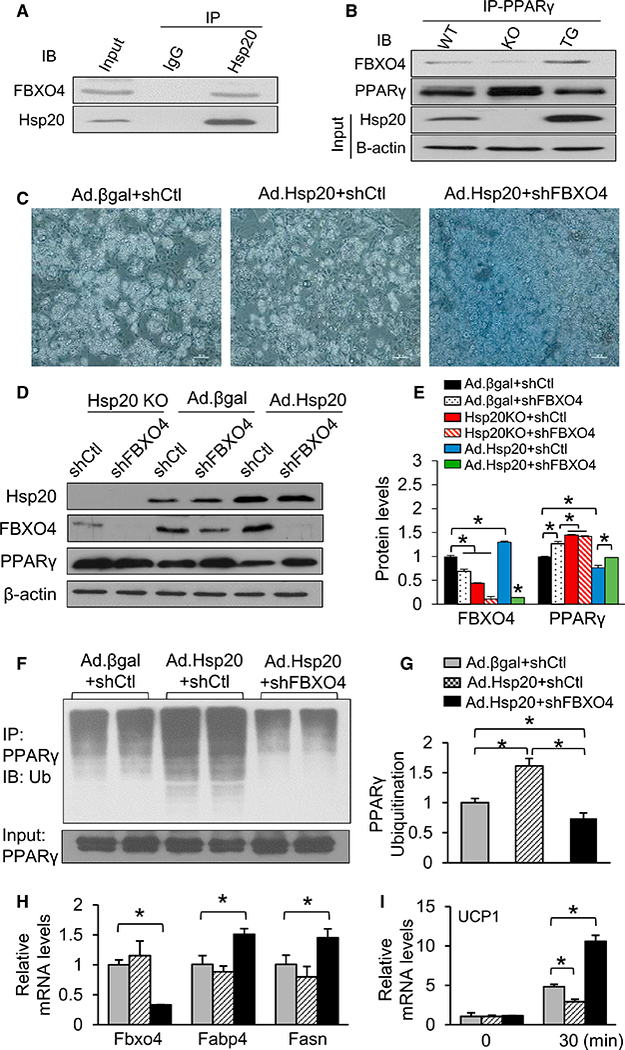
(A and B) Co-immunoprecipitation of FBXO4 with Hsp20(A)and with PPARγ (B) in WT and KO as well as TG adipocytes differentiated ex vivo.
(C) Representative images of 3T3-L1 cells infected with Ad.β-gal or Ad.Hsp20, followed by FBXO4 shRNA knockdown (n = 4 per cell line).
(D and E) Western blot analysis (D) and quantification (E) of FBXO4 and PPARγ in adipocytes treated with the indicated adenovirus and shRNAs.
(F and G) Cells from (C) were treated with MG132 (10 μM) for 4 hr, and protein extracts were immunoprecipitated with anti-PPARγ and immunoblotted with anti-Ub (F); quantification was shown in (G).
(H) mRNA levels of lipogenic genes in cells from (C).
(I) mRNA levels of Ucp1 in cells from (C) after being treated with CL316,243 for 30 min.
*p < 0.05. Data are represented as the mean ± SEM. See also Figure S7.
Increased Efficacy of Rosiglitazone (TZD) on Metabolic Control in Hsp20-KO Mice
We reasoned that, if the physiological control of adipocyte metabolism by Hsp20 hinges on reducing PPARγ levels, then Hsp20-KO mice should experience increased metabolic control following treatment with TZDs; namely, PPARγ agonists. Therefore, we treated HFD-fed WT and KO mice with rosiglitazone (a TZD) or PBS for 3 weeks. As expected, rosiglitazone treatment significantly increased body weight in WT mice compared with the PBS group (Figures 7A and 7B). Consistent with our previous observations (Figures 2B and 4B), untreated HFD-KO mice exhibited a significant increase in body weight (BW) gain compared with untreated WT controls (Figures 7A and 7B). Rosiglitazone treatment promoted a similar BW gain in WT and KO mice (Figures 7A and 7B), which represents a significant reduction in the relative efficacy of rosiglitazone in promoting BW in KO compared with WT mice (112% versus 297%, p < 0.01, t test). We also performed an IPGTT and IPITT to assess the effect of rosiglitazone on whole-body glucose homeostasis. In fact, rosiglitazone significantly improved glucose tolerance (Figure 7C) and insulin sensitivity in WT mice (Figure 7D). Consistent with our prediction, the beneficial effect of rosiglitazone on glucose homeostasis was significantly enhanced in Hsp20-KO mice (Figures 7C and 7D). We then measured the expression of genes in iWAT that were involved in lipogenesis, thermogenesis, and inflammation, processes known to be regulated by PPARγ agonists. Rosiglitazone treatment led to similar changes in the expression of lipogenic genes in both genotypes, except for a significant upregulation in the expression of Acaca when comparing the WT-HFD,TZD with the KO-HFD,TZD groups (Figure 7E). Rosiglitazone also affected the expression of thermogenic genes in both groups. However, the expression levels of Ucp1 and Cidea were significantly higher in KO-TZD compared with WT-TZD controls (Figure 7E), suggesting that a reduction in Hsp20 activity enhances activation of the thermogenic program by rosiglitazone, which is consistent with the relatively lower BW gain exhibited by the KO-TZD mice. Likewise, the expression of genes related to inflammation (i.e., Cxcl1 and Cxcl7) was reduced to the same extent after drug treatment in both groups of mice, whereas others, like Ccl2 or IL1b, were reduced only in WT mice (Figure 7E). We then examined PPARγ expression and its phosphorylation state in iWAT. As expected, and consistent with previous ex vivo data (Figure S6A), total PPARγ protein expression levels were significantly increased in iWAT of KO mice irrespective of TZD treatment (Figures 7F and 7G). Given the link between phosphorylation of PPARγ at S273 with glucose homeostasis (Choi et al., 2010), we also measured phospho-PPARγ levels. Consistent with the improved glycemic control (Figures 7F and 7G), the relative levels of phospho-PPARγ (S273 and S112) were significantly lower in the TZD treated-KO mice compared with the WT controls (Figures 7F and 7G). Taken together, these data demonstrate that loss of Hsp20 augments the efficacy of rosiglitazone by improving glycemic control and activating the thermogenic program in iWAT of obese mice.
Figure 7. Ablation of Hsp20 Improves the Efficacy of TZD Metabolic Control.
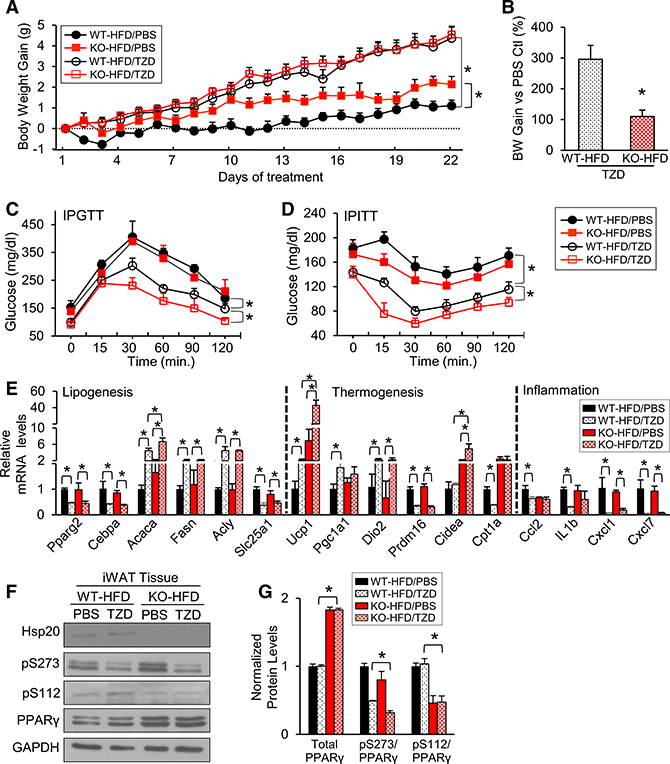
(A) HFD-fed WT and KO mice were injected i.p. with vehicle (PBS + DMSO) or rosiglitazone (10 mg/kg, resolved in PBS + DMSO) for 3 weeks. Body weight was measured daily.
(B) Body weight gain was calculated as the difference between each day and day 0 (n = 6–10 per group).
(C and D) IPGTT (C) and IPITT (D) analyses were conducted in mice from (A).
(E) mRNA levels of lipogenic, thermogenic, and inflammation genes were measured in iWAT of mice from (A) using RT-PCR.
(F and G) Western blot analysis (F) and quantification (G) of total PPARγ and phospho-PPARγ at Ser-112 and Ser-273 in iWAT of mice from (A).
*p < 0.05. Data are represented as the mean ± SEM.
DISCUSSION
Regulation of adipose tissue expansion and thermogenesis is usually investigated as two distinct aspects of adipose biology. Our data highlight the importance of Hsp20 activity as a common regulator of those two programs and illustrate the extent to which they are connected at the molecular and physiological levels. Before our study, the only evidence linking Hsp20 to adipocyte function was its induction during differentiation of human adipose-derived stem cells in vitro (DeLany et al., 2005). We now uncover a previously unrecognized role of Hsp20 in lipogenesis of WAT. Here, mice lacking Hsp20 have shown increased PPARγ activity in WAT and corresponding increases in WAT mass and adipocyte size. Interestingly, we observed increased food intake in Hsp20-KO mice; such a phenotype may be caused by increased energy expenditure, triggering signals in the brain to increase food intake, or Hsp20-KO causes leptin resistance, which may be attributed to the higher leptin concentration observed in KO mice. Importantly, loss of Hsp20 also results in a significant increase in the thermogenic capacity of iWAT. Moreover, the striking changes in chemokine gene expression in iWAT suggest that signaling from Hsp20 could serve as a trigger of immune cell chemo-attraction. Hence, the convergence of lipogenesis and thermogenesis identified here not only suggests an angle for targeting metabolic diseases but also provides a perspective for understanding basic adipose tissue biology.
Despite increased adiposity, Hsp20-KO mice demonstrate improved glucose tolerance and enhanced systemic insulin sensitivity. Although the animals studied here have a global Hsp20 deletion, our ex vivo and in vivo results demonstrate that Hsp20 activity in adipose tissues contributes significantly to that phenotype. First, the gene expression changes in adipose tissue of KO mice were cell-autonomous, including enhanced thermogenic capacity and a decreased inflammatory response, both of which might contribute to the increased insulin sensitivity observed in KO mice. Indeed, iWAT transplantation demonstrates improved glucose tolerance and increased insulin sensitivity in recipient mice (Tran et al., 2008). Regarding the inflammatory response, it has been widely reported that chronic tissue inflammation can be a major cause of insulin resistance and that macrophage-mediated pro-inflammatory effects are an underlying mechanism (Donath and Shoelson, 2011). Here we show that the expression of pro-inflammatory genes in iWAT was reduced in KO mice, and this was accompanied by decreased numbers of pro-inflammatory M1 macrophages as well as by increased numbers of M2 macrophages in iWAT. Interestingly, one of the important chemokines released by adipose tissue is MCP-1 (CCL2), and PPARγ activation is known to inhibit adipose tissue MCP-1 expression and secretion (Nguyen et al., 2012). We found decreased levels of MCP-1 in adipose tissue from KO mice, consistent with the activated PPARγ phenotype; this could provide an explanation for the reduced inflammation. Second, minimal differences in these key metabolic pathways were observed in other metabolically active organs (liver and skeletal muscle) in the same time frame (data not shown). These data strongly suggest that the phenotypes seen in WAT are unlikely to be secondary to those organs. Of note, the lipokine C16:1n7-palmitoleate (Cao et al., 2008) was also found to be upregulated in adipose tissue of KO mice, which could contribute to the phenotype exhibited by KO mice, considering its role in systemic metabolic regulation.
PPARγ primarily functions as a master regulator of adipogenesis and is a critical factor associated with various metabolic diseases (Tontonoz et al., 1994). Subsequent studies established that PPARγ activation by rosiglitazone also drives browning of WAT (Petrovicet al., 2010). Here we demonstrate that the contribution of Hsp20 activity to lipogenesis and thermogenesis is mediated by PPARγ. Furthermore, we elucidate that Hsp20 modulates the ubiquitination of PPARγ via an unreported E3 ligase, FBXO4, as assessed with a series of co-immunoprecipitations combined with loss-of-function strategies. However, although we demonstrated that Hsp20 could interact with FBXO4 (Figure 6A), the exact mechanism of how Hsp20 regulates FBXO4 is not investigated in the current study. Given that FBXO4-mediated ubiquitination requires phosphorylation of FBXO4 by GSK3β (Lin et al., 2006), which is positively regulated by Hsp20 (Fan et al., 2008), it is very likely that Hsp20 activates GSK3β and, subsequently, phosphorylates and activates FBXO4.
Critically, the regulation of PPARγ by Hsp20 is clearly endorsed by the increased efficacy of the PPARγ agonist rosiglitazone in improving whole-body metabolic control in KO mice, an improvement supported by changes in gene expression that are consistent with enhanced PPARγ activity specifically in iWAT. Interestingly, mice with adipocyte-specific overexpression of Hsp20 do not exhibit reduced fat mass, even during an HFD challenge, as expected from the phenotype displayed by KO mice. This apparent discrepancy implies that the relatively low endogenous levels of Hsp20 detected in adipocytes of WT mice are sufficient to modulate all Hsp20-dependent adipocyte functions. On the other hand, the low levels of Hsp20 in adipocytes possess great potential for pharmacological inhibition using doses that are otherwise subthreshold for other tissues.
In summary, this study unveils an interesting molecular mechanism whereby an Hsp20-FBXO4-ubiquitin-dependent pathway controls adipocyte function by restraining PPARγ activity in response to β-adrenergic signaling. Thus, drugs able to achieve meaningful suppression of Hsp20 activity in tissues with low Hsp20 expression, such as WAT, may exhibit a beneficial therapeutic index for the treatment of type 2 diabetes when administered either alone or in combination with PPARγ or adrenergic agonists.
EXPERIMENTAL PROCEDURES
Mice
Procedures involving mice were approved by the Institutional Animal Care and Use Committee (IACUC) of the University of Cincinnati and were conducted according to NIH guidelines. Hsp20-KO mice and adipose tissue-specific overexpression mice were generated by the Division of Developmental Biology at Cincinnati Children’s Hospital Medical Center. Male C57BL/6J mice and FVB/NJ mice were purchased from The Jackson Laboratory and allowed to acclimate in the animal facility for 2 weeks before being enrolled in studies. Unless specified, male mice at age 8–10 weeks were used for experiments. Mice were housed under a 12-hr light-dark cycle at constant temperature (23°C) and given regular CD (7022, Envigo).
Indirect Calorimetry
Micewere housed in chamberswith integrated control ofambient temperature and simultaneous measurement offood intake, locomotoractivity, and energy expenditure by indirect calorimetry (TSE Systems, Chesterfield, MO, USA). Mice were monitored at 23°C or 29°C to compare energy balance at standard room temperature or at thermoneutrality, respectively.
Lipidomics Assay
The lipidomics assay was performed by the Lipidomics Research Program at the University of Cincinnati. For details regarding the lipidomics assay, see the Supplemental Experimental Procedures.
Statistics
Student’s t test was used for single comparisons. Two-way ANOVA (repeated measurements) was used for IPGTT and IPITT. The correlation between energy expenditure, oxygen consumption, and carbon dioxide production and lean mass for WT and KO mice was analyzed by linear regression to assess intercept using GraphPad Prism 7 for Mac (GraphPad, La Jolla, California, USA). Analysis of fatty acid methyl esters was based on areas calculated with Shimadzu Class VP 4.3 software, and data are expressed as weight percent of total fatty acids (mg fatty acid/100 mg fatty acids). Unless specified, data are represented as mean ± SEM. *p < 0.05 and not significant (n.s.) p > 0.05.
Supplementary Material
Highlights.
Hsp20 knockout mice demonstrate enhanced thermogenic capacity
Ablation of Hsp20 increases adiposity and improves glucose homeostasis
PPARγ accumulation is dependent on decreased FBOX4 activity upon Hsp20 deletion
Hsp20 knockout mice show increased responsiveness to rosiglitazone
ACKNOWLEDGMENTS
The authors gratefully acknowledge that the plasmid containing the 5.4-kb adiponectin promoter was a gift from Prof. Philipp E. Scherer (University of Texas Southwestern Medical Center). We thank Drs. Yueh-Chiang Hu, Yinhuai Chen, and Alexandra Dias for helpful discussions and technical advice. This work was funded by University of Cincinnati funds (to D.P.-T.), an American Heart Association (AHA) Established Investigator Award (17EIA33400063), and NIH grants HL-087861 and GM-112930 (to G.-C.F.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and seven figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.05.065.
DATA AND SOFTWARE AVAILABILITY
The accession number for the RNA-seq data reported in this paper is GEO: GSE94436.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Ahmadian M, Suh JM, Hah N, Liddle C, Atkins AR, Downes M, and Evans RM (2013). PPARγ signaling and metabolism: the good, the bad and the future. Nat. Med 19, 557–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartelt A, Bruns OT, Reimer R, Hohenberg H, Ittrich H, Peldschus K, Kaul MG, Tromsdorf UI, Weller H, Waurisch C, et al. (2011). Brown adipose tissue activity controls triglyceride clearance. Nat. Med 17, 200–205. [DOI] [PubMed] [Google Scholar]
- Cao Y (2013). Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab. 18, 478–489. [DOI] [PubMed] [Google Scholar]
- Cao H, Gerhold K, Mayers JR, Wiest MM, Watkins SM, and Hotamisligil GS (2008). Identification of a lipokine, a lipid hormone linking adipose tissue to systemic metabolism. Cell 134, 933–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JH, Banks AS, Estall JL, Kajimura S, Boström P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Blüher M, et al. (2010). Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature 466, 451–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig EA, Gambill BD, and Nelson RJ (1993). Heat shock proteins: molecular chaperones of protein biogenesis. Microbiol. Rev 57, 402–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng Y-H, Doria A, et al. (2009). Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med 360, 1509–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLany JP, Floyd ZE, Zvonic S, Smith A, Gravois A, Reiners E, Wu X, Kilroy G, Lefevre M, and Gimble JM (2005). Proteomic analysis of primary cultures of human adipose-derived stem cells: modulation by Adipogenesis. Mol. Cell. Proteomics 4, 731–740. [DOI] [PubMed] [Google Scholar]
- Donath MY, and Shoelson SE (2011). Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol 11, 98–107. [DOI] [PubMed] [Google Scholar]
- Dutchak PA, Katafuchi T, Bookout AL, Choi JH, Yu RT, Mangelsdorf DJ, and Kliewer SA (2012). Fibroblast growth factor-21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones. Cell 148, 556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan GC, Zhou X, Wang X, Song G, Qian J, Nicolaou P, Chen G, Ren X, and Kranias EG (2008). Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ. Res 103, 1270–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorenko A, Lishko PV, and Kirichok Y (2012). Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151, 400–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feldmann HM, Golozoubova V, Cannon B, and Nedergaard J (2009). UCP1 ablation induces obesity and abolishes diet-induced thermogenesis in mice exempt from thermal stress by living at thermoneutrality. Cell Metab. 9, 203–209. [DOI] [PubMed] [Google Scholar]
- Hu E, Kim JB, Sarraf P, and Spiegelman BM (1996). Inhibition of adipogenesis through MAP kinase-mediated phosphorylation of PPARγ. Science 274, 2100–2103. [DOI] [PubMed] [Google Scholar]
- Kajimura S, Seale P, Kubota K, Lunsford E, Frangioni JV, Gygi SP, and Spiegelman BM (2009). Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 460, 1154–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimura S, Spiegelman BM, and Seale P (2015). Brown and beige fat: Physiological roles beyond heat generation. Cell Metab. 22, 546–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilroy G, Kirk-Ballard H, Carter LE, and Floyd ZE (2012). The ubiquitin ligase Siah2 regulates PPARγ activity in adipocytes. Endocrinology 153, 1206–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J-H, Park KW, Lee E-W, Jang W-S, Seo J, Shin S, Hwang KA, and Song J (2014). Suppression of PPARγ through MKRN1-mediated ubiquitination and degradation prevents adipocyte differentiation. Cell Death Differ. 27, 594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KW, Kwak SH, Koo YD, Cho Y-K, Lee HM, Jung HS, Cho YM, Park YJ, Chung SS, and Park KS (2016). F-box only protein 9 is an E3 ubiquitin ligase of PPARγ. Exp. Mol. Med 48, e234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DI, Barbash O, Kumar KGS, Weber JD, Harper JW, Klein-Szanto AJP, Rustgi A, Fuchs SY, and Diehl JA (2006). Phosphorylation-dependent ubiquitination of cyclin D1 by the SCF(FBX4-alphaB crystallin) complex. Mol. Cell 24, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindgren EM, Nielsen R, Petrovic N, Jacobsson A, Mandrup S, Cannon B, and Nedergaard J (2004). Noradrenaline represses PPAR (peroxisomeproliferator-activated receptor) gamma2 gene expression in brown adipocytes: intracellular signalling and effects on PPARgamma2 and PPARgamma1 protein levels. Biochem. J 382, 597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MTA, Chen A, Lu WJ, Fan W, Li PP, Oh DY, and Patsouris D (2012). Regulation of chemokine and chemokine receptor expression by PPARγ in adipocytes and macrophages. PLoS ONE 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peirce V, Carobbio S, and Vidal-Puig A (2014). The different shades of fat. Nature 510, 76–83. [DOI] [PubMed] [Google Scholar]
- Petrovic N, Walden TB, Shabalina IG, Timmons JA, Cannon B, and Nedergaard J (2010). Chronic peroxisome proliferator-activated receptorγ (PPARgamma) activation of epididymally derived white adipocyte cultures reveals a population of thermogenically competent, UCP1-containing adipocytes molecularly distinct from classic brown adipocytes. J. Biol. Chem 285, 7153–7164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, Rosenbaum M, Zhao Y, Gu W, Farmer SR, and Accili D (2012). Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell 150, 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen ED, and Spiegelman BM (2014). What we talk about when we talk about fat. Cell 156, 20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tontonoz P, Hu E, and Spiegelman BM (1994). Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 79, 1147–1156. [DOI] [PubMed] [Google Scholar]
- Tran TT, Yamamoto Y, Gesta S, and Kahn CR (2008). Beneficial effects of subcutaneous fat transplantation on metabolism. Cell Metab. 7, 410–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gu H, Huang W, Peng J, Li Y, Yang L, Qin D, Essandoh K, Wang Y, Peng T, and Fan GC (2016). Hsp20-mediated activation of exosome biogenesis in cardiomyocytes improves cardiac function and angiogenesis in diabetic mice. Diabetes 65, 3111–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe M, Takahashi H, Saeki Y, Ozaki T, Itoh S, Suzuki M, Mizushima W, Tanaka K, and Hatakeyama S (2015). The E3 ubiquitin ligase TRIM23 regulates adipocyte differentiation via stabilization of the adipogenic activator PPARγ. eLife 4, e05615. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.