

The invention of methods for the streamlined construction of small organic molecules is essential to the advancement of organic chemistry. Given the ubiquity of C–N bonds in biologically active molecules, improved methods for the synthesis of amines is an important challenge.1 The difunctionalization of olefins provides direct access to complex molecular frameworks that otherwise require multiple synthetic operations to assemble. The carboamination of alkenes is an important subset of such reactions, resulting in concurrent introduction of carbon and nitrogen functionalities into accessible alkene frameworks.2
To date, most carboamination methods have exploited a two-component approach, delivering cyclic products from preassembled starting materials.2a–2g As a consequence of their two-component natures, these methods are limited in their modularities. To address this, we envisioned a three-component alkene carboamination could be realized by harnessing the reactivities and selectivities of radical intermediates.3 We became interested in the proclivity of copper salts to generate electrophilic alkyl radicals from functionalized tertiary alkyl halides, such as α-halocarbonyl compounds.4 We reasoned that combination of such electrophiles with nucleophilic amines in the presence of an olefin could deliver the carboamination product in a regio- and chemo-selective fashion (Scheme 1a). These electrophiles are resistant to undesirable SN1/SN2 reactivity, reducing the tendency to couple this electrophilic component directly with the amine nucleophile. Furthermore, addition of these radicals to olefins is well-documented in related methodologies and in atom transfer radical polymerization (ATRP).5,6
Scheme 1.

Three-Component Carboamination Reactions
Concurrent with our investigations, two radical couplings utilizing alkyl nitriles2h and dialkyl peroxides,2i,2j were reported (Scheme 1b). The philicities of the radicals generated in these reactions significantly influence the scope of the transformations, wherein favorable matching of the alkene and alkyl radical polarities is essential.7 Efficient reactivity is accomplished by employing electronically activated alkenes and further enhanced with superstoichiometric loadings of the other coupling partners and of strong oxidants. An alternative approach to the three-component carboamination of alkenes via Pd-catalysis was also reported.2k A preinstalled bidentate directing group is utilized to avoid β-hydride elimination and to promote oxidation, thereby enabling difunctionalization.
In the context of our mechanistic hypothesis, oxidation of a radical addition intermediate (I) would afford a cationic species. Typically, such oxidations require electronically activated alkenes, which facilitate oxidation of the resultant radical addition intermediate.2h–2j Although this tactic enables the desired transformation, it imposes significant limitations on the substrate scope. We reasoned use of an α-halocarbonyl compound as the electrophilic source of carbon would reveal access to a geometric relationship between the incident carbonyl group and the newly formed alkyl radical that could assist with oxidation of this intermediate.8 Such an oxidation event would afford an oxocarbenium ion intermediate (II), which could undergo opening via nucleophilic attack by the amine to furnish the carboamination product.9 Through this assisted oxidation, we envisioned a platform for olefin carboamination. Indeed, we report herein a carboamination reaction of unprecedented generality.
Experimentation began by exploring the reactivity of ethyl α-bromo isobutyrate (1), styrene, and N-methylaniline as model reaction components in the presence of various Cu catalysts. Product 2a could be formed under a variety of conditions, with the optimized parameters as follows: 5.0 mol % Cu(OTf)2, 5.0 mol % 2,2′-bipyridine (bpy), 1.1 equiv K3PO4, 2.0 equiv 1, 1.0 equiv styrene, 1.0 equiv N-methylaniline in DCE (0.50 M) at 80 °C for 24 h under N2. Product 2a can be obtained in an 87% isolated yield on 0.20 mmol scale (see Tables S1–S10 for optimization studies). The reaction is scalable, providing 86% and 95% yields on 1.0 and 10 mmol scales, respectively.
Under the optimized conditions, the carboamination of a variety of vinylarenes with 1 and various amines can be accomplished (Table 1). Table S11 provides a summary of conditions for all substrate combinations. Depending on the degree of substitution of the nucleophile and of the alkene substrate employed, either acyclic or cyclic products can be obtained. Notably, electron poor styrenes, which are unreactive in related carboamination methods,(2h–2j) are suitable substrates for this reaction (2b–d). Electron-neutral and -rich alkenes are also excellent substrates, providing the carboaminated products in good to excellent yields (2a, 2e–h, 2l). Steric hindrance proximal to the alkene does not significantly affect reactivity (2e–h). In cases where reactivity of the alkene substrate is low, it was generally found that use of a mild excess of this reagent leads to improved yields, such as with 2c.
Table 1.
Scope of Vinylarenes and Aminesa
 |
1 (2.0 equiv), alkene (1.0 equiv), amine (1.0 equiv), K3PO4 (1.1 equiv), Cu(OTf)2 (5 mol %), bpy (5 mol %), DCE (0.50 M), 80 °C.
With 3.0 equiv alkene.
This carboamination reaction tolerates a range of functionalities on both the alkene and amine substrates, retaining handles for further functionalizations of the products. Vinylarenes bearing pendant aliphatic alkenes undergo carboamination in a site-selective manner, leaving the aliphatic olefin intact (2e–h). Functionalities such as trifluoromethyl groups (2b), nitro groups (2c, 2i, 2n), halides (2d, 2j, 2m), ethers (2e–f, 2l, 2p–q), and heterocycles (2j, 2l, 2p) are also tolerated under the reaction conditions.
Several classes of functionalized bromides can be used in addition to esters (Table 2): imides (2r), amides (2s), ketones (2t), malonates (2w), and sulfones (2z–aa) provide modest to good yields of the products. The Weinreb amide-type electrophile produces 2s in good yield, retaining the amide moiety for facile elaboration of the carbonyl group. In addition to methyl-bearing esters, those substituted with fluorine atoms (2u–v) are effective for this transformation, providing access to products of pharmacokinetic value given the unique properties of the difluoromethyl moiety.10 A differentially substituted electrophile was also reactive (2×), providing the product in good yield with moderate diastereoselectivity.
Table 2.
Scope of Electrophilesa
 |
Importantly, secondary electrophiles are also amenable to the three-component coupling (2w, 2y–aa) including a malonate (2w), which provides access to products containing a free methylene unit, following monodecarboxylation.11,12 Given this generality, products with any degree of substitution at the α-carbon can be accessed. The diastereoselectivity with secondary α-bromoesters is moderate (2y, 2:1), but the analogous secondary sulfone affords the product in high d.r. (2z–aa, 10:1). The additional steric encumbrance in the corresponding sulfoxonium intermediate may be responsible for enhanced diastereoselectivity.13 It was found that each electrophile class requires a different polyamine ligand to achieve significant reactivity. We attribute this phenomenon to the unique range of reduction potentials of each electrophile class and oxidation potentials of the corresponding radical addition intermediates. Although more reducing catalysts may rapidly activate an electrophile, they may be slow to oxidize the radical intermediate. Thus, a common Cu catalyst of fixed redox reactivity does not achieve high reactivity across electrophile classes.6a Ongoing work in our laboratory aims to understand these ligand effects and to identify a general Cu catalyst for this transformation.
The scope of the amine component in this carboamination reaction is broad. In addition to N-methylaniline, secondary arylamine nucleophiles bearing electron-withdrawing (2i, 2m, 2n) and donating (2v, 2z–aa) groups perform well in the reaction. Products derived from electron-neutral and rich primary nucleophiles generally close in situ to form γ-lactams (2d, 2o–p, 2×), whereas those derived from electron withdrawn primary nucleophiles remain acyclic (2m–n). Products 2fand 2h indicate steric properties of the alkene may also influence whether in situ closure occurs. Diaryl and heterocyclic amines (2j–l) also react smoothly to give the corresponding products in good yield. Arylamines are excellent substrates in the reaction; however, primary alkylamines also perform well (2o–p). Benzophenone imine is also reactive, providing access to the corresponding N–H γ-lactam following acidic deprotection (2q, see SI).14
To expand reaction scope, we focused on enabling use of internal and aliphatic alkenes as substrates. These represent challenging substrate classes, as the radical additions are significantly slower compared to terminal vinylarenes.7 Aliphatic alkenes are challenging, as subsequent oxidation is less favorable. However, we hypothesized the oxocarbenium intermediate could promote this key step. Indeed, only slightly modified reaction conditions elicit reactivity from these olefin classes (Table 3). Typically, higher temperatures and loadings of the alkene are required to consume the amine nucleophile. Under these reaction conditions, unfunctionalized α-olefins can be employed with aryl- and alkyl-amine nucleophiles (2ab–ac). Functional groups sensitive to basic conditions are tolerated, including primary alkyl bromides (2ad), terminal epoxides (2ae–af), ketones (2ag), and acetoxy groups (2ah). 1,1-Disubstituted alkenes are also viable components, enabling formation of highly hindered C–N bonds (2ai–aj). An exocyclic 1,1-disubstituted olefin produced the corresponding spirocyclic lactam with good yield and d.r. (2aj, 77%, 10:1 d.r.). Finally, cyclopropyl substituted aliphatic alkenes can be used to form the ζ-amino carbonyl products in moderate yields (2ak–al).
Table 3.
Scope of Aliphatic and Internal Alkenesa
 |
See SI for experimental details.
1:1 d.r.
10:1 d.r.
>20:1 d.r.
From (Z) alkene.
From (E) alkene.
In addition to terminal alkenes, more hindered internal alkenes readily participate in the reaction, providing highly substituted products in good yields and with excellent d.r. (2am–ao). Cyclic internal alkenes afford the trans carboamination product in >20:1 d.r. (2am), whereas acyclic internal alkenes provide the cis product in >20:1 d.r. (2an–ao). For the latter, the reactions are diastereoconvergent; regardless of starting configuration of the alkene, the cis γ-lactam product is obtained selectively. Given this scope, substitution at each position on the lactam core is easily controlled. Historically, such substituted γ-lactams require many synthetic operations to assemble; however, this protocol provides access to a customizable γ-lactam core in a single step from readily obtainable, inexpensive materials.1a
In an effort to understand the mechanism of this transformation a series of radical clock experiments were conducted to probe the fate of the radical addition intermediate. A cyclopropyl moiety adjacent to an olefin (3) was found to open under the reaction conditions, providing the corresponding carboamination product (2al) in 50% isolated yield (eq 1).
 |
(1) |
When N,N-diallylaniline (4) was subjected to the reaction conditions, 5-exo-trig cyclization was observed, affording the carbobromination product (2ap) in 69% isolated yield and 1.5:1 d.r. instead of the expected carboamination product (2ap′, eq 2).15 Our proposal invokes oxidation of radical addition intermediates to form an oxocarbenium species. We propose formation of this intermediate facilitates this key step in cases where this oxidation is more difficult, such as with aliphatic alkenes or electron poor vinylarenes.16 For both radical clock experiments, either ring-opening or radical cyclization occurs, making the oxocarbenium ion geometrically difficult to form. For 3, the radical is benzylic and oxidation to the cation is facile, even without the intermediacy of the oxocarbenium ion; thus, carboamination can still occur. However, for 4, unassisted oxidation of the radical intermediate would produce an unstable primary carbocation. Consequently, the rate of bromine atom transfer from Cu becomes competitive with the productive reaction pathway. Products 2ad and 2ap eliminate the possibility of an atom-transfer radical addition mechanism followed by SN2 amination.
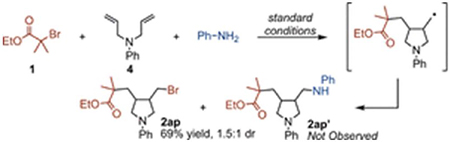 |
(2) |
These radical trap experiments demonstrate the importance of the carbonyl moiety to enable the carboamination of less reactive substrates. However, results observed with 3 indicate carboamination of vinylarene substrates could occur without proceeding through an oxocarbenium ion intermediate. Therefore, the viability of this intermediate was further established by a thorough consideration of the stereochemical outcomes from the internal alkene substrates. For 2am, the system is conformationally locked, and opening of the purported oxocarbenium would deliver only the trans product (eq 3).
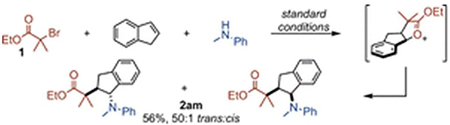 |
(3) |
For the carboamination of β-methylstyrene, the system may rotate freely, and the thermodynamically more stable trans oxocarbenium structure may form. Stereoinvertive opening of the oxocarbenium followed by lactamization delivers selectively the cis product 2ao (eq 4). Together, these probes indicate oxocarbenium ion intermediates form when geometrically permitted, though are not required for product formation if the radical addition intermediate is oxidizable by Cu(II), as is the case for the benzylic radical preceding the formation of 2al (eq 1).8,17
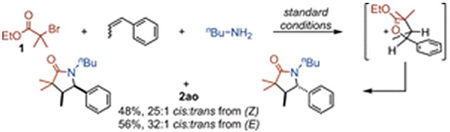 |
(4) |
In conclusion, we developed a three-component carboamination reaction that couples readily available alkenes, functionalized alkyl halides, and amines. Yields are good to excellent, and scope of the reaction in all three components is broad. Olefin classes traditionally challenging to functionalize through transition metal catalysis are exceptionally reactive in this system, which we attribute to the intermediacy of an oxocarbenium species. The molecules constructed through this method represent examples of the potential of base metal catalysis to enable the rapid difunctionalization of olefins. Future work will focus on developing diastereoselective conditions for differentially substituted electrophiles, expanding nucleophile scope, and developing an asymmetric protocol.
Supplementary Material
Acknowledgment
The authors thank the NIH (1R35GM125029), the Sloan Research Foundation, and the University of Illinois for their generous support of this work.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.7b10529.
• NMR files (ZIP)
• Experimental procedures and spectra (PDF)
The authors declare no competing financial interest.
References
- (1).(a) Caruano J; Muccioli GG; Robiette R Org. Biomol. Chem 2016, 14, 10134–10156. [DOI] [PubMed] [Google Scholar]; (b) Carey JS; Laffan D; Thomson C; Williams MT Org. Biomol. Chem 2006, 4, 2337–2347. [DOI] [PubMed] [Google Scholar]; (c) Vitaku E; Smith DT; Njardarson JT J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (d) Roughley SD; Jordan AM J. Med. Chem 2011, 54, 3451–3479. [DOI] [PubMed] [Google Scholar]
- (2).For reviews, see: Wolfe JP Top. Heterocycl. Chem 2013, 32, 1–37.Garlets ZJ; White DR; Wolfe JP Asian J. Org. Chem 2017, 6, 636–653For relevant methods, see:. Ney JE; Wolfe JP Angew. Chem., Int. Ed 2004, 43, 3605–3608.White DR; Hutt JT; Wolfe JP J. Am. Chem. Soc 2015, 137, 11246–11249.Zeng W; Chemler SR J. Am. Chem. Soc 2007, 129, 12948–12949.Liwosz TW; Chemler SR J. Am. Chem. Soc 2012, 134, 2020–2023.Piou T; Rovis T Nature 2015, 527, 86–90.Liu Y-Y; Yang X-H; Song R-J; Luo S; Li J-H Nat. Commun 2017, 8, 14720–14725.Qian B; Chen S; Wang T; Zhang X; Bao HJ Am. Chem. Soc 2017, 139, 13076–13082.Zhu N; Wang T; Ge L; Li Y; Zhang X; Bao H Org. Lett 2017, 19, 4718–4721.Liu Z; Wang Y; Wang Z; Zeng T; Liu P; Engle KM J. Am. Chem. Soc 2017, 139, 11261–11270.Choi GJ; Knowles RR J. Am. Chem. Soc 2015, 137, 9226–9229.Wang D; Wu L; Wang F; Wan X; Chen P; Lin Z; Liu GJ Am. Chem. Soc 2017, 139, 6811–6814.Um C; Chemler SR Org. Lett 2016, 18, 2515–2518.
- (3).For general books and reviews, see: Radicals in Organic Synthesis; Renaud P, Sibi MP, Eds.; Wiley-VCH: New York, 2001.Giese B Radicals in Organic Synthesis: Formation of Carbon−Carbon Bonds; Pergamon: Oxford, 1986.Hart DJ Science 1984, 223, 883–887.Sibi MP; Manyem S; Zimmerman J Chem. Rev 2003, 103, 3263–3295.Zimmermann J; Sibi MP Top. Curr. Chem 2006, 263, 107–162.Hoffmann RW Chem. Soc. Rev 2016, 45, 577–583.Matyjaszewski K; Xia J Chem. Rev 2001, 101, 2921–2990.Tang S; Liu K; Liu C; Lei A Chem. Soc. Rev 2015, 44, 1070–1082For redox catalysis involving radicals, see:. Studer A; Curran DP Angew. Chem., Int. Ed 2016, 55, 58–102.Romero NA; Nicewicz DA Chem. Rev 2016, 116, 10075–10166.Prier CK; Rankic DA; MacMillan DW C. Chem. Rev 2013, 113, 5322–5363.Shaw MH; Twilton J; MacMillan DW C. J. Org. Chem 2016, 81, 6898–6926.Skubi KL; Blum TR; Yoon TP Chem. Rev 2016, 116, 10035–10074.Narayanam JMR; Stephenson CR J. Chem. Soc. Rev 2011, 40, 102–113.Tucker JW; Stephenson CR J. J. Org. Chem 2012, 77, 1617–1622.Douglas JJ; Sevrin MJ; Stephenson CR J. Org. Process Res. Dev 2016, 20, 1134–1147For powerful approaches to alkene functionalization via radical additions, see:. Margrey KA; Nicewicz DA Acc. Chem. Res 2016, 49, 1997–2006.Gentry EC; Knowles RR Acc. Chem. Res 2016, 49, 1546–1556For carboamination reactions involving aryl radicals from aryl diazonium precursors, see:. Kindt S; Wicht K; Heinrich MR Org. Lett 2015, 17, 6122–6125.Blank O; Wetzel A; Ullrich D; Heinrich MR Eur. J. Org. Chem 2008, 2008, 3179–3189.Citterio A; Minisci F; Albinati A; Bruckner S Tetrahedron Lett 1980, 21, 2909–2910.Hari DP; Hering T; König B Angew. Chem., Int. Ed 2014, 53, 725–728.Fehler SK; Heinrich MR Synlett 2015, 26, 580–603.
- (4).(a) Chen X; Liu X; Mohr JT J. Am. Chem. Soc 2016, 138, 6364–6367. [DOI] [PubMed] [Google Scholar]; (b) Xu T; Hu X Angew. Chem., Int. Ed 2015, 54, 1307–1311. [DOI] [PubMed] [Google Scholar]; (c) Fisher DJ; Burnett GL; Velasco R; Alaniz JR J. Am. Chem. Soc 2015, 137, 11614–11617. [DOI] [PubMed] [Google Scholar]; (d) Gietter AAS; Gildner PG; Cinderella AP; Watson DA Org. Lett 2014, 16, 3166–3169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Nishikata T; Noda Y; Fujimoto R; Sakashita T J. Am. Chem. Soc 2013, 135, 16372–16375. [DOI] [PubMed] [Google Scholar]; (b) Tang S; Liu K; Liu C; Lei A Chem. Soc. Rev 2015, 44, 1070–1082. [DOI] [PubMed] [Google Scholar]
- (6).(a) Matyjaszewski K Macromolecules 2012, 45, 4015–4039. [Google Scholar]; (b) Soejima T; Satoh K; Kamigaito MJ Am. Chem. Soc 2016, 138, 944–954. [DOI] [PubMed] [Google Scholar]
- (7).(a) Fischer H; Radom L Angew. Chem., Int. Ed 2001, 40, 1340–1371. [DOI] [PubMed] [Google Scholar]; (b) Giese B; He J; Mehl W Chem. Ber 1988, 121, 2063–2066. [Google Scholar]
- (8).Kochi JK; Bemis A; Jenkins CL J. Am. Chem. Soc 1968, 90, 4616–4625. [Google Scholar]
- (9).Formation of analogous intermediates was described by Kochi in the analysis of the mechanism of oxidative substitution catalyzed by Cu(II).8 Kochi found through deuterium labeling experiments α- and β-carbons of homobenzylic radicals scrambled upon oxidation by Cu(II), ostensibly due to intermediacy of a symmetric, cationic intermediate that undergoes nucleophilic attack by an exogeneous nucleophile.
- (10).Gillis EP; Eastman KJ; Hill MD; Donnelly DJ; Meanwell NA J. Med. Chem 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- (11).Krapcho AP; Glynn GA; Grenon BJ Tetrahedron Lett 1967, 8, 215–217. [DOI] [PubMed] [Google Scholar]
- (12).Lee A; Michrowska A; Sulzer-Mosse S; List B Angew. Chem., Int. Ed 2011, 50, 1707–1710. [DOI] [PubMed] [Google Scholar]
- (13).Lucchini V; Modena G; Pasquato LJ Chem. Soc., Chem. Commun 1992, 293–294. [Google Scholar]
- (14).(a) Wuts PGM; Greene TW Protection for the Amino Group. In Greene’s Protective Groups in Organic Synthesis; Wiley-VCH: New York, 2014. [Google Scholar]; (b) Zhu Y; Buchwald SL J. Am. Chem. Soc 2014, 136, 4500–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Clark AJ Eur. J. Org. Chem 2016, 2016, 2231–2243. [Google Scholar]
- (16).(a) Larsen CH; Ridgway BH; Shaw JT; Smith DM; Woerpel KA J. Am. Chem. Soc 2005, 127, 10879–10884. [DOI] [PubMed] [Google Scholar]; (b) Larsen CH; Ridgway BH; Shaw JT; Woerpel KA J. Am. Chem. Soc 1999, 121, 12208–12209. [Google Scholar]; (c) Huan F; Chen Q-Y; Guo YJ Org. Chem 2016, 81, 7051–7063. [DOI] [PubMed] [Google Scholar]
- (17).For additional mechanistic information and a proposed catalytic cycle, see Supporting Information.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.