

Abstract
In recent years the synthesis of amines and other nitrogen containing motifs has been a major area of research in organic chemistry due to their being widely represented in biologically active molecules. Current strategies rely on a multistep approach and require one reactant to be activated prior to the carbon-nitrogen bond formation. This leads to reaction inefficiency and functional group intolerance. As such, a general approach to the synthesis of nitrogen-containing compounds from readily available and benign starting materials is highly desirable. Here we present a Pd-catalyzed oxidative amination reaction, where the addition of the nitrogen occurs at the less substituted carbon of a double bond, in what is known as anti-Markovnikov selectivity. Alkenes are shown to react with imides in the presence of a palladate catalyst to generate the terminal imide via trans-aminopalladation. Subsequently, olefin isomerization occurs to afford the thermodynamically favored products. Both the scope of the transformation and mechanistic investigations are reported.
Alkene amination reactions, including hydro- and oxidative amination, are atom-economical approaches to the synthesis of ubiquitous C–N bonds.1–7 Oxidative amination, also known as the aza-Wacker oxidation, differs from hydroamination reactions in that it retains the degree of unsaturation in the product, allowing for further elaboration of this functionality.3–7 As terminal amines are prevalent in pharmaceuticals, especially distal to polar functionalities,8 the development of an anti-Markovnikov selective aza-Wacker oxidation of unactivated alkenes would represent a significant advance. Such a process would constitute a novel approach to the remote, anti-Markovnikov amination of organic molecules, and represent a new, powerful disconnection in organic synthesis. Additionally, when performed aerobically, this transformation would couple two easily accessible starting materials and would generate only an equivalent of H2O as waste.
The primary challenge in generating the anti-Markovnikov product is biasing the aminometalation step to afford the branched [M]–C and terminal N–C bonds. Due to both steric repulsion and the inherent electronic bias of nucleophile addition to alkenes, known as Markovnikov’s rule, this selectivity is not generally favored for an intermolecular aminometalation.6,7 However, several strategies have been successfully employed to reverse this inherent selectivity. One approach uses activated alkenes that can form π-benzyl or [M]-enolate intermediates (Figure 1a).9–16 A second approach utilizes an allylic C–H activation followed by nucleophilic attack at the terminal carbon on the resulting π-allyl.17–20 Alternatively, proximal Lewis basic groups can direct functionalization of the alkene to afford the favored metallacycle (Figure 1b).21–27 Finally, in stoichiometric investigations, the combination of a palladate complex and sterically hindered amine nucleophiles, promoted a trans-aminopalladation to functionalize the terminal carbon and produce the anti-Markovnikov constitutional isomer (Figure 1c).28,29 However, conditions that afford the anti-Markovnikov aza-Wacker product, with simple aliphatic alkenes, have not been reported in catalytic amination reactions.
Figure 1: Current strategies for anti-Markovnikov oxidative amination of simple alkenes.
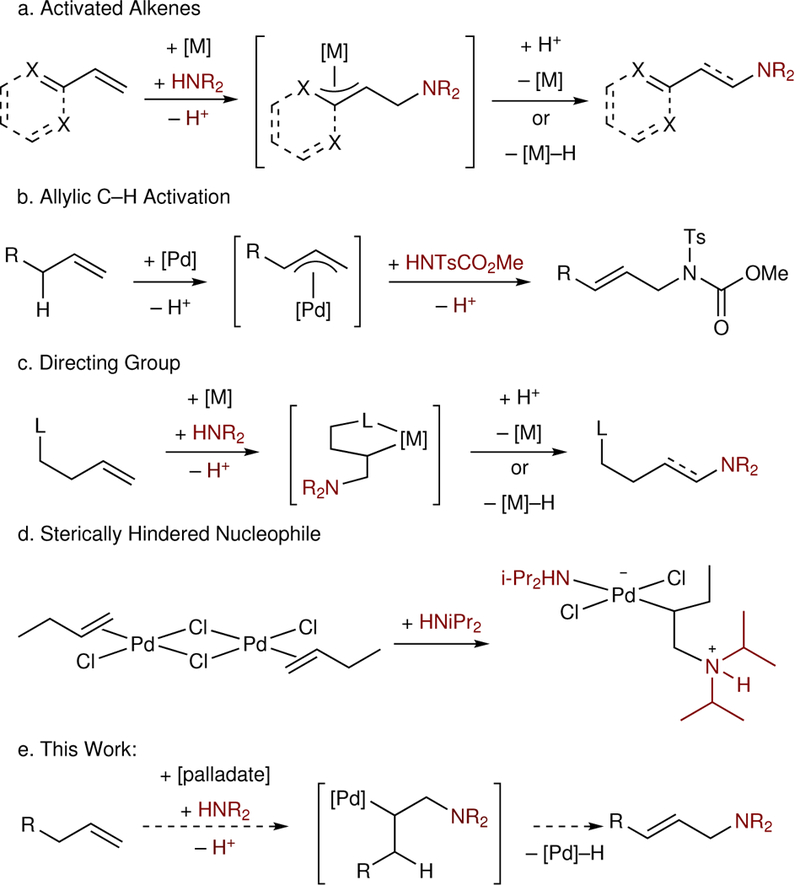
a, Use of activated alkenes gives rise to stabilized alkyl-Pd intermediates, providing a basis for anti-Markovnikov selectivity. b, Allylic C–H bond activation can afford a π-allyl intermediate, which can be intercepted by a nucleophile to afford allylic amines. c, A tethered directing group can direct functionalization at the terminal position through formation of a more stable metallacyclic intermediate. d, Use of a palladate catalyst with sterically encumbered nucleophiles can kinetically favor anti-Markovnikov functionalization in stoichiometric studies and in the present work.
We envisioned utilizing a palladate catalyst, which could promote a selective trans-nucleopalladation by saturating the coordination sites with excess halide, as seen in the Wacker oxidation (Figure 1d).7,30 During the trans-aminopalladation step, a relatively large nucleophile would be kinetically biased to approach the alkene to form the less hindered C–N bond, leading to the desired anti-Markovnikov product.28,29,31–33 While the trans-aminopalladation with cationic palladium catalysts are known to occur to afford the Markovnikov constitutional isomer,34,35 we hypothesized that the electronic differentiation between the double bond carbons, which leads to Markovnikov’s rule, would be minimized with a less electrophilic anionic palladate catalyst, leading to a more sterically biased transformation.28 The successful development of an anti-Markovnikov selective aminopalladation of simple alkenes would have implications that reach far beyond the aza-Wacker reaction, as aminopalladation is the regioselectivity-determining step in many olefin difunctionalization reactions.34,35 Thus, the principles we have learned from the studies reported herein could find future applications in other anti-Markovnikov aminofunctionalizations of simple alkenes, enabling single-step access to a wide class of products.36,37
Results and Discussion
Reaction discovery and optimization.
Our initial investigation focused on developing an anti-Markovnikov selective oxidative amination of homoallyl benzene. As seen in Table 1 and further elaborated in the Supplementary Section B, employing known oxidative amination conditions affords the expected Markovnikov isomer exclusively (88% yield).4 However, in the presence of 10 mol % Pd(OAc)2, addition of either 40 mol % Bu4NCl (Table 1, Entry 2) or 20 mol % Bu4NOAc (Table 1, Entry 6) the regioselectivity is reversed to favor the anti-Markovnikov products, in 95% (3.0:1 a-M:M) and 59% (1.3:1 a-M:M) total yields, respectively. The in situ generation of a palladate complex is supported by the independent synthesis and crystallographic characterization of [PdCl4](Bu4N)2 (see Supplementary Section I). The significant increase in reactivity of the Bu4N+ salts, relative to Li+ or Cs+, can be attributed to the increased solubility of the resulting palladate complexes under the reaction conditions. Similarly, no product was observed when Na2PdCl4 or K2PdCl4 were used directly, with or without added acetate sources. Known aza-Wacker conditions typically form the enimide, where the double bond remains unmoved and the nitrogen replaces one of the hydrogen atoms, as the exclusive product; however, using the palladate catalyst, mixtures of olefin isomers are observed with the (E)-styrenyl isomer (2a) as the major product.4,5 When both additives are present, the regioselectivity is improved, affording 86% combined yield (4:1 a-M:M), with a 62% yield of 2a (Table 1, entry 8). Excitingly, further optimization lead to the conditions employed in Table 1, Entry 9, which afforded 2a in 60% GC and 57% isolated yield as a single isomer; the combined yield of all isomers was 75% (8:1 a-M:M) (Table 1, Entry 9).
Table 1.
Optimization of Oxidative Amination Reaction on 1a.
 |
| Entry | Additive (mol %) | Total Yield (%)a | Yield 2a (%)a | a-M/Ma |
|---|---|---|---|---|
| 1 | None | 20 | < 1 | 0.06 |
| 2 | Bu4NCl (40 mol %) | 95 | 68 | 3.0 |
| 3 | LiCl (40 mol %) | 32 | 18 | 1.4 |
| 4 | CsCl (40 mol %) | 13 | 2 | 0.2 |
| 5 | Bu4NI (40 mol %) | 80 | 24 | 2.3 |
| 6 | Bu4NOAc (20 mol %) | 59 | 26 | 1.3 |
| 7 | NaOAc (20 mol %) | 15 | 0 | <0.01 |
| 8b | Bu4NCl (10 mol %), Bu4NOAc (20 mol %) | 86 | 62 | 4.0 |
| 9b | Bu4NCl (15 mol %), Bu4NOAc (25 mol %) | 75 | 60 | 8.0 |
| 10b | Bu4NCl (20 mol %), Bu4NOAc (30 mol %) | 24 | 21 | 130 |
| 11c | None | 88 | 0 | NA |
In situ yield determined by gas chromatographic analysis and comparison to an internal standard.
5 mol % Pd(OAc)2.
Literature conditions for oxidative amination used: Pd(OAc)2 (5 mol %), PhCN (1 M)
(6 equiv), 60 °C, 1 atm O2
Reaction generality.
With optimized conditions identified, we investigated the anti-Markovnikov amination of other olefinic substrates. Substitution on the aryl group of homoallylbenzene showed a minimal effect on yield and regioselectivity (2a-2e). The functionalization of allylbenzene (1f) afforded 2f with significantly higher selectivity (47:1) and moderate yield (47%). Interestingly, it was observed that electron poor p-CF3-allylbenzene (1j) gave 2j in a much higher selectivity (57:1 a-M:M) than electron rich p-MeO-allylbenzene (1g) for 2g (6:1 a-M:M). Overall, the selectivity is generally lower for the homologated products (2a-2e) compared to the allyl benzenes (2f-2j), with greater quantities of Markovnikov and double bond isomers observed. Increasingly distal functionality, such as a phenyl ring three methylene units away, 1k, affords the primary amine derivative 2k in modest yield and good (8:1 a-M:M) selectivity. However, when substrates lack an inductively withdrawing aryl ring (1l), reactivity and regioselectivity are poor.
These studies suggest that proximal electron-withdrawing groups improve the selectivity for the anti-Markovnikov isomer. Given this observation, we became interested in investigating homoallylic alcohols as substrates. The alcohol would presumably increase the regioselectivity by acting as an electron withdrawing group and would, via formation of the ketone, favor a single isomeric product.34,38–42 Moreover, the reaction would afford the thermodynamic γ-aminoketone product – a common motif and intermediate in biologically active molecules and their synthetic precursors.8 Excitingly, when homoallylic alcohol 1m is combined with phthalimide in the presence of 5 mol % Pd(OAc)2 and 20 mol % Bu4NCl, a 77% yield of the γ-aminoketone 2m is observed. The regioselectivity is also significantly enhanced, favoring the anti-Markovnikov product in a 14:1 ratio. It is notable that this aerobic oxidation reaction can be conducted at ambient pressures, as high pressure is a common requirement for other aza-Wacker processes.5 The reaction is also readily scalable, providing 2m in 75% yield on 0.5 mmol scale and 77% yield on 5.0 mmol scale. Given the presence of the electron withdrawing oxygen atom, Bu4NOAc is no longer required to achieve excellent levels of selectivity in most cases. Importantly, the thermodynamically driven isomerization process indeed occurred to exclusively generate the ketone product; i.e., no enimide or allyl imide products are observed.
Under the modified conditions identified for these alcohol-containing substrates, both electron-withdrawing and donating groups were well tolerated. It was found that substituting the aryl ring with electron withdrawing substituents (1q-1u) generally gave higher yields and regioselectivities of 2q-2u compared to unsubstituted or electron-rich substrates 2m-2p; for example 2t bearing a p-CF3 substituent was formed in 72% yield and 19:1 a-M:M selectivity while 2n, bearing a p-MeO, was afforded in 56% yield and 14:1 a-M:M. Steric hindrance on the aryl ring has a deleterious effect on the yield, but results in an increase in the regioselectivity of the reaction. For example, the o-tolyl (2o) and mesityl (2p) products were obtained with improved selectivities (20:1 and 18:1 a-M:M, respectively) as compared with 2m (14:1 a-M:M) in albeit lower isolated yields (64% for 2o and 31% for 2p). Overall, the reaction offers very good functional group tolerance. For example, ethers (2b, 2g, 2n), chlorides (2d, 2i, 2s), trifluoromethyl groups (2t and 2u), and heterocycles (2v and 2w) were all well tolerated. Additionally, a triflate (2r), an α,β-unsaturated ketone (2x), and a silyl ether (2ab) were unaffected under the reaction conditions. Substrates with free primary alcohols do not afford any of the desired products, as the primary alcohol oxidizes under the reaction conditions.
Interestingly, in the case of alkyl substitution α to the alcohol, we observed high yield but significantly diminished anti-Markovnikov selectivity under the conditions optimized for the α-aryl alcohols. This is likely due to the diminished inductive effect of alkyl substituents relative to aryl groups. As with the simple alkenes, the addition of Bu4NOAc (5 mol %) restores the regioselectivity. It was observed that the size of the aliphatic group had an impact on regioselectivity, with larger substituents affording a more selective transformation (2y-2ab).
The increase in regioselectivity observed with substrates bearing a homoallylic alcohol suggest that it may be acting as a directing group during the aminopalladation step.21–27 Although this was not observed in related reactions, it has been proposed in the Wacker oxidation of β-substituted homoallylic alcohols43 but not unsubstituted homoallylic alcohols or ethers.44 When 1ac was subjected to the optimized reaction conditions, 2ac was afforded in 58% yield as a 3.6:1 mixture of Z/E isomers and 9:1 mixture of 2ac to all other constitutional and stereoisomers. These experiments indicate that the coordination of the alcohol to the catalyst is not necessary for an anti-Markovnikov selective transformation, though we cannot eliminate the possibility that it is participating in the reaction for these substrates. Similarly, inductively withdrawing imides and amides at the allylic or homoallylic position promoted the anti-Markovnikov selective oxidative amination. These substrates, lacking the thermodynamic sink of a styrene or carbonyl, afford the enimides 2ad and 2ae with the allylic substrate and the allyl imide 2af with the homoallylic substrate. It is important to note that the resulting terminal phthalimides are easily deprotected. 2a has been shown to react with NH2NH2 to afford 2a’ in 85% yield (Table 2).45 Additionally, under similar condition we were able to remove the phthalimide from 2s to afford cyclic imine 6s in 78% yield (Table 2).
Table 2.
Anti-Markovnikov oxidative amidation of homoallylic alcohols.
 |
Bu4NOAc (15 mol %), Bu4NCl (25 mol %)
Pd(OAc)2 (10 mol %), Bu4NOAc (10 mol %), Bu4NCl (40 mol %)
Bu4NCl (20 mol %)
Bu4NOAc (5 mol %), Bu4NCl (20 mol %)
Bu4NCl (15 mol %)
Bu4NCl (25 mol %)
Next, we sought to explore the scope of the reaction. Cyclic, other acidic amine nucleophiles, including succinimide, saccharine, and 4-nitrophthalimide were all effective under the reaction conditions, affording 3m-5m in good to very good yields.
Mechanistic Investigations.
The dramatic selectivity difference between this transformation and that of other oxidative amination reactions4,5 led us to probe whether this reaction is, in fact, going through an aminopalladation mechanism or if it is going through π-allyl Pd intermediates formed via an allylic C–H activation event, as has been demonstrated in related reactions.
To gain mechanistic insight into the nature of the catalytic cycle, we performed kinetic analyses on the optimized reaction conditions for homoallyl benzene. The concentrations of additives were kept constant, relative to the Pd for all the investigations, as a certain concentration is required to form the active catalyst and then additional AcO– has an inhibitory effect on the rate (Figure 2a). Interestingly, results indicate that the reaction is zero order in nucleophile, first order in olefin, and 1.4 order in [Pd]. The non-integer order in [Pd] is likely due to an equilibrium between monomeric and dimeric palladate complexes, with both being competent catalysts for the reaction at low concentrations and generating less active complexes at high concentrations. A similar effect has previously been reported by Henry.46 The addition of Bu4NOAc first shifts the equilibrium towards the dimeric species and thus reduces the concentration of active [Pd] catalysts.47 To support this conclusion, we determined the order in catalyst in the absence of additional Bu4NOAc and found an order of 1.1 (Figure 2b). The oxidative amination of homoallylic alcohols have similar orders in reagents (see Supplementary Section C): zero order in phthalimide, first order in homoallyl alcohol, and first order in [Pd]. The order in [Pd] suggests that a monomeric palladate complexes is the active catalysts for the homoallylic alcohol system where Bu4NOAc is not added to the reaction. These order experiments are consistent with either coordination of the olefin or allylic C–H activation being the turnover limiting step.
Figure 2: Mechanistic investigation of the anti-Markovnikov oxidative amination through reagent order determination and Hammett plot analysis.

a, Determination of the order in all reagents for the anti-Markovnikov selective oxidative amination of 1a, showing first order kinetics for alkene, non-integer 1.4 order for catalyst, and zero order for nucleophile. b, Determination in the order of [Pd] when no Bu4NOAc is added, indicating first order in catalyst with lower acetate equivalence and implicating palladium oligomerization. c, Hammett investigation for the effect of electronics on the aryl ring on the rate of the oxidative amination reaction, demonstrating the rate enhancement of electron withdrawing groups, even several bonds from the reactive alkene. Error bars represent the standard deviation of the measured values across multiple independent runs.
As the optimized conditions are related to the Jeffery’s conditions for the Heck reaction, which under similar conditions are thought to involve Pd nanoparticles, we sought to determine if the catalyst was homogeneous or heterogeneous.48 When the reaction is run in the presence of Hg0 the product is still generated, albeit in lower yield (12–14%). However, an induction period is observed for the Heck reaction, and no induction period was observed in our oxidative amination reaction.48,49 These studies suggest that the PdII catalytic intermediate is homogeneous, though Pd0 species may be stabilized as transient nanoparticles prior to oxidation by O2.
The requirement for the olefin to bear an electron withdrawing group was investigated by performing a Hammett study. A ρ-value of 0.878 was observed for a series of homoallylbenzene derivatives, indicating that electron withdrawing groups increase the rate of the oxidative amination reaction. This is consistent with either aminopalladation or C–H activation mechanism. In the first, reducing the electrostatic repulsion between the olefin and electron-rich anionic palladate complex would accelerate the rate of the ligand exchange. A similar effect was reported by Hartwig in computationally comparing the ∆∆G≠ for of styrene derivatives undergoing ligand exchange with a neutral Pd(II) complex, where m-MeO-styrene was predicted to have a lower barrier than p-Me-styrene.50 Alternatively, electron-withdrawing groups would stabilize the anionic charge build-up and therefore accelerate the rate of C–H allylic activation via deprotonation.
The order in reagents and Hammett investigations demonstrate that the alkene and the catalysts are both involved during the rate determining step but does not allow us to distinguish between aminopalladation or C–H activation, as this would be consistent with the turnover limiting step being either olefin coordination, for the aminopalladation mechanism,6,7 or allylic C–H activation,17–19 with C–N bond formation occurring after the rate determining step (Figure 3a). To eliminate one of the two possible mechanisms, we isotopically labeled the substrate at the allylic position. Subjecting substrate 1a-d2 to the reaction conditions allows us to distinguish between the two mechanistic pathways. If the reaction were proceeding through the aminometalation pathway, it is expected that 2a-d2 would be the primary product of the reaction, where one of the deuterium atoms migrates to C3. Additionally, as no C–H bond cleavage would occur until after the turnover-limiting step, no kinetic isotope effect is expected. Alternatively, if the reaction was proceeding through an allylic C–H activation pathway, 2a-d1 would be formed, with only a single deuterium atom in the product, as the second would have been deprotonated during the C–H activation step. Further, if allylic C–H activation is the turnover limiting step, a primary kinetic isotope effect would be observed. Subjecting 1a-d2 to the reaction conditions afforded 2a-d2 selectively and a kH/kD of 1.0 was observed, consistent with the reaction occurring via aminopalladation and not C–H activation (Figure 3b).
Figure 3: Deuterium labelling studies as probes to distinguish between multiple mechanistic pathways.

a, Potential C–H activation and aminopalladation mechanisms. b, Isotopic labeling experiments to test the two possible mechanisms, showing full deuterium retention and no kinetic isotope effect as evidence for aminopalladation and against C–H activation. c, Possible outcomes for cis- and trans-aminopalladation pathways. d, Isotopic labeling study to probe the mechanism of aminopalladation, showing predominately deuterium retention at the terminal carbon and thereby indicating an anti-aminopalladation pathway (see Supplementary Figure 32).
Next, we sought to distinguish between cis- and trans-aminopalladation pathways. As the stereochemical outcome of this transformation cannot be determined by examining the stereochemistry of the products after the olefin isomerization has occurred, we chose to investigate styrene as a substrate because it cannot undergo an olefin isomerization and can only afford the enimide product (Figure 3c). Subjecting (Z)-β-deuterostyrene (1ag-d1) to the standard reaction conditions affords 2ag-d1, with 79% deuterium α to the phthalimide and 13% x to the phenyl ring (see Supplementary Section E). As shown in Figure 3d, the major isomer is indeed consistent with the reaction occurring via trans-aminopalladation followed by β-hydride elimination. The minor isomer, where the deuterium has migrated, may be the result of initial trans-aminopalladation/β-hydride elimination to afford the cis-diastereomer and subsequent isomerization to the trans product by the Pd–D. A kinetic isotope effect could also account for some of the preference toward hydride elimination if a cis-aminopalladation was occurring; however, if this were the case the cis-diastereomer (Z-2ag-d1) would be the expected product.
Combining the mechanistic information garnered from the kinetic data and the isotope labeling studies, the catalytic cycle shown in Figure 4 is proposed. Either the Pd(0) or the Pd(II) palladate complex is the catalyst resting state, with loss of either an anionic ligand being required during or prior to the turnover limiting olefin coordination. Outer-sphere nucleophilic attack by the phthalimide and olefin isomerization of the Pd(0) all occur between the turn over limiting step and the catalyst resting state, as supported by the absence of a kinetic isotope effect. As the reaction does not work in the absence of –OAc, we propose that it is required to act as a catalytic base under the reaction conditions, both serving to deprotonate the phthalimide and to undergo reductive elimination from Pd(H)OAc to generate AcOH and Pd(0). The two equivalents of H+ are eventually used in the aerobic oxidation of Pd(0) to Pd(II) to generate H2O2 or H2O and regenerate the –OAc.
Figure 4: A catalytic cycle proposal based upon the mechanistic studies undertaken.

The order in reagents implicates the involvement of the alkene and catalyst at or before the rate determining step, while excluding the involvement of the phthalimide. This suggests that alkene binding through associative ligand dissociation is the rate determining step, and that nucleophilic attack upon the bound olefin is fast relative to this. The requirement of some catalytic quantity of acetate suggests its involvement in this process, and its function as a catalytic base is proposed, to generate a nucleophilic anionic phthalimide. Subsequent β-hydride elimination and olefin isomerization generates the most stable olefin isomer, and the resultant Pd–H is then oxidized aerobically to regenerate the palladate.
Conclusion
In conclusion, we have demonstrated a palladate catalyst promotes an anti-Markovnikov selective aza-Wacker oxidation. Additionally, under the reaction conditions, olefin isomerization occurs to translocate the unit of unsaturation to the most thermodynamically favored position in the molecule. Further, we have demonstrated that this reaction occurs through a trans-aminopalladation mechanism with rate determining olefin coordination. This report represents a major advance in oxidative amination technology, and constitutes a unique approach to conceptualizing remote amination disconnections in organic synthesis. Our current efforts seek to develop an in-depth mechanistic understanding of the regioselectivity-determining step, as well as exploring the intermediacy and capture of alkylpalladium species for the development of alkene difunctionalization reactions.
Supplementary Material
Acknowledgements
The authors would like to thank the University of Illinois and the NIH (R35-GM125029) for their generous support.
Footnotes
Data availability
Synthetic procedures, NMR spectra and characterization for all new compounds, kinetic plots, deuterium labelling data, and X-ray diffraction data, are available within this Article and its Supplementary Information. X-Ray structural data for the tetrachloropalladate have also been deposited with the Cambridge Crystallographic Data Centre under numbers 1548343and are available from CCDC in cif format. Data are also available from the corresponding author upon request.
References
- 1.Muller TE, Hultzsch KC, Yus M, Foubelo F & Tada M Hydroamination: Direct addition of amines to alkenes and alkynes. Chem. Rev 108, 3795–3892 (2008). [DOI] [PubMed] [Google Scholar]
- 2.Nishina N & Yamamoto Y Late transition metal-catalyzed hydroamination. Top. Organomet. Chem 43, 115–144 (2012). [Google Scholar]
- 3.Timokhin VI, Anastasi NR & Stahl SS Dioxygen-Coupled Oxidative Amination of Styrene. J. Am. Chem. Soc 125, 12996–12997 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Brice JL, Harang JE, Timokhin VI, Anastasi NR & Stahl SS Aerobic Oxidative Amination of Unactivated Alkenes Catalyzed by Palladium. J. Am. Chem. Soc 127, 2868–2869 (2005). [DOI] [PubMed] [Google Scholar]
- 5.Rogers MM, Kotov V, Chatwichien J & Stahl SS Palladium-Catalyzed Oxidative Amination of Alkenes: Improved Catalyst Reoxidation Enables the Use of Alkene as the Limiting Reagent. Org. Lett 9, 4331–4334 (2007). [DOI] [PubMed] [Google Scholar]
- 6.McDonald RI, Liu G & Stahl SS Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev 111, 2981–3019 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kočovský P & Bäckvall JE The syn/anti-Dichotomy in the Palladium-Catalyzed Addition of Nucleophiles to Alkenes. Chem. Eur. J 21, 36–56 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Smith BR, Eastman CM & Njardarson JT Beyond C, H, O, and N! Analysis of the Elemental Composition of U.S. FDA Approved Drug Architectures. J. Med. Chem 57, 9764–9773 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Baker R & Halliday DE Ligand effects in the rhodium catalysed reaction of butadiene and amines. Tetrahedron Lett 13, 2773–2776 (1972). [Google Scholar]
- 10.Hosokawa T, Takano M, Kuroki Y & Murahashi S-I Palladium(II)-catalyzed amidation of alkenes. Tetrahedron Lett 33, 6643–6646 (1992). [Google Scholar]
- 11.Beller M, Trauthwein H, Eichberger M, Breindl C & Müller TE Anti-Markovnikov reactions. Part 6. Rhodium-catalyzed amination of vinylpyridines. Hydroamination versus oxidative amination. Eur. J. Inorg. Chem 1121–1132 (1999). [Google Scholar]
- 12.Utsunomiya M, Kuwano R, Kawatsura M & Hartwig JF Rhodium-catalyzed anti-Markovnikov hydroamination of vinylarenes. J. Am. Chem. Soc 125, 5608–5609 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Timokhin VI & Stahl SS Brønsted Base-Modulated Regioselectivity in the Aerobic Oxidative Amination of Styrene Catalyzed by Palladium. J. Am. Chem. Soc 127, 17888–17893 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Lee JM et al. Hydrogen-Bond-Directed Highly Stereoselective Synthesis of Z-Enamides via Pd-Catalyzed Oxidative Amidation of Conjugated Olefins. J. Am. Chem. Soc 128, 12954–12962 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Goldfogel MJ, Roberts CC & Meek SJ Intermolecular Hydroamination of 1,3-Dienes Catalyzed by Bis(phosphine)carbodicarbene-Rhodium Complexes. J. Am. Chem. Soc 136, 6227–6230 (2014). [DOI] [PubMed] [Google Scholar]
- 16.Dong JJ, Browne WR & Feringa BL Palladium-Catalyzed anti-Markovnikov Oxidation of Terminal Alkenes. Angew. Chem. Int. Ed 54, 734–744 (2014). [DOI] [PubMed] [Google Scholar]
- 17.Reed SA & White MC Catalytic Intermolecular Linear Allylic C−H Amination via Heterobimetallic Catalysis. J. Am. Chem. Soc 130, 3316–3318 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu G, Yin G & Wu L Palladium-Catalyzed Intermolecular Aerobic Oxidative Amination of Terminal Alkenes: Efficient Synthesis of Linear Allylamine Derivatives. Angew. Chem. Int. Ed 47, 4733–4736 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Yin G, Wu Y & Liu G Scope and Mechanism of Allylic C−H Amination of Terminal Alkenes by the Palladium/PhI(OPiv)2 Catalyst System: Insights into the Effect of Naphthoquinone. J. Am. Chem. Soc 132, 11978–11987 (2010). [DOI] [PubMed] [Google Scholar]
- 20.Pattillo CC et al. Aerobic Linear Allylic C–H Amination: Overcoming Benzoquinone Inhibition. J. Am. Chem. Soc 138, 1265–1272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hoveyda AH, Evans DA & Fu GC Substrate-directable chemical reactions. Chem. Rev 93, 1307–1370 (1993). [Google Scholar]
- 22.Delcamp JH, Brucks AP & White MC A General and Highly Selective Chelate-Controlled Intermolecular Oxidative Heck Reaction. J. Am. Chem. Soc 130, 11270–11271 (2008). [DOI] [PubMed] [Google Scholar]
- 23.Grünanger CU & Breit B Branched-Regioselective Hydroformylation with Catalytic Amounts of a Reversibly Bound Directing Group. Angew. Chem. Int. Ed 47, 7346–7349 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Weiner B, Baeza A, Jerphagnon T & Feringa BL Aldehyde selective Wacker oxidations of phthalimide protected allylic amines: a new catalytic route to beta3-amino acids. J. Am. Chem. Soc 131, 9473–9474 (2009). [DOI] [PubMed] [Google Scholar]
- 25.Choi PJ, Sperry J & Brimble MA Heteroatom-Directed Reverse Wacker Oxidations. Synthesis of the Reported Structure of (−)-Herbaric Acid. J. Org. Chem 75, 7388–7392 (2010). [DOI] [PubMed] [Google Scholar]
- 26.Ensign SC, Vanable EP, Kortman GD, Weir LJ & Hull KL Anti-Markovnikov Hydroamination of Homoallylic Amines. J. Am. Chem. Soc 137, 13748–13751 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Gurak JA Jr., Yang KS, Liu Z & Engle KM Directed, Regiocontrolled Hydroamination of Unactivated Alkenes via Protodepalladation. J. Am. Chem. Soc 138, 5805–5808 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Äkermark B et al. Palladium-promoted addition of amines to isolated double bonds. J. Organomet. Chem 72, 127–138 (1974). [Google Scholar]
- 29.Pryadun R, Sukumaran D, Bogadi R & Atwood JD Amine Attack on Coordinated Alkenes: An Interconversion from Anti-Markovnikoff to Markovnikoff Products. J. Am. Chem. Soc 126, 12414–12420 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Keith JA & Henry PM The Mechanism of the Wacker Reaction: A Tale of Two Hydroxypalladations. Angew. Chem. Int. Ed 48, 9038–9049 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Wenzel TT Oxidation of olefins to aldehydes using a palladium–copper catalyst. J. Chem. Soc., Chem. Commun 862–864 (1993). [Google Scholar]
- 32.Ogura T, Kamimura R, Shiga A & Hosokawa T Reversal of Regioselectivity in Wacker-Type Oxidation of Simple Terminal Alkenes and Its Paired Interacting Orbitals (PIO) Analysis. Bull. Chem. Soc. Jap 78, 1555–1557 (2005). [Google Scholar]
- 33.Dong G, Teo P, Wickens ZK & Grubbs RH Primary Alcohols from Terminal Olefins: Formal Anti-Markovnikov Hydration via Triple Relay Catalysis. Science 333, 1609–1612 (2011). [DOI] [PubMed] [Google Scholar]
- 34.Liu G & Stahl SS Highly Regioselective Pd-Catalyzed Intermolecular Aminoacetoxylation of Alkenes and Evidence for cis-Aminopalladation and SN2 C−O Bond Formation. J. Am. Chem. Soc 128, 7179–7181 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Martínez C et al. Palladium-Catalyzed Intermolecular Aminoacetoxylation of Alkenes and the Influence of PhI(OAc)2 on Aminopalladation Stereoselectivity. J. Org. Chem 78, 6309–6315 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mann S, Benhamou L & Sheppard T Palladium(II)-Catalysed Oxidation of Alkenes. Synthesis 47, 3079–3117 (2015). [Google Scholar]
- 37.Yin G, Mu X & Liu G Palladium(II)-Catalyzed Oxidative Difunctionalization of Alkenes: Bond Forming at a High-Valent Palladium Center. Acc. Chem. Res 49, 2413–2423 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Jeffery T Palladium-catalysed vinylation of organic halides under solid–liquid phase transfer conditions. J. Chem. Soc., Chem. Commun 1287–1289 (1984). [Google Scholar]
- 39.Cheng J, Qi X, Li M, Chen P & Liu G Palladium-Catalyzed Intermolecular Aminocarbonylation of Alkenes: Efficient Access of β-Amino Acid Derivatives. J. Am. Chem. Soc 137, 2480–2483 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Larock RC, Leung W-Y & Stolz-Dunn S Synthesis of aryl-substituted aldehydes and ketones via palladium-catalyzed coupling of aryl halides and non-allylic unsaturated alcohols. Tetrahedron Lett 30, 6629–6632 (1989). [Google Scholar]
- 41.Bouquillon S, Ganchegui B, Estrine B, Hénin F & Muzart J Heck arylation of allylic alcohols in molten salts. J. Organomet. Chem 634, 153–156 (2001). [Google Scholar]
- 42.Werner EW, Mei T-S, Burckle AJ & Sigman MS Enantioselective Heck Arylations of Acyclic Alkenyl Alcohols Using a Redox-Relay Strategy. Science 338, 1455–1458 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nokami J et al. A new synthetic method for γ-butyrolactols by the paladium-catalyzed regioselective oxidation of 1-alken-4-ols. Tetrahedron Lett 29, 5181–5184 (1988). [Google Scholar]
- 44.Yatagai H, Yamamoto Y & Maruyama K A new procedure for the stereoselective synthesis of (Z)-2-alkenylsilanes and -tins and their application to erythro-selective synthesis of β-alkyl alcohol derivatives. J. Am. Chem. Soc 102, 4548–4550 (1980). [Google Scholar]
- 45.Olsen DK, Torian BE, Morgan CD & Braun LL Preparation and stereochemistry of 4-aryl-3-butenylamines. A novel synthesis of an oxazolo[2,3-a]isoindole. J. Org. Chem 45, 4049–4052 (1980). [Google Scholar]
- 46.Winstein S, McCaskie J, Lee H-B & Henry PM Oxidation of olefins by palladium(II). 9. Mechanism of the oxidation of olefins by the dimeric species, disodium dipalladium hexaacetate, in acetic acid. J. Am. Chem. Soc 98, 6913–6918 (1976). [Google Scholar]
- 47.Carrow BP & Hartwig JF Ligandless, Anionic, Arylpalladium Halide Intermediates in the Heck Reaction. J. Am. Chem. Soc 132, 79–81 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Vries AHM et al. A practical recycle of a ligand-free palladium catalyst for Heck reactions. Adv. Synth. Catal 344, 996–1002 (2002). [Google Scholar]
- 49.Reetz MT & Westermann E Phosphane-Free Palladium-Catalyzed Coupling Reactions: The Decisive Role of Pd Nanoparticles. Angew. Chem. Int. Ed 39, 165–168 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Hanley PS & Hartwig JF Intermolecular Migratory Insertion of Unactivated Olefins into Palladium--Nitrogen Bonds. Steric and Electronic Effects on the Rate of Migratory Insertion. J. Am. Chem. Soc 133, 15661–15673 (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.