

Abstract
ERAAP is an intracellular protease that plays a central role in determining the repertoire of peptides displayed by cells by MHCI molecules, and dysfunctions in ERAAP are linked to a variety of diseases. There is therefore great interest in developing probes that can image ERAAP in cells. In this report we present a fluorescent probe, termed Ep, that can image ERAAP activity in live cells. Ep is composed of an 10 amino acid ERAAP substrate that has a donor quencher pair conjugated to it, composed of BODIPY and dinitro-toluene. Ep undergoes a 20 fold increase in fluorescence after ERAAP cleavage, and was able to image ERAAP activity in cell culture via fluorescent microscopy. In addition, we used Ep to develop a high throughput screen for ERAAP inhibitors, and screened an electrophile library containing 1,460 compounds. From this Ep based screen we identified aromatic alkyne-ketones as lead fragments that can irreversibly inhibit ERAAP activity. We anticipate numerous applications of Ep given its unique ability to image ERAAP within cells.
ER aminopeptidase associated with antigen processing (ERAAP) is an intracellular amino-peptidase, located in the endoplasmic reticulum, which cleaves peptides presented by the major histocompatibility complex (MHC) class I molecules and determines which subset of peptides are displayed on cell surfaces and recognized by killer T cells.1–3 ERAAP thus plays a key role in shaping the immune response and its polymorphic variants have been implicated in a number of autoimmune diseases.4–6 Down-regulation of ERAAP results in alterations in the peptides that are displayed on cell surfaces. The altered peptide repertoire is recognized as foreign, because killer T cells are not tolerant towards the new peptides.7–9 Therapeutics that can down regulate ERAAP therefore have great potential as cancer vaccines and increasing the immune response towards tumors.10, 11 Downregulation of ERAAP is also implicated in the development of auto-immune disorders, such as ankylosing spondylitis and psoriasis.4, 5
It is therefore of great interest in developing probes that can image ERAAP in cells and also in the development of new inhibitors for ERAAP. However, developing imaging probes for ERAAP has been challenging because it is an intracellular amino-peptidase that cleaves large peptides, and developing membrane permeable probes that can turn-on fluorescence after ERAAP mediated cleavage remains a challenge. In addition, there are numerous other amino-peptidases within cells that will also potentially cleave fluorescent probes designed to specifically detect ERAAP. For example, the only fluorescent probe currently commercially available for measuring ERAAP activity is leucine-7-amino-5-methylcoumarin (L-Amc), which is cleaved by all amino-peptidases, and also lacks the optimal emission wavelength for cellular imaging. Although recent progress has been made on developing ERAP1 (human ortholog of mouse ERAAP) imaging probes, using two photon emitting dyes,12 there is still a great need for fluorescent probes that can detect ERAAP via conventional fluorescent microscopy.
In this report we present a fluorescent ERAAP probe, composed of a known ERAAP-substrate, the N-terminally extended KK-SIINFEKL decapeptide. The modified peptide has a donor quencher pair conjugated to it and is termed Ep. Ep emits at 510 nm after ERAAP cleavage, and can thus image ERAAP activity in cells. In addition, Ep was specific for ERAAP over other cellular amino-peptidases and has negligible activity in cells that lack ERAAP. Finally, Ep has high sensitivity for ERAAP and was used to develop a high-throughput-screen (HTS) for ERAAP inhibitors, that allowed us to identify a novel electrophile fragment, alkyne-ketone that can inhibit ERAAP.
The chemical structure of Ep is shown in Figure 1a, it is composed of the modified decapeptide K(DNP)-K-S-I-I-N-F-E-C(BODIPY)-L. Ep contains the fluorescent dye BODIPY and the fluorescent quencher dinitrophenyl (DNP), conjugated to the KK-SIINFECL peptide. Ep should initially have low fluorescence because the DNP group will quench the BODIPY fluorescence. However, in the presence of ERAAP the fluorescence of EP should increase, because ERAAP will cleave the N-terminal amino acid modified by the DNP group, due to its amino-peptidase activity. The diffusion of DNP away from the BODIPY should increase the quantum yield of Ep. The peptide KK-SIINFECL was selected as the ERAAP substrate for Ep because of its similarity to the well characterized ERAAP substrate KK-SIINFEKL, which is trimmed by ERAAP to SIINFEKL, the immunodominant H-2Kb-binding peptide.3, 13 SIINFECL is identical to SIINFEKL except that its lysine residue is replaced with a cysteine, which was needed for conjugation to BODIPY. Importantly, Serwold et al. demonstrated that the internal lysine in SIINFEKL containing peptides can be modified to other residues without affecting its ability to bind the Kb MHC I molecule.14, 15 Based on this design, Ep was synthesized in one step using the commercially synthesized peptide K(DNP)K-SIINFECL, which contained the fluorescent quencher, DNP (Dinitrophenyl).16–18 The BODIPY (4,4-Difluoro-3a,4a-diaza-s-indacene), a widely used fluorophore for fluorescent imaging in the life sciences, was conjugated to this peptide via a reaction between the cysteine side-chain and iodoacetylamide BODIPY (see supporting information for reaction scheme and procedures). The final Ep was purified by reverse-phase HPLC and its synthesis was confirmed by ESI-HRMS (see Figure S1).
Figure 1. Ep is a peptide-based fluorescent probe that can detect ERAAP.

a: Ep is based on the peptide sequence (KK-SIINFECL) and its fluorescence is quenched through a FRET pair between DNP and BODIPY. ERAAP cleaves the N-terminal amino acid of Ep and releases the quencher, causing a dramatic increase in fluorescence. b: ERAAP activity was imaged in wildtype fibroblasts and in ERAAP knockout fibroblasts. c: A high throughput screening assay for identifying ERAAP inhibitors was developed in a 96-well fluorescent plate reader format. A 1460-electrophile library was screened and we identified para-bromo-, ortho-fluoro-phenyl ketone alkyne as a lead electrophile fragment that can irreversibly inhibit ERAAP.
Ep needs to be initially non-fluorescent because of DNP mediated quenching, but then increase its fluorescence after ERAAP cleavage. We therefore investigated the fluorescence of Ep in its intact state, and its fluorescence after cleavage by Ep. The quantum yield of Ep is extremely low in aqueous solution, about 1%, due to efficient fluorescence quenching. However, upon addition of ERAAP the fluorescence quantum yield of Ep dramatically increases, by a factor of 22 due to ERAAP mediated cleavage, resulting in bright green fluorescence (see Figure S2 in SI). Figure 2 demonstrates the Ep can sensitively detect ERAAP and was able to detect ERAAP at concentrations as low as 0.1 μg/mL, and linearly responded to ERAAP concentrations within the sub μg/mL range.
Figure 2. Ep can detect ERAAP activity.
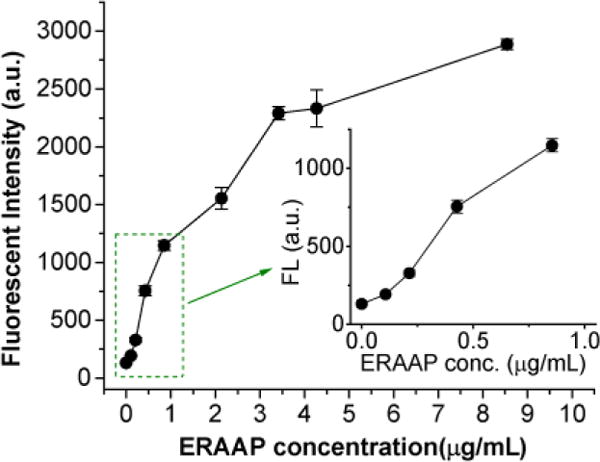
The fluorescence intensity of Ep (at 510 nm) changes in response to different concentrations of ERAAP after incubation in 1x PBS for 10 min. (inset: response curve in the range below 1 μg/mL of ERAAP).
We performed HPLC experiments to determine if Ep was sequentially trimmed by ERAAP at the N terminus to release the DNP residue. Ep was incubated with ERAAP and HPLC was performed on the reaction mixture, after 10 min, and the absorption at 350 nm (DNP) and 490 nm (BODIPY) was measured. Figure 3 demonstrates that Ep by itself gives only one peak at 350 nm and 490 nm, which overlap. However, after incubation with ERAAP, the original peak decreased and several new peaks emerged corresponding to the products of the enzymatic reaction. We studied the kinetics of the enzymatic trimming reaction between Ep and ERAAP using florigenic assay, by plotting the initial reaction velocities against Ep concentrations. Ep underwent typical Michealis–Menten hydrolysis kinetics (see Figure S3 in SI), and fitting of the kinetic data to a Lineweaver–Burk plot gave the kinetics parameters Km = 6.7 μM, Vmax = 1.14 nM·s−1, kcat = Vmax/[ERAAP] = 0.13 s−1, and corresponding to a catalytic efficiency (kcat/Km) = 1.94 × 104 M−1·s−1. The Km and kcat of Ep for ERAAP are comparable to other known substrates of ERAAP, and suggests that it will be able to compete with endogenous cellular substrates for ERAAP in cells.19
Figure 3.
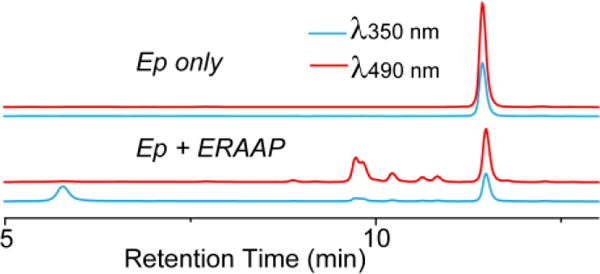
HPLC analysis of Ep before and after trimming by ERAAP.
To further understand the binding mechanism of Ep with ERAAP, we performed molecular docking of Ep with ERAAP, using the solved crystal structure of ERAP1 (human ortholog of mouse ERAAP, PDB 2DY0). ERAP1 has a large and long binding pocket that contains two critical binding sites at its terminal two ends. The Zn-associated catalytic site is at one end of the binding pocket and binds the N-terminus of the target peptide and performs the trimming. The other peptide binding site is the regulatory site (~30 Å away from catalytic center) which binds the C-terminus of the peptide and could function as a molecular ruler to control the trimming selectivity. The middle part of the binding pocket has a large volume and high conformational flexibility.19–22
We decided to dock only the first 4 and last 3 amino acids residues of Ep (tetrapeptide and tripeptide) to the active site and regulatory site, respectively, due to the challenges associated with docking with a flexible protein. Figure 4a shows the optimized conformation of the tetrapeptide, K(DNP)KSI in the catalytic site. We found that the N-terminal amine group and the first amide bond were located close to the Zn center and the carbonyl group was stabilized via hydrogen bonding with Tyr438 and His353. The bulky DNP side chain of N-terminal Lys(DNP) fell into the hydrophobic sub-pocket S1 through hydrophobic interactions, the rest of the peptide chain extended backward from the catalytic site. We compared this docking result with the crystal structure of the ERAP1-Bestatin complex (PDB 2DY0). Bestatin is a widely used Zn metallo-aminopeptidase inhibitor which tightly binds into the catalytic site and we found that the tetrapeptide K(DNP)KSI and Bestatin shared very similar conformations in the catalytic site (Figure 4a and 4b). The results above demonstrated that the N-terminus of Ep has great affinity to the ERAP1 catalytic site, which triggers the trimming process. The docking result of the C-terminal tripeptide EC(BODIPY)L with the ERAP1 regulatory site is shown in Figure 4c. The carboxylate end of the tripeptide was strongly anchored in the ERAP1 regulatory site by direct and water mediated hydrogen bonds with Tyr684, Lys685, Arg807 and Arg841, and the BOIDPY residue on the side chain of the cysteine fell into the non-polar domain surrounded by Leu733 and Leu734. This result indicates that the C-terminus of Ep can form strong binding with the ERAP1 regulatory site. It is also in agreement with the reported binding preference of the ERAP1 regulatory site towards hydrophobic side chains.20
Figure 4.

a: Optimized confirmations of the tetrapeptide (K(DNP)KSI) in the ERAP1 catalytic site. b: Crystal structure of Bestatin in the ERAP1 catalytic site (PDB: 2DY0). c: Optimized confirmations of the tetrapeptide (K(Dnp)KSI) in the ERAP1 regulatory site.
There is great interest in being able to image ERAAP activity in cells and we therefore investigated if Ep could allow this. For these experiments we incubated Ep with a fibroblast cell line generated from B6 wildtype (WT) mouse and from an ERAAP knockout (ERAAP-KO) mouse. We treated WT and ERAAP-KO fibroblast cells with 10 or 20 μM of Ep for 2h or 3h, and then imaged via fluorescent microscopy. Figure 5 demonstrates that Ep can image ERAAP activity in live cells. For example, strong fluorescence was detected in WT cells, in contrast, ERAAP-KO cells did not show any observable fluorescence signal, in all Ep concentrations and incubation times investigated. These results demonstrate that Ep can detect ERAAP in cells in a sensitive and specific manner.
Figure 5.
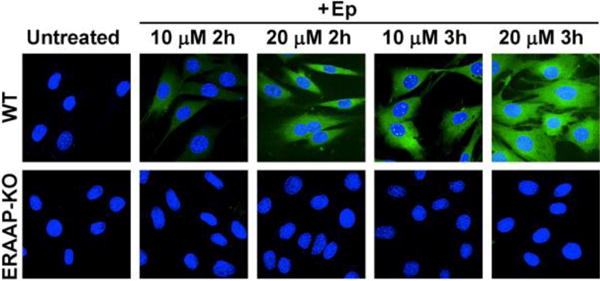
Fluorescence imaging of wildtype (WT) and ERAAP knockout (ERAAP-KO) fibroblast cells with Ep. WT cells generate green fluorescence after Ep incubation, whereas ERAAP-KO fibroblasts generate minimal fluorescence.
The downregulation of ERAP1 (human ortholog of ERAAP) triggers potent anticancer immune responses and can rescue mice from experimental models of cancer. Therefore, there is great interest in developing ERAAP inhibitors for cancer immunotherapy.10, 11 However, developing ERAAP inhibitors has been challenging because of the lack of assays available for detecting ERAAP with sensitivity in a high throughput manner. Current ERAAP assays have low selectivity and sensitivity, and are therefore challenging to use for the development of HTS assays for identifying ERAAP (ERAP1) inhibitors. We therefore investigated if Ep had the sensitivity needed to develop an HTS assay for identifying ERAAP inhibitors. We determined the Z factor of Ep, with regards to developing ERAAP inhibitors, by measuring its fluorescence in the presence and in absence of ERAAP. In a 96-well plate format, this assay gave a Z’-factor of 0.79, which demonstrates that it is suitable for performing HTS screens (see Figure S5 in SI). Ep was used to screen a 1460- compound electrophile library (Enamine Ltd). We chose an electrophile library for screening because these fragments can serve as leads for the development of irreversible inhibitors.
The results of our HTS are shown in Figure 6 as an activity scatter-plot (Figure 6a). We identified 8 compounds that could reproducibly inhibit ERAAP activity. Three compounds (A-C) were chosen for further study because of their low IC50 values in comparison to the other members of the library (Figure 6b). Of these, compound A, (1-(4-bromo-2-fluorophenyl)prop-2-yn-1-one, exhibited the lowest IC50 (23 μM), whereas compounds B and C had IC50 values of 92 μM and 175 μM, respectively (Figure 6C). Although this value is still high coeeeempared with several other reported ERAP1 inhibitors, which have sub-micromolar range IC50s,23, 24 we anticipate that the efficiency of A-C can considerably be improved after further optimization. Steady-state time dependent inhibition studies with A demonstrated it could generate an almost 100% of inhibition of ERAAP after a 4.5 h pre-incubation (see Figure S6 in SI), suggesting that it is an irreversible inhibitor. Compound A is a halogen substituted phenyl ketone alkyne (Figure 6d), and its small size and low electrophilicity suggest that it is a suitable pharmacophore for developing irreversible ERAAP inhibitors.
Figure 6. Ep based high throughput screen identifies electrophiles that can inhibit ERAAP activity.

a: Scatter plot describing the results of the 1460-compound screen for inhibition of ERAAP activity. b: Validation of the top 8 hits for ERAAP inhibition. Compounds A, B and C showed significantly higher inhibition efficiency (≥62%) than the other 5 hit compounds. c: Dose-dependent inhibition curves of compounds A, B and C. Compound A had an IC50 of 23 μM, which was the lowest among all three compounds. d: the structure of compound A, a promising pharmacophore for developing irreversible ERAAP inhibitors.
In conclusion, in this report we present a new peptide based fluorescent probe for ERAAP, termed Ep, which can image ERAAP activity in cells and in a plate reader format. We anticipate numerous applications for Ep given the great interest in understanding the biology of ERAAP and in developing new ERAAP inhibitors.
Supplementary Material
Acknowledgments
Support from NIH grants R37060040 and RO1AI130210.
Footnotes
Electronic Supplementary Information (ESI) available: [detail procedures for Ep synthesis, kinetics, quantum yield, docking and Ep-based high throughput screening are described].
Notes and references
- 1.Serwold T, Gonzalez F, Kim J, Jacob R, Shastri N. Nature. 2002;419:480–483. doi: 10.1038/nature01074. [DOI] [PubMed] [Google Scholar]
- 2.Saric T, Chang SC, Hattori A, York IA, Markant S, Rock KL, Tsujimoto M, Goldberg AL. Nat Immunol. 2002;3:1169. doi: 10.1038/ni859. [DOI] [PubMed] [Google Scholar]
- 3.York IA, Chang SC, Saric T, Keys JA, Favreau JM, Goldberg AL, Rock KL. Nat Immunol. 2002;3:1177. doi: 10.1038/ni860. [DOI] [PubMed] [Google Scholar]
- 4.Strange A, Capon F, Spencer CCA, Knight J, Weale ME, Allen MH, Barton A, Band G, Bellenguez C, Bergboer JGM, et al. Nat Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reeves E, Colebatch-Bourn A, Elliott T, Edwards CJ, James E. Proc Natl Acad Sci USA. 2014;111:17594–17599. doi: 10.1073/pnas.1408882111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirino Y, Bertsias G, Ishigatsubo Y, Mizuki N, Tugal-Tutkun I, Seyahi E, Ozyazgan Y, Sacli FS, Erer B, Inoko H, et al. Nat Genet. 2013;45:202. doi: 10.1038/ng.2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammer GE, Gonzalez F, Champsaur M, Cado D, Shastri N. Nat Immunol. 2005;7:103. doi: 10.1038/ni1286. [DOI] [PubMed] [Google Scholar]
- 8.Yan J, Parekh VV, Mendez-Fernandez Y, Olivares-Villagómez D, Dragovic S, Hill T, Roopenian DC, Joyce S, Van Kaer L. J Exp Med. 2006;203:647–659. doi: 10.1084/jem.20052271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagarajan NA, Gonzalez F, Shastri N. Nat Immunol. 2012;13:579–586. doi: 10.1038/ni.2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cifaldi L, Lo Monaco E, Forloni M, Giorda E, Lorenzi S, Petrini S, Tremante E, Pende D, Locatelli F, Giacomini P, et al. Cancer Res. 2011;71:1597. doi: 10.1158/0008-5472.CAN-10-3326. [DOI] [PubMed] [Google Scholar]
- 11.James E, Bailey I, Sugiyarto G, Elliott T. J Immunol. 2013;190:5839. doi: 10.4049/jimmunol.1300220. [DOI] [PubMed] [Google Scholar]
- 12.Xu S, Liu HW, Hu XX, Huan SY, Zhang J, Liu YC, Yuan L, Qu FL, Zhang XB, Tan W. Anal Chem. 2017;89:7641–7648. doi: 10.1021/acs.analchem.7b01561. [DOI] [PubMed] [Google Scholar]
- 13.York IA, Brehm MA, Zendzian S, Towne CF, Rock KL. Proc Natl Acad Sci U S A. 2006;103:9202. doi: 10.1073/pnas.0603095103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Serwold T, Gaw S, Shastri N. Nat Immunol. 2001;2:644. doi: 10.1038/89800. [DOI] [PubMed] [Google Scholar]
- 15.Brouwenstijn N, Serwold T, Shastri N. Immunity. 2001;15:95–104. doi: 10.1016/s1074-7613(01)00174-1. [DOI] [PubMed] [Google Scholar]
- 16.Farber SA, Pack M, Ho SY, Johnson LD, Wagner DS, Dosch R, Mullins MC, Hendrickson HS, Hendrickson EK, Halpern ME. Science. 2001;292:1385–1388. doi: 10.1126/science.1060418. [DOI] [PubMed] [Google Scholar]
- 17.Hendrickson HS, Hendrickson EK, Johnson ID, Farber SA. Anal Biochem. 1999;276:27–35. doi: 10.1006/abio.1999.4280. [DOI] [PubMed] [Google Scholar]
- 18.Wijewickrama GT, Kim JH, Kim YJ, Abraham A, Oh YS, Ananthanarayanan B, Kwatia M, Ackerman SJ, Cho W. J Biol Chem. 2006;281:10935–10944. doi: 10.1074/jbc.M512657200. [DOI] [PubMed] [Google Scholar]
- 19.Chang SC, Momburg F, Bhutani N, Goldberg AL. Proc Natl Acad Sci U S A. 2005;102:17107–17112. doi: 10.1073/pnas.0500721102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gandhi A, Lakshminarasimhan D, Sun Y, Guo HC. Sci Rep. 2011;1:186. doi: 10.1038/srep00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kochan G, Krojer T, Harvey D, Fischer R, Chen L, Vollmar M, von Delft F, Kavanagh KL, Brown MA, Bowness P, et al. Proc Natl Acad Sci U S A. 2011;108:7745–7750. doi: 10.1073/pnas.1101262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nguyen TT, Chang SC, Evnouchidou I, York IA, Zikos C, Rock KL, Goldberg AL, Stratikos E, Stern LJ. Nat Struct Mol Biol. 2011;18:604–613. doi: 10.1038/nsmb.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zervoudi E, Saridakis E, Birtley JR, Seregin SS, Reeves E, Kokkala P, Aldhamen YA, Amalfitano A, Mavridis IM, James E, et al. Proc Natl Acad Sci USA. 2013;110:19890. doi: 10.1073/pnas.1309781110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kokkala P, Mpakali A, Mauvais FX, Papakyriakou A, Daskalaki I, Petropoulou I, Kavvalou S, Papathanasopoulou M, Agrotis S, Fonsou TM, et al. J Med Chem. 2016;59:9107–9123. doi: 10.1021/acs.jmedchem.6b01031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.