

Supplemental Digital Content is available in the text.
Key Words: adoptive cell transfer, intra-arterial, melanoma, immunotherapy
Abstract
Adoptive cell transfer therapy for cancer has existed for decades and is experiencing a resurgence in popularity that has been facilitated by improved methods of production, techniques for genetic modification, and host preconditioning. The trafficking of adoptively transferred lymphocytes and infiltration into the tumor microenvironment is sine qua non for successful tumor eradication; however, the paradox of extremely poor trafficking of lymphocytes into the tumor microenvironment raises the issue of how best to deliver these cells to optimize entry into tumor tissue. We examined the route of administration as a potential modifier of both trafficking and antitumor efficacy. Femoral artery cannulation and tail vein injection for the intra-arterial (IA) and IV delivery, respectively, were utilized in the B16-OVA/OT-I mouse model system. Both IV and IA infusions showed decreased tumor growth and prolonged survival. However, although significantly increased T-cell tumor infiltration was observed in IA mice, tumor growth and survival were not improved as compared with IV mice. These studies suggest that IA administration produces increased early lymphocyte trafficking, but a discernable survival benefit was not seen in the murine model examined.
Coupling the recent success of a variety of immunotherapy strategies and the increasing availability of cellular immunotherapy production facilities, adoptive cell transfer (ACT) is a compelling field for preclinical investigation and clinical translation. ACT is the process where the patient’s T cells are harvested, activated ex vivo, and infused back into the patient. As a therapeutic modality, this has been studied since the 1980s.1 The field has primarily focused on how to generate evermore powerful and efficient T cells to increase antitumor responses. To this end various sources of T cells including tumor-infiltrating lymphocytes (TILs),2 lymph nodes,3 and peripheral blood mononuclear cells4 have been evaluated for efficacy. In addition, studies have examined how best to activate these cells and examined how the addition of different cytokines to ex vivo cultures may alter the T-cell populations to maintain cytotoxic responses.5,6 In order to optimize all of these variables, trials have been performed utilizing genetic engineering to make T cells that overcome all of these issues and maximize clinical effect.7–9 These studies led to the emergence of chimeric antigen receptor–modified T cells and have invigorated the field.10 One consideration of the host in preparing these treatments is that there is a defined lymphocyte carrying capacity in patents, thus preconditioning with nonmyeloblative chemotherapy and or whole body radiation has become standard before ACT as it “creates space” for the newly transferred lymphocytes.11 Another concern is that regardless of the source, culturing conditions, or genetic modification of the T cells, entry into the tumor is an absolute requirement for cell-mediated killing.12 Previous studies have clearly shown that T-cell trafficking into tumors is relatively poor13,14 and that the early level of T-cell trafficking in the tumor is important for optimal tumor control.15 Therefore, a major unanswered question in the field is how to more effectively deliver adoptively transferred cells to the tumor microenvironment to potentiate antitumor effects.
Standard protocols for ACT involve infusion of the cellular product through a venous catheter. As all venous return must pass through the right heart and pulmonary vasculature, ACT cells delivered into the venous system may become trapped in the capillary beds of the lungs thereby decreasing their availability to traffic to tumor sites.16 One mechanism to increase the early delivery of T cells to the tumor microenvironment is delivery of the cells through the regional arterial blood supply (Fig. 1). In rare clinical instances of intra-arterial (IA) ACT, improved antitumor responses were noted and were theoretically related to enhanced delivery of T cells to the tumor bed.17,18 To potentially leverage this mechanism of improved delivery, clinical trials of ACT via the hepatic artery to treat liver metastasis have been attempted.19,20 Although anecdotal reports suggest that IA ACT may better direct T cells into the tumor and improve tumor control, this observation is not well established. The actual benefit of this IA approach has yet to be defined, but preclinical models of ACT may offer insight into this technique.
FIGURE 1.
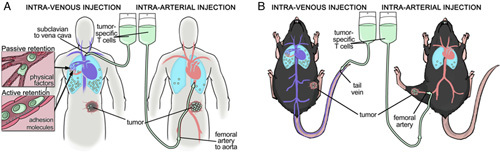
A, In patients adoptive transfer via the regional arterial supply of the tumor (right) was hypothesized to increase exposure of adoptively transferred cells (green) to the tumor vasculature and avoid trapping in the lungs as seen with intravenous delivery (left). Adoptively transferred lymphocyte retention in the pulmonary capillaries is multifactorial (inset). B, Experimental design to examine this hypothesis in a murine model of cancer.
The B16 murine melanoma model expressing ovalbumin as a novel tumor antigen can be effectively treated with antigen specific CD8 OT-1 T cells and is a useful model for the study of lymphocyte trafficking. Although this model system can be considered too artificial for examination of antitumor efficacy owing to the strong foreign antigen expression and uniform T-cell population, it is ideal for lymphocyte homing studies. By eliminating tumor cell antigen expression variability and having a clonal T-cell population, mechanisms of trafficking and delivery can be singularly addressed in the absence of confounding variables. We therefore evaluated regional IA delivery of T cells compared with standard IV delivery in this preclinical mouse model to recapitulate human protocols.
MATERIALS AND METHODS
Tumor Model
For all experiments, the B16-OVA/OT-I mouse model system was utilized. All experimental protocols were approved by the Roswell Park Cancer Institutional Animal Care and Use Committee. To generate tumors, 8–12-week-old female wild-type C57BL/6 mice (Charles Rivers) were injected with 1×106 B16F10-OVA cells (ATCC, Manassas, VA) subcutaneously in the distal hind leg. Tumors were established for 7 days before treatment.
ACT
Spleens, peripheral lymph nodes and mesenteric lymph nodes were harvested from Tcr transgenic OT-1 mice (Taconic Biosciences) and disaggregated into single cell suspensions. The CD8 T cells were then activated with plate-bound CD3 antibody (145-2C11; BD Pharmingen) for 2 days and then expanded with 100 units/mL IL2 (Peprotech) for 3 days. Cells were harvested and utilized for ACT into C57BL/6 wild-type mice. Experimental groups consisted of no treatment (control), sham IA injection, IV ACT and IA ACT. Treatment mice were injected with 1×106, 5×106, or 10×106 OT-1 cells depending on the experiment. Three perpendicular axes of the tumors were measured by a single investigator every 2–3 days with digital calipers (Thermo Fisher Scientific, Grand Island, NY). Mice with tumors >1.5 cm in greatest dimension were sacrificed in accordance with institutional policy.
For IA ACT, a mouse model for isolated limb perfusion was modified.21 Briefly, mice were anesthetized with isoflurane (Piramal Pharma, Mumbai, India). Hair from the ipsilateral hind leg was removed with depilatory cream. A longitudinal incision was made on the thigh exposing the femoral vessels. The superficial femoral artery was identified distal to the take-off of the profunda femoris artery and 6-0 silk suture (Ethicon Inc., Somerville, NJ) was passed around the artery proximally and distally. A 0.36 mm outer diameter catheter (Scientific Commodities Inc., Lake Havasu City, AZ) was then threaded distally through a small arteriotomy. The silk suture was then tied in place to secure the catheter without femoral vein injury. Although the mouse was still under anesthesia, T cells were delivered via bolus dosing or via peristaltic pump (Cole Palmer Instrument Co, Vernon Hills, IL) infusion at 30 μL/min for 10 minutes. IV ACT was performed through bolus injection of cells via the tail vein.
Integrin Blockade
C57BL/6 mice with established tumors were injected IV through the tail vein with αL-integrin blocking antibody 50 μg/mouse (CD11a Clone 2D7; BD Biosciences) or isotype control antibody (Rat IgG2a, κ; BD Biosciences) 20 minutes before IV ACT. IV ACT was performed with 10×106 OT-1 T cells, and mice sacrificed after 3 hours to examine early lymphocyte trafficking. Liver, lung, spleen, and tumor were harvested, and T-cell infiltration was evaluated by flow cytometry. Experiments were performed in triplicate and results are reported as mean number of T cells±SD.
Cell Trafficking
Cell Tracker Orange (Invitrogen, Carlsbad, CA) and carboxyfluorescein succinimidyl ester (Invitrogen) were used to label OT-1 T cells. Cell Tracker Orange–labeled cells were injected IA, and carboxyfluorescein succinimidyl ester–labeled cells were injected IV into wild-type C57BL/6 mice with established B16-OVA tumors. Mice were sacrificed at 1 and 24 hours postinjection, and the liver, spleen, lymph nodes, lungs, and tumor were harvested. Tissues were manually disaggregated using a MediMachine (BD Biosciences) analyzed by flow cytometry or flash frozen in OCT compound, cryosectioned in 9 µm sections, stained with 4’,6-diamidino-2-phenylindole, mounted and examined by immunofluorescence microscopy for localization within tumor tissue.
Statistical Analyses
For tumor growth and lymphocyte trafficking experiments, comparison between groups was performed using the Student t test with statistical significance accepted for P<0.05. Kaplan-Meier survival curves were generated through standard methods with statistical significance determined by log-rank test and P<0.05.
RESULTS
Activated T cells are Retained in the Lungs Following IV Administration
To identify factors that may lead to the decreased availability of adoptively transferred cells to enter sites of tumor, the adoptive transfer of OT-1 T cells into B16-OVA tumor bearing mice via tail vein injection was examined. Enumerating the adoptively transferred cells by FACS demonstrated that the vast majority of administered cells localize to the lung in the short term, using a 3-hour time point, to allow cells to complete the initial intravastation into tissues, but not traffic through tissues and exit back into circulation (Fig. 2). At this same time point, only 0.4% of adoptively transferred cells can be detected in the tumor. Since the mechanism of retention of the adoptively transferred cells in the lung could be either an active or passive process, the blockade of firm adhesion was investigated. As the prototypical mediator of firm adhesion, integrin blockade before adoptive transfer significantly decreased the T-cell retention in the lung by ~50%. However, there was still a marked retention of T cells in the lung as compared with other organs indicating that T cells are retained in the lung via “active” cell adhesion processes as well as “passive” processes. As this route of administration demonstrated significant differences at peripheral sites, infiltration into sites of tumor were investigated using IA or IV ACT delivery.
FIGURE 2.
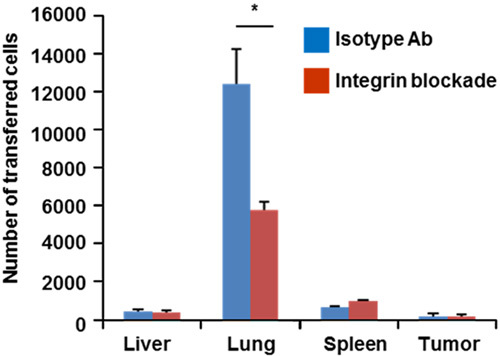
Retention of adoptively transferred lymphocytes 3 hours after IV injection. 1×107 cells were injected into recipient mice. Blocking integrin interactions before transfer partially abrogates lung retention suggesting both active and passive mechanisms. Transferred cells recovered from subcutaneous tumor only account for 0.4% of total cells recovered (*P<0.05). Ab indicates antibody.
IA Administration of T Cells Demonstrates Increased Tumor Infiltration
The adoptive transfer of OT-1 T cells into mice bearing B16-OVA tumor in the distal thigh via both IA injection into the ipsilateral femoral artery and IV tail vein injection showed that IA injected cells qualitatively appeared to be more numerous by immunofluorescence microscopy (Fig. 3). Irrespective of route of administration, the T cells appeared to infiltrate the tumor and were not confined to the periphery. Moreover, of note, the adoptively transferred lymphocytes were vastly outnumbered by the tumor with the bulk consisting of melanoma cells as opposed to stroma. Quantitative analysis using flow cytometry confirmed a statistically significant increase in accumulation of IA delivered cells as compared with IV at the critical sites of tumor and lymph node at 24 hours (Fig. 4 and Supplemental Fig. 1, Supplemental Digital Content 1, http://links.lww.com/JIT/A506). The number of IA ACT cells recovered from the tumor and lymph node was 1.6 and 3.1 times higher, respectively, than recovered IV ACT cells at 24 hours. However, a greater number of ACT cells were also noted in the liver and lung at 24 hours following IA versus IV cell transfer indicative of increased accumulation at competing sites. Although the IA approach would have been anticipated to be favorable for immediate tumor infiltration, the 1 hour time point after adoptive transfer favored the IV approach, suggesting that this time point may be too early to reflect the sum of subsequent trafficking that produces clinical effects. To determine if these observed differences in adoptive cell trafficking were related to clinical outcomes, growth and survival studies were performed.
FIGURE 3.
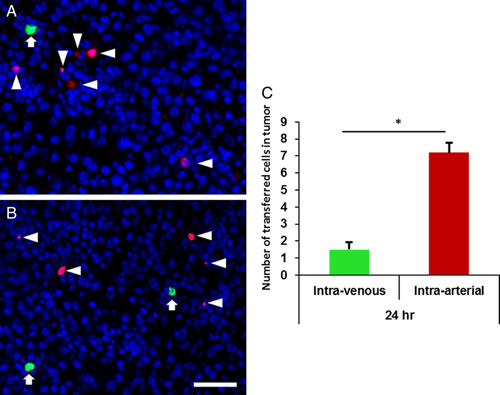
A–C, Representative photomicrographs of immunofluorescence images of tumor tissue 24 hours after adoptive transfer with 1×107 green CFSE-labeled cells IV (arrows) and 1×107 orange CTO-labeled cells intra-arterial (arrow heads). DAPI-stained nuclei of endogenous cells within the tumor appear as blue background. CFSE indicates carboxyfluorescein succinimidyl ester; CTO, Cell Tracker Orang; DAPI, 4’,6-diamidino-2-phenylindole. *P<0.05.
FIGURE 4.
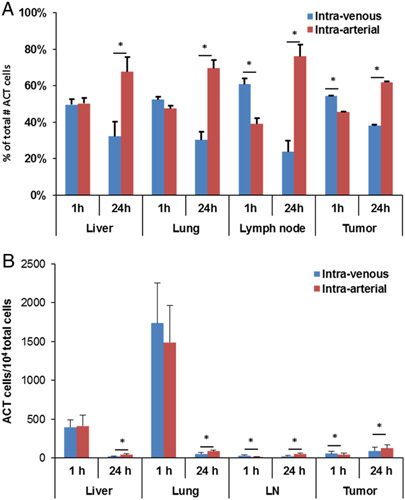
Percentage of total adoptively transferred cells (A) and total number of adoptively transferred cell/104 total cells (B) identified by flow cytometry in various tissues after ACT. Intra-arterial injection resulted in increased tumor infiltration of T cells and lymph node accumulation at 24 hours (*P<0.05). ACT indicates adoptive cell transfer; LN, lymph node.
Similar Tumor Growth Control and Survival Between IA and IV ACT
To determine the clinical effect of the lymphocyte trafficking differences noted in the IA versus IV ACT groups, we analyzed tumor growth and survival. B16-OVA tumor bearing mice underwent no treatment (control), IA ACT, IV ACT, or sham IA control. The sham IA control group was performed using the femoral artery cannulation as described, but did not infuse T cells. This control group was necessary to ensure that ligation of the femoral artery did not result in differential tumor growth from ischemia or surgical manipulation. No difference was seen between the no treatment control group and sham IA injection group (data not shown).
Delivery of 5×106 OT-1 T cells via IA or IV ACT uniformly produced significant reductions in tumor growth compared with untreated controls (Fig. 5). However, comparing IA to IV ACT, no significant differences were noted in tumor growth between groups. Given the differences noted in trafficking between these groups, corresponding differences in antitumor efficacy would have been anticipated. As IA cells were delivered through a bolus in this experiment and those preceding, with significant retention in the lungs for both IA and IV groups, it was hypothesized that a bolus IA injection may result in the tumor capillary bed being overwhelmed by T cells. In clinical practice cells would not be given as a bolus, but rather via a controlled infusion, so it was postulated that a 10 minute infusion of T cells would allow for increased uptake of cells through the tumor capillary bed and possibly improved tumor growth control. OT-1 T cells were infused via IA at 3 doses: 1×106, 5×106, and 10×106 and compared with our standard IV ACT at the same doses. These manipulations would not only mimic clinical conditions, but also determine if the variable of infusion time and cell number infused could influence tumor growth. IA ACT via 10 minute infusion did not result in improved tumor growth curves at any dosage as compared with IV ACT (Fig. 6). Of note, regardless of the route of administration there was a direct association between the number of cells infused and reduction in tumor growth with mice receiving 10×106 ACT cells having the greatest tumor control. Although not directly compared to IA bolus dosing in these experiments, the tumor volume measurements were consistent with the prior observations indicating that bolus versus slow infusion had no measurable influence on clinical outcome in our model system.
FIGURE 5.
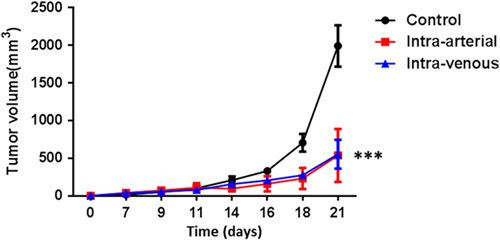
Treatment of established B16 melanoma tumor with 5×106 T cells given IA (given as a bolus) or IV. IA compared with IV ACT shows no difference in tumor growth no treatment control. However, both forms of ACT show improvement as compared with no treatment control (day 0, tumor inoculation; day 7, T-cell adopted transfer; ***P<0.0001). ACT indicates adoptive cell transfer; IA, intra-arterial.
FIGURE 6.
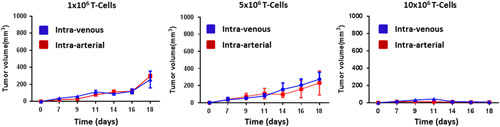
Treatment of established B16 melanoma tumor with various doses of T cells given IA (infusion over 10 min) or IV. IA compared with IV ACT shows no improvement in tumor growth curves at given doses (day 0, tumor inoculation; day 7, T-cell adoptive transfer; n=5). IA indicates intra-arterial.
Both IV and IA ACT significantly improved survival compared with no treatment controls (Fig. 7). All mice in the control group died within 14 days, whereas all mice in the IV and IA groups survived at least 20 days with approximately half having long-term survival (>50 d) with no evidence of tumor. As regards potential differences in survival between IA and IV ACT groups, which may be independent of tumor growth control during immunotherapy treatments, no significant difference was noted. In both IA and IV ACT groups, long-term survival was evident by day 20 after adoptive transfer with no late deaths.
FIGURE 7.
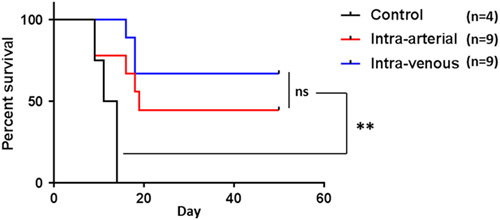
Treatment of established B16 melanoma tumor with 10×106 T cells given IA or IV. IA and IV ACT both significantly improve survival over no treatment control (**P<0.001). However, No difference was seen in IA ACT versus IV ACT (P=0.3). ACT indicates adoptive cell transfer; IA, intra-arterial; ns, not significant.
DISCUSSION
The rationale for IA delivery of adoptively transferred lymphocytes for tumor immunotherapy is based on several observations suggesting that IA may increase the therapeutic efficacy of ACT.17,18,22 By delivering cells directly into the arterial supply of the tumor, retention and/or sequestration in the capillary beds of the lungs and spleen may be avoided and allow the adoptively transferred cells an immediate opportunity for tumor infiltration. In the initial 24 hours after adoptive transfer of activated lymphocytes, it has been clinically demonstrated that there is rapid uptake into the lungs with clearance over the course of several days.16 It is also apparent that the liver and spleen appear to be large “sinks” for the adoptively transferred cells. As the degree of tumor infiltration by antitumor lymphocytes is directly related to the clinical response to virtually all forms of immunotherapy, the critical examination of IA cellular immunotherapy is highly relevant especially in the era of chimeric antigen receptor–modified T-cell therapy and burgeoning adoptive immunotherapy protocols. The retention of the majority of adoptively transferred lymphocytes at sites outside the tumor limits the number of cells available for tumor infiltration. Our model clearly demonstrated an improvement in tumor infiltration of adoptively transferred cells when given by the IA route; however, a clinical benefit beyond the IV route was not found. The limited clinical and animal data concerning IA ACT may offer some degree of context to these findings.
Cognizant of the potential importance of directed delivery of cells to the tumor bed, rare case reports exist of the clinical superiority with IA delivery. In 1992, a patient with metastatic melanoma to the liver was found to have an improved response to IA delivery versus IV delivery of lymphokine-activated killer (LAK) cells.17 The study involved 4 separate adoptive transfers with LAK cells with the first and second treatments solely via the IV route. In the third treatment, 33% of the LAK cells were administered via the right hepatic artery with the remainder given IV. Partial response was noted in the right liver, but progression of disease in the left liver. The fourth treatment, therefore, consisted of 33% of LAK cells delivered via the right hepatic artery, 33% delivered via the left hepatic artery and the remainder given IV. Follow-up imaging demonstrated responses in both lobes of the liver suggesting improvement with directed infusion to tumor sites. A similar experience with a single patient with regionally metastatic melanoma of the head and neck was reported in 2003.18 This patient underwent multiple infusions of TILs with the first through third treatments delivered IV resulting in partial response of the melanoma lesions on the patient’s face and upper neck. Subsequent catheter-directed IA infusion of TIL into the thyrocervical artery, that supplied these tumors, lead to a significantly improved response. Eventual loss of HLA-A2 expression rendered future cellular immunotherapy ineffective in this patient.
Although these clinical case reports offer some insight into the feasibility and efficacy of regionally delivered ACT, a preclinical animal model previously reported also showed potential benefit of IA delivery, but had significant limitations. In a melanoma model metastatic to bone, tumor-draining lymph node cells were delivered IA compared with IV.22 IV ACT was unable to clear visible tumor deposits from the femur of mice with metastatic melanoma. However, when injecting the same number of cells IA, all mice were cleared of visible tumor. Unfortunately, a major limitation of applying this study clinically was that the IA injections were via the cardiac left ventricle, a procedure that is not feasible in humans and likely lacked the specificity of catheter-directed infusion.
Our current study utilized a mouse model that mimics clinical application with the only major difference being the ligation of the superficial femoral artery required in the mouse model, which would be unnecessary in clinical catheter-based treatments. To account for this difference and the potential for antitumor effects from ischemia, a sham procedure was performed which showed no effect on tumor growth compared with no treatment control mice. Ischemia as a contributing factor to clinical outcome was unlikely as sufficient collateral flow from the profunda femoris ensured adequate blood supply to the lower leg and tumor in this model. With adoptive transfer, IA delivery was clearly associated with increased accumulation of injected T cells in the tumor microenvironment at 24 hours. Coupled with data demonstrating that adoptive lymphocyte retention in the lungs is both an active and passive process, directed infusion likely bypassed this potential “sink.” It is interesting to note that, the number of transferred lymphocytes recovered was also higher in the lymph nodes after IA ACT suggesting better seeding of a critical reservoir that could support lymphocyte persistence. The increased presence of IA ACT lymphocytes in the lung and liver at 24 hours as compared with IV ACT potentially reflects a delayed redistribution as the majority of cells ultimately return through the venous system.
Although favorable lymphocyte distribution was noted with IA ACT, the clinical outcomes of tumor growth inhibition and survival were no different than IV administration. Even when accounting for variables associated with infusion time and number of cells infused, no difference in tumor growth could be detected. A possible explanation for this disconnect between trafficking and clinical outcome may be specific to our model as the adoptively transferred cells were all antigen specific and highly potent. Perhaps clinical differences would be noted with a mixed population of effector cells with various potencies as would be seen clinically. Moreover, our model is likely homogeneous in terms of adhesion molecule presence on the tumor vasculature which may not be the situation in humans, especially those that may have had prior treatments which may favor improved adhesion molecule expression and an IA approach (eg, radiation therapy). Unlike clinical case reports, a limitation of our model is that it only examined a single adoptive transfer and any potential “priming” of lymphocyte trafficking from prior adoptive transfers would not be achieved. Inherent to any mouse model, including the one presented here, is the anatomic differences and size of blood vessels as compared with humans that could be a confounding variable for clinical outcome.
In summary, it appears that IA delivery of anticancer lymphocytes during adoptive immunotherapy can enhance infiltration into tumor sites. As tumor cell vasculature is dysfunctional and deficient in adhesions molecules,23,24 any technique to overcome these limitations may be beneficial. Further exploration of this approach in clinical trials is warranted as it represents a low-cost, low-risk intervention to potentially augment responses.
CONFLICTS OF INTEREST/FINANCIAL DISCLOSURES
This study was supported by P30 CA016056 from Cancer Center Support Grant at Roswell Park Cancer Institute. All authors have declared there are no financial conflicts of interest with regard to this work.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.
REFERENCES
- 1.Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. N Engl J Med. 1988;319:1676–1680. [DOI] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Yang JC, Sherry RM, et al. Durable complete responses in heavily pretreated patients with metastatic melanoma using T-cell transfer immunotherapy. Clin Cancer Res. 2011;17:4550–4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang LX, Huang WX, Graor H, et al. Adoptive immunotherapy of cancer with polyclonal, 108-fold hyperexpanded, CD4+ and CD8+ T cells. J Transl Med. 2004;2:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yee C, Thompson JA, Byrd D, et al. Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effect of transferred T cells. Proc Natl Acad Sci USA. 2002;99:16168–16173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hinrichs CS, Spolski R, Paulos CM, et al. IL-2 and IL-21 confer opposing differentiation programs to CD8+ T cells for adoptive immunotherapy. Blood. 2008;111:5326–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Q, Carr AL, Donald EJ, et al. Synergistic effects of IL-12 and IL-18 in skewing tumor-reactive T-cell responses towards a type 1 pattern. Cancer Res. 2005;65:1063–1070. [PubMed] [Google Scholar]
- 7.Morgan RA, Dudley ME, Wunderlich JR, et al. Cancer regression in patients after transfer of genetically engineered lymphocytes. Science. 2006;314:126–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med. 2013;5:177ra38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Singh N, Shi J, June CH, et al. Genome-editing technologies in adoptive t cell immunotherapy for cancer. Curr Hematol Malig Rep. 2017;12:522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maus MV, June CH. Making better chimeric antigen receptors for adoptive T-cell therapy. Clin Cancer Res. 2016;22:1875–1884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maine GN, Mule JJ. Making room for T cells. J Clin Invest. 2002;110:157–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mukai S, Kjaergaard J, Shu S, et al. Infiltration of tumors by systemically transferred tumor-reactive T lymphocytes is required for antitumor efficacy. Cancer Res. 1999;59:5245–5249. [PubMed] [Google Scholar]
- 13.Carlos TM. Leukocyte recruitment at sites of tumor: dissonant orchestration. J Leukoc Biol. 2001;70:171–184. [PubMed] [Google Scholar]
- 14.Fisher DT, Chen Q, Skitzki JJ, et al. IL-6 trans-signaling licenses mouse and human tumor microvascular gateways for trafficking of cytotoxic T cells. J Clin Invest. 2011;121:3846–3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skitzki J, Craig RA, Okuyama R, et al. Donor cell cycling, trafficking, and accumulation during adoptive immunotherapy for murine lung metastases. Cancer Res. 2004;64:2183–2191. [DOI] [PubMed] [Google Scholar]
- 16.Fisher B, Packard BS, Read EJ, et al. Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. J Clin Oncol. 1989;7:250–261. [DOI] [PubMed] [Google Scholar]
- 17.Keilholz U, Schlag P, Tilgen W, et al. Regional administration of lymphokine-activated killer cells can be superior to intravenous application. Cancer. 1992;69:2172–2175. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg SA, Yang JC, Robbins PF, et al. Cell transfer therapy for cancer: lessons from sequential treatments of a patient with metastatic melanoma. J Immunother. 2003;26:385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okuno K, Takagi H, Nakamura T, et al. Treatment for unresectable hepatoma via selective hepatic arterial infusion of lymphokine-activated killer cells generated from autologous spleen cells. Cancer. 1986;58:1001–1006. [DOI] [PubMed] [Google Scholar]
- 20.Katz SC, Burga RA, McCormack E, et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin Cancer Res. 2015;21:3149–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim M, Camoriano M, Muhitch JB, et al. A novel mouse model of isolated limb perfusion for extremity melanoma. J Surg Res. 2012;178:294–298. [DOI] [PubMed] [Google Scholar]
- 22.Ruttinger D, Li R, Urba WJ, et al. Regression of bone metastases following adoptive transfer of anti-CD3-activated and IL-2-expanded tumor vaccine draining lymph node cells. Clin Exp Metastasis. 2004;21:305–312. [DOI] [PubMed] [Google Scholar]
- 23.Fisher DT, Muhitch JB, Kim M, et al. Intraoperative intravital microscopy permits the study of human tumour vessels. Nat Commun. 2016;7:10684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fisher DT, Chen Q, Appenheimer MM, et al. Hurdles to lymphocyte trafficking in the tumor microenvironment: implications for effective immunotherapy. Immunol Invest. 2006;35:251–277. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.immunotherapy-journal.com.