

Supplemental Digital Content is available in the text.
Key Words: congenital dyserythropoietic anemia, KLF1, hydrops fetalis, sex reversal, CD44, CD71, DRAQ5, alpha spectrin, HPFH
Abstract
We identified a child with KLF1-E325K congenital dyserythropoietic anemia type IV who experienced a severe clinical course, fetal anemia, hydrops fetalis, and postnatal transfusion dependence only partially responsive to splenectomy. The child also had complete sex reversal, the cause which remains undetermined. To gain insights into our patient’s severe hematologic phenotype, detailed analyses were performed. Erythrocytes from the patient and parents demonstrated functional abnormalities of the erythrocyte membrane, attributed to variants in the α-spectrin gene. Hypomorphic alleles in SEC23B and YARS2 were also identified. We hypothesize that coinheritance of variants in relevant erythrocyte genes contribute to the clinical course in our patient and other E325K-linked congenital dyserythropoietic anemia IV patients with severe clinical phenotypes.
Congenital dyserythropoietic anemia (CDA) type IV has been linked to a heterozygous, gain-of-function mutation of the erythroid transcription factor KLF1; E325K.1–5 This report describes a child with CDA type IV due to KLF1 E325K who presented with fetal anemia and nonimmune hydrops fetalis, postnatal transfusion dependence, and only partial response to splenectomy. She also exhibited complete 46 XY sex reversal. Detailed laboratory and genetic analyses provided insights into our patient’s severe hemolytic anemia phenotype.
CASE REPORT
The patient, now 10 years of age, was the second child born to a nonconsanguineous Ashkenazi Jewish couple. Genetic amniocentesis revealed a fetal karyotype of 46 XY. Pregnancy surveillance identified hydrops fetalis and fetal anemia, hemoglobin 4 g/dL, at 25-week gestation. Hemolytic anemia from blood group incompatibility was excluded. The fetus received 2 in utero transfusions before an elective Cesarean section at 33-week gestation. Birth weight was 1360 g. The infant was hydropic with splenomegaly; there were no skeletal deformities. Despite the karyotype results, the neonate was phenotypically female with a normal vagina, uterus, and what appeared to be ovaries were seen on ultrasound. There was severe anemia and pRBC transfusions were administered. She required transfusion support approximately once every 4 to 6 weeks for the first several years of life. No genetic cause for the hemolytic anemia nor for the sex reversal was identified despite extensive laboratory testing. Complete androgen insensitivity syndrome was excluded by clinical testing. Splenectomy was performed at 4 years of age because of the concerns for iron accumulation and only a modest response to deferasiriox. Transfusion requirement abated, although moderate to severe anemia requiring occasional transfusion persists.
Presplenectomy, postsplenectomy, and recent blood counts are shown in Table 1. The peripheral smear presplenectomy showed spherocytes, spiculated erythrocytes, and nucleated red blood cells, with some clover leaf nuclei. After splenectomy, there was marked normoblastosis, up to 10-fold in excess of the total leukocyte count, with prominent large, round macrocytes (Fig. 1A). The fetal hemoglobin level was 34.6%. Bone marrow morphology showed a predominantly erythroid marrow (83% erythroid precursors, 3% lymphocytes, 15% myeloid cells). Of the erythroid precursors, the majority were orthochromatic with 3% binucleate forms and 3% cells with clover leaf nuclei. There were some tight clusters of orthochromatic erythroblasts, similar to that described in early fetal erythropoiesis (Fig. 1B). Delayed enucleation of orthochromatic normoblasts was suggested by the asynchrony of the nuclear versus cytoplasmic maturation, with centrally spaced nuclei. Peripheral blood smears from both parents were normal.
TABLE 1.
Hematologic Findings Before and After Splenectomy

FIGURE 1.

Laboratory studies. A, Peripheral blood smears. Peripheral blood smears from the proband after splenectomy shows spherocytes, unusually large round macrocytes, spherocytes, spiculated red blood cell and nucleated red blood cells; magnification ×1000. B, Bone marrow smear. Tight clusters of orthochromic erythroblasts are seen in bone marrow smears from the proband; a clover leaf form noted at the upper edge and binucleate form middle right. C, Osmotic gradient ektacytometry. Red cell deformability studies by osmotic gradient ektacytometry shows reduced deformability in the proband (low DI max); the rightward shift suggests presence of large erythrocytes with low MCHC. Ektacytometry of erythrocytes from the father and mother shows a pattern consistent with mild spherocytosis. D, Eosin-5′-maleimide-binding (EMA) binding. EMA studies of erythrocytes from the proband and both parents demonstrated decreased fluorescence consistent with a defect in the erythrocyte membrane. Patterns in dominant spherocytosis (HS) are shown for comparison. E, Isoelectric focusing of hemoglobin. Isoelectric focusing shows the presence of embryonic hemoglobin in erythrocytes from the proband. F, Sanger sequencing. Sanger sequencing confirmed heterozygosity for the G to A substitution in the KLF1 gene and wild-type status for the mother and father (not shown).
Osmotic gradient ektacytometry performed postsplenectomy demonstrated a unique pattern indicating presence of erythrocytes with low mean corpuscular hemoglobin concentration in the proband; erythrocytes from the parents revealed a pattern consistent with mild hereditary spherocytosis (Fig. 1C).6 Eosin-5′-maleimide-binding (EMA) studies of erythrocytes from the proband and both parents demonstrated decreased fluorescence consistent with a defect in the erythrocyte membrane (Fig. 1D). Erythrocyte enzyme analyses, including erythrocyte pyruvate kinase activity, were normal.
The severity of anemia, the elevated nucleated red blood cell (nRBC) count, high fetal hemoglobin (HbF) and elevated mean corpuscular volume suggested that the high HbF is not likely due to the known disorders of familial hereditary persistence of high fetal hemoglobin. Rather the findings were similar to the observations described in the first 2 cases of CDA type IV due to KLF1 E325K.1,2 CD44 expression on red cells was used as a screening tool and additional confirmatory tests were performed. CD44 expression (CD71− population) was absent on mature erythrocytes from the proband (Fig. 2B) compared with its expression on mature erythrocytes from the parents and a control. CD44 was also reduced or absent in CD71+ cells from the proband (reticulocytes and nucleated red blood cells [CD45−, DRAQ5+, CD71+ cells) in contrast to its presence on reticulocytes and nRBC from a thalassemia major patient with elevated numbers of circulating nRBC and on cells in a normal control bone marrow (Fig. 2C). CD44 expression was present in patient’s lymphocytes but was decreased compared with control values (not shown). Sanger sequencing of the KLF1 gene, performed as described,2 revealed the proband was heterozygous for the E325K KLF1 variant associated with CDA type IV (NM_006563:c.G973A:p.E325K) (Fig. 1E). Neither parent carried the E325K variant (Fig. 1F; data on father not shown), suggesting this occurred de novo or 1 parent is mosaic.
FIGURE 2.
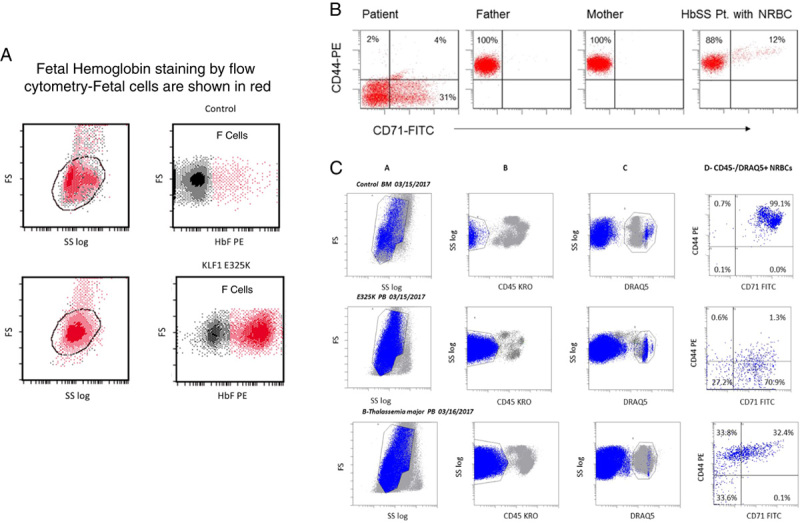
A, Fetal hemoglobin staining by flow cytometry—2 distinct population of cells can be seen suggesting a heterocellular pattern of staining for fetal hemoglobin (shown in red). B, CD44 staining of peripheral blood red blood cell (RBC). CD71 was used to mark reticulocytes and immature red cells. Red cells from the proband are deficient in CD44 expression and as well the immature RBC containing reticulocytes and nucleated red blood cell (nRBC). Normal staining in the parents and in a sickle cell anemia patient with high reticulocytes and nRBC are shown for comparison. C, CD44 staining in nRBC was further assessed by using the nuclear stain DRAQ5. CD45 was used to mark white blood cell/myeloid fraction (column A). In a normal control marrow, erythroid precursors (CD45−, DRAQ5+, CD71+—column D) showed the expected positive staining for CD44 (top row) and in a child with β-thalassemia major with high nRBC (bottom row). In contrast the nRBC in the proband (middle row) show markedly decreased staining with CD44.
Identification of the KLF1 E325K mutation prompted detailed hemoglobin analyses based on previous cases. Isoelectric focusing of erythrocyte hemoglobin yielded a pattern identical to that published by Arnaud and colleagues, indicating the presence of embryonic hemoglobins (Fig. 1F). Presence of ζ-globin was confirmed by proteomic analysis of low–molecular-weight bands on the sodium dodecyl sulfate polyacrylamide gel electrophoresis of red cell membranes (not shown). By flow cytometry the distribution of HbF was heterocellular (Fig. 2A).
Additional genetic variants contributing to the patient’s clinical course were sought. With parental consent; patient and parental samples were subjected to whole exome sequencing (WES) to identify other coinherited mutations in red cell genes. Data were processed using the SeattleSeq annotation platform (http://snp.gs.washington.edu/SeattleSeqAnnotation137) to identify the pathologic variants. Erythrocyte membrane protein genes were initially analyzed due to the abnormal erythrocyte EMA binding and ektacytometry results in the proband and her parents. Numerous nonsynonymous coding sequence variants in the α-spectrin gene (SPTA1) were found in the proband (Supplemental Table 1, Supplemental Digital Content 1, http://links.lww.com/JPHO/A231).7
The proband inherited several variant SPTA1 alleles from the father and mother (Supplemental Table 1, Supplemental Digital Content 1, http://links.lww.com/JPHO/A231) including the αLELY allele (NM_003126:c.5572C>G:p.L1858V), which may result in reduced α-spectrin membrane content.8,9 Another variant allele was the αBughill allele (NM_003126:c.2909C>A:p.A970D), inherited from the mother. This allele is associated with recessive hereditary spherocytosis, is associated with a mild hereditary spherocytosis “carrier state,”10 and is frequently inherited with the αLEPRA mutation. The αLEPRA allele was not detected in either parent or the proband. No functional studies of the αBughill allele have been performed to inform its contribution to spectrin function, but αBughill homozygous patients without the αLEPRA mutation exhibit mild spectrin deficiency and the hereditary spherocytosis carrier state.10 No predicted pathogenic mutations were found in the coding regions or intron/exon junctions of the β-spectrin, ankyrin-1, band 3, protein 4.2 or other erythrocyte membrane genes. These studies do not exclude deep intronic or regulatory element mutations or small genic deletions.
Other red cell disease genes were examined for potential variants contributing to the proband’s phenotype. Hypomorphic variants with significantly elevated CADD scores were noted in SEC23B (NM_001172745:c.1467C>G:p.H489Q), the CDA type II gene, and in YARS2 (NM_001040436:c.572G>T:p.G191V0), a pathogenic variant in a gene linked to a type of sideroblastic anemia.
DISCUSSION
Phenotypic features and the clinical course of 5 cases of KLF1 E325K-associated CDA type IV have been reported (Table 2). Review reveals 2 distinctive clinical courses, one characterized by a mild course, with childhood anemia or transfusion dependence in infancy that resolves spontaneously, and the other characterized by a more severe course. Severely affected patients present with fetal anemia and hydrops fetalis followed by postnatal transfusion dependence; splenectomy is palliative but not curative. To gain insight into our patient’s severe hematologic phenotype and the sex reversal phenotype, detailed laboratory and genetic analyses were performed.
TABLE 2.
Clinical and Laboratory Findings in Cases of KLF1 E325K-associated Congenital Dyserythropoietic Anemia Type IV

Abnormal EMA binding and ektacytometry of erythrocytes from the patient and parents identified functional abnormalities of the erythrocyte membrane. Analyses of WES data, while finding numerous membrane protein gene variants, did not provide a specific genetic diagnosis for the mild HS/HS carrier phenotype in either parent. The mother’s mild clinical picture could be attributed to the αBugHill allele as previously described,10,15 or other as yet unidentified variants. It is possible that the numerous membrane protein gene variants, for example in SPTA1, in cis or trans may lead to a cumulative effect on the protein of interest, with variants interacting to produce a clinical phenotype as described in other proteins.16
Other variant alleles in 2 other red blood cell disease-associated genes, SEC23B and YARS2, that may have contributed to the severity of our patient’s clinical course were also identified. Hypomorphic mutations in SEC23B have been associated with mild phenotypes in CDA type II.17 Similarly, a wide spectrum of phenotypic and genotypic variability have been described in the YARS2 mitochondrial myopathy, lactic acidosis, and sideroblastic anemia syndrome, including isolated anemia presenting in adulthood.18 Taken together, we hypothesize that coinheritance of variants in relevant erythrocyte genes contribute to the clinical course in our patient and other E325K-linked CDA IV patients with severe clinical phenotypes.
In 2 of the cases with severe phenotypic features, the patients were phenotypic males with urogenital anomalies.2 One patient had micropenis with hypospadias and had growth retardation unresponsive to human growth hormone while our case had sex reversal. In both cases, the etiology of these findings remains unexplained. A child with a KLF1 null phenotype due to 2 different loss of function mutations who presented with anemia and hydrops fetalis and severe postnatal growth retardation was recently described.19 This patient did not exhibit urogenital anomalies.
The KLF1 E325K mutation has a strong dominant negative effect, like the mutation in the homologous amino acid, E339D, in the neonatal anemia nan mouse. This mutation not only alters affinity of KLF1 for its cognate binding sites across the genome, it also binds to degenerate KLF1 motifs not normally occupied by KLF1 in a promiscuous manner. Together, these alterations lead to numerous changes in erythroid gene expression, altering expression of direct KLF1 target genes and inducing ectopic expression of genes not typically expressed.20,21 KLF1 mutant erythrocytes may have defects in proteins involved in growth and differentiation, maintenance of membrane integrity, hemoglobins and heme biosynthesis, iron homoeostasis, and regulation of metabolism.22
The role of KLF1 in erythropoiesis has recently been further refined.7 In normal erythropoiesis, there is an orderly, progressive reduction in CD44 expression from proerythroblast to the orthochromatic erythroblast along with changes in the distribution of CD44 within the cell.23,24 Similar to reported cases, loss of CD44 appears near total in mature erythrocytes (CD71− fraction, Fig. 2B). Circulating orthochromatic erythroblasts (normoblasts; CD45−, DRAQ5+, CD71+ fraction, Fig. 2C) are predominantly negative for CD44, a finding not noted in the previously published reports. The loss of CD44 in early erythroid precursors could play an important role in KLF1 E325K-mediated disruption of the transition from fetal to adult erythropoiesis in the KLF1 mutant cases.25 In addition, emerging data show that CD44, along with several other genes, for example SDF1/CXCLR4, VCAM/VLA-4, are involved in the hematopoietic stem cell and bone marrow niche interaction.25,26 The presence of trace amounts of embryonic hemoglobin, the unusually large round macrocytes, the enucleation defect (Fig. 1) and the heterocellular pattern of fetal hemoglobin (Fig. 2) taken together suggest the persistence of a clone of fetal erythroid cells.
The exact cause of the 46 XY sex reversal in our patient is not yet determined. Our patient did not have any other congenital anomalies, associated with syndromic gonadal dysgenesis cases.27 In the WES studies, an evaluation of exons and exon-intron boundary regions of the following genes linked to male sex reversal syndromes failed to identify a pathogenic mutation—SRY, AMH. AMHR, ATRX1, DAX1, DHH, DMRT1, FOG2, GATA4, HOXB2, HOXB3, MAP3K, NR5A1, SOX9, and WT1 (genes are from OMIM).
Clinical management of KLF1 E325K-assoiated CDA IV remains empiric. Severe anemia is treated with transfusion support. An attempt to treat anemia in one case with erythropoietin failed.2 We did not prescribe erythropoietin, but in vitro erythroid cultures from our patient failed to grow, while there was normal growth of granulocyte, macrophage colonies (not shown, studies kindly done by James Palis, Rochester). It is important to monitor for iron overload and treat when indicated.
Poor somatic growth was described in 1 patient that was unresponsive to growth hormone and thyroid supplementation.2 This is less likely attributable to end organ effects from iron accumulation, as transfusion dependency abated after splenectomy at age 4 and one would have expected ferritin levels to have normalized soon thereafter. In our case, ferritin values normalized 9 months after splenectomy without chelation and remain normal to date. In cases with growth and endocrine changes, imaging studies with magnetic resonance imaging (T2* or superconducting quantum interference device) may be necessary to exclude tissue iron accumulation. Although our patient can maintain a hemoglobin of 9 to 10 g/dL and shows no evidence of growth retardation, there is a mild high receding forehead and mid face dysplasia reminiscent of Cooley anemia with skull radiographs and computerized tomography showing marked expansion of the diploic space. Thus of the 5 cases included in this review, all 3 karyotypic males with CDA IV seem to have a more severe, often transfusion-dependent course compared with the milder course in the 2 females with the same mutation. Because of ongoing health concerns in our patient, hematopoietic stem cell transplantation, an option for severely affected KLF1 E325K CDA IV patients, was considered but deferred for now as the only sibling was not a full match on HLA typing.
Summary
A comparison of the first 5 cases with KLF1 E325K mutation suggests that the spectrum of clinical effects of this mutation can vary from moderate to severe, transfusion-dependent anemia. The presence of modifier alleles in non-KLF1 genes associated with congenital anemia may lead to the wide variability in clinical phenotypes observed. The concurrent presence of urogenital anomalies in 2 of the karyotypic males remains unexplained.
Supplementary Material
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.jpho-online.com.
ACKNOWLEDGMENTS
Dr Senthil Sundaram provided invaluable help to the primary author in getting started with exome sequencing studies and the use of various annotation Websites. A special thanks to Dr Laurence Boxer (University of Michigan) for his encouragement and sharing in the care of this child. The authors acknowledge the contributions of James Hoyer (Mayo Laboratories for Isoelectric focusing studies of hemoglobin), James Palis (Rochester, NY for in vitro erythroid colony studies), Douglas Whitten (Michigan State University, Lansing Michigan for proteomic studies), and Adele Kruger for assistance with WES analyses. The authors also acknowledge the contribution of the child and her parents to these investigative studies.
Footnotes
Y.R.: designed diagnostic strategies and wrote the manuscript; G.G., M.G., and S.B.: executed and interpreted laboratory studies; P.G.G.: performed genetic analyses and assisted in writing the manuscript; R.M.J.: supervised laboratory testing, assisted in genetic analyses, and assisted in writing the manuscript.
Presented in part in abstract form at the American Society of Hematology annual meeting [ASH Annual Meeting Abstracts. 2011;118(21):2101].
Supported by the Georgie Ginopolis Chair Award (Y.R.); the Melissa Ann Krinsky Research Fund, the David Carr Memorial Fund and NIDDK RO1DK104046.
The authors declare no conflict of interest.
REFERENCES
- 1.Wickramasinghe SN, Illum N, Wimberley PD. Congenital dyserythropoietic anaemia with novel intra-erythroblastic and intra-erythrocytic inclusions. Br J Haematol. 1991;79:322–330. [DOI] [PubMed] [Google Scholar]
- 2.Arnaud L, Saison C, Helias V, et al. A dominant mutation in the gene encoding the erythroid transcription factor KLF1 causes a congenital dyserythropoietic anemia. Am J Hum Genet. 2010;87:721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaffray JA, Mitchell WB, Gnanapragasam MN, et al. Erythroid transcription factor EKLF/KLF1 mutation causing congenital dyserythropoietic anemia type IV in a patient of Taiwanese origin: review of all reported cases and development of a clinical diagnostic paradigm. Blood Cells Mol Dis. 2013;51:71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de-la-Iglesia-Inigo S, Moreno-Carralero MI, Lemes-Castellano A, et al. A case of congenital dyserythropoietic anemia type IV. Clin Case Rep. 2017;5:248–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singleton BK, Fairweather VSS, Lau W, et al. A novel EKLF mutation in a patient with dyserythropoietic anemia: the first association of EKLF with disease in man [abstract]. Blood. 2009;114:162. [Google Scholar]
- 6.Johnson RM, Ravindranath Y. Osmotic scan ektacytometry in clinical diagnosis. J Pediatr Hematol Oncol. 1996;18:122–129. [DOI] [PubMed] [Google Scholar]
- 7.Gnanapragasam MN, Bieker JJ. Orchestration of late events in erythropoiesis by KLF1/EKLF. Curr Opin Hematol. 2017;24:183–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marechal J, Wilmotte R, Kanzaki A, et al. Ethnic distribution of allele alpha LELY, a low-expression allele of red-cell spectrin alpha-gene. Br J Haematol. 1995;90:553–556. [DOI] [PubMed] [Google Scholar]
- 9.Wilmotte R, Marechal J, Morle L, et al. Low expression allele alpha LELY of red cell spectrin is associated with mutations in exon 40 (alpha V/41 polymorphism) and intron 45 and with partial skipping of exon 46. J Clin Invest. 1993;91:2091–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tse WT, Gallagher PG, Jenkins PB, et al. Amino-acid substitution in alpha-spectrin commonly coinherited with nondominant hereditary spherocytosis. Am J Hematol. 1997;54:233–241. [DOI] [PubMed] [Google Scholar]
- 11.Tang W, Cai SP, Eng B, et al. Expression of embryonic zeta-globin and epsilon-globin chains in a 10-year-old girl with congenital anemia. Blood. 1993;81:1636–1640. [PubMed] [Google Scholar]
- 12.Parsons SF, Jones J, Anstee DJ, et al. A novel form of congenital dyserythropoietic anemia associated with deficiency of erythroid CD44 and a unique blood group phenotype [In(a-b-), Co(a-b-)]. Blood. 1994;83:860–868. [PubMed] [Google Scholar]
- 13.Agre P, Smith BL, Baumgarten R, et al. Human red cell Aquaporin CHIP. II. Expression during normal fetal development and in a novel form of congenital dyserythropoietic anemia. J Clin Invest. 1994;94:1050–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mitchell WB, Gnanapragasam MN, Jaffray JA, et al. Case Report of Erythroid Transcription Factor EKLF Mutation Causing a Rare Form of Congenital Dyserythropoetic Anemia in a Patient of Taiwanese Origin. ASH Annual Meeting Abstracts. 2011;118:2154. [Google Scholar]
- 15.Agre P, Asimos A, Casella JF, et al. Inheritance pattern and clinical response to splenectomy as a reflection of erythrocyte spectrin deficiency in hereditary spherocytosis. N Engl J Med. 1986;315:1579–1583. [DOI] [PubMed] [Google Scholar]
- 16.Yoo YJ, Sun L, Poirier JG, et al. Multiple linear combination (MLC) regression tests for common variants adapted to linkage disequilibrium structure. Genet Epidemiol. 2017;41:108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Russo R, Langella C, Esposito MR, et al. Hypomorphic mutations of SEC23B gene account for mild phenotypes of congenital dyserythropoietic anemia type II. Blood Cells Mol Dis. 2013;51:17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Riley LG, Menezes MJ, Rudinger-Thirion J, et al. Phenotypic variability and identification of novel YARS2 mutations in YARS2 mitochondrial myopathy, lactic acidosis and sideroblastic anaemia. Orphanet J Rare Dis. 2013;8:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Magor GW, Tallack MR, Gillinder KR, et al. KLF1-null neonates display hydrops fetalis and a deranged erythroid transcriptome. Blood. 2015;125:2405–2417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gillinder KR, Ilsley MD, Nebor D, et al. Promiscuous DNA-binding of a mutant zinc finger protein corrupts the transcriptome and diminishes cell viability. Nucleic Acids Res. 2017;45:1130–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Planutis A, Xue L, Trainor CD, et al. Neomorphic effects of the neonatal anemia (Nan-Eklf) mutation contribute to deficits throughout development. Development. 2017;144:430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perkins A, Xu X, Higgs DR, et al. Kruppeling erythropoiesis: an unexpected broad spectrum of human red blood cell disorders due to KLF1 variants. Blood. 2016;127:1856–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen K, Liu J, Heck S, et al. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc Natl Acad Sci USA. 2009;106:17413–17418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu J, Zhang J, Ginzburg Y, et al. Quantitative analysis of murine terminal erythroid differentiation in vivo: novel method to study normal and disordered erythropoiesis. Blood. 2013;121:e43–e49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou HS, Carter BZ, Andreeff M. Bone marrow niche-mediated survival of leukemia stem cells in acute myeloid leukemia: Yin and Yang. Cancer Biol Med. 2016;13:248–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zoller M. CD44, hyaluronan, the hematopoietic stem cell, and leukemia-initiating cells. Front Immunol. 2015;6:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biason-Lauber A. Control of sex development. Best Pract Res Clin Endocrinol Metab. 2010;24:163–186. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.jpho-online.com.