

Abstract
Although cigarette smoke is known to alter immune responses, whether and how CD4 T cells are affected is not well-described. We aimed to characterize how exposure to cigarette smoke extract impacts CD4 T cell effector generation in vitro under Th1-polarizing conditions. Our results demonstrate that cigarette smoke directly acts on CD4 T cells to impair effector expansion by decreasing division and increasing apoptosis. Furthermore, cigarette smoke enhances Th1-associated cytokine production and increases expression of the transcription factor T-bet, the master regulator of Th1 differentiation. Finally, we show that exposure to cigarette smoke extract during priming impairs the ability of effectors to form memory cells. Our findings thus demonstrate that cigarette smoke simultaneously enhances effector functions but promotes terminal differentiation of CD4 T cell effectors. This study may be relevant to understanding how smoking can both aggravate autoimmune symptoms and reduce vaccine efficacy.
Keywords: CD4 T cell, Th1 effector, T-bet, cytokines, memory, cigarette smoke
1. Introduction
Despite aggressive anti-smoking campaigns, about 15 percent of adults in the United States smoke cigarettes and roughly 500,000 deaths in the United States and 6.4 million deaths world-wide are annually attributed to exposure to cigarette smoke [1] [2]. Not surprisingly, many studies have established cigarette smoking as a risk factor in a number of inflammatory diseases [3] and many autoimmune conditions [4]. These outcomes in turn have been correlated with the ability of cigarette smoke and cigarette smoke extract exposure to impact diverse aspects of both innate and adaptive immune cells [5, 6]. Despite such analysis, detailed studies on how T cell activation, differentiation, and function are altered by cigarette smoke are lacking. This is an important area of investigation given that T cell responses are critical for orchestrating clearance of respiratory pathogens including influenza A virus [7], as well as in promoting autoimmune conditions associated with smoking including Crohn’s disease [8].
The CD4 T cell subset is critical for effective immune responses against pathogens. CD4 T cells provide ‘help’ to optimize B cell and CD8 T cell functions, and directly combat microbial invaders through the production of cytokines [9]. As they can respond to epitopes of conversed proteins shared between viral strains [10], CD4 T cells are especially important in providing immunity against influenza virus and related pathogens that are able to mutate external proteins to avoid detection by neutralizing antibody [11]. The ability of CD4 T cells to mount effective responses is dependent not only on the magnitude of the effector populations generated from naïve precursors, but also on their polarization into specialized effector subsets. For example, optimal responses against influenza and other intracellular pathogens require T helper type 1 (Th1) polarization that is typified by strong production of the cytokines interferon gamma (IFNγ) and tumor necrosis factor (TNF) [12]. Strong Th1 responses have also been correlated with promoting inflammatory bowel disease in human studies [13] and animal models [14].
Here, we use antigen presenting cells (APC) and peptide to activate CD4 T cells, and cigarette smoke extract (CSE), a well-established in vitro model of smoke exposure, to determine how CSE impacts Th1 effector expansion, function, and memory fate. We find that CSE, even when present for only the final 2 days of a 4-day culture period, reduces the yield of effector cells in a dose-dependent manner by causing a decrease in CD4 T cell division while increasing apoptosis in activated cells. Surprisingly, our results also show that CSE at the same time enhances Th1-associated cytokine production, especially boosting the number of multi-cytokine producing cells. Enhanced Th1 function driven by CSE exposure correlates with increased expression of the transcription factor T-bet which is critical for directing Th1 programming [15]. As exposure to cigarette smoke is known to impact APC function in relation to T cell priming [16–18], we also employed an APC-free system to trigger CD4 T cell activation using antibody-mediated T cell receptor and CD28 stimulation. We observed identical impacts of CSE using both the APC-dependent and the APC-independent model, indicating that CSE acts directly on the responding CD4 T cells rather than by altering signals delivered by the APC during priming.
Finally, we tested the extent to which CSE signals during priming impacts the ability of effector cells to transition to memory. Most memory cells arise from effector cells, and signals during T cell activation appear to ‘program’ memory fate [19, 20]. Our results indicate that CSE exposure during priming reduces the ability of effectors to survive the contraction phase of the immune response and to transition to memory, even in the absence of continuing CSE exposure.
In summary, our data support a model in which cigarette smoke pushes CD4 T cell effectors towards a more terminally-differentiated state that is characterized by enhanced Th1 cytokine production, increased expression of T-bet, and reduced potential to survive to memory. These studies have relevance for understanding how cigarette smoke can impact not only quantitative, but also qualitative aspects of CD4 T cell responses against respiratory pathogens as well as their ability to promote autoimmunity. Our work also supports the hypothesis that cigarette smoke can have a strong negative influence on the outcome of vaccination in terms of the efficiency of memory T cell generation.
2. Materials and Methods
2.1. Mice
HNT T cell receptor (TcR) transgenic mice on a BALB/c background were used as donors to obtain CD4 T cells. The HNT TcR recognizing aa 126–138 (HNTNGVTAACSHE) of A/Puerto Rico/8/34 influenza hemagglutinin protein [21]. Some experiments utilized naïve CD4 T cells obtained from OT-II TcR transgenic mice on the C57BL/6 background that recognize aa 323–339 (ISQAVHAAHAEINEAGR) of chicken ovalbumin17. All mice were used between 4–8 weeks of age and were bred at the University of Central Florida Vivarium at Lake Nona. All experimental animal procedures were conducted in accordance with the University of Central Florida’s Animal Care and Use Committee guidelines.
2.2. Naïve CD4 T cell isolation and generation of effectors
Naïve CD4 T cells were obtained from pooled spleen and peripheral lymph nodes of donor mice as previously described [22]. Briefly, organs were made into a single cell suspension by gentle pressing through a stainless steel wire mesh with a sterile rubber-tipped plunger from a 3mL syringe. The cells were then passed over nylon wool followed by Percoll gradient separation (Sigma-Aldrich) to isolate small resting cells. Naïve CD4 T cells were then isolated by positive selection using CD4+ MACS beads (Miltenyi Biotec). Resulting cells were consistently >95% CD4+ and displayed a naïve phenotype (CD44low, CD62Lhigh).
Naïve CD4 T cells were activated to generate effector cells by co-culturing with irradiated APC, either A20 B cells (H-2d) or B cell blasts prepared from T cell-depleted C57BL/6 splenocytes (H-2b) isolated by removing CD90.2+ cells by MACS separation and stimulating for two days with LPS and dextran sulfate (both 25 μg/mL). Cognate peptide was added to cultures of equal numbers of naïve T cells and irradiated APC (both 2x105 cells/ml) in RPMI media supplemented with 2mM L-Glutamine, 7.5% fetal bovine serum (HyClone), 10mM Hepes (Invitrogen), 50 μM 2-mercaptoethanol (Sigma-Aldrich) 100 IU penicillin, and 100 μg/ml streptomycin (Invitrogen). Exogenous recombinant murine IL-2 (11 ng/mL, Peprotech), and Th1-polarizing reagents (2 ng/ml recombinant IL-12, Peprotech, 15 μg/mL anti-IL-4 clone 11B11, BioXcell) were added at the initiation of cultures. Cells were fed with fresh media and IL-2 on the second of culture and harvested on day 4 of culture.
In some experiments, Th1 effector cells were generated via anti-CD3/anti-CD28 co-stimulation. Tissue culture well plates (Falcon) were coated overnight with anti-CD3 antibody (clone 2c11, BioXcell) at 10 μg/mL in PBS. Plates were washed thoroughly with PBS prior to the addition of 4 x 105 CD4 T cells/mL with anti-CD28 (clone 37.51, 2 μg/ml, BioXcell), IL-2, and Th1-polarizing factors as above. Cells were removed from CD3 coated wells on day 2 of culture, transferred to uncoated wells, and fed with fresh media containing IL-2 as previously described [23]. Effector cells were harvested and analyzed on day 4 of culture.
In some experiments, 6-Carboxyfluorescein succinimidyl ester (CFSE) was used to detect cell division in culture. Naïve CD4 T cells were incubated with 1μM CFSE (Invitrogen) in a 37 °C shaking water bath for 12 min and washed thoroughly with cold RPMI media.
In conditions with CSE, the CSE was added at the initiation of culture, and/or at 48 hours from frozen aliquots stored at −80 °C that were not reused. At least three replicates per condition were included in each experiment.
2.3. CSE Preparation
Cigarette smoke extract was prepared as we have previously described [24, 25]. Briefly, research-reference cigarettes (1R3F, 1.16 mg nicotine and 15 mg tar) were obtained from the Kentucky Tobacco Research and Development Center, University of Kentucky (Lexington, KY). A single cigarette was lit after removing the filter. The smoke was collected with a nozzled 10ml glass syringe and the smoke contents were bubbled through 25ml of serum-free RPMI medium. This CSE stock medium was filtered through a 0.22-μm filter, aliquoted out into 1.5ml microcentrifuge tubes, and stored at −80°C. Each aliquot was designated as 100% and was used only once after thawing. The extract is estimated to represent approximately 1/4 of the average smoker’s plasma concentration of soluble cigarette smoke contents [26]. The CSE was diluted to the desired concentrations in culture medium when used. CSE was standardized between lots by plating naïve CD4 T cells with irradiated APC and peptide in Th1 conditions with increasing concentrations of CSE and assessing expansion on day 4.
2.4. Flow cytometry
Cytokine and surface staining was performed as previously described17. Briefly, cell suspensions were washed, resuspended in FACS buffer (PBS plus 0.5% BSA and 0.02% sodium azide), and incubated on ice with anti-FcR (2.4G2, BioXcell) followed by saturating concentrations of the following fluorochrome-labeled antibodies for surface staining: anti-Thy1.2 (53-2.1), anti-CD4 (RM4.5), anti-CD25 (PC61.5), anti-CD127 (A7R34), anti-CD69 (H1.2F3) (BD Pharmingen, eBioscience, or BioLegend).
Intracellular staining was performed by stimulating effector cells with 10 ng/ml PMA and 50 ng/ml ionomycin (Sigma-Aldrich). After 2 hours, 10 μg/ml brefeldin A (Sigma-Aldrich) was added. After a further 2 hours the cells were surface stained and fixed for 20 min in 4% paraformaldehyde followed by permeabilization with a 10 minute incubation in 0.1% saponin buffer (PBS plus 1% FBS, 0.1% sodium azide, and 0.1% saponin). Cytokines were detected by the addition of fluorescently labeled anti–IFNγ (XMG1.2), TNF (MP6-XT22), anti-IL-4 (11B11), and anti-IL-17 (eBio17B7) antibodies or appropriate isotype controls (eBioscience) for 20 min.
Transcription factor staining was performed using a transcription factor staining buffer kit as per manufacturer’s instructions (eBioscience) and T-bet levels were detected using fluorescently labeled anti-T-bet antibody (4B10) (eBioscience).
Cell death was measured by flow cytometry using Annexin V Apoptosis Detection Kit with 7-AAD as per manufacturer’s instructions (BioLegend). All FACS analyses was performed using a FACSCanto flow cytometer (BD Biosciences) and FlowJo (Tree Star) analysis software.
2.5. In-vitro primed memory cell generation
In vitro-primed memory cells were obtained by first thoroughly washing effector obtained after 4 days of Th1 culture and incubating the cells on ice for 2 hours. The live cells were then isolated from the cultures by Lympholyte separation (Cedarlane) and equal numbers of the live cells originally obtained from cultures containing CSE or not were re-plated in fresh media for 3 days in the absence of CSE and exogenous cytokines. Live cells recovered after the 3-day rest period were isolated by Lympholyte separation and enumerated.
2.6. Statistical analysis
All statistics were analyzed by GraphPad Prism. Unpaired two-tailed student t test was utilized to determine the significance by which two normally distributed groups differed. One-way ANOVA analysis was used to compare multiple means. A P value of <0.05 was deemed significant. All error bars represent the standard deviation. In all figures, significance is indicated as * P<0.05, **P<.005, and ***P<.001.
3. Results
3.1. CSE reduces proliferation and increases apoptosis of CD4 T cell effectors
To test the impact of CSE on CD4 T cell activation, function, and memory development, we first stimulated naïve CD4 T cells obtained from HNT T cell receptor (TcR) transgenic mice with irradiated APC and cognate peptide. IL-12 and blocking antibody against IL-4 were added at the initiation of culture to promote Th1 differentiation [27]. The cultures were analyzed after 4 days. Experiments were repeated with donor cells obtained from both male and female mice with identical results (not shown).
We first titrated the amount of CSE added at the initiation of culture and assessed the recovery of the CD4 T cells. As shown in replicate experiments presented in Figure 1A and B, CSE concentrations of higher than 10% resulted in minimal recovery of effector cells, while cells treated in 7.5–10% CSE averaged around 50–70% recovery compared to control cultures not exposed to CSE. Reduced expansion in the presence of CSE correlated with decreased division as detected by the loss of CFSE label (Figure 1C). Importantly, no CFSEhigh cells were detected in control cultures or with 7.5% CSE added, with uniform patterns of CFSE dilution (Figure 1C), indicating efficient stimulation of the CD4 T cells in both conditions. We also observed increased apoptosis in effector cells cultured with CSE as determined by staining with Annexin-V and 7ADD (Figure 1D and E). We saw similar results using different TcR transgenic cells on a C57BL/6 background (OT-II) (not shown), indicating that these findings are not specific to one particular mouse strain or TcR transgenic model. As these results are in agreement with previous studies findings of decreased effector generation from murine and human CD4 T cells cultured in non-polarizing conditions at 3 days [28], we used 7.5% CSE for all subsequent experiments.
Figure 1. Exposure to CSE during priming reduces CD4 T cell division and increases apoptosis.

Triplicate wells of equal numbers of naïve HNT cells and irradiated APC were cultured with cognate peptide under Th1-polarizing conditions for 4 days in the presence of stated concentrations of CSE. The number of resulting effector cells was determined in replicate experiments with a broad (A) and narrower (B) titration of CSE (2 of 5 similar experiments). (C) In some experiments naïve CD4 T cells were labeled with CFSE prior to culture. Shown is representative CFSE staining from effectors at 4 days of culture in the presence of 0% (black) or 7.5% (red) CSE versus CFSE staining of undivided naïve cells not exposed to antigen. Day 4 effector cells were analyzed for apoptotic phenotype by staining for Annexin V and 7-AAD. Shown are (D) representative staining of effectors cultured with or without CSE and (E) summary of 4 wells per condition (one of 2 experiments).
3.2. CSE enhances Th1 cytokine production
We next assessed whether CSE exposure during activation impacts cytokine production potential from Th1-polarized effectors. To ensure that cells were similarly activated with and without exposure to CSE we first assessed the size of cells by forward scatter, and surface expression of the activation markers CD25 and CD69. As expected, effectors cultured with or without CSE displayed similar forward scatter that was much greater than naïve cells (Figure 2A and B). CD25 and CD69 were also highly expressed by effectors compared to naïve cells regardless of CSE addition (Figure 2A and B). Curiously, effectors generated in the presence of CSE expressed significantly more CD69 and less CD25 (Figure 2A and B).
Figure 2. Exposure to CSE during priming enhances Th1 polarization.

Naïve CD4 T cells were stimulated with APC and peptide under Th1 conditions in the presence or absence of CSE. Effector cells were harvested on day 4 of culture and analyzed for forward scatter (FSC), CD25, and CD69 with (A) representative staining and (B) mean fluorescence intensity (MFI) analysis from triplicate cultures shown. Values from naïve CD4 T cells are shown as grey histograms in (A) and as dotted line in (B). Effector cells were assessed for their capacity to produce of IFNγ and TNF by intracellular cytokine staining. Shown is (C) representative staining in the absence or presence of stimulation by PMA and Ionomycin by effectors generated in the presence or absence of CSE, as well as (D) the frequency of cells from each condition capable of co-producing TNF and IFNγ and (E) the MFI of IFNγ+ cells from triplicate wells (one of 4 experiments). Expression of T-bet in effectors was determined by intracellular staining with MFI analysis and representative staining shown (F and G). Representative staining of re-stimulated cells for production of IL-4 (H) and IL-17 (I) (one of 2 experiments).
We next used intracellular cytokine staining (ICCS) to determine the capacity of the cells to produce the key Th1-associated factors TNF and IFNγ. Cytokine production was markedly enhanced by CSE (Figure 2C). While the frequency of IFNγ+ single-positive cells was not dramatically increased by CSE, the mean fluorescence intensity of IFNγ+ cells was significantly higher, indicating more cytokine production per cell (Figure 2D). Most notably, CSE significantly increased the number of cells co-producing IFNγ and TNF (Figure 2E). In line with increased Th1 cytokine production, levels of the transcription factor T-bet, the master regulator of Th1 function [15], were significantly increased in effector cells activated in the presence of CSE (Figure 2F and G). We did not observe any IL-4+ cells (Figure 2H) or IL-17+ cells (Figure 2I) in any experimental conditions. These findings indicate that the presence of CSE does not skew the Th1-polarizing milieu towards Th2 or Th17 development but instead amplifies key aspects of the Th1 phenotype.
3.3. Delayed CSE exposure reduces effector expansion and increases Th1 function
We next tested whether and how delaying the exposure of developing effectors to CSE by 48 hours would impact its effect. This was done to test two aspects of how CSE acts. First, antigen presentation is limited in this culture system to the first 48 hours, after which the irradiated APC are no longer detectable [23]. Thus, adding CSE at 48 hours occurs at a time point when cognate interactions can no longer occur. Second, we sought to test whether the ability of CSE to modulate effector expansion and function was temporally restricted to the period of initial activation during which Th1-polarization is also largely programmed [29], or whether exposure to CSE at later time points could also impact effector function.
Even when first added 48 hours after the initiation of culture, CSE reduced effector recovery (Figure 3A) which again correlated with less division (Figure 3B) and increased apoptosis (Figure 3C and D) of the effector cells. It is important to note that addition of CSE at 48 hours had a reduced impact on these criteria than addition at the initiation of culture. Th1 cytokine production was also enhanced by late CSE exposure (Figure 4A) including more IFNγ and TNF double-positive cells (Figure 4B) and higher T-bet expression (Figure 4C and D). These results indicate that CSE can impact the development of Th1 cells even when present only during the latter antigen-independent phase of effector generation.
Figure 3. Delayed exposure to CSE reduces effector expansion.
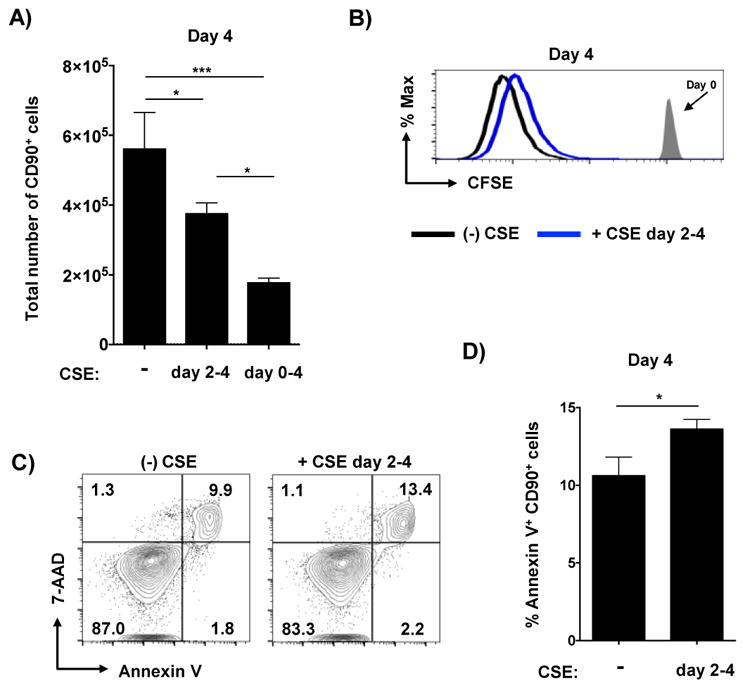
CSE was introduced to Th1 cultures at either day 0 or day 2 and (A) the total number of effector cells recovered at day 4 is shown from quadruplicate wells (one of 3 experiments). (B) Representative analysis of CFSE dilution from effectors either not exposed to CSE (black) or exposed to CSE for only from days 2–4 of culture. Effector cells were analyzed for apoptotic cells with (C) representative staining and (D) analysis from triplicate wells shown (one of 2 experiments).
Figure 4. Delayed exposure to CSE enhances Th1-polarization.
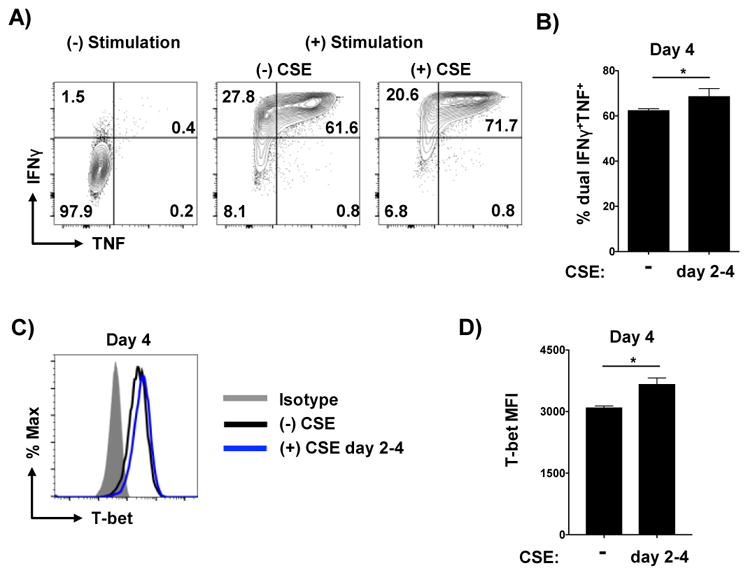
Effectors generated without exposure of CSE or CSE added only from days 2–4 of culture only were analyzed for Th1 cytokine production by intracellular staining with (A) representative staining and (B) analysis of dual cytokine producers from triplicate conditions shown. T-bet levels in effectors was also determined with (C) representative staining and (D) MFI analysis from triplicate wells shown (one of 2 experiments).
3.4. CSE directly impacts CD4 T cells to modulate Th1 function
CSE could affect effector programming either by acting on APC to alter signals delivered by them to CD4 T cells, or act directly on the CD4 T cells, or both. While the results presented in Figures 3 and 4 indicate that CSE can impact CD4 T cells in the absence of APC, the delayed addition of CSE to cultures complicates analysis. To determine the extent to which direct impacts of CSE on CD4 T cells are responsible for altered effector development, we cultured naïve CD4 T cells with plate-bound anti-CD3 and soluble anti-CD28 antibody to stimulate their activation in Th1-polarizing conditions. We observed that CSE introduced at the initiation of culture reduced effector accumulation (Figure 5A). Effectors cultured in CSE expressed similar levels of CD25 and increased CD69 (Figure 5B), similar to the pattern observed in effectors primed by APC and peptide shown in Figure 2. Reduced effector accumulation again correlated with decreased effector division (Figure 5C) and increased apoptosis (Figure 5D and E). Furthermore, CSE drove increased capacity to produce TNF and IFNγ (Figure 5F), particularly enhancing double-positive cells (Figure 5G). This increased effector function again correlated with enhanced T-bet expression by effector cells stimulated in the presence of CSE (Figure 5H and I). These results, which mirror those observed with APC-dependent stimulation, clearly demonstrate the ability of CSE to directly stimulate CD4 T cells to modulate Th1 function.
Figure 5. CSE acts directly on CD4 T cells to reduce expansion and increase Th1-polarization.

Naïve CD4 T cells were stimulated with plate-bound anti-CD3 and soluble anti-CD28 antibodies in the presence or absence of CSE. The recovery of effectors at day 4 (A), CFSE dilution (B), FSC and CD69 MFI values (C), analysis of apoptosis (D and E), Th1 cytokine production (F and G), and T-bet expression (H and I) is shown from one of 3 experiments.
3.5 CSE reduces memory potential in Th1 effectors
Finally, we tested whether CSE exposure impacts the ability of CD4 T cell effectors to survive the contraction phase and to form memory cells. We hypothesized that effector cells exposed to CSE may adopt a more terminal state, given previous results that found higher IFNγ production in CD4 T cell effectors to correlate with reduced long-term survival capacity [30]. Furthermore, recent studies have found CD4 T cell effectors expressing higher levels of T-bet to be less fit in terms of their memory potential in vivo [31], and our results show that CSE exposure drives increased T-bet expression (Figures 2,4, and 5). These findings led us to hypothesize that effectors exposed to CSE are more terminally-differentiated than control cells, and thus less fit to form memory.
To test this hypothesis, we first assessed levels of the alpha chain of the IL-7 receptor on effector cells after 4 days of culture. Our work in an influenza infection model found increased level of IL-7 receptor on CD4 T cell effectors to be a strong positive predictor of their capacity to compete for entry into and survival within the memory niche [32]. Effector cells generated in the presence of CSE, primed both by APC and cognate peptide and by anti-CD3 and anti-CD28, expressed slightly but significantly reduced CD127 compared to effector cells not exposed to CSE (Figure 6A–D). Although the CSE-induced reduction is relatively minor, it is similar in scale to differences in CD127 expression that we have correlated with profoundly reduced CD4 T cell memory generation in vivo following pathogen challenge [32].
Figure 6. Exposure to CSE reduces memory potential in Th1-polarized effector cells.

Effectors were generated by stimulation with either APC and peptide or with anti-CD3 and anti-CD28 antibodies. On day 4, live effector cells were isolated from cultures and analyzed for expression of the α chain of the IL-7 receptor (CD127). Representative staining and MFI analysis from triplicate wells is shown for cells exposed or not to CSE from cultures stimulated with APC and peptide (A and B) or by antibody-mediated activation (C and D). Equal numbers of live effectors exposed to CSE were thoroughly washed and re-plated in media alone for 3 additional days to form in-vitro generated memory cells. Shown is the ratio of memory cells recovered from effectors not exposed to CSE versus from effectors exposed to CSE during priming using both methods of CD4 T cell activation (E and F). Results representative from experiments repeated twice for both APC and peptide and antibody-mediated effector generation.
We next directly tested the relative per-cell efficiency of cells exposed or not to CSE to transition from activated effectors to resting memory cells. To do this, we used a well-characterized in vitro model for interrogating the ability of effector cells to survive contraction in which we have previously shown effector cells to adopt virtually identical phenotypic, functional, and gene-expression signatures as those identified in long-lived memory cells isolated from mice [22, 33]. Live effector cells were isolated after 4 days of culture in Th1 cultures with or without CSE and washed thoroughly. The cells were then plated in fresh media, without CSE, for a further 3 days, after which live effectors were again recovered and enumerated. Significantly more in vitro-generated memory cells were recovered from cells not exposed to CSE than from effectors that were generated in the presence of CSE. Similar results were seen using effectors generated with APC and peptide (Figure 6E) and with anti-CD3 and anti-CD28 antibody stimulation (Figure 6F). These experiments indicate that the presence of CSE during effector generation drives the differentiation of terminal effectors with reduced capacity to survive long-term, even in the absence of continuing CSE exposure.
4. Discussion
Surprisingly little is known about how exposure to cigarette smoke impacts the ability of CD4 T cells to respond to antigen, form functional effector populations, or how it affects the ability of effector cells to successfully transition to memory. This is at least in part due to the fact that cigarette smoke has been found to have wide ranging impacts on many different cell types in vivo. For example, CSE has been shown to impact human and murine APC function, resulting in their decreased ability to stimulate T cell activation [18]. We used reductionist models to assess the extent to which CSE is able to directly impact CD4 T cell effector generation using both APC-dependent and APC-independent approaches to activate naïve CD4 T cells in defined culture conditions. We focused on understanding the impact of CSE during Th1 polarization given the key role of Th1 responses in immunity against respiratory pathogens, such as IAV, and in certain autoimmune conditions associated with smoking, such as inflammatory bowel disease. Some of our findings are in agreement with a previous study in non-Th-polarizing conditions demonstrating a direct negative impact of CSE on CD4 T cell expansion [28], which we show here on a per-cell level to be impacted both by reduced capacity for cell division and by increased rates of effector cell death. Our results further indicate that exposure to CSE even for only the final 48 hours of culture reduces the efficiency of effector generation.
Our results clearly demonstrate a strong ability of CSE to impact the functional potential of CD4 T cells. CSE enhances Th1 cytokine production from effector cells, particularly the ability of cells to co-produce IFNγ and TNF. The increase in Th1 functional potential correlated with increased expression of T-bet, the ‘master regulator’ needed to promote full Th1-polarization [15]. Our results are consistent with and help frame a mechanism for a recent report in a murine model showing that when compared to control cells not exposed to smoke, CD4 T cells exposed to cigarette smoke demonstrate increased efficiency to promote colitis in an adoptive transfer model through an IFNγ- and T-bet-dependent mechanism [34]. Th1-polarized cells have been found to contribute to symptoms of chronic obstructive pulmonary disease (COPD) associated with smoking [35], again supporting a link between cigarette smoke exposure and deleterious Th1 responses. In the setting of infectious disease, we speculate that cigarette smoke may also contribute to enhanced morbidity through increasing Th1 cytokine production in some situations. This hypothesis is supported by findings that adoptive transfer of IFNγ-deficient Th1-polarized effector CD4 T cells specific for influenza to wild-type mice promote improved recovery and less weight loss than an equal number of wild-type effectors recognizing the same viral epitope [12]. This suggests that exaggerated Th1 cytokine production in the lung can contribute to immunopathology. Worsened lung immunopathology in the presence of strong IFNγ signaling has recently been correlated with impaired responses from innate lymphoid cells (ILC) [36], which are an important subset needed for repair following the resolution of infection [37]. A potential mechanism by which enhanced Th1 cytokine production induced by exposure to smoke could exacerbate damage associated with respiratory infection is thus through the inhibiting ILC functions. Reduced ILC activity has also recently been found to contribute to COPD [38].
On the other hand, there may be situations in which increased Th1 cytokine production induced by cigarette smoke could have a beneficial impact given the important role of IFNγ in cellular immunity against diverse pathogens. This raises the possibility that select compounds within CSE might be of use as Th1-stimulating adjuvants. Further studies will be required to test this hypothesis and to identify and characterize individual factors within CSE that stimulate enhanced Th1 polarization. Interestingly, hydroquinone, which is present at high concentrations in cigarette smoke, has been shown to inhibit IFNγ production from T cells [39]. This highlights the inherent complexity in understanding how cigarette smoke, and its individual constituents, impact lymphocyte function during immune responses in vivo. Indeed, further studies using the reductionist approach described here are required to define in detail the extent to which cigarette smoke similarly or differentially impacts the development of other CD4 T cell subsets including Th2, Th17, Treg, and Th0 cells.
Our studies demonstrate, for the first time, reduced memory formation from Th1 effector cells exposed to CSE during priming. Although we used an in vitro approach here, we have shown that effectors undergo a virtually identical and rapid transition to memory in vivo [22, 40]. Furthermore, our results correlate impaired memory potential in effectors exposed to CSE with increased IFNγ production, reduced CD127 expression, and enhanced T-bet levels. All of these phenotypic criteria have been found to mark a terminally-differentiated effector state in CD4 T cells [30–32]. We did not see an impact of CSE on expression of the transcription factor Eomesodermin, differential expression of which has also been linked to distinct memory fates [41] (not shown). Our results are consistent with findings correlating decreased frequencies of memory phenotype CD4 T cells in children with increasing levels of serum cotinine, a major metabolite of nicotine that was used as a marker of smoke exposure [42]. On the other hand, smoking has been correlated with an increased proportion of memory phenotype CD4 T cells in adults [43]. While the root of this discrepancy in human studies is not yet clear, it might relate to the more immature status of the immune system in children. A reductionist approach, such as we have described here, may provide novel insight into how cigarette smoke differentially impacts distinct subsets of human CD4 T cells, and whether it has distinct effects on young versus aged T cells.
Further work is also required to investigate the intracellular signaling pathways underlying impaired memory formation programmed by CSE. Regulation of lung-resident CD4 T cell memory cells, that are likely exposed to the highest level of smoke and that play a crucial role in immunity against influenza [44, 45], is particularly important to address. Recent findings correlating higher T-bet levels with reduced capacity to form lung-resident memory CD4 T cells [46] support that the actions of CSE we have described may likely also impact this subset.
In summary, we present data highlighting the ability of cigarette smoke to modulate distinct programs during CD4 T cell activation through direct effects on the responding cells. Although we have employed a reductionist model and focused on Th1 development, these observations have broad implications for how exposure to cigarette smoke could impact CD4 T cell responses in vivo. Enhanced Th1 cytokine production could, depending on situational circumstances, improve CD4 T cell responses against pathogens and/or contribute to enhanced collateral damage and impaired resolution of infection. Our findings are also relevant for understanding the etiology of autoimmune symptoms linked with smoking such as inflammatory bowel disease that are driven in part by T cell responses [47]. Finally, the ability of CSE to promote terminal differentiation in CD4 T cell effectors might help to explain impaired high affinity antibody production following human papillomavirus vaccination [48] and reports of decreased influenza vaccine effectiveness in smokers [49].
Highlights.
Cigarette smoke extract reduces division and increases apoptosis of Th1 effectors.
Cigarette smoke extract enhances Th1 cytokine production and T-bet expression.
Cigarette smoke extract mediates its impact directly on responding CD4 T cells.
Cigarette smoke extract drives terminal differentiation and restricts memory fate.
Acknowledgments
This work was supported by funding from the University of Central Florida and from the State of Florida Crohn’s funding appropriation (to K.K.M.), and NIH grant R21AI11745701A1 to T.M.S. J.T. was supported by funding from the Office of Undergraduate Research and Academic Advancement Programs at the University of Central Florida.
Abbreviations
- 7-AAD
7-Aminoactinomycin D
- APC
antigen presenting cell
- CFSE
carboxyfluorescein succinimidyl ester
- CSE
cigarette smoke extract
- IFNγ
- interferon gamma
- T-bet
T-box transcription factor TBX21
- Th1
T helper type 1
- TNF
tumor necrosis factor alpha
Footnotes
Conflict of interest
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jamal A, Phillips E, Gentzke AS, Homa DM, Babb SD, King BA, Neff LJ. Current Cigarette Smoking Among Adults - United States, 2016. MMWR Morb Mortal Wkly Rep. 2018;67:53–59. doi: 10.15585/mmwr.mm6702a1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Collaborators GBDT. Smoking prevalence and attributable disease burden in 195 countries and territories, 1990–2015: a systematic analysis from the Global Burden of Disease Study 2015. Lancet. 2017;389:1885–1906. doi: 10.1016/S0140-6736(17)30819-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crotty Alexander LE, Shin S, Hwang JH. Inflammatory Diseases of the Lung Induced by Conventional Cigarette Smoke: A Review. Chest. 2015;148:1307–1322. doi: 10.1378/chest.15-0409. [DOI] [PubMed] [Google Scholar]
- 4.Perricone C, Versini M, Ben-Ami D, Gertel S, Watad A, Segel MJ, Ceccarelli F, Conti F, Cantarini L, Bogdanos DP, Antonelli A, Amital H, Valesini G, Shoenfeld Y. Smoke and autoimmunity: The fire behind the disease. Autoimmun Rev. 2016;15:354–374. doi: 10.1016/j.autrev.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Qiu F, Liang CL, Liu H, Zeng YQ, Hou S, Huang S, Lai X, Dai Z. Impacts of cigarette smoking on immune responsiveness: Up and down or upside down? Oncotarget. 2017;8:268–284. doi: 10.18632/oncotarget.13613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zavitz CC, Gaschler GJ, Robbins CS, Botelho FM, Cox PG, Stampfli MR. Impact of cigarette smoke on T and B cell responsiveness. Cell Immunol. 2008;253:38–44. doi: 10.1016/j.cellimm.2008.04.012. [DOI] [PubMed] [Google Scholar]
- 7.Kohlmeier JE, Woodland DL. Immunity to respiratory viruses. Annu Rev Immunol. 2009;27:61–82. doi: 10.1146/annurev.immunol.021908.132625. [DOI] [PubMed] [Google Scholar]
- 8.Cosnes J. Tobacco and IBD: relevance in the understanding of disease mechanisms and clinical practice. Best Pract Res Clin Gastroenterol. 2004;18:481–496. doi: 10.1016/j.bpg.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 9.Swain SL, McKinstry KK, Strutt TM. Expanding roles for CD4(+) T cells in immunity to viruses. Nat Rev Immunol. 2012;12:136–148. doi: 10.1038/nri3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Powell TJ, Strutt T, Reome J, Hollenbaugh JA, Roberts AD, Woodland DL, Swain SL, Dutton RW. Priming with cold-adapted influenza A does not prevent infection but elicits long-lived protection against supralethal challenge with heterosubtypic virus. J Immunol. 2007;178:1030–1038. doi: 10.4049/jimmunol.178.2.1030. [DOI] [PubMed] [Google Scholar]
- 11.McKinstry KK, Strutt TM, Swain SL. Hallmarks of CD4 T cell immunity against influenza. J Intern Med. 2011;269:507–518. doi: 10.1111/j.1365-2796.2011.02367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown DM, Dilzer AM, Meents DL, Swain SL. CD4 T cell-mediated protection from lethal influenza: perforin and antibody-mediated mechanisms give a one-two punch. J Immunol. 2006;177:2888–2898. doi: 10.4049/jimmunol.177.5.2888. [DOI] [PubMed] [Google Scholar]
- 13.Parronchi P, Romagnani P, Annunziato F, Sampognaro S, Becchio A, Giannarini L, Maggi E, Pupilli C, Tonelli F, Romagnani S. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn's disease. Am J Pathol. 1997;150:823–832. [PMC free article] [PubMed] [Google Scholar]
- 14.Matsuoka K, Inoue N, Sato T, Okamoto S, Hisamatsu T, Kishi Y, Sakuraba A, Hitotsumatsu O, Ogata H, Koganei K, Fukushima T, Kanai T, Watanabe M, Ishii H, Hibi T. T-bet upregulation and subsequent interleukin 12 stimulation are essential for induction of Th1 mediated immunopathology in Crohn's disease. Gut. 2004;53:1303–1308. doi: 10.1136/gut.2003.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH. A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell. 2000;100:655–669. doi: 10.1016/s0092-8674(00)80702-3. [DOI] [PubMed] [Google Scholar]
- 16.Vassallo R, Tamada K, Lau JS, Kroening PR, Chen L. Cigarette smoke extract suppresses human dendritic cell function leading to preferential induction of Th-2 priming. J Immunol. 2005;175:2684–2691. doi: 10.4049/jimmunol.175.4.2684. [DOI] [PubMed] [Google Scholar]
- 17.Robbins CS, Franco F, Mouded M, Cernadas M, Shapiro SD. Cigarette smoke exposure impairs dendritic cell maturation and T cell proliferation in thoracic lymph nodes of mice. J Immunol. 2008;180:6623–6628. doi: 10.4049/jimmunol.180.10.6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Givi ME, Folkerts G, Wagenaar GT, Redegeld FA, Mortaz E. Cigarette smoke differentially modulates dendritic cell maturation and function in time. Respir Res. 2015;16:131. doi: 10.1186/s12931-015-0291-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Masopust D, Kaech SM, Wherry EJ, Ahmed R. The role of programming in memory T-cell development. Curr Opin Immunol. 2004;16:217–225. doi: 10.1016/j.coi.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 20.Dhume K, McKinstry KK. Early programming and late-acting checkpoints governing the development of CD4 T-cell memory. Immunology. 2018 doi: 10.1111/imm.12942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Scott B, Liblau R, Degermann S, Marconi LA, Ogata L, Caton AJ, McDevitt HO, Lo D. A role for non-MHC genetic polymorphism in susceptibility to spontaneous autoimmunity. Immunity. 1994;1:73–83. doi: 10.1016/1074-7613(94)90011-6. [DOI] [PubMed] [Google Scholar]
- 22.McKinstry KK, Golech S, Lee WH, Huston G, Weng NP, Swain SL. Rapid default transition of CD4 T cell effectors to functional memory cells. J Exp Med. 2007;204:2199–2211. doi: 10.1084/jem.20070041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jelley-Gibbs DM, Lepak NM, Yen M, Swain SL. Two distinct stages in the transition from naive CD4 T cells to effectors, early antigen-dependent and late cytokine-driven expansion and differentiation. J Immunol. 2000;165:5017–5026. doi: 10.4049/jimmunol.165.9.5017. [DOI] [PubMed] [Google Scholar]
- 24.Fu YY, Nergard JC, Barnette NK, Wang YL, Chai KX, Chen LM. Proteasome inhibition augments cigarette smoke-induced GM-CSF expression in trophoblast cells via the epidermal growth factor receptor. PLoS One. 2012;7:e43042. doi: 10.1371/journal.pone.0043042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen LM, Nergard JC, Ni L, Rosser CJ, Chai KX. Long-term exposure to cigarette smoke extract induces hypomethylation at the RUNX3 and IGF2-H19 loci in immortalized human urothelial cells. PLoS One. 2013;8:e65513. doi: 10.1371/journal.pone.0065513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gangl K, Reininger R, Bernhard D, Campana R, Pree I, Reisinger J, Kneidinger M, Kundi M, Dolznig H, Thurnher D, Valent P, Chen KW, Vrtala S, Spitzauer S, Valenta R, Niederberger V. Cigarette smoke facilitates allergen penetration across respiratory epithelium. Allergy. 2009;64:398–405. doi: 10.1111/j.1398-9995.2008.01861.x. [DOI] [PubMed] [Google Scholar]
- 27.Zhu J, Yamane H, Paul WE. Differentiation of effector CD4 T cell populations (*) Annu Rev Immunol. 2010;28:445–489. doi: 10.1146/annurev-immunol-030409-101212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandez CP, Morrow K, Velasco C, Wyczechowska DD, Naura AS, Rodriguez PC. Effects of cigarette smoke extract on primary activated T cells. Cell Immunol. 2013;282:38–43. doi: 10.1016/j.cellimm.2013.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Iezzi G, Scotet E, Scheidegger D, Lanzavecchia A. The interplay between the duration of TCR and cytokine signaling determines T cell polarization. Eur J Immunol. 1999;29:4092–4101. doi: 10.1002/(SICI)1521-4141(199912)29:12<4092::AID-IMMU4092>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 30.Wu CY, Kirman JR, Rotte MJ, Davey DF, Perfetto SP, Rhee EG, Freidag BL, Hill BJ, Douek DC, Seder RA. Distinct lineages of T(H)1 cells have differential capacities for memory cell generation in vivo. Nat Immunol. 2002;3:852–858. doi: 10.1038/ni832. [DOI] [PubMed] [Google Scholar]
- 31.Marshall HD, Chandele A, Jung YW, Meng H, Poholek AC, Parish IA, Rutishauser R, Cui W, Kleinstein SH, Craft J, Kaech SM. Differential expression of Ly6C and T-bet distinguish effector and memory Th1 CD4(+) cell properties during viral infection. Immunity. 2011;35:633–646. doi: 10.1016/j.immuni.2011.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McKinstry KK, Strutt TM, Bautista B, Zhang W, Kuang Y, Cooper AM, Swain SL. Effector CD4 T-cell transition to memory requires late cognate interactions that induce autocrine IL-2. Nat Commun. 2014;5:5377. doi: 10.1038/ncomms6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McKinstry KK, Strutt TM, Swain SL. The effector to memory transition of CD4 T cells. Immunol Res. 2008;40:114–127. doi: 10.1007/s12026-007-8004-y. [DOI] [PubMed] [Google Scholar]
- 34.Lee G, Jung KH, Shin D, Lee C, Kim W, Lee S, Kim J, Bae H. Cigarette Smoking Triggers Colitis by IFN-gamma(+) CD4(+) T Cells. Front Immunol. 2017;8:1344. doi: 10.3389/fimmu.2017.01344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caramori G, Casolari P, Barczyk A, Durham AL, Di Stefano A, Adcock I. COPD immunopathology. Semin Immunopathol. 2016;38:497–515. doi: 10.1007/s00281-016-0561-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Califano D, Furuya Y, Roberts S, Avram D, McKenzie ANJ, Metzger DW. IFN-gamma increases susceptibility to influenza A infection through suppression of group II innate lymphoid cells. Mucosal Immunol. 2018;11:209–219. doi: 10.1038/mi.2017.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim BS, Artis D. Group 2 Innate Lymphoid Cells in Health and Disease. Cold Spring Harb Perspect Biol. 2015 doi: 10.1101/cshperspect.a016337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N, Mori M, Pham TH, Ward CK, Criner GJ, Marchetti N, Mustelin T, Erjefalt JS, Kolbeck R, Humbles AA. Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity. 2015;42:566–579. doi: 10.1016/j.immuni.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 39.Choi JM, Cho YC, Cho WJ, Kim TS, Kang BY. Hydroquinone, a major component in cigarette smoke, reduces IFN-gamma production in antigen-primed lymphocytes. Arch Pharm Res. 2008;31:337–341. doi: 10.1007/s12272-001-1161-1. [DOI] [PubMed] [Google Scholar]
- 40.McKinstry KK, Strutt TM, Swain SL. Regulation of CD4+ T-cell contraction during pathogen challenge. Immunol Rev. 2010;236:110–124. doi: 10.1111/j.1600-065X.2010.00921.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, Gapin L, Ryan K, Russ AP, Lindsten T, Orange JS, Goldrath AW, Ahmed R, Reiner SL. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat Immunol. 2005;6:1236–1244. doi: 10.1038/ni1268. [DOI] [PubMed] [Google Scholar]
- 42.Vardavas CI, Plada M, Tzatzarakis M, Marcos A, Warnberg J, Gomez-Martinez S, Breidenassel C, Gonzalez-Gross M, Tsatsakis AM, Saris WH, Moreno LA, Kafatos AG, Group HHS. Passive smoking alters circulating naive/memory lymphocyte T-cell subpopulations in children. Pediatr Allergy Immunol. 2010;21:1171–1178. doi: 10.1111/j.1399-3038.2010.01039.x. [DOI] [PubMed] [Google Scholar]
- 43.Nakata A, Takahashi M, Irie M, Fujioka Y, Haratani T, Araki S. Relationship between cumulative effects of smoking and memory CD4+ T lymphocyte subpopulations. Addict Behav. 2007;32:1526–1531. doi: 10.1016/j.addbeh.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 44.Zens KD, Chen JK, Farber DL. Vaccine-generated lung tissue-resident memory T cells provide heterosubtypic protection to influenza infection. JCI Insight. 2016;1 doi: 10.1172/jci.insight.85832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Strutt TM, Dhume K, Finn CM, Hwang JH, Castonguay C, Swain SL, McKinstry KK. IL-15 supports the generation of protective lung-resident memory CD4 T cells. Mucosal Immunol. 2018;11:668–680. doi: 10.1038/mi.2017.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zens KD, Chen JK, Guyer RS, Wu FL, Cvetkovski F, Miron M, Farber DL. Reduced generation of lung tissue-resident memory T cells during infancy. J Exp Med. 2017;214:2915–2932. doi: 10.1084/jem.20170521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Parkes GC, Whelan K, Lindsay JO. Smoking in inflammatory bowel disease: impact on disease course and insights into the aetiology of its effect. J Crohns Colitis. 2014;8:717–725. doi: 10.1016/j.crohns.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 48.Namujju PB, Pajunen E, Simen-Kapeu A, Hedman L, Merikukka M, Surcel HM, Kirnbauer R, Apter D, Paavonen J, Hedman K, Lehtinen M. Impact of smoking on the quantity and quality of antibodies induced by human papillomavirus type 16 and 18 AS04-adjuvanted virus-like-particle vaccine - a pilot study. BMC Res Notes. 2014;7:445. doi: 10.1186/1756-0500-7-445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Godoy P, Castilla J, Soldevila N, Mayoral JM, Toledo D, Martin V, Astray J, Egurrola M, Morales-Suarez-Varela M, Dominguez A, Cases C S. Controls in Pandemic Influenza Working Group. Smoking may increase the risk of influenza hospitalization and reduce influenza vaccine effectiveness in the elderly. Eur J Public Health. 2018;28:150–155. doi: 10.1093/eurpub/ckx130. [DOI] [PubMed] [Google Scholar]