

Summary
Stress adaptation is essential for neuronal health. While the fundamental role of mitochondria in neuronal development has been demonstrated, it is still not clear how adult neurons respond to alterations in mitochondrial function and how neurons sense, signal, and respond to dysfunction of mitochondria and their interacting organelles. Here, we show that neuron-specific, inducible in vivo ablation of the mitochondrial fission protein Drp1 causes ER stress, resulting in activation of the integrated stress response to culminate in neuronal expression of the cytokine Fgf21. Neuron-derived Fgf21 induction occurs also in murine models of tauopathy and prion disease, highlighting the potential of this cytokine as an early biomarker for latent neurodegenerative conditions.
Keywords: Alzheimer’s disease, autophagy, biomarker, endoplasmic reticulum, heme, metabolism, mitochondria, neurodegeneration, tau, unfolded protein response
Graphical Abstract
Highlights
-
•
Neuronal Drp1 ablation is sensed by branches of the integrated stress response (ISR)
-
•
Activation of the ISR induces catabolic cytokine Fgf21 in the brain
-
•
Brain Fgf21 induced in neurodegeneration models may be a potential biomarker
Restelli et al. show that deletion of mitochondrial fission protein Drp1 in adult mouse neurons activates multiple stress-sensing pathways. These converge on the integrated stress response, resulting in neuron-specific expression of metabolic cytokine Fgf21. Cerebral induction of Fgf21 also occurs in mechanistically independent mouse models of protein misfolding-associated neurodegeneration.
Introduction
In neurons, mitochondrial fission, which in mammals depends mainly on dynamin-related protein Drp1, facilitates both axonal mitochondrial transport and mitophagic elimination (Oettinghaus et al., 2016, Pernas and Scorrano, 2016, Twig et al., 2008, Wai and Langer, 2016). Not surprisingly, Drp1 is therefore required for brain development and mature neuronal function (Berthet et al., 2014, Ishihara et al., 2009, Kageyama et al., 2012, Oettinghaus et al., 2016, Shields et al., 2015, Wakabayashi et al., 2009). In several clinically and pathologically distinct neurodegenerative diseases, mitochondrial morphology is disrupted (reviewed by DuBoff et al., 2013), substantiating the link among mitochondrial dynamics, function, and brain pathophysiology. Given this, brain mitochondrial dysfunction could represent an important marker to identify subjects at risk of developing neurodegenerative disorders, before neurological symptoms manifest; however, no surrogate marker of brain mitochondrial dysfunction is available.
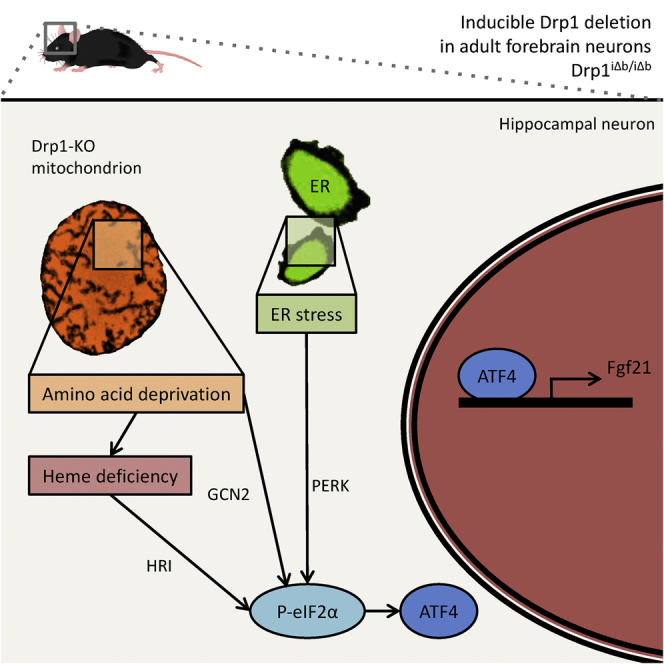
Mitochondria of peripheral organs (e.g., liver and muscle) can convey disruption of their function by mounting cell-wide stress responses (Cornu et al., 2014, Guridi et al., 2015, Hagiwara et al., 2012, Keipert et al., 2014, Kim et al., 2013a, Touvier et al., 2015, Tyynismaa et al., 2010). A common trait of these models is the expression, causally linked to mitochondrial dysfunction, of the metabolic cytokine fibroblast growth factor 21 (Fgf21). This cytokine exerts a plethora of tissue-specific effects and is subject to complex pharmacology and pharmacokinetics, aspects of which continue to be matter of debate (reviewed by Kharitonenkov and DiMarchi, 2017). In addition to being canonically produced by liver and white adipose tissue (WAT) in response to starvation, Fgf21 can be secreted by other organs, such as pancreas and brown adipose tissue (Degirolamo et al., 2016, Fisher and Maratos-Flier, 2016). Moreover, Fgf21 has been established as a marker of mitochondrial myopathies (Suomalainen et al., 2011, Tezze et al., 2017). Whether Fgf21 is also produced by the CNS in response to mitochondrial dysfunction is unknown. Here we capitalize on our previously generated mouse model of inducible mitochondrial fission ablation in mature forebrain neurons (Oettinghaus et al., 2016), as well as on bona fide mouse models of neurodegeneration, to demonstrate that various forms of neuronal cell stress, including mitochondrial dysfunction, are sensed by the integrated stress response (ISR). In each of these models, ISR activation leads to neuron-specific Fgf21 expression, highlighting the potential of this cytokine as biomarker for neurodegenerative conditions.
Results
Neuronal Drp1 Ablation Activates the ISR, Resulting in Fgf21 Induction
We previously generated a model of inducible ablation of the mitochondrial fission protein Drp1 in the adult mouse forebrain (Drp1flx/flx; CaMKIIα::CreERT2, tamoxifen-inducible Drp1 deletion from brain, Drp1iΔb/iΔb). In this model, neuronal Drp1 ablation compromised mitochondrial respiration and shifted cellular metabolism toward glycolysis (Oettinghaus et al., 2016); in addition, systemic catabolic changes occurred, which prompted further investigation.
Because mitochondrial dysfunction in several peripheral organs can lead to the induction of the cytokine Fgf21 (Kharitonenkov and DiMarchi, 2015, Kim et al., 2013a, Suomalainen et al., 2011), we checked whether the same was true for a brain-specific mitochondrial defect. Starting 7 weeks post-tamoxifen injection (PTI, i.e., after Drp1 ablation), we found elevated Fgf21 plasma levels (Figure 1A), prompting us to investigate its source organ or organs. Subsequent analyses revealed that as early as 4 weeks after Drp1 ablation, Fgf21 transcription was induced specifically in hippocampus and cortex but not in: the cerebellum (where the Cre-driving CaMKIIα promoter is inactive); the canonical Fgf21 sources (liver and adipose tissue); or the skeletal muscle (Figure 1B). These results establish brain as the Fgf21 tissue source in our model. mRNA fluorescence in situ hybridization identified neurons as the primary Fgf21 producers in the Drp1-ablated mouse brain, shown in Figures 1C and 1D for the hippocampal CA1 region (Figures S1A–S1C detail the histological distribution of hippocampal pyramidal neurons for reference; for comparison, Figures S1D and S1E show the distribution of astrocytes and microglia). These observations are significant from two perspectives: we demonstrate that Fgf21 can be produced by neurons in response to mitochondrial dysfunction (Bookout et al., 2013, Suomalainen et al., 2011, Fon Tacer et al., 2010) and that Fgf21 produced in the brain can cross the blood-brain barrier (BBB) to become detectable in plasma (Kharitonenkov and DiMarchi, 2015).
Figure 1.

Neuronal Drp1 Ablation Induces ISR-Driven Brain Fgf21 Expression
(A) FGF21 plasma levels in cardiac blood of Drp1-ablated and control mice. Positive control represents overnight-starved animals. Data represent average + SEM of at least 3 animals.
(B) qRT-PCR analysis of different mouse tissues at indicated time points PTI. Fgf21 mRNA ct values normalized against 18S rRNA ct values. Data represent average + SEM of at least 4 animals.
(C and D) Representative Fgf21 mRNA-fluorescence in situ hybridization (FISH) analysis of the hippocampal CA1 region of a Drp1-ablated mouse at 10 weeks PTI (C). Fgf21 mRNA hybridization signals are in green; nuclei are DAPI stained. Scale bar, 20 μm. Quantification of hybridization signals (D).
(E) Representative western blots of Drp1-ablated and control hippocampal lysates separated by SDS-PAGE and immunoblotted using indicated antibodies.
(F and G) Quantification of phosphorylated elF2α band intensity normalized to total elF2α (F) and Atf4 intensity normalized to actin (G). Data represent average + SEM of at least 4 animals.
(H) qRT-PCR analysis of Chop expression in various mouse brain regions. Data represent average + SEM of at least 4 animals.
The ISR Is Activated upon Neuronal Drp1 Ablation
We next wished to understand how mitochondrial dysfunction led to neuronal Fgf21 transcription. Among the transcription factors known to control Fgf21 expression, only Atf4 was upregulated in Drp1-ablated brains (Figure S2A). Atf4 translation is activated by phosphorylated eukaryotic translation initiation factor 2α (eIF2α), a crucial node in the ISR, responding to different forms of stress via decreased global translation and activation of transcriptional programs aimed at damage control (Donnelly et al., 2013). Western blot analysis confirmed Atf4 increase and eIF2α phosphorylation in Drp1iΔb/iΔb hippocampi (Figures 1E–1G). The canonical Atf4 target gene Chop (Ddit3 or Gadd153) was accordingly upregulated in Drp1iΔb/iΔb hippocampus and cortex (Figure 1H). Furthermore, proteomics revealed that Atf4 target genes were significantly enriched in Drp1iΔb/iΔb brains (Figure S2B).
Four eIF2α kinases are capable of sensing specific ISR-triggering stress conditions: Perk responds to endoplasmic reticulum (ER) stress; Gcn2 acts as sensor for amino acid deficiency activated by uncharged tRNAs; Pkr senses double-stranded RNAs to signal viral infections and transduces the mitochondrial unfolded protein response (mtUPR) (Rath et al., 2012); and HRI becomes activated in the absence of heme (Donnelly et al., 2013). In vivo, the eIF2α-phosphorylating arm or arms of the ISR could not be detected by direct immunoblot (due to paucity of their expression and the mixed cellular nature of brain lysates). However, we investigated the specific upstream stress or stresses triggering the Fgf21-inducing cascade as a proxy for their respective ISR arm activation.
The close apposition of ER and mitochondria favors ER stress upon loss of proteins involved in mitochondrial and ER morphology regulation (de Brito and Scorrano, 2008, Muñoz et al., 2013). Drp1 was originally identified as a modulator of mitochondrial, as well as ER shape (Pitts et al., 1999, Yoon et al., 1998), suggesting that its deletion might also affect the ER. Electron microscopy (EM) of hippocampi 4 weeks PTI revealed that the ER was round and swollen, unlike the flat ER cisternae in the Drp1flx/flx counterparts (Figure 2A). In addition, the ER stress marker Bip/GRP78 was upregulated in Drp1iΔb/iΔb brains at later stages (Figures 2B and 2C), pointing to a link between neuronal Drp1 deletion and ER stress activation. As a proof of principle, when we intraventricularly injected wild-type mice with the ER stress inducer tunicamycin, we noticed not only an activated unfolded protein response (UPR), as reflected by eIF2α phosphorylation and Atf4 elevation, but also an increase in Fgf21 mRNA (Figures 2D–2F), leading to an elevation in plasma Fgf21 levels (Figure 2G). Altogether, these data indicate that Drp1iΔb/iΔb brains mount an UPR and that brain ER stress is sufficient to induce local Fgf21 expression and plasma Fgf21 elevation.
Figure 2.

ER Stress Is Present and Sufficient for Fgf21 Induction upon Drp1 Ablation
(A) Representative transmission electron microscopy (TEM) images of ER (marked by black arrows) of hippocampal neurons of 4-week-Drp1-ablated and control mice. Scale bar, 1 μm.
(B) Bip and actin western blot on hippocampal lysates of mice of the indicated genotypes at 10 weeks PTI. This experiment shares a loading control with Figure 1E.
(C) Quantification of Bip band intensity normalized to actin. Data represent average + SEM of at least 4 animals.
(D–G) Markers of ER stress and Fgf21 levels in brain tissue and plasma following intraventricular tunicamycin (TUM) injection.
(D) Fgf21 mRNA in TUM- and Sham-injected hippocampi.
(E and F) Representative western blot (E) and quantification (F) of TUM- and Sham-injected hippocampal lysates probed for the indicated proteins. Data represent average + SEM of at least 3 animals.
(G) Fgf21 plasma levels as determined by ELISA. Data represent average + SEM of at least 5 animals.
The ER Stress Component Is Dispensable for Fgf21 Induction
To understand whether ER stress was necessary to induce Fgf21 expression, we capitalized on the orally bioavailable chemical chaperone tauroursodeoxycholic acid (TUDCA), which reduces mitochondria-induced ER stress in flies and mice (Debattisti et al., 2014, Muñoz et al., 2013). Co-treatment of Drp1iΔb/iΔb mice with TUDCA and tamoxifen resulted in lower Bip and P-eIF2α levels, confirming TUDCA bioavailability and activity in the brain. However, eIF2α phosphorylation and expression of Fgf21, as well as of the canonical Atf4 target gene Chop, remained elevated in TUDCA-treated Drp1iΔb/iΔb mice (Figures 3A–3E).
Figure 3.

ER Stress-Independent ISR Branches Contribute to Fgf21 Induction
(A–E) Analysis of TUDCA-treated Drp1-ablated and control mice.
(A) Representative western blots of hippocampal lysates of the indicated genotype 10 weeks PTI, probed for Bip, P-eIF2α, total eIF2α, Atf4, and actin.
(B and C) Quantification of Bip (B) and phosphorylated elF2α (C) band intensities normalized to actin (Bip) or total eIF2α, respectively.
(D and E) Hippocampal Fgf21 (D) and Chop (E) mRNA expression determined by qRT-PCR, normalized to 18S rRNA ct values. Data represent average + SEM of at least 4 animals.
(F) Free amino acids in the indicated brain regions 10 weeks PTI. Data represent average ± SEM of at least 4 animals.
(G) Fold change in selected hits of a total proteomics screen 10 weeks PTI, plotted as a function of the respective q value.
(H) Fold change in selected, iron-related hits of a proteomics screen 10 weeks PTI.
Asterisks in (E) denote q values (i.e., p values adjusted for multiple testing): ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001.
Overall, although ER stress is present in Drp1iΔb/iΔb brains and can cause Fgf21 expression, its inhibition alone is not sufficient to abolish Fgf21 in our model, suggesting that one or more of the alternative ISR branches contribute to the observed phenotype.
Impaired Amino Acid and Iron Metabolism Contributes to ISR-Driven Fgf21 Induction
To understand which mechanism accounted for Fgf21 production, we explored the role of the mitochondrial bioenergetics defect caused by Drp1 ablation (Oettinghaus et al., 2016). However, at 10 weeks PTI, protein levels of Hsp60 and mRNA levels of Hsp10, ClpP, and Yme1l, markers of the mtUPR, were normal in Drp1iΔb/iΔb brains (Figures S3A–S3C). We confirmed this by an unbiased proteomic analysis on pooled hippocampi and cortices from mice 10 weeks PTI: mtUPR was not in place in Drp1iΔb/iΔb mice, with three of the five acknowledged mtUPR genes even significantly decreased (Figure S3D).
Alternatively, ISR could result from a defect in amino acid biosynthesis or catabolism, both crucially located at the mitochondrial level. Total amino acid content was moderately decreased in the hippocampus, whereas amino acids were significantly increased in the cortex of Drp1iΔb/iΔb mice (Figure 3F). In addition, aminoacyl-tRNA synthetases were significantly enriched hits of the proteomics analysis of Drp1iΔb/iΔb brains. Even more remarkable was the signature of the subcellular distribution of the altered aminoacyl-tRNA synthetases: 80% of cytosolic aminoacyl-tRNA synthetases were significantly upregulated in Drp1iΔb/iΔb brains, while their mitochondrial counterparts were mostly downregulated (Figure 3G). Because heme biosynthesis depends on amino acid metabolism, as well as on mitochondrial function (Lane et al., 2015), we next wanted to check its potential involvement in the phenotype. Ferrochelatase, a rate-limiting mitochondria-localized component of the heme biosynthetic pathway, was downregulated in Drp1iΔb/iΔb mice, as was mitochondrial matrix iron metabolism regulator frataxin (Figure 3H). Accordingly, cytosolic iron storage protein ferritin H was upregulated, and iron importer transferrin receptor was downregulated in Drp1iΔb/iΔb brains. Finally, heme-containing hemoglobin was also strongly downregulated in Drp1iΔb/iΔb brains (Figure 3H), reflecting the global changes in iron metabolism caused by Drp1 ablation. Altogether, these data suggest that both amino acid metabolism and heme biosynthesis are altered upon Drp1 ablation and may contribute to ISR activation in Drp1iΔb/iΔb mice.
Brain-Derived Fgf21 Is Induced in Two Etiologically Distinct Mouse Models of Neurodegeneration
We next wished to investigate the possibility that Fgf21 was induced in clinically relevant models of brain pathologies affecting mitochondrial dynamics and function. To this end, we examined tau-mutated P301L mice, a model of frontotemporal dementia (FTD) (Lewis et al., 2000), as well as the tg37-Rocky Mountain Laboratory (RML) prion inoculation model (Mallucci et al., 2003, Moreno et al., 2013). Both models display ISR that responds to PERK inhibition (Moreno et al., 2013, Radford et al., 2015), as well as defects in mitochondrial function and transport (Choi et al., 2014, Chou et al., 2011). In both models, the ER stress and ISR activation markers Bip, P-eIF2α, and Atf4 were upregulated (Figures 4A–4C, 4E, and 4F). Fgf21 mRNA was elevated in hippocampi of P301L, as well as of prion-injected mice at different time points, also before the onset of symptoms (Figures 4D and 4H). Prion-inoculated mice orally treated with PERK inhibitor GSK2606414 showed a demonstrable decrease in Fgf21 expression, confirming that Fgf21 induction is ER stress dependent (Figure 4H). Thus, Fgf21 transcription is induced in etiologically distinct mouse models of neurodegeneration not caused by a primary mitochondrial defect. Our data collectively indicate that Fgf21 might be an early biomarker for neurodegenerative processes associated with mitochondrial dysfunction and ISR activation.
Figure 4.

Brain-Derived Fgf21 Is Induced in Mouse Models of Neurodegeneration
Data from P301L tau mutant mice (A–D) and RML prion inoculation disease model (E–H).
(A) Western blot of hippocampi of 9-month-old P301L mutant mice and age-matched controls.
(B and C) Quantification of Bip (B) and Atf4 (C) band intensities, normalized to their respective loading control.
(D) Hippocampal Fgf21 mRNA expression of 9-month-old P301L mutant mice and age-matched controls, determined by qRT-PCR. Data represent average + SEM of at least 4 animals.
(E) Western blot of hippocampi of mice intracerebrally inoculated with RML prions at 10 weeks postinoculation (wpi).
(F and G) Quantification of Bip (F) and Atf4 (G) band intensities normalized to their respective loading control.
(H) Fgf21 transcript levels in hippocampi of normal brain homogenate (NBH) or RML prion-inoculated mice at 6, 8, and 10 wpi. Data represent average + SEM of at least 8 animals.
Discussion
Here, we report that Drp1 deletion from adult forebrain neurons in vivo triggers the activation of a multi-branched stress response culminating in brain Fgf21 expression. Impaired mitochondrial function is linked to Fgf21 production in various peripheral tissues, but not in brain (Cornu et al., 2014, Guridi et al., 2015, Hagiwara et al., 2012, Keipert et al., 2014, Kim et al., 2013a, Kim et al., 2013b, Touvier et al., 2015, Tyynismaa et al., 2010). In our system, Drp1 deletion impairs mitochondrial dynamics and ER morphology and induces ER stress (Figure 2). This could be due to alterations in mitochondria-ER contacts affecting the two organellar membranes, modifying membrane curvature and activating ER stress sensor PERK (Volmer et al., 2013), or due to direct Drp1 action on ER shape (Yoon et al., 1998). Lack of rescue by TUDCA treatment (Figures 3A–3E) indicates that additional stress pathways converging on eIF2α are activated in the brain upon Drp1 deletion. In particular, heme biosynthesis and amino acid metabolism, two mitochondria-related pathways, are affected by brain Drp1 ablation (Figures 3F–3H). Heme biosynthesis is a partially mitochondria-resident pathway that requires iron to be actively imported into the organelles, as well as amino acids as precursors. Therefore, given the compromised bioenergetic state of Drp1-ablated mitochondria, which could affect organellar iron import, it is plausible that intramitochondrial steps of heme biosynthesis are impaired. This is corroborated by the increase in cytosolic ferritin H and the decrease in transferrin receptor (a signature of high cytosolic iron) (Muckenthaler et al., 2008). The stark decrease of two hemoglobin subunits, which would bind heme, is also in line with the heme deprivation hypothesis (Figure 3H) (Chen, 2007, Han et al., 2001).
Different brain regions display differential sensitivity to Drp1 deletion, reflecting their individual metabolic needs and responses (Berthet et al., 2014, Kageyama et al., 2012, Oettinghaus et al., 2016, Shields et al., 2015). One explanation can lie in the ability of Atf4 to act with other transcription factors in a combinatorial manner based on the nature of the stress (Kilberg et al., 2009). Total amino acid concentration varied drastically between hippocampus and cortex (Figure 3F), which could be linked to the presence, in the anterior piriform cortex, of the body’s amino acid deficiency sensor (Anthony and Gietzen, 2013). It is therefore conceivable that the differential sensitivity to Drp1 ablation is linked to the different degrees of amino acid concentration changes within the brain.
The importance of Fgf21 as a proxy of brain mitochondrial defects is reinforced by the finding that Fgf21 is expressed in brains of prion-inoculated mice, as well as of mice carrying the FTD-associated P301L tau mutation (Figures 4D and 4H). Similar to Drp1-deleted mice, both models display defects in mitochondrial function and transport, as well as ER stress-dependent ISR activation that can be reversed by PERK blockage (Chou et al., 2011, Radford et al., 2015). Although dysregulated iron and heme have also been linked to tau accumulation and prion diseases (Belaidi and Bush, 2016, Singh, 2014), ISR activation in these models is exclusively PERK dependent, which makes them an ideal model to study the reversal of Fgf21 expression by GSK2606414.
Fgf21 is regulated by circadian and nutritional factors (Fisher and Maratos-Flier, 2016, Kharitonenkov and DiMarchi, 2017), potentially limiting the specificity of its detection in vivo as a biomarker of neuronal mitochondrial dysfunction. In particular, liver Fgf21 production is modulated by diverse dietary manipulations beyond fasting (Fisher and Maratos-Flier, 2016). For these reasons, the exploration of Fgf21 levels in patients' samples was beyond the scope of this work. Nevertheless, in light of our findings and of Fgf21 being a biomarker of mitochondrial myopathies, we encourage clinicians to explore this possibility in existing, appropriately controlled patient cohorts. Of course, confounding factors such as diabetes, obesity, or liver disease shall be carefully considered. To this end, cerebrospinal fluid (CSF) analyses might prove relevant, although at least upon its acute peripheral injection in mice, this cytokine can also cross the BBB (Douris et al., 2015, Hsuchou et al., 2007).
Future work grounded in the discoveries reported here will test whether Fgf21 is induced in other diseases sharing mitochondrial and ER stress components, like Parkinson’s disease and disorders of the amyotrophic lateral sclerosis/FTD spectrum (Stoica et al., 2014, Wang et al., 2016a, Wang et al., 2016b). While we do not expect Fgf21 to aid in the diagnostic discrimination of different neurodegenerative entities, we can predict that our discovery of a secreted, brain-derived factor induced in etiologically diverse CNS diseases would represent a much-needed progress in presymptomatic screening, within a combined framework of validated biomarkers and predictive algorithms.
In conclusion, our work shows that neurons sense dysregulated mitochondrial dynamics and ER stress to activate a multi-branched stress response culminating in the release of Fgf21. The same Fgf21 mitokine is produced in the brains of bona fide tauopathy and prion disease mouse models, highlighting its potential as a marker of brain mitochondrial dysfunction.
Experimental Procedures
For more details, refer to the Supplemental Experimental Procedures.
Mice
Drp1iΔb/iΔb and tau mutant (P301L) mice were described previously (Oettinghaus et al., 2016, Lewis et al., 2000, Radford et al., 2015). For the prion disease model, hemizygous tg37 mice inoculated with RML prion were used (Mallucci et al., 2003, Moreno et al., 2013).
Western Blot
Lysates of perfused mouse brains were analyzed as previously described (Oettinghaus et al., 2016). Antibodies are listed in Supplemental Experimental Procedures.
Real-Time PCR
Organs were collected from PBS-perfused mice and RNA was isolated (RNeasy kits 74104, 74704, and 74804). RT-PCR was performed using the High Capacity cDNA Reverse Transcription Kit (Invitrogen, 4368814). Real-time PCR was performed using TaqMan assays (Life Technologies) on a 7900HT Real-Time PCR System (Applied Biosystems). TaqMan assays are listed in Supplemental Experimental Procedures. Cross-threshold (ct) values were normalized to 18S ct values.
Fluorescent Detection of Fgf21 mRNA
This analysis was performed as a service by Advanced Cell Diagnostics with the RNAscope technology, following the standard manufacturer’s protocol.
ELISA
Blood was collected between 9 and 10 a.m., and plasma Fgf21 levels were analyzed according to the manufacturer’s instructions (BioVendor, RD291108200R).
Quantitative Proteomics
Details on this analysis were described previously (Oettinghaus et al., 2016). Data are available via ProteomeXchange: PXD004890.
Statistics
Data analysis was performed with GraphPad Prism 6. For statistical analyses, two-tailed Student’s t test and one-way ANOVA with Sidak’s multiple comparison corrections were used. Unless otherwise specified, asterisks denote p values of an unpaired, two-tailed Student’s t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Acknowledgments
The authors thank M. Dürrenberger (Microscopy Center Biozentrum, University of Basel), the staff of the Microscopy Core Facility at the Department of Biomedicine (University of Basel), and M. Baumann (Institute of Pathology, Basel University Hospitals) for their expert help with experimental procedures. This work was supported by the SNSF (31003A_127308), Novartis Foundation for Medical-Biological Research, Desirée and Nils Yde Foundation (420-14), Bangerter-Rhyner Foundation (8472/HEG-DSV), Nora van Meeuwen-Haefliger Foundation, and Mach-Gaensslen Foundation (to S.F.); Forschungsfonds of Basel University (to L.M.R. and B.O.); ERC (grant UPR Neuro; 282280), MRC, Alzheimer's Society, and ARUK/DRI (to G.M.); and Telethon Italy (GGP12162 and GPP10005), AIRC Italy (IG15748), ERC ERMITO, FP7 CIG CristOpa (PCIG13-GA-2013-618697), and MIUR FIRB Automed (RBAP11Z3YA_005 to L.S.).
Author Contributions
L.M.R., B.O., M.H., M.L., C.A., L.S., C.S., A.N., and A.S. performed experiments. L.M.R., B.O., J.H., M.T., A.S., and A.E. analyzed and interpreted experimental data. G.M., L.S., and S.F. conceived the project and coordinated and supervised research. L.M.R., B.O., L.S., and S.F. wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: August 7, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures and three figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.07.023.
Supplemental Information
References
- Anthony T.G., Gietzen D.W. Detection of amino acid deprivation in the central nervous system. Curr. Opin. Clin. Nutr. Metab. Care. 2013;16:96–101. doi: 10.1097/MCO.0b013e32835b618b. [DOI] [PubMed] [Google Scholar]
- Belaidi A.A., Bush A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: targets for therapeutics. J. Neurochem. 2016;139(Suppl 1):179–197. doi: 10.1111/jnc.13425. [DOI] [PubMed] [Google Scholar]
- Berthet A., Margolis E.B., Zhang J., Hsieh I., Zhang J., Hnasko T.S., Ahmad J., Edwards R.H., Sesaki H., Huang E.J., Nakamura K. Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J. Neurosci. 2014;34:14304–14317. doi: 10.1523/JNEUROSCI.0930-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bookout A.L., de Groot M.H., Owen B.M., Lee S., Gautron L., Lawrence H.L., Ding X., Elmquist J.K., Takahashi J.S., Mangelsdorf D.J., Kliewer S.A. FGF21 regulates metabolism and circadian behavior by acting on the nervous system. Nat. Med. 2013;19:1147–1152. doi: 10.1038/nm.3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.J. Regulation of protein synthesis by the heme-regulated eIF2α kinase: relevance to anemias. Blood. 2007;109:2693–2699. doi: 10.1182/blood-2006-08-041830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H.S., Choi Y.G., Shin H.Y., Oh J.M., Park J.H., Kim J.I., Carp R.I., Choi E.K., Kim Y.S. Dysfunction of mitochondrial dynamics in the brains of scrapie-infected mice. Biochem. Biophys. Res. Commun. 2014;448:157–162. doi: 10.1016/j.bbrc.2014.04.069. [DOI] [PubMed] [Google Scholar]
- Chou J.L., Shenoy D.V., Thomas N., Choudhary P.K., Laferla F.M., Goodman S.R., Breen G.A. Early dysregulation of the mitochondrial proteome in a mouse model of Alzheimer’s disease. J. Proteomics. 2011;74:466–479. doi: 10.1016/j.jprot.2010.12.012. [DOI] [PubMed] [Google Scholar]
- Cornu M., Oppliger W., Albert V., Robitaille A.M., Trapani F., Quagliata L., Fuhrer T., Sauer U., Terracciano L., Hall M.N. Hepatic mTORC1 controls locomotor activity, body temperature, and lipid metabolism through FGF21. Proc. Natl. Acad. Sci. USA. 2014;111:11592–11599. doi: 10.1073/pnas.1412047111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Brito O.M., Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. doi: 10.1038/nature07534. [DOI] [PubMed] [Google Scholar]
- Debattisti V., Pendin D., Ziviani E., Daga A., Scorrano L. Reduction of endoplasmic reticulum stress attenuates the defects caused by Drosophila mitofusin depletion. J. Cell Biol. 2014;204:303–312. doi: 10.1083/jcb.201306121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degirolamo C., Sabbà C., Moschetta A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat. Rev. Drug Discov. 2016;15:51–69. doi: 10.1038/nrd.2015.9. [DOI] [PubMed] [Google Scholar]
- Donnelly N., Gorman A.M., Gupta S., Samali A. The eIF2α kinases: their structures and functions. Cell. Mol. Life Sci. 2013;70:3493–3511. doi: 10.1007/s00018-012-1252-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douris N., Stevanovic D.M., Fisher F.M., Cisu T.I., Chee M.J., Nguyen N.L., Zarebidaki E., Adams A.C., Kharitonenkov A., Flier J.S. Central fibroblast growth factor 21 browns white fat via sympathetic action in male mice. Endocrinology. 2015;156:2470–2481. doi: 10.1210/en.2014-2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuBoff B., Feany M., Götz J. Why size matters—balancing mitochondrial dynamics in Alzheimer’s disease. Trends Neurosci. 2013;36:325–335. doi: 10.1016/j.tins.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Fisher F.M., Maratos-Flier E. Understanding the physiology of FGF21. Annu. Rev. Physiol. 2016;78:223–241. doi: 10.1146/annurev-physiol-021115-105339. [DOI] [PubMed] [Google Scholar]
- Fon Tacer K., Bookout A.L., Ding X., Kurosu H., John G.B., Wang L., Goetz R., Mohammadi M., Kuro-o M., Mangelsdorf D.J., Kliewer S.A. Research resource: comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 2010;24:2050–2064. doi: 10.1210/me.2010-0142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guridi M., Tintignac L.A., Lin S., Kupr B., Castets P., Rüegg M.A. Activation of mTORC1 in skeletal muscle regulates whole-body metabolism through FGF21. Sci. Signal. 2015;8:ra113. doi: 10.1126/scisignal.aab3715. [DOI] [PubMed] [Google Scholar]
- Hagiwara A., Cornu M., Cybulski N., Polak P., Betz C., Trapani F., Terracciano L., Heim M.H., Rüegg M.A., Hall M.N. Hepatic mTORC2 activates glycolysis and lipogenesis through Akt, glucokinase, and SREBP1c. Cell Metab. 2012;15:725–738. doi: 10.1016/j.cmet.2012.03.015. [DOI] [PubMed] [Google Scholar]
- Han A.P., Yu C., Lu L., Fujiwara Y., Browne C., Chin G., Fleming M., Leboulch P., Orkin S.H., Chen J.J. Heme-regulated eIF2α kinase (HRI) is required for translational regulation and survival of erythroid precursors in iron deficiency. EMBO J. 2001;20:6909–6918. doi: 10.1093/emboj/20.23.6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsuchou H., Pan W., Kastin A.J. The fasting polypeptide FGF21 can enter brain from blood. Peptides. 2007;28:2382–2386. doi: 10.1016/j.peptides.2007.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N., Nomura M., Jofuku A., Kato H., Suzuki S.O., Masuda K., Otera H., Nakanishi Y., Nonaka I., Goto Y. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- Kageyama Y., Zhang Z., Roda R., Fukaya M., Wakabayashi J., Wakabayashi N., Kensler T.W., Reddy P.H., Iijima M., Sesaki H. Mitochondrial division ensures the survival of postmitotic neurons by suppressing oxidative damage. J. Cell Biol. 2012;197:535–551. doi: 10.1083/jcb.201110034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keipert S., Ost M., Johann K., Imber F., Jastroch M., van Schothorst E.M., Keijer J., Klaus S. Skeletal muscle mitochondrial uncoupling drives endocrine cross-talk through the induction of FGF21 as a myokine. Am. J. Physiol. Endocrinol. Metab. 2014;306:E469–E482. doi: 10.1152/ajpendo.00330.2013. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A., DiMarchi R. FGF21 revolutions: recent advances illuminating FGF21 biology and medicinal properties. Trends Endocrinol. Metab. 2015;26:608–617. doi: 10.1016/j.tem.2015.09.007. [DOI] [PubMed] [Google Scholar]
- Kharitonenkov A., DiMarchi R. Fibroblast growth factor 21 night watch: advances and uncertainties in the field. J. Intern. Med. 2017;281:233–246. doi: 10.1111/joim.12580. [DOI] [PubMed] [Google Scholar]
- Kilberg M.S., Shan J., Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab. 2009;20:436–443. doi: 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K.H., Jeong Y.T., Oh H., Kim S.H., Cho J.M., Kim Y.N., Kim S.S., Kim D.H., Hur K.Y., Kim H.K. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat. Med. 2013;19:83–92. doi: 10.1038/nm.3014. [DOI] [PubMed] [Google Scholar]
- Kim K.H., Jeong Y.T., Kim S.H., Jung H.S., Park K.S., Lee H.Y., Lee M.S. Metformin-induced inhibition of the mitochondrial respiratory chain increases FGF21 expression via ATF4 activation. Biochem. Biophys. Res. Commun. 2013;440:76–81. doi: 10.1016/j.bbrc.2013.09.026. [DOI] [PubMed] [Google Scholar]
- Lane D.J., Merlot A.M., Huang M.L., Bae D.H., Jansson P.J., Sahni S., Kalinowski D.S., Richardson D.R. Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta. 2015;1853:1130–1144. doi: 10.1016/j.bbamcr.2015.01.021. [DOI] [PubMed] [Google Scholar]
- Lewis J., McGowan E., Rockwood J., Melrose H., Nacharaju P., Van Slegtenhorst M., Gwinn-Hardy K., Paul Murphy M., Baker M., Yu X. Neurofibrillary tangles, amyotrophy and progressive motor disturbance in mice expressing mutant (P301L) tau protein. Nat. Genet. 2000;25:402–405. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- Mallucci G., Dickinson A., Linehan J., Klöhn P.C., Brandner S., Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302:871–874. doi: 10.1126/science.1090187. [DOI] [PubMed] [Google Scholar]
- Moreno J.A., Halliday M., Molloy C., Radford H., Verity N., Axten J.M., Ortori C.A., Willis A.E., Fischer P.M., Barrett D.A., Mallucci G.R. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci. Transl. Med. 2013;5:206ra138. doi: 10.1126/scitranslmed.3006767. [DOI] [PubMed] [Google Scholar]
- Muckenthaler M.U., Galy B., Hentze M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008;28:197–213. doi: 10.1146/annurev.nutr.28.061807.155521. [DOI] [PubMed] [Google Scholar]
- Muñoz J.P., Ivanova S., Sánchez-Wandelmer J., Martínez-Cristóbal P., Noguera E., Sancho A., Díaz-Ramos A., Hernández-Alvarez M.I., Sebastián D., Mauvezin C. Mfn2 modulates the UPR and mitochondrial function via repression of PERK. EMBO J. 2013;32:2348–2361. doi: 10.1038/emboj.2013.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oettinghaus B., Schulz J.M., Restelli L.M., Licci M., Savoia C., Schmidt A., Schmitt K., Grimm A., Morè L., Hench J. Synaptic dysfunction, memory deficits and hippocampal atrophy due to ablation of mitochondrial fission in adult forebrain neurons. Cell Death Differ. 2016;23:18–28. doi: 10.1038/cdd.2015.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas L., Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 2016;78:505–531. doi: 10.1146/annurev-physiol-021115-105011. [DOI] [PubMed] [Google Scholar]
- Pitts K.R., Yoon Y., Krueger E.W., McNiven M.A. The dynamin-like protein DLP1 is essential for normal distribution and morphology of the endoplasmic reticulum and mitochondria in mammalian cells. Mol. Biol. Cell. 1999;10:4403–4417. doi: 10.1091/mbc.10.12.4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radford H., Moreno J.A., Verity N., Halliday M., Mallucci G.R. PERK inhibition prevents tau-mediated neurodegeneration in a mouse model of frontotemporal dementia. Acta Neuropathol. 2015;130:633–642. doi: 10.1007/s00401-015-1487-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rath E., Berger E., Messlik A., Nunes T., Liu B., Kim S.C., Hoogenraad N., Sans M., Sartor R.B., Haller D. Induction of dsRNA-activated protein kinase links mitochondrial unfolded protein response to the pathogenesis of intestinal inflammation. Gut. 2012;61:1269–1278. doi: 10.1136/gutjnl-2011-300767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shields L.Y., Kim H., Zhu L., Haddad D., Berthet A., Pathak D., Lam M., Ponnusamy R., Diaz-Ramirez L.G., Gill T.M. Dynamin-related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell Death Dis. 2015;6:e1725. doi: 10.1038/cddis.2015.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh N. The role of iron in prion disease and other neurodegenerative diseases. PLoS Pathog. 2014;10:e1004335. doi: 10.1371/journal.ppat.1004335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoica R., De Vos K.J., Paillusson S., Mueller S., Sancho R.M., Lau K.F., Vizcay-Barrena G., Lin W.L., Xu Y.F., Lewis J. ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat. Commun. 2014;5:3996. doi: 10.1038/ncomms4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suomalainen A., Elo J.M., Pietiläinen K.H., Hakonen A.H., Sevastianova K., Korpela M., Isohanni P., Marjavaara S.K., Tyni T., Kiuru-Enari S. FGF-21 as a biomarker for muscle-manifesting mitochondrial respiratory chain deficiencies: a diagnostic study. Lancet Neurol. 2011;10:806–818. doi: 10.1016/S1474-4422(11)70155-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tezze C., Romanello V., Desbats M.A., Fadini G.P., Albiero M., Favaro G., Ciciliot S., Soriano M.E., Morbidoni V., Cerqua C. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. 2017;25:1374–1389.e6. doi: 10.1016/j.cmet.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touvier T., De Palma C., Rigamonti E., Scagliola A., Incerti E., Mazelin L., Thomas J.L., D’Antonio M., Politi L., Schaeffer L. Muscle-specific Drp1 overexpression impairs skeletal muscle growth via translational attenuation. Cell Death Dis. 2015;6:e1663. doi: 10.1038/cddis.2014.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twig G., Hyde B., Shirihai O.S. Mitochondrial fusion, fission and autophagy as a quality control axis: the bioenergetic view. Biochim. Biophys. Acta. 2008;1777:1092–1097. doi: 10.1016/j.bbabio.2008.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyynismaa H., Carroll C.J., Raimundo N., Ahola-Erkkilä S., Wenz T., Ruhanen H., Guse K., Hemminki A., Peltola-Mjøsund K.E., Tulkki V. Mitochondrial myopathy induces a starvation-like response. Hum. Mol. Genet. 2010;19:3948–3958. doi: 10.1093/hmg/ddq310. [DOI] [PubMed] [Google Scholar]
- Volmer R., van der Ploeg K., Ron D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA. 2013;110:4628–4633. doi: 10.1073/pnas.1217611110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T., Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol. Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- Wakabayashi J., Zhang Z., Wakabayashi N., Tamura Y., Fukaya M., Kensler T.W., Iijima M., Sesaki H. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J. Cell Biol. 2009;186:805–816. doi: 10.1083/jcb.200903065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Wang L., Lu J., Siedlak S.L., Fujioka H., Liang J., Jiang S., Ma X., Jiang Z., da Rocha E.L. The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 2016;22:869–878. doi: 10.1038/nm.4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W., Wang X., Fujioka H., Hoppel C., Whone A.L., Caldwell M.A., Cullen P.J., Liu J., Zhu X. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat. Med. 2016;22:54–63. doi: 10.1038/nm.3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon Y., Pitts K.R., Dahan S., McNiven M.A. A novel dynamin-like protein associates with cytoplasmic vesicles and tubules of the endoplasmic reticulum in mammalian cells. J. Cell Biol. 1998;140:779–793. doi: 10.1083/jcb.140.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.