

Abstract
Key points
Dietary Na restriction, through the mineralocorticoid aldosterone, acts on epithelial Na channels via both fast (24 h) and slow (5–7 days) mechanisms in the kidney.
The fast effect entails increased proteolytic processing and trafficking of channel protein to the apical membrane. It is rapidly reversible by the mineralocorticoid receptor antagonist eplerenone and is largely lost when tubules are studied ex vivo.
The slow effect does not require increased processing or surface expression, is refractory to acute eplerenone treatment, and is preserved ex vivo.
Both slow and fast effects contribute to Na retention in vivo.
Increased Na+ reabsorption in the proximal tubule also promotes Na conservation under conditions of chronic dietary Na restriction, reducing Na+ delivery to the distal nephron.
Abstract
Changes in the activity of the epithelial Na channel (ENaC) help to conserve extracellular fluid volume. In rats fed a low‐salt diet, proteolytic processing of ENaC increased within 1 day, and was almost maximal after 3 days. The rapid increase in the abundance of cleaved αENaC and γENaC correlated with decreased urinary Na+ excretion and with increased ENaC surface expression. By contrast, ENaC activity, measured ex vivo in isolated cortical collecting ducts, increased modestly after 3 days and required 5 days to reach maximal levels. The mineralocorticoid receptor antagonist eplerenone reversed the increase in cleaved γENaC and induced natriuresis after 1 or 3 days but failed to alter either ENaC currents or Na+ excretion after 7 days of Na restriction. We conclude that Na depletion, through aldosterone, stimulates ENaC via independent fast and slow mechanisms. In vivo, amiloride‐induced natriuresis increased after 1 day of Na depletion. By contrast, hydrochlorothiazide (HCTZ)‐induced natriuresis decreased gradually over 7 days, consistent with increased ability of ENaC activity to compensate for decreased Na+ reabsorption in the distal convoluted tubule. Administration of amiloride and HCTZ together increased Na+ excretion less in Na‐depleted compared to control animals, indicating decreased delivery of Na+ to the distal nephron when dietary Na is restricted. Measurements of creatinine and Li+ clearances indicated that increased Na reabsorption by the proximal tubules is responsible for the decreased delivery. Thus, Na conservation during chronic dietary salt restriction entails enhanced transport by both proximal and distal nephron segments.
Keywords: low‐Na diet, surface expression, eplerenone, amiloride, HCTZ, Li clearance
Key points
Dietary Na restriction, through the mineralocorticoid aldosterone, acts on epithelial Na channels via both fast (24 h) and slow (5–7 days) mechanisms in the kidney.
The fast effect entails increased proteolytic processing and trafficking of channel protein to the apical membrane. It is rapidly reversible by the mineralocorticoid receptor antagonist eplerenone and is largely lost when tubules are studied ex vivo.
The slow effect does not require increased processing or surface expression, is refractory to acute eplerenone treatment, and is preserved ex vivo.
Both slow and fast effects contribute to Na retention in vivo.
Increased Na+ reabsorption in the proximal tubule also promotes Na conservation under conditions of chronic dietary Na restriction, reducing Na+ delivery to the distal nephron.
Introduction
In response to dietary salt restriction, the kidneys conserve Na+ by increasing reabsorption of the ion from the glomerular filtrate. This is accomplished, in part, by secretion of the mineralocorticoid aldosterone from the adrenal cortex. The epithelial Na+ channel (ENaC) is a principal target for aldosterone in the kidneys; the density of conducting channels increases in response to either a low‐Na diet or administration of the hormone (Pácha et al. 1993).
In addition to increasing channel function, as assessed ex vivo, aldosterone produces biochemical changes in ENaC. The expression of the αENaC subunit increases at both the mRNA and protein levels (Asher et al. 1996; Masilamani et al. 1999b). The α and γENaC subunits also undergo aldosterone‐dependent proteolytic cleavage (Masilamani et al. 1999b; Ergonul et al. 2006). These changes correlate with increases in surface expression of the channel measured by immunocytochemistry (Masilamani et al. 1999b) or biotinylation of plasma membrane proteins (Frindt et al. 2008; Frindt & Palmer, 2009).
The correlation between biochemical and functional measurements is not perfect. We recently reported that, during acute administration of aldosterone to rats, surface expression increased more rapidly than channel activity (Frindt & Palmer, 2015). Conversely, in mice, deletion of the gene for serum and glucocorticoid induced kinase 1 (Sgk1) suppressed increases in surface expression but not that of channel activity in response to chronic hormone treatment (Yang et al. 2017).
In the present study, we investigate the time courses of biochemical and electrophysiological responses to dietary Na restriction. We also correlate these effects with changes in overall urinary Na+ excretion and the renal responses to the diuretics amiloride and hydrochlorothiazide (HCTZ), which target ENaC and the NaCl cotransporter (NCC), respectively. We conclude that aldosterone induces both rapid and slow effects on ENaC via different mechanisms. Furthermore, increased distal nephron Na+ transport combines with diminished delivery of Na+ to the aldosterone‐sensitive segments to minimize urinary Na+ losses.
Methods
Ethical approval
All procedures using animals were approved by the Institutional Animal Care and Use Committee of Weill Cornell Medical College and conform with the standards as described by Grundy (Grundy, 2015).
Animals
Sprague–Dawley rats (Charles River Laboratories, Kingston, NY, USA) weighing from 150 to 250 g were raised free of viral infections and fed either a Na‐deficient diet containing 0.004% Na by weight or a matched diet supplemented with 1% NaCl (MP Biomedicals, Solon, OH, USA). For urine collection, animals were kept in metabolic cages with free access to drinking water but without food. After completion of urine collection, blood was taken from the abdominal aorta under anaesthesia using ketamine (90 mg kg–1) plus xylazine (4 mg kg–1). Animals were killed under anaesthesia by thoracotomy followed by cutting the outflow tract from the heart. The kidneys were removed for electrophysiological and biochemical analysis.
Eplerenone (AstraZeneca, Mölndal, Sweden) was administered by oral gavage at a dose of 30 mg kg–1 bw. HCTZ and amiloride (Sigma‐Aldrich, St Louis, MO, USA) were dissolved in DMSO (dose–response relationships) or polyethylene glycol 300 (PEG‐300) (maximal dose experiments) and injected s.c. in a volume of 1 mL kg–1. Controls were treated with 1 mL kg–1 of the appropriate vehicles. Electrolyte excretion was similar with the two solvents but, in two animals, DMSO produced a slight discoloration of the urine, prompting the switch to PEG‐300. Data from animals with discolored urine were excluded. Li+ was administered through osmotic minipumps (model 2001D; Alzet, Cupertino, CA, USA) implanted s.c. at 16.00 h the day before the experiment. Pumps were filled with 750 mm LiCl dissolved in H2O. The following morning, rats were challenged with diuretics and urine was collected for 80 min.
Losartan, propranolol and dopamine (Sigma‐Aldrich) were dissolved in PEG‐300 at concentrations of 10, 5 and 5 mg mL–1 respectively and administered s.c. at final doses of 10, 5 and 5 mg kg–1 bw. The doses for losartan (Wang et al. 1997) and propranolol (Lees, 1968) were based on previous studies demonstrating natriuresis in rats. That of dopamine was determined empirically as the lowest dose producing consistent natriuresis.
Analytical
Na, K and Li concentrations in the collected urine and plasma were measured using a flame photometer (model IL 943; Instrumentation Laboratory, Bedford, MA, USA). Creatinine was measured using a colorimetric assay kit (BioAssay Systems, Hayward, CA, USA). HCTZ concentrations in urine were measured with an enzyme‐linked immunosorbent assay kit (Neogen, Lexington, KY, USA) in accordance with the manufacturer's instructions.
Electrophysiology
Whole‐cell amiloride‐sensitive currents were measured in principal cells of isolated, split‐open cortical collecting ducts (CCDs) as described previously (Frindt & Palmer, 2012, 2015).
In situ biotinylation
Biotinylation of membrane proteins was performed in accordance with a protocol described previously (Frindt et al. 2017). Briefly, kidneys were perfused with an impermeant biotinylating reagent (Sulfo‐NHS biotin; Campbell Science, Rockville, IL, USA) in situ. After quenching the biotinylation reaction with Tris, the perfused kidney was removed and microsomes were prepared from it. Microsomes were solubilized with 3% Triton X‐100 and surface proteins from equal amounts of microsomal protein were isolated using neutravidin beads.
Western blots
Whole‐kidney microsomes were prepared for western blot analysis by mincing one kidney and disrupting the tissue with a Dounce homogenizer. The homogenate was filtered through a nylon cell strainer (70 μm mesh) to remove intact tissue and centrifuged at 100,000 g for 90 min to obtain a microsomal pellet. This was suspended in 3 mL of lysis buffer and frozen for subsequent analysis. In some experiments, the medulla and the superficial cortex were dissected from kidney slices and processed as described above. In other experiments, medullary rays were dissected from kidney slices with fine forceps under a microscope. The tubule bundles typically contained four CCD together with proximal straight tubules and ascending thick limbs but no connecting tubules (CNTs) or other segments from the superficial cortex. Twenty to 30 rays were dissected from each rat kidney and transferred to 200 μL of sucrose lysis buffer containing protease and phosphatase inhibitors and 1% SDS. The tissue was disrupted by sonication for 40 s. After measurement of the protein concentration, samples were prepared for electrophoresis as described previously (Frindt et al. 2017). Western blot analysis showed abundant expression of ENaC, presumably from the CCDs, and no detectable expression of NCC, confirming the absence of distal convoluted tubules (DCTs) in the preparation (Fig. 6).
Figure 6. Time course of the effects of low‐Na diet on proteins from renal cortical medullary rays.
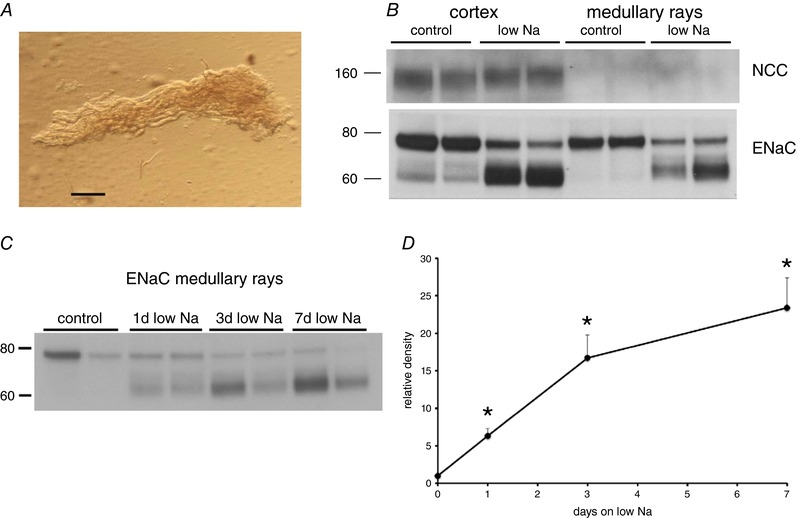
A, photomicrograph of a cortical medullary ray (scale bar = 0.2 mm). Further dissection revealed that the ray contained four CCDs, several proximal straight tubules and ascending thick limbs. Twenty to 30 such rays were dissected from each rat kidney, lysed and prepared for western blot. B, western blots for γENaC and NCC in samples from superficial cortex and cortical medullary rays of rats on control‐Na diet and on a low‐Na diet for 3 days. Equal amounts of protein from a different animal were loaded into each lane. The medullary rays show abundant expression of γENaC, presumably from cortical collecting ducts. NCC protein was abundant in the cortex, presumably from DCTs. No NCC could be detected in the medullary rays. C, western blot of tissue from medullary rays isolated from kidneys of rats fed a control diet or a low‐Na diet for 1, 3 or 7 days. D, quantitative analysis of γENaC expression. Data represent the mean ± SEM for four animals at each time point. *Statistical difference of the indicated low‐Na group relative to controls (0.001 < P < 0.002). [Color figure can be viewed at http://wileyonlinelibrary.com]
Samples were electrophoresed on 4–12% Bis‐Tris gels (Invitrogen, Carldbad, CA, USA) and the proteins were transferred electrophoretically to polyvinylidene fluoride membranes. After blocking, membranes were incubated overnight at 4°C with primary antibodies. Anti‐rabbit IgG conjugated with alkaline phosphatase was used as a secondary antibody. Bound antibody was visualized on autoradiography film (HyBlot CL; Denville Scientific, Holliston, MA, USA) using a chemiluminescence substrate (Western Breeze; Invitrogen). Semiquantitative densitometry of protein bands was performed with background subtraction using Photoshop CS5 (Adobe Systems Inc., San Jose, CA, USA). After films were developed, blots were stained with Coomassie blue to assess protein loading in individual lanes, which varied by less than 10%.
Antibodies
Polyclonal antibodies against the β and γ subunits of rat ENaC were based on short peptide sequences in the carboxy termini as described previously (Masilamani et al. 1999a; Ergonul et al. 2006). Anti‐sera were purified using peptide‐linked agarose bead affinity columns (Sulfolink Kit; Pierce Biotechnology, Rockford, IL, USA). The antibody against the N‐terminus of mouse αENaC (Sorensen et al. 2013) was a gift from Professor Johannes Loffing (University of Zürich, Zürich, Swizerland). The antibody against NCC (Nguyen et al. 2013) was a gift from Professor Alicia McDonough (University of Southern California, Los Angeles, CA, USA).
Statistical analysis
Statistical significance between two groups was assessed by unpaired Student's t test. P < 0.05 was considered statistically significant.
Results
Previous studies of the rat kidney showed increased activity of epithelial Na+ channels in the aldosterone‐sensitive distal nephron in response to dietary Na restriction (Frindt, 1990; Pácha, 1993), accompanied by enhanced proteolytic processing and trafficking of the ENaC subunits to the apical membrane (Frindt, 2008; Frindt, 2016). In the present study, we tested the hypothesis that these two measures of channel upregulation were correlated with each other and with the overall reduction in Na+ excretion with respect to the time that the animals were Na‐deprived.
Figure 1 shows typical whole‐cell recordings from principal cells of the rat CCD after 3, 5 and 7 days on a low‐Na diet. ENaC current (I Na) was assessed as the component blocked by 10 μm amiloride. Previous studies showed that, in animals consuming the matched control diet containing 1% NaCl, these currents were undetectable (Frindt et al. 1990; Frindt et al. 2001), in agreement with the lack of net Na+ reabsorption by isolated‐perfused rat CCDs under control conditions (Tomita et al. 1985; Reif et al. 1986). After 1 day of Na depletion, amiloride‐sensitive currents remained within the drift of the system (4 ± 5 pA cell–1). With 3 days of Na depletion, I Na was clearly detectable in some cases but was small; the largest current in any cell was 140 pA, and the mean was 30 pA cell–1. By contrast, with 5 days of Na depletion, I Na was consistently large, often exceeding 500 pA cell–1. Current magnitudes were similar after 7 days on the low‐Na diet.
Figure 1.
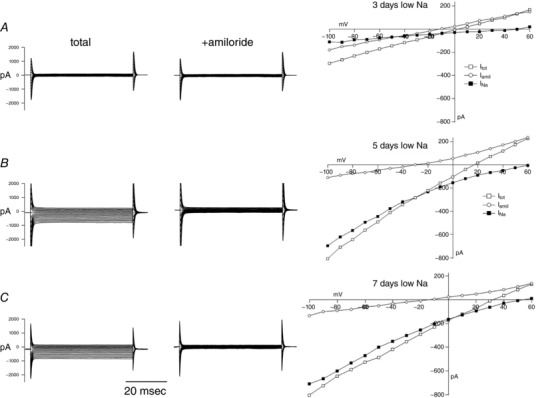
Time course of the development of ENaC currents with dietary Na restriction
Whole‐cell currents in principal cells of CCDs from rats on a low‐Na diet for (A) 3 days, (B) 5 days and (C) 7 days. Left: current traces in response to voltage jumps to –100 to +60 mV from a holding potential of 0 mV in the absence and presence of 10 μm amiloride in the bath solution. Right: I–V relationships in the absence and presence of amiloride, as well as the difference current (I Na).
Figure 2 A illustrates the biochemical responses under similar conditions, exemplified by the appearance of the cleaved (60–65 kDa) form of the γENaC subunit. Here, the response was robust after a single day of Na depletion. A similar time course characterized the appearance of the cleaved (25–30 kDa) form of αENaC. βENaC does not undergo proteolytic cleavage in response to Na depletion or aldosterone, although a highly glycosylated form of this subunit appears as a diffuse band above the main 85–90 kDa form, again after a single day on a low‐Na diet. Figure 2 B summarizes and quantifies these changes.
Figure 2. Western blots of microsomes from kidneys of rats on a control diet or a low‐Na diet for 1, 3, and 7 days.
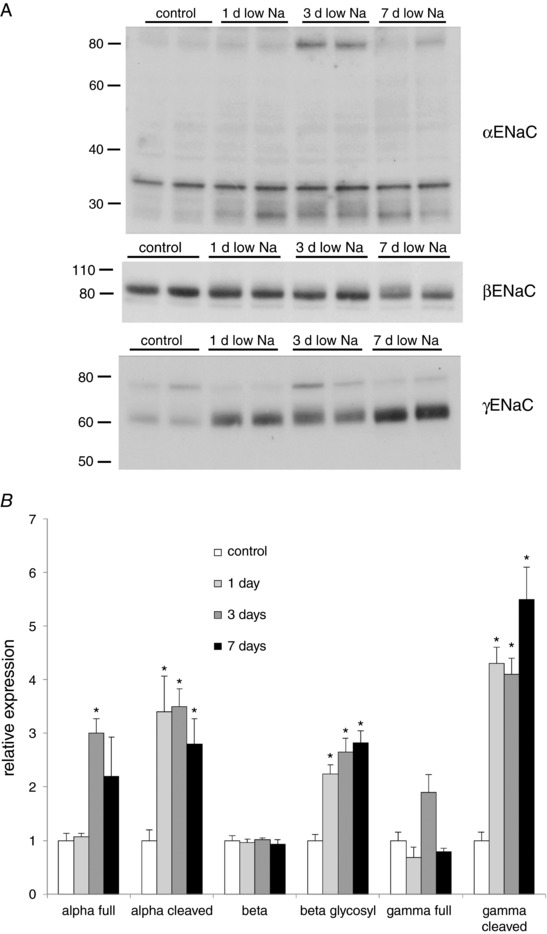
A, typical blots. Each lane presents a different animal. Equal amounts (30 μg) of protein were loaded onto each lane. Blots were stained with antibodies against αENaC, βENaC and γENaC. Bands at 75–85 kDa represent full‐length forms of the proteins. Cleaved forms of αENaC and γENaC appear as ∼25 and 65 kDa bands, respectively. Glycosylated βENaC runs as a diffuse band just above the major full‐length species. The 33 kDa band recognized by the anti‐αENaC antibody has not been identified and does not vary with salt intake. B, quantitation by densitometry. Data represent the mean ± SEM for four animals for each condition. *Statistical difference of the indicated low‐Na group relative to controls (range 0.001 < P < 0.01).
Thus, the biochemical response of ENaC to Na restriction occurs much earlier than does the increase in ENaC current. In light of this discrepancy, we investigated which of these events correlated best with the ability of the kidneys to conserve Na. Figure 3 shows that Na+ excretion falls from ∼0.9 μmol min–1 on control diet to very low levels (∼0.01 μmol min–1) after 1 day of low‐Na intake. This is much faster than the electrical response, assessed as I Na (Fig. 3 A), but matches the biochemical response, plotted as the relative abundance of cleaved γENaC (Fig. 3 B). Here, we use the expression of cleaved γENaC as an indicator of ENaC processing because this measurement has a better signal‐to‐noise ratio than that of the cleaved form of αENaC or the glycosylated form of βENaC.
Figure 3.

Time courses of ENaC processing and currents relative to decreased Na excretion with dietary Na restriction
A, amiloride‐sensitive current (I Na) and urinary Na+ excretion as a function of time on low‐Na diet. I Na was measured at cell potentials of –100 mV as shown in Fig. 1. Data represent the mean ± SEM for 15–24 cells from five to eight different tubules and two to five different animals at each time point. Na excretion (U Na V) was measured from urine collections of ∼2 h in the morning. Data represent the mean ± SEM for five to 12 animals at each time point. B, expression of cleaved γENaC and urinary Na+ excretion as a function of time on low‐Na diet. Relative amounts of cleaved γENaC were measured using western blots as shown in Fig. 2. Data represent the mean ± SEM for four animals at each time point.
Although the appearance of the mature ENaC subunits is associated with increased surface expression of the channels (Frindt et al. 2016), a lag between the proteolytic processing of the subunits and their insertion into the apical membrane could explain the discrepancy between processing and channel activity. Figure 4 compares the surface expression of γENaC measured as the biotinylated fraction of the protein after 1 day and 7 days on a low‐Na diet. The amount of cleaved γENaC at the surface after 1 day was ∼50% of that after 7 days. This indicates that surface expression may be slightly slower than cleavage, which reached 80% of its final level within 1 day. However, the time course of changes in surface expression was still much faster than that of I Na.
Figure 4. Total and surface expression of the cleaved form of γENaC as a function of time on low‐Na diet.
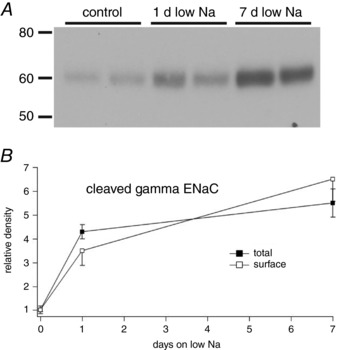
A, western blots of biotinylated proteins. Each lane represents an eluate from neutravidin beads corresponding to 1 mg of total membrane protein. B, comparison of total and surface fractions. Total cleaved γENaC was measured in microsomal fractions that were then used to isolate biotinylated proteins. Cleaved γENaC was measured in blots such as that shown in (A). Data represent the mean ± SEM for four animals at each time point.
The discordance between biochemical and electrophysiological measurements could be spatial rather than temporal. For example, Nesterov et al. (2012) showed that ENaC is regulated differently in the late DCT/CNT compared with the CCD of mice. In our studies, I Na was measured in the CCD, whereas the biochemical information comes from the whole kidney. The test this possibility, we compared the processing of γENaC in cortex and medulla from control rats and animals on low Na for 1 day. If the collecting duct responds more slowly than the more proximal ENaC‐expressing segments then we would expect to see a smaller change in the medulla where the ENaC come exclusively from medullary collecting ducts As shown in Fig. 5, this was not the case. One day of low Na was sufficient to induce significant and similar cleavages of γENaC in both cortex and medulla.
Figure 5. Comparison of the effects of low‐Na diet on γENaC and NCC protein from renal cortex and medulla.
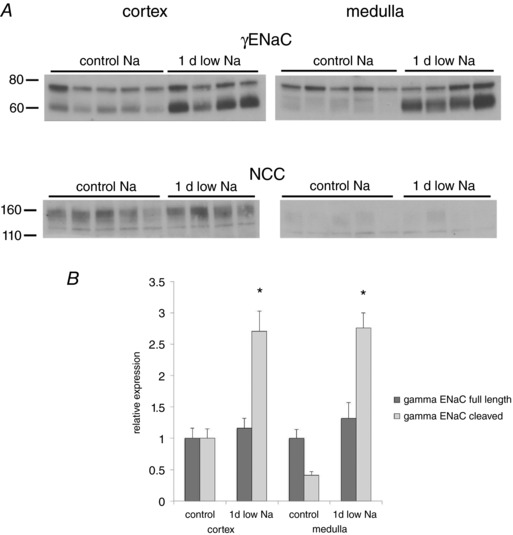
A, changes in γENaC and NCC after 1 day of a low‐Na diet in tissue from the outer cortex and from the medulla. NCC protein was abundant in the cortex but not in the medulla, consistent with expression mainly in the DCT. B, quantitative analysis of γENaC expression. Significant increases in the cleaved form of the subunit occurred in both cortex and medulla. Data represent the mean ± SEM for four low‐Na and five control rats. *Statistical difference of the indicated low‐Na group relative to controls [P = 0.003 (cortex), P = 0.0001 (medulla)].
To assess this point further, we analysed ENaC protein in medullary rays dissected from the renal cortex. The ENaC protein in this preparation comes from the cortical collecting ducts, the same part of the nephron used for electrophysiological measurements but not other segments that express ENaC. We chose to use this approach rather than the isolation of individual CCDs because a large number of long ducts could be obtained in a relatively short time without the need for treating the tissue with collagenase. Figure 6 A shows a photomicrograph of one of these rays. Further dissection of this tissue revealed the presence of four CCDs, as well as proximal straight tubules and thick ascending limbs; there were no CNTs evident. The preparation was further characterized by western blotting (Fig. 6 B). These blots showed abundant γENaC protein but no detectable NCC, indicating the absence of distal convoluted tubules. As shown in Fig. 6 C and D, the fractional increase in the abundance of the cleaved form of γENaC in this preparation was quite large, reaching ∼20‐fold after 7 days on low Na. After single day of Na restriction cleaved γENaC was already 6‐fold greater than controls, and almost maximal levels were achieved after 3 days, when the electrophysiological response was still small. Therefore, the dissociation between biochemical and electrophysiological responses remained even when the same part of the nephron was assayed.
These results raise the conundrum of why we do not detect significant ENaC current in tubules from animals on low Na for periods less than 5 days, even though surface expression is increased, and urinary Na excretion is minimal. One explanation is that, under these conditions, ENaC activation is labile and does not survive the removal of the kidneys from the animal and/or the time required (at least 1 h) to isolate and open the tubules and make the electrophysiological recordings. To test for reversibility of activation, we examined the effects of the mineralocorticoid receptor (MR) antagonist (MRA) eplerenone on the channels under different conditions. We administered the drug in a single dose of 30 mg kg–1 and measured Na+ and K+ excretion during two consecutive 2 h intervals. In rats on a low‐Na diet for 1 day, eplerenone produced a large natriuresis, especially during the second 2 h period after drug administration (Fig. 7 A). After 3 days on low Na, the effect was smaller and not statistically significant. After 7 days of low Na, the natriuresis was completely absent. Consistent with the last result, ENaC activity measured ex vivo was not significantly affected by eplerenone treatment (Fig. 7 B). However, the drug significantly reversed the biochemical effects at each time point (Fig. 8).
Figure 7. Effect of eplerenone on Na+ excretion and INa .
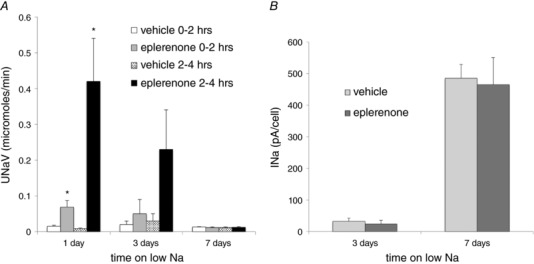
A, single dose of eplerenone (30 mg kg–1 p.o.) or vehicle was administered to rats on a low‐Na diet for 1, 3 or 7 days. Na+ excretion was measured from 0 to 2 h and from 2 to 4 h after the gavage. Data represent the mean ± SEM for four rats at each time point. *Statistical difference of the indicated low‐Na group relative to controls (0.01 < P < 0.03). B, I Na measured in CCDs isolated 4 h after administration of eplerenone or vehicle in CCDs isolated from rats on a low‐Na diet for 3 or 7 days. Data represent the mean ± SEM for 11–28 measurements on three to five animals for each condition.
Figure 8. Effect of eplerenone on expression of γENaC.
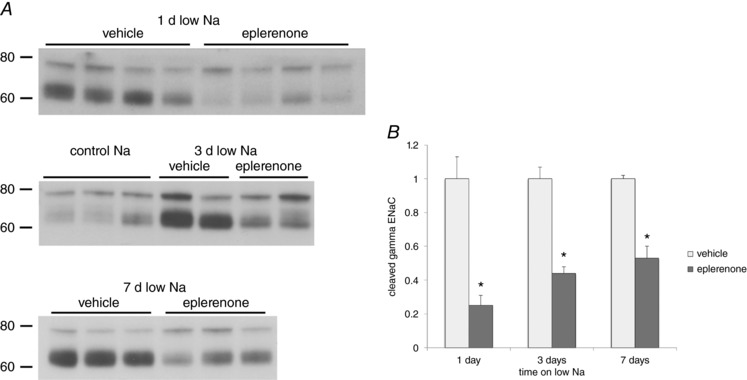
A, western blots of kidney microsomes from rats on control‐Na or a low‐Na diet for 1, 3 or 7 days and treated with eplerenone or vehicle. B, quantitation of the cleaved form of γENaC from blots such as those shown in (A). Data represent the mean ± SEM for four rats under each condition. *Statistical difference of the eplerenone‐treated group relative to vehicle (0.0007 < P < 0.002).
This suggests two distinct processes of ENaC regulation. One involves processing and trafficking of ENaC subunits to the apical membrane. This effect is relatively rapid, being evident after 1 day of Na depletion, and is labile. It is rapidly reversed by administration of an MRA and apparently does not survive the process of isolating the tubules for ex vivo measurement. This would explain the inability to detect significant channel activity in isolated tubules after ≤3 days of Na depletion. The second mechanism of activation becomes evident after 5 or more days of salt deprivation. It appears to be independent of channel processing and trafficking and is not readily reversed by the MR antagonist. This does not necessarily mean that this effect is independent of the MR because it might be mediated by proteins induced through MR activation that have a long half‐lives. A 5 h treatment with an MR antagonist would be insufficient to reverse the abundance of these proteins.
To assess the roles of these processes in regulating Na+ transport in vivo, we used acute administration of amiloride to block ENaC. We reasoned that the increase in Na+ excretion would reflect the amount of Na+ reabsorbed through the channels. We first defined the dose–response relationship in rats on control diet as shown in Fig. 9 A. A maximal natriuresis was produced by injection (s.c.) of 0.6 mg kg–1 of the diuretic. This also had a maximal K‐sparing effect. The finding that K+ excretion under these conditions decreased to almost zero suggests that ENaC activity is completely inhibited. However, the large natriuresis observed could include some inhibition of NCC as a result of hyperkalaemia and dephosphorylation of the cotransporter (Frindt et al. 2017). We then compared the responses of rats on a low‐Na diet for 1, 3 and 7 days to a maximal dose of 1 mg kg–1 (Fig. 9B). The amiloride‐induced Na+ excretion increased after 1 day of low Na, although the change was not statistically significant (P = 0.1) and then remained fairly constant. This suggests that ENaC is activated in vivo within 1 day of dietary Na depletion.
Figure 9. Effects of amiloride on Na+ and K+ excretion in animals on control and low‐Na diets.
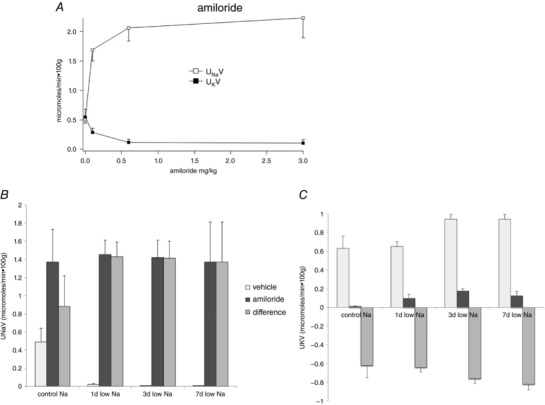
A, dose–response relationships for changes in U Na V and U K V in response to a single injection of amiloride in rats on normal laboratory chow. B and C, effects of amiloride (1 mg kg–1) on U Na V (B) and U K V (C) in animals on control diet and on a low‐Na diet for 1, 3 or 7 days. Data represent the mean ± SEM for four to 12 animals.
A more quantitative interpretation of these results is complicated by the response to ENaC blockade probably depending both on channel activity and on delivery of Na+ to the ENaC‐expressing part of the nephron. In particular, previous studies showed increased expression of the NCC in the DCT in response to dietary Na restriction or aldosterone administration (Kim et al. 1998; van der Lubbe et al. 2012). We therefore also tested the responses to the NCC blocker HCTZ. A dose–response relationship to single injections of the drug indicated a biphasic natriuretic response (Fig. 10 A). This could indicate that two different processes are affected by the diuretic, although, in mice lacking NCC, an even larger dose of HCTZ had no acute effect on Na+ excretion (Leviel et al. 2010). We chose a dose of 15 mg kg–1, which had an almost maximal natriuretic effect and also a maximal kaliuretic effect in control animals. We measured HCTZ concentrations in the urine under these conditions in two sets of animals and found values of 180 ± 50 μg mL–1 for collections between 0 and 30 min after injection, and 560 ± 60 μg mL–1 for 0–60 min. To estimate concentrations of the drug in tubular fluid, we used the average urine flow of 45 μL min–1, and assumed that glomerular filtration rate (GFR) was 1 mL min–1 (see below) and also that 15% of filtered fluid was delivered to the distal nephron. This predicts concentrations of 150 and 500 μm respectively in the tubular fluid in the DCT. This should be adequate to complete block NCC (Monroy et al. 2000).
Figure 10. Effects of HCTZ on Na+ and K+ excretion in animals on control and low‐Na diets.
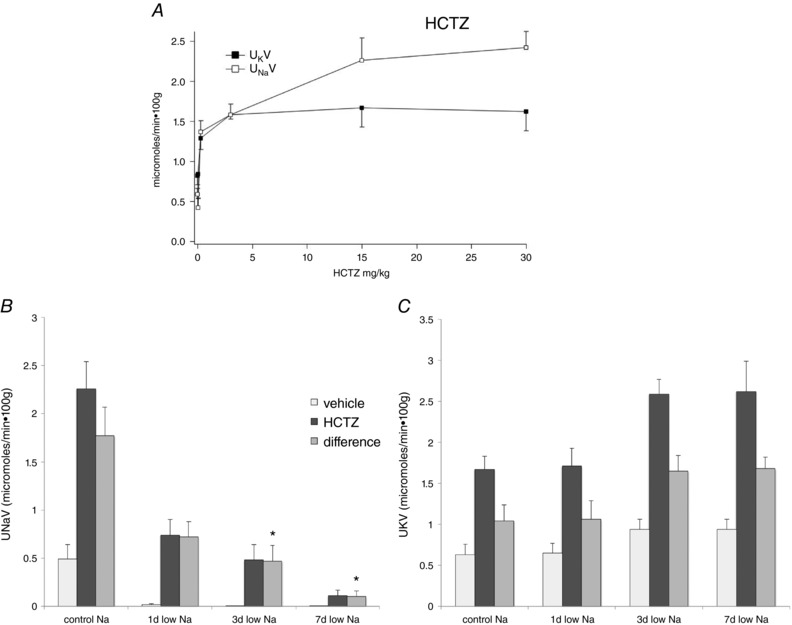
A, dose–response relationships for changes in U Na V and U K V in response to a single injection of HCTZ in rats on standard laboratory chow. B and C, effects of HCTZ (15 mg kg–1) on U Na V (B) and U K V (C) in animals on control diet and on a low‐Na diet for 1, 3 or 7 days. Data represent the mean ± SEM for four to 12 animals. *Statistical difference for the HCTZ‐induced changes in excretion (difference) of the indicated low‐Na group relative to controls [P = 0.005 (3 days), P = 0.0003 (7 days)].
When animals were fed a low‐Na diet, the natriuretic response to HCTZ gradually decreased; after 7 days, the effect was more or less abolished. However, the drug‐induced kaliuresis persisted and was, if anything, elevated (Fig. 10 B). The most probable explanation is that the Na+ that escapes the DCT in the presence of HCTZ is reabsorbed in downstream nephron segments, presumably by ENaC. Under this interpretation, the capacity of ENaC increases continuously over 7 days of low‐Na intake. This suggests that both the rapid biochemical response and the slower response assessed by electrophysiology contribute to ENaC activity in vivo.
Although these interactions between delivery and reabsorption prevented us from quantitatively dissecting the individual contributions of NCC and ENaC to Na+ reabsorption, we reasoned that we could use the two diuretics together to assess the role of the entire distal nephron in Na conservation. As expected, the combination induces a rapid and very large natriuresis. As shown in Fig. 11 A, the effect was maximal during the initial 1 h after administration of the cocktail, with only a slight difference between the 0–30 min period and the 30–60 min period, and then declined significantly during the subsequent hour, presumably as a result of a reduction in drug concentration and/or a compensatory response. We assumed that Na+ excretion under these conditions represents the delivery of Na+ to the distal nephron, with minimal distal reabsorption. This apparent distal delivery was ∼6% of the filtered load of Na+ (Table 1). When animals were fed a low‐Na diet for 1 day, there was a 13% decrease in excretion that was not statistically significant. However, with 7 days on a low‐Na diet, Na+ excretion in the presence of HCTZ + amiloride declined substantially to 60% of control values (Fig. 11 B). This suggests that Na+ delivery to the distal segments is diminished under these circumstances.
Figure 11. Effects of amiloride + HCTZ on Na+ excretion in animals on control and low‐Na diets.
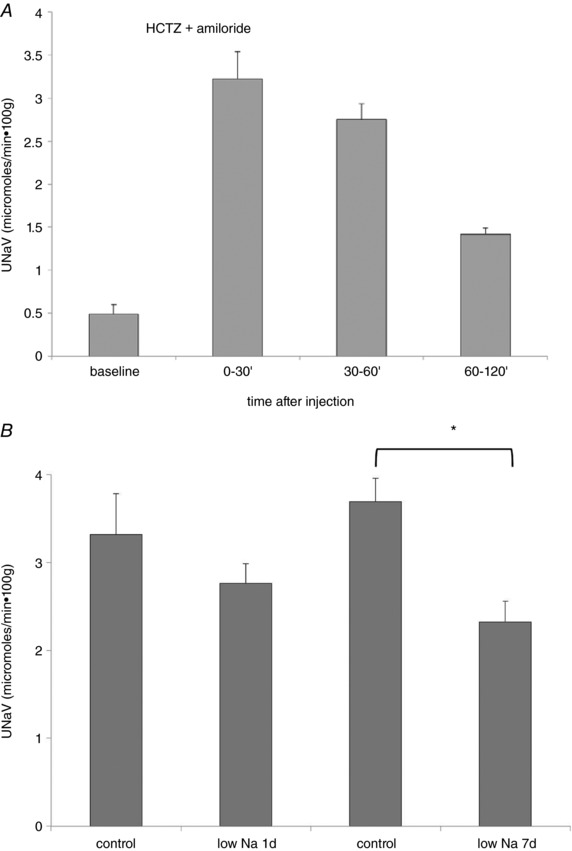
A, time course of U Na V in response to a single injection of amiloride (1 mg kg–1) + HCTZ (15 mg kg–1) in rats on control‐Na diet. Excretion was almost constant for 1 h and declined during the second hour. B, response of animals on control diet and on a low‐Na diet for 1 or 7 days. Data represent the mean ± SEM for eight to 16 animals. *Statistical difference of the 7‐day low‐Na group relative to controls (P = 0.0002).
Table 1.
Effects of Na depletion on Na, creatinine and Li clearance after HCTZ + amiloride administration
| Control | Low Na | P | |
|---|---|---|---|
| GFR (mL min–1) | 0.84 ± 0.05 | 0.76 ± 0.05 | 0.3 |
| Distal Na delivery (μmol min–1) | 7.00 ± 0.51 | 3.67 ± 0.56 | 0.005 |
| FENa | 0.058 ± 0.003 | 0.033 ± 0.002 | 0.0006 |
| CLi (mL min–1) | 0.31 ± 0.02 | 0.24 ± 0.02 | 0.04 |
| FELi | 0.356 ± 0.004 | 0.308 ± 0.011 | 0.006 |
Data are given as the mean ± SEM for four animals. P values compare low‐Na vs. control animals.
To test whether decreased distal delivery was secondary to decreased GFR, we measured creatinine clearance under the two conditions. There was no significant difference, with values of 1.05 ± 0.10 mL min–1 in animals on control diet and 1.02 ± 0.07 mL min–1 in animals on a low‐Na diet for 7 days. This implies that diminished distal delivery is achieved through an increase in reabsorption of Na+ in proximal tubules and/or Henle's loop.
To test this more directly, we used the Li+ clearance technique (Thomsen et al. 1969; Shirley et al. 1983). This approach is considered to accurately assess proximal reabsorption under conditions in which Li+ transport through ENaC is blocked (i.e. in the presence of amiloride) (Fransen et al. 1992). We therefore repeated the experiment of Fig. 11 when infusing animals with LiCl. The results are shown in Table 1. In these experiments, GFR was slightly reduced under low‐Na conditions, although the difference was not statistically significant. Both Li clearance and the fractional excretion of Li were reduced in the Na‐depleted rats. These results are consistent with the idea that proximal tubule Na+ reabsorption is increased under conditions of chronic Na deprivation, and that this reduces delivery of Na+ to the distal convoluted tubules and beyond.
The Na/H exchanger NHE3 is the major Na+ transporter in the proximal tubule. We tested whether the increased proximal tubular Na+ reabsorption resulted from enhanced expression of NHE3 protein using western blotting. As shown in Fig. 12, there was no detectable difference in NHE3 protein in kidney microsomes from rats on the control diet and animals on a low‐Na diet for 7 days.
Figure 12. NHE3 protein expression in rats on control‐Na diet and on low‐Na diet for 7 days.
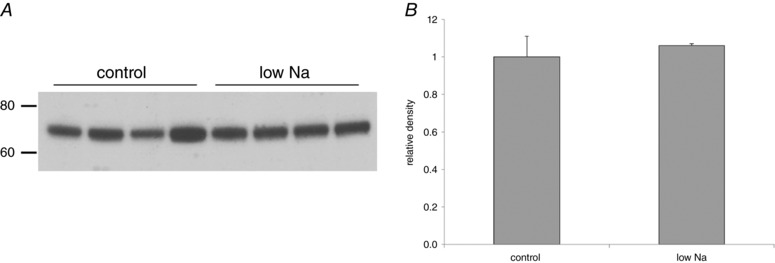
A, western blot. Equal amounts (30 μg) of protein from kidney microsomes of a different animal were loaded into each lane. B, quantitation. No difference between the two groups of animals was detected. Data represent the mean ± SEM for four animals under each condition.
This indicates that pre‐existing NHE3 exchangers are activated under these conditions. To further explore the mechanisms involved, we repeated the measurements of amiloride‐ and thiazide‐independent Na+ excretion at the same time as acutely perturbing pathways that mediate increased proximal tubule reabsorption without changing protein expression. We used the AT1R blocker losartan to inhibit effects of angiotensin II, the β‐adrenergic receptor blocker propranolol to inhibit effects through the sympathetic nervous system, and exogenous dopamine to abolish effects of endogenous dopamine release. All of these agents increased basal Na excretion (Table 2). However none of them prevented the decrease in distal Na delivery (Fig. 13).
Table 2.
Effects of natriuretic agents on Na+ excretion in μmol min–1 ·100 g
| Control period | Treatment period | P | |
|---|---|---|---|
| Vehicle (8) | 0.71 ± 0.16 | 0.10 ± 0.04 | |
| Losartan (8) | 0.71 ± 0.13 | 0.28 ± 0.06 | .02 |
| Vehicle (6) | 0.52 ± 0.16 | 0.28 ± 0.06 | |
| Propranolol (5) | 0.23 ± 0.04 | 0.91 ± 0.26 | .02 |
| Vehicle (4) | 1.04 ± 0.18 | 0.21 ± 0.05 | |
| Dopamine (4) | 0.92 ± 0.15 | 0.67 ± 0.17 | .005 |
Control period was 1 h before administration of vehicle or drug. Treatment period was 60–120 min after administration (losartan, dopamine) or 0–60 min after administration (propranolol). Data represent the mean ± SEM. The number of animals is shown in parentheses. P values compare drug‐treated vs. vehicle during the treatment period.
Figure 13. Effects of losartan, propranolol and dopamine on Na+ excretion in the presence of HCTZ + amiloride.
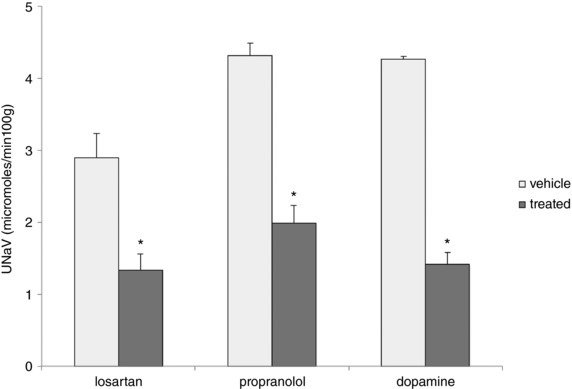
Rats on either a control‐Na or low‐Na diet were treated with losartan (10 mg kg–1), propranolol (5 mg kg–1) or dopamine (10 mg kg–1) and then challenged with amiloride (1 mg kg–1) + HCTZ (15 mg kg–1). The diuretics were administered either 1 h after losartan or dopamine, or at the same time as propranolol. Na+ excretion was measured for 1 h after administration of amiloride + HCTZ. Data represent the mean ± SEM for four animals (propranolol and dopamine) or seven animals (losartan) under each condition. *Statistical difference of the low‐Na group relative to controls (losartan, P = 0.009; propranolol, P = 0.0001; dopamine, P = 0.0001).
Discussion
The main finding of the present study is that dietary Na restriction can upregulate ENaC through two distinct mechanisms. One of these is defined by the processing of the channel protein through increased proteolytic cleavage and glycosylation. The other is revealed through electrophysiological measurements of amiloride‐sensitive currents ex vivo. These two processes are probably both mediated by aldosterone. However, they can be temporally dissociated and are largely independent of each other.
Nephron specificity
Because the electrophysiological measurements were made on CCDs, whereas biochemical data came mainly from whole kidney extracts, it is possible that the two effects reflect events in different parts of the kidney. Indeed, there is evidence for variations in the speed of responses to aldosterone along the nephron. Loffing et al. (2001) found more rapid changes in ENaC translocation in response to hormone administration in the more proximal parts of the aldosterone‐sensitive portion of the nephron (late DCT and CNT) compared to the CCD. Genetic ablation experiments suggested that upregulation of ENaC in the CNT is sufficient for Na conservation. Deletion of ENaC from only the collecting duct did not affect adaptation to a low‐Na diet (Rubera et al. 2003), whereas elimination of MR from the late CNT and CD resulted in salt wasting with reduced Na intake (Ronzaud et al. 2007).
Furthermore, Nesterov et al. (2012) showed that ENaC activity in the DCT/CNT was to a great extent constitutive, whereas that in the CCD was highly dependent on aldosterone. Consistent with this idea, the proportion of γENaC in the cleaved form under control conditions was lower in tissue dissected from the medulla (where ENaC comes from the MCD) than from the outer cortex (where it comes mainly from the CNT and DCT) (Fig. 5) and was still lower in the medullary rays (where it derives from the CCD) (Fig. 6).
It is therefore possible that the observed discrepancies arise in part because the channels are simultaneously processed and activated first in the more proximal parts of the ASDN and later in the collecting duct. However, our results suggest that this axial heterogeneity does not entirely explain the different time courses of biochemical and functional assays. Medullary rays dissected from the renal cortex contain several different nephron segments, although only the CCDs express ENaC. The delay of functional activity with respect to biochemical changes persists (Fig. 6) even when comparing results from the same segment.
ENaC processing and trafficking occur rapidly
Increased cleavage of the γENaC subunit occurs relatively rapidly; the response is almost complete after 24 h of sodium depletion. In agreement with previous results (Frindt et al. 2016), this processing of ENaC protein is associated with increases in its surface expression. Cleavage of αENaC and increased glycosylation of βENaC occur over the same time course. These observations agree with previous findings showing increased processing and trafficking of the channels within 5 h of administration of aldosterone (Frindt & Palmer, 2015) and within 2 h of an acute increase in plasma K+ (Frindt et al. 2017).
The time course of these events correlates well with the overall response of the kidneys to diminish Na+ excretion when dietary Na intake is reduced. In addition, after 24 h of Na depletion, treatment of the animals for 5 h with the mineralocorticoid receptor antagonist eplerenone substantially reversed both the urinary Na+ excretion and the cleavage of γENaC. This suggests that aldosterone, the major mineralocorticoid in terrestrial vertebrates, mediates these responses.
Channel activity assessed ex vivo occurs more slowly
By contrast to the biochemical events, the activity of ENaC measured ex vivo in the CCD using electrophysiology appears more slowly. Amiloride‐sensitive whole‐cell currents were small even after 3 days of Na depletion. These currents subsequently increased to near maximal levels 5 days after the change in diet. The discrepancy in the two responses mirrors that observed with administration of aldosterone; the hormone induced biochemical changes much more rapidly than the electrophysiological changes (Frindt & Palmer, 2015).
The electrophysiological responses also developed much more slowly than did the fall in Na+ excretion. However, these changes did correlate with another in vivo indicator of channel activity: the decreased natriuretic response to HCTZ. Between 3 and 7 days of Na restriction, the increased Na+ excretion induced by the diuretic more or less disappeared, whereas increased K+ excretion persisted. Our interpretation is that, within 1 day of reducing Na intake, ENaC activity is sufficient to reabsorb all of the Na+ that normally reaches the late distal nephron but, when Na+ reabsorption through the HCTZ‐sensitive cotransporter is blocked, the delivery of Na+ downstream exceeds the ENaC transport capacity. With more chronic decreases in intake, ENaC activity further increases such that it can reabsorb both the basal and HCTZ‐induced Na delivery. The decreased HCTZ‐induced Na+ excretion could also reflect in part decreased delivery to the DCT. However, the finding that K+ excretion in response to the diuretic was maintained with Na depletion indicates that substantial Na+ was delivered to the ENaC‐expressing segments even under these conditions.
This suggests ‘early’ and ‘late’ responses to Na depletion. The early response involves increased expression of ENaC protein at the apical surface. The late response entails additional, as yet unidentified, changes in the channels that further increase and stabilize their activity. Under these conditions, this activity can be readily detected after removing the kidneys from the animal and isolating individual nephron segments.
We consider that these slower effects also depend on aldosterone. The biphasic time course of the response to aldosterone administration is consistent with this idea. In addition, Nesterov et al. (2012) showed that activation of ENaC currents after 7 days on a low‐Na diet required the presence of aldosterone. Although eplerenone did not reverse the increases in I Na induced by Na depletion, this could be explained if these effects are mediated by a more permanent change, possibly through an unidentified aldosterone‐induced protein with a long half‐life. Previous work also identified ‘early’ and ‘late’ responses of the toad urinary bladder to aldosterone, based in part on sensitivity to butyrate (Truscello et al. 1983; Asher & Garty, 1988). Analogous to our results, the late response to the hormone was preserved in vesicles isolated from the bladder, whereas the early response was not (Asher & Garty, 1988).
The slow responses to Na depletion might be mediated by occupancy of glucocorticoid receptors by aldosterone (Gaeggeler et al. 2005). Previous measurements of plasma aldosterone in Na‐depleted rats indicated a modest increase in plasma levels of the hormone over the first 3 days and then a steeper increase to 1260 ng dL–1 (35 nm) after 7 days. Assuming a K d of the GR for aldosterone of 160 nm (Grossmann et al. 2004) and negligible competition from corticosterone as a result of 11β‐HSD activity, the GR could be as much as 20% occupied by aldosterone under these conditions. This mechanism cannot be ruled out, although we consider it improbable for two reasons. First, similar activation of ENaC currents could be observed with substantially lower aldosterone levels (Palmer et al. 1993). Second, dexamethasone, a selective GR agonist, induced expression of αENaC in rat kidney but did not activate currents (Frindt & Palmer, 2012).
Either the early or the late response is sufficient to minimize Na excretion
As discussed above, the early response to Na depletion, realized within 24 h, is sufficient to almost completely eliminate Na+ from the urine. Less obviously, the late response may also be sufficient. After 7 days on the low‐Na diet, administration of eplerenone largely reverses the increase in channel processing but does not cause natriuresis. This suggests that a late effect, largely independent of channel processing, also enhances channel activity to an extent that it can transport almost all of the Na+ reaching the ENaC‐expressing parts of the nephron. Observations of mice lacking the aldosterone‐induced protein Sgk1 are consistent with this idea (Yang et al. 2017). In Sgk1–/– animals, ENaC processing and trafficking are severely impaired, although the mice are able to fully conserve Na and ENaC activity measured ex vivo is not different from that of Sgk1+/+ controls. This suggests that the two responses occur in parallel and are, at least under chronic conditions, redundant. It also implies that the early but not the late response requires this kinase.
Chronic Na depletion decreases Na delivery to the distal nephron
Dietary Na depletion had little effect on GFR, as assessed by creatinine clearance. However, the low‐Na diet apparently did increase proximal tubule Na+ reabsorption and reduce Na+ delivery to the distal nephron, at least after 7 days. Two observations support this idea. First, Na+ excretion in the presence of acute maximal doses of HCTZ and amiloride was lower in rats on a low‐Na diet. Assuming that these diuretics abolish reabsorption by NCC in the DCT and by ENaC in downstream segments, excretion under these circumstances will reflect the amount of Na+ escaping the proximal tubule and the loop of Henle. We observed a statistically significant decrease in this quantity only under chronic conditions. However we cannot rule out a smaller change that occurs more rapidly. Even a 10% decrease in delivery could be physiologically important but very difficult to document. Second, chronic Na depletion reduced fractional excretion of Li+. When measured in the presence of amiloride, Li+ handling is considered to be a fairly good indicator of proximal tubule function (Fransen et al. 1992). Thus these measurements imply enhanced Na+ reabsorption in this segment. A similar conclusion was reached recently based on measurements and mathematical modeling of Na+ handling by the mouse kidney (Udwan et al. 2017)
We have not identified the mechanism underlying the increased proximal Na+ transport. NHE3 protein expression was not elevated, suggesting that the effect involves activation of existing transporters. The differences in the responses to HCTZ + amiloride administration were not affected by an angiotensin II receptor antagonist (losartan), a β‐adrenergic receptor antagonist (propranolol) or dopamine. These systems are all capable of altering proximal tubule reabsorption (Weinstein, 1992) and all of these agents increased baseline Na+ excretion. We tested them only under acute conditions and we cannot rule out more chronic effects mediated by these pathways. Physical factors associated with extracellular volume depletion, particularly reduced hydrostatic pressure or increased oncotic pressure in peritubular capillaries, may also contribute to increased net reabsorption through paracellular pathways.
Decreased distal Na+ delivery may explain an apparent conundrum raised in Figs 9 and 10. Although biochemical (Kim et al. 1998) and functional (Ellison et al. 1989) measurements indicate that Na depletion upregulates NCC, as well as ENaC, assessment of the amount of Na reabsorbed by these transporters in vivo through application of diuretics indicated only a modest increase in the response to amiloride and a reduced response to HCTZ. This suggests that, particularly under chronic conditions, the upregulation of NCC and ENaC serves to increase the fractional reabsorption of Na+ delivered to these segments, rather than the total amount of Na+ that they transport.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
Experiments were carried out in the laboratory of L. G. Palmer in the Department of Physiology and Biophysics at the Weill‐Cornell Medical College. LGP, GF and KB conceived and designed the work. GF, LY and LGP acquired, analysed and interpreted the data. LGP drafted the manuscript. All authors edited and critically revised the manuscript, and approved the final version submitted for publication. All authors are accountable for all aspects of the work, and qualify as authors. All persons qualified as authors are listed as such.
Funding
This work was supported by grants 5R01‐DK111380 and 1R01‐DK099284 from the US National Institutes of Health.
Biography
Gustavo Frindt acquired his life‐long interest in the kidney as a medical student in Santiago Chile. In 1970, he began his career in research as a fellow in the laboratory of Moe Burg at NIH, where he learned to dissect and perfuse individual nephron segments and studied the effects of anti‐diuretic hormone on Na+ reabsorption by the collecting duct. Later, he joined the faculty of the Department of Physiology and Biophysics at Cornell University Medical College, where he has collaborated with Erich Windhager and Lawrence Palmer to study the mechanisms of renal handling of H2O, Na+ and K+.

Edited by: Kim Barrett and Robert Fenton
References
- Asher C & Garty H (1988). Aldosterone increases the apical Na+ permeability of toad bladder by two different mechanisms. Proc Natl Acad Sci U S A 85, 7413–7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher C, Wald H, Rossier BC & Garty H (1996). Aldosterone‐induced increase in the abundance of Na+ channel subunits. Am J Physiol Cell Physiol 271, C605–C611. [DOI] [PubMed] [Google Scholar]
- Ellison DH, Velazquez H & Wright FS (1989). Adaptation of the distal convoluted tubule of the rat. Structural and functional effects of dietary salt intake and chronic diuretic infusion. J Clin Invest 83, 113–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ergonul Z, Frindt G & Palmer LG (2006). Regulation of maturation and processing of ENaC subunits in the rat kidney. Am J Physiol Renal Physiol 291, F683–F693. [DOI] [PubMed] [Google Scholar]
- Fransen R, Boer WH, Boer P & Koomans HA (1992). Amiloride‐sensitive lithium reabsorption in rats: a micropuncture study. J Pharmacol Exp Ther 263, 646–650. [PubMed] [Google Scholar]
- Frindt G, Ergonul Z & Palmer LG (2008). Surface expression of epithelial Na channel protein in rat kidney. J Gen Physiol 131, 617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frindt G, Gravotta D & Palmer LG (2016). Regulation of ENaC trafficking in rat kidney. J Gen Physiol 147, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frindt G, Masilamani S, Knepper MA & Palmer LG (2001). Activation of epithelial Na channels during short‐term Na deprivation. Am J Physiol Renal Physiol 280, F112–F118. [DOI] [PubMed] [Google Scholar]
- Frindt G & Palmer LG (2009). Surface expression of sodium channels and transporters in rat kidney: effects of dietary sodium. Am J Physiol Renal Physiol 297, F1249–F1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frindt G & Palmer LG (2012). Regulation of epithelial Na+ channels by adrenal steroids: Mineralocorticoid and glucocorticoid effects. Am J Physiol Renal Physiol 302, F20–F26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frindt G & Palmer LG (2015). Acute effects of aldosterone on the epithelial Na channel in rat kidney. Am J Physiol Renal Physiol 308, F572–F578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frindt G, Sackin H & Palmer LG (1990). Whole‐cell currents in rat cortical collecting tubule: low‐Na diet increases amiloride‐sensitive conductance. Am J Physiol Renal Physiol 258, F562–F567. [DOI] [PubMed] [Google Scholar]
- Frindt G, Yang L, Uchida S, Weinstein AM & Palmer LG (2017). Responses of distal nephron Na+ transporters to acute volume depletion and hyperkalemia. Am J Physiol Renal Physiol 313, F62–F73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaeggeler HP, Gonzalez‐Rodriguez E, Jaeger NF, Loffing‐Cueni D, Norregaard R, Loffing J, Horisberger JD & Rossier BC (2005). Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone‐stimulated sodium transport in a novel renal cell line. J Am Soc Nephrol 16, 878–891. [DOI] [PubMed] [Google Scholar]
- Grossmann C, Scholz T, Rochel M, Bumke‐Vogt C, Oelkers W, Pfeiffer AF, Diederich S & Bahr V (2004). Transactivation via the human glucocorticoid and mineralocorticoid receptor by therapeutically used steroids in CV‐1 cells: a comparison of their glucocorticoid and mineralocorticoid properties. Eur J Endocrinol 151, 397–406. [DOI] [PubMed] [Google Scholar]
- Grundy D (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim GH, Masilamani S, Turner R, Mitchell C, Wade JB & Knepper MA (1998). The thiazide‐sensitive Na‐Cl cotransporter is an aldosterone‐induced protein. Proc Natl Acad Sci U S A 95, 14552–14557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lees P (1968). The influence of beta‐adrenoceptive receptor blocking agents on urinary function in the rat. Br J Pharmacol 34, 429–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leviel F, Hubner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R & Eladari D (2010). The Na+‐dependent chloride‐bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120, 1627–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffing J, Zecevic M, Feraille E, Asher C, Rossier BC, Firestone GL, Pearce D & Verrey F (2001). Aldosterone induces rapid apical translocation of ENaC in early portion of renal collecting system: possible role of SGK. Am J Physiol Renal Physiol 280, F675–F682. [DOI] [PubMed] [Google Scholar]
- Masilamani S, Kim GH, Mitchell C, Wade JB & Knepper MA (1999a). Aldosterone‐mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104, R19–R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masilamani S, Kim GH, Mitchell C, Wade JB & Knepper MA (1999b). Aldosterone‐mediated regulation of ENaC alpha, beta, and gamma subunit proteins in rat kidney. J Clin Invest 104, R19–R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroy A, Plata C, Hebert SC & Gamba G (2000). Characterization of the thiazide‐sensitive Na+‐Cl− cotransporter: a new model for ions and diuretics interaction. Am J Physiol Renal Physiol 279, F161–F169. [DOI] [PubMed] [Google Scholar]
- Nesterov V, Dahlmann A, Krueger B, Bertog M, Loffing J & Korbmacher C (2012). Aldosterone‐dependent and ‐independent regulation of the epithelial sodium channel (ENaC) in mouse distal nephron. Am J Physiol Renal Physiol 303, F1289–F1299. [DOI] [PubMed] [Google Scholar]
- Nguyen MT, Lee DH, Delpire E & McDonough AA (2013). Differential regulation of Na+ transporters along nephron during ANG II‐dependent hypertension: distal stimulation counteracted by proximal inhibition. Am J Physiol Renal Physiol 305, F510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pácha J, Frindt G, Antonian L, Silver R & Palmer LG (1993). Regulation of Na channels of the rat cortical collecting tubule by aldosterone. J Gen Physiol 102, 25–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer LG, Antonian L & Frindt G (1993). Regulation of the Na‐K pump of the rat cortical collecting tubule by aldosterone. J Gen Physiol 102, 43–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif MC, Troutman SL & Schafer JA (1986). Sodium transport by rat cortical collecting tubule. Effects of vasopressin and desoxycorticosterone. J Clin Invest 77, 1291–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronzaud C, Loffing J, Bleich M, Gretz N, Grone HJ, Schutz G & Berger S (2007). Impairment of sodium balance in mice deficient in renal principal cell mineralocorticoid receptor. J Am Soc Nephrol 18, 1679–1687. [DOI] [PubMed] [Google Scholar]
- Rubera I, Loffing J, Palmer LG, Frindt G, Fowler‐Jaeger N, Sauter D, Carroll T, McMahon A, Hummler E & Rossier BC (2003). Collecting duct specific gene inactivation of aENaC in the mouse kidney does not impair sodium and potassium balance. J Clin Invest 112, 554–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirley DG, Walter SJ & Thomsen K (1983). A comparison of micropuncture and lithium clearance methods in the assessment of renal tubular function in rats with diabetes insipidus. Pflugers Arch 399, 266–270. [DOI] [PubMed] [Google Scholar]
- Sorensen MV, Grossmann S, Roesinger M, Gresko N, Todkar AP, Barmettler G, Ziegler U, Odermatt A, Loffing‐Cueni D & Loffing J (2013). Rapid dephosphorylation of the renal sodium chloride cotransporter in response to oral potassium intake in mice. Kidney Int 83, 811–824. [DOI] [PubMed] [Google Scholar]
- Thomsen K, Schou M, Steiness I & Hansen HE (1969). Lithium as an indicator of proximal sodium reabsorption. Pflugers Arch 308, 180–184. [DOI] [PubMed] [Google Scholar]
- Tomita K, Pisano JJ & Knepper MA (1985). Control of sodium and potassium transport in the cortical collecting tubule of the rat. Effects of bradykinin, vasopressin, and deoxycorticosterone. J Clin Invest 76, 132–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truscello A, Geering K, Gaggeler HP & Rossier BC (1983). Effects of butyrate on histone deacetylation and aldosterone‐dependent Na+ transport in the toad bladder. J Biol Chem 258, 3388–3395. [PubMed] [Google Scholar]
- Udwan K, Abed A, Roth I, Dizin E, Maillard M, Bettoni C, Loffing J, Wagner CA, Edwards A & Feraille E (2017). Dietary sodium induces a redistribution of the tubular metabolic workload. J Physiol 595, 6905–6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Lubbe N, Lim CH, Meima ME, van Veghel R, Rosenbaek LL, Mutig K, Danser AH, Fenton RA, Zietse R & Hoorn EJ (2012). Aldosterone does not require angiotensin II to activate NCC through a WNK4‐SPAK‐dependent pathway. Pflugers Arch 463, 853–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CT, Zou LX & Navar LG (1997). Renal responses to AT1 blockade in angiotensin II‐induced hypertensive rats. J Am Soc Nephrol 8, 535–542. [PubMed] [Google Scholar]
- Weinstein AM (1992). Sodium and chloride transport: proximal nephron In The Kidney, ed. Selden DW. & Giebish G, pp. 1925–1974. Raven Press, New York, NY. [Google Scholar]
- Yang L, Frindt G, Lang F, Kuhl D, Vallon V & Palmer LG (2017). SGK1‐dependent ENaC processing and trafficking in mice with high dietary K intake and elevated aldosterone. Am J Physiol Renal Physiol 312, F65–F76. [DOI] [PMC free article] [PubMed] [Google Scholar]