

Abstract
Key points
A healthy mitochondrial pool is dependent on the removal of dysfunctional organelles via mitophagy, but little is known about how mitophagy is altered with ageing and chronic exercise.
Chronic contractile activity (CCA) is a standardized exercise model that can elicit mitochondrial adaptations in both young and aged muscle, albeit to a lesser degree in the aged group.
Assessment of mitophagy flux revealed enhanced targeting of mitochondria for degradation in aged muscle, in contrast to previous theories.
Mitophagy flux was significantly reduced as an adaptation to CCA suggesting that an improvement in organelle quality reduces the need for mitochondrial turnover.
CCA enhances lysosomal capacity and may ameliorate lysosomal dysfunction in aged muscle.
Abstract
Skeletal muscle exhibits deficits in mitochondrial quality with age. Central to the maintenance of a healthy mitochondrial pool is the removal of dysfunctional organelles via mitophagy. Little is known on how mitophagy is altered with ageing and chronic exercise. We assessed mitophagy flux using colchicine treatment in vivo following chronic contractile activity (CCA) of muscle in young and aged rats. CCA evoked mitochondrial biogenesis in young muscle, with an attenuated response in aged muscle. Mitophagy flux was higher in aged muscle and was correlated with the enhanced expression of mitophagy receptors and upstream transcriptional regulators. CCA decreased mitophagy flux in both age groups, suggesting an improvement in organelle quality. CCA also reduced the exaggerated expression of TFEB evident in aged muscle, which may be promoting the age‐induced increase in lysosomal markers. Thus, aged muscle possesses an elevated drive for autophagy and mitophagy which may contribute to the decline in organelle content observed with age, but which may serve to maintain mitochondrial quality. CCA improves organelle integrity and reduces mitophagy, illustrating that chronic exercise is a modality to improve muscle quality in aged populations.
Keywords: mitochondria, exercise, aging
Key points
A healthy mitochondrial pool is dependent on the removal of dysfunctional organelles via mitophagy, but little is known about how mitophagy is altered with ageing and chronic exercise.
Chronic contractile activity (CCA) is a standardized exercise model that can elicit mitochondrial adaptations in both young and aged muscle, albeit to a lesser degree in the aged group.
Assessment of mitophagy flux revealed enhanced targeting of mitochondria for degradation in aged muscle, in contrast to previous theories.
Mitophagy flux was significantly reduced as an adaptation to CCA suggesting that an improvement in organelle quality reduces the need for mitochondrial turnover.
CCA enhances lysosomal capacity and may ameliorate lysosomal dysfunction in aged muscle.
Introduction
During the natural course of advancing age, the loss of muscle mass and function, commonly referred to as sarcopenia, remains an incompletely described phenomenon (Leon, 2017; Sakuma et al. 2017). Implicated in the maintenance of skeletal muscle with ageing is the presence of a healthy population of mitochondria (Romanello & Sandri, 2016; Rygiel et al. 2016; Alway et al. 2017). These organelles are widely recognized to produce the bulk of cellular energy, regulate metabolism, act as mediators for apoptosis and participate in a plethora of cellular signalling cascades. With engagement in habitual aerobic exercise, expansion of the mitochondrial reticulum and improved integrity of the organelles in skeletal muscle has been well described to occur in young, healthy individuals (Holloszy, 1967; Hoppeler et al. 1985; Menzies et al. 2013).
Although controversial, ageing muscle often presents with a population of compromised mitochondria. Observed impairments include reduced organelle content (Chabi et al. 2008; Ljubicic et al. 2009; O'Leary et al. 2013; St‐Jean‐Pelletier et al. 2017), impaired transcription of PGC‐1α (Carter et al. 2018), altered synthesis of mitochondrial proteins (Rooyackers et al. 1996; Miller et al. 2012), poor respiratory function (Conley et al. 2000; Ljubicic et al. 2009), higher reactive oxygen species (ROS) emission (Chabi et al. 2008; Ljubicic et al. 2009), loss of mitochondrial membrane potential (ΔΨm; Chabi et al. 2008), decreased calcium retention (Gouspillou et al. 2014), increased mtDNA mutational load (Melov et al. 1995) and greater release of pro‐apoptotic proteins (Chabi et al. 2008; Gouspillou et al. 2014). These changes may potentially contribute to the observed decline in muscle performance with ageing, but it remains unclear if these changes are due solely to ageing or are a product of changes in physical activity (Kent‐Braun & Ng, 2000; Picard et al. 2011; Joseph et al. 2012; Hepple, 2014; Leduc‐Gaudet et al. 2015; St‐Jean‐Pelletier et al. 2017).
Furthermore, while beneficial, exercise training may not produce the same magnitude of mitochondrial adaptation in aged muscle when compared to younger cohorts (Carter et al. 2015; Hood et al. 2016), and controversy on this topic remains (Robinson et al. 2017). Previous work has detailed that the signalling towards nuclear genes encoding mitochondrial proteins (NuGEMPs) is blunted, suggesting that mitochondrial biogenesis remains low in aged muscle (Ljubicic & Hood, 2009). While intact mitochondrial biogenesis is essential to produce/maintain healthy organelles, the removal of damaged/dysfunctional organelles also has a critical role in the health of mitochondria and skeletal muscle. This selective removal of mitochondria is termed mitophagy, and is part of the larger, evolutionarily conserved autophagy pathway. Our understanding of autophagy and mitophagy and the role they play in skeletal muscle health with exercise and ageing remains in its infancy.
Autophagy is an intracellular recycling mechanism, whereby damaged or redundant cellular components are engulfed in a double membrane structure called the autophagosome, which is ultimately delivered to the lysosome for digestion of its contents by pH‐sensitive enzymes. Through the use of genetic ablation of critical autophagy genes, it has been shown that intact autophagy is necessary to preserve muscle mass (Masiero et al. 2009), and the degeneration of muscle that occurs in the absence of autophagy is reminiscent of the maladaptive changes that occur with ageing (Carnio et al. 2014). Indeed, the literature often discusses the concept that autophagy declines with ageing, contributing to deleterious organ and tissue health through aggregation of cellular debris (Brunk & Terman, 2002; Terman et al. 2006, 2010; Rubinsztein et al. 2011; Kroemer et al. 2015; Sakuma et al. 2016; Sebastián et al. 2016). However, many of these ideas are developed from observations noted in lower organisms or in non‐muscle tissue, and whether the same conclusions can be drawn for skeletal muscle remains unknown. In aged skeletal muscle, it has been documented that the expression of many autophagy proteins increases (O'Leary et al. 2013; Sakuma et al. 2016). Furthermore, there is an increased presence of the mitophagy receptor Parkin on isolated organelles (O'Leary et al. 2013). However, we now appreciate that the documentation of changes in upstream autophagy proteins levels provides a less than complete picture of the process of autophagy within cells. A difficultly in assessing autophagy and mitophagy in vivo is that these are highly dynamic processes, and static measures are often subject to discrepancies in interpretation (Klionsky, 2016; Yoshii & Mizushima, 2017). Thus, measures of autophagosome flux are required, and an accepted technique for this obligates the use of an autophagy‐inhibited condition for all experimental conditions, along with the corresponding vehicle controls, to allow for the calculation of flux using isolated organelles (Ju et al. 2010). However, no studies to date have yet to capture the dynamic process of autophagy/mitophagy flux in ageing skeletal muscle with exercise.
In addition, very little information has been garnered about the effects of chronic exercise on autophagy and mitophagy, particularly in aged muscle. Recent evidence has demonstrated that autophagy and mitophagy flux increase subsequent to a single bout of acute exercise in young, untrained animals (Grumati et al. 2011; He et al. 2012; Vainshtein et al. 2015b; Laker et al. 2017). This is likely to be a precipitating factor for the eventual remodelling of the mitochondrial network that would occur with repeated, successive bouts of exercise. However, how autophagy and mitophagy flux adapt to exercise following training has not been conclusively described. Two studies have begun to examine mitophagy with chronic exercise training in a young cohort (Lira et al. 2013; Ju et al. 2016) and both of these studies have concluded that autophagy flux was higher following exercise training. However, only one study used an autophagy inhibitor to capture the dynamic turnover of autophagosomes through autophagy, and neither study isolated mitochondria to examine the rate at which mitochondria may be targeted for mitophagy following training. Thus, the purposes of our study were (1) to examine autophagy and mitophagy autophagosome flux in aged muscle, and (2) to understand how flux may adapt following chronic contractile activity (CCA) in skeletal muscle of young and aged animals.
Methods
Animals
All animal procedures were conducted in strict accordance with the standards set by the Canadian Council on Animal Care and with the approval of York University Animal Care Committee (YUACC). Young (5–6 months) and aged (35–36 months) male Fisher 344 Brown Norway F1 Hybrid rats were obtained from the National Institute of Ageing (NIA, Bethesda, MD, USA). Upon arrival, animals were acclimated to the facility in accordance with YUACC protocol guidelines. Food and water were provided ad libitum and food intake for each age group was monitored for 2 weeks subsequent to the acclimation period. Animal characteristics are described in Table 1.
Table 1.
Young and aged animal characteristics
| Young | Aged | |||||
|---|---|---|---|---|---|---|
| CON | CCA | CON | CCA | |||
| Body mass (g) | 396.3 ± 9.1 | 461 ± 13.8* | ||||
| Food intake (g day−1) | 16.13 ± 0.54 | 15.14 ± 0.5 | ||||
| Heart mass/BM (mg g−1) | 2.45 ± 0.03 | 2.84 ± 0.08* | ||||
| Epididymal fat/BM (mg g−1) | 5.33 ± 0.46 | 10.72 ± 0.87* | ||||
| TA mass/BM (mg g−1) | 1.82 ± 0.03 | 1.87 ± 0.07 | 0.91 ± 0.07* | 0.95 ± 0.06* | ||
| EDL mass/BM (mg g−1) | 0.45 ± 0.004 | 0.45 ± 0.01 | 0.26 ± 0.02* | 0.28 ± 0.01* | ||
BM, body mass; EDL, extensor digitorum longus; TA, tibialis anterior. * P < 0.05 aged vs. young (n = 11–16 per group). No effect of colchicine was noted on these measures.
Chronic contractile activity
Animals were sedated with isoflurane anesthesia and surgical implantation of a stimulator unit was performed as previously described (Adhihetty et al. 2007; Ljubicic et al. 2009). Oral temperature of the animals was continuously monitored during the procedure using a CODA monitor (Kent Scientific, Torrington, CT, USA) and a heating pad was used to keep body temperature stable at ∼37°C. The fur was shaved over the caudal rib cage as well as over the left hindlimb. Antiseptic (Povidone‐iodine 10%) was applied to the exposed skin and all surgical procedures were performed under aseptic conditions. The first incision (1–2 cm) was made vertically between the last rib and pelvic girdle. A second incision was made horizontally over the cephalic aspect of the left hindlimb. Blunt dissection liberated the skin from underlying tissues and a subcutaneous passage was made from the vertical incision to the horizontal incision. The abdominal musculature was lifted with forceps and cut. The opening was large enough to accommodate insertion of the stimulator unit. The sterile stimulator unit (purchased from JC Jarvis, Liverpool, UK; Jarvis & Salmons, 1991; Salmons & Jarvis, 1991; Mayne et al. 1993) was inserted into the abdominal cavity and placed caudally toward the ilium with the Dacron mesh facing towards the incision. The wires were passed through the subcutaneous tunnel to the left hindlimb. The deep musculature of the abdominal wall was closed with the Dacron mesh incorporated into the suture line with sterile 5.0 silk suture. The superficial musculature was closed with separate sutures and the skin was stapled together. Each muscle and skin layer received antibiotic treatment. At the hindlimb, the biceps femoris was lifted with forceps and cut. Gentle blunt dissection was used to locate the peroneal nerve. The loop ends of the electrodes were affixed to the deep musculature, with one wire flanking each side of the peroneal nerve. Using a digital stroboscope, the stimulation unit was turned on and the tibialis anterior (TA) and extensor digitorum longus (EDL) muscles were palpated to ensure muscle recruitment. The stimulation unit was turned off and the muscle incision at the left hindlimb was carefully closed with sutures so as not to disrupt the electrodes. The skin was stapled closed and antibiotic was applied to both layers. Animals were given pain medication (Meloxicam, 2 mg kg–1) subcutaneously for three consecutive days with the dose decreasing by half each day. Antibiotic (amoxicillin) was also provided in the drinking water for 7 days (0.3 mg L–1). Animals were closely monitored for mobility and behaviour. After one week of recovery, the CCA protocol began. The left TA and EDL were stimulated for 3 h per day (09.00 to 12.00) for nine consecutive days with 21 h of recovery between bouts, while the contralateral limb served as an internal control (CON). To inhibit autophagy, sterile solutions of colchicine (COL; 0.4 mg kg day–1; Sigma, Oakville, ON, Canada) or vehicle (VEH; 0.9% saline) were injected into the intraperitoneal cavity during the last 3 days of CCA. Animals were randomly assigned to the treatment groups. No signs of distress as a result of the injections were observed. Moreover, this duration of colchicine treatment is sufficient to inhibit autophagy for the measurement of flux (Ju et al. 2010; Mofarrahi et al. 2013; Vainshtein et al. 2015a, b ), yet will not induce myopathy, which is generally observed with treatments lasting 10 days or greater at the same dosage (Ching et al. 2013). Animals were anaesthetized with isoflurane between 08.30 and 09.00, 21 h after the last stimulation period, and relevant tissues were removed and weighed. Animals were killed by exsanguination after median sternotomy.
Mitochondrial isolations
IMF mitochondria were isolated from the distal 2/3 of the TA as previously described (Chabi et al. 2008; Ljubicic et al. 2009). Briefly, tissue was minced and subjected to mechanical homogenization, differential centrifugation and protease treatment (nagarse; 0.025 ml g–1; Sigma) to liberate the organelles from the dense myofibril network. Mitochondrial pellets were resuspended in ice‐cold buffer (100 mm KCl, 10 mm MOPS, 0.2% BSA) supplemented with two phosphatase inhibitor cocktails (Sigma) as well as protease inhibitors (Roche, Mississauga, ON, Canada). Samples were stored at –80°C until later use.
Protein extraction and Western blotting
The proximal one‐third of the TA was snap frozen in liquid nitrogen upon removal and stored at –80°C. The tissue was pulverized to a fine powder at the temperature of liquid nitrogen. Protein extracts were made by combining a small amount of powder with extraction buffer (20 mm HEPES, 2 mm EGTA, 1% Triton‐X 100, 50% glycerol and 50 mm β‐glycerophosphate) supplemented with protease (Roche) and phosphatase (Sigma) inhibitor cocktails, rotating end‐over‐end for 1 h at 4°C followed by sonication on ice. The solution was spun in a microfuge at 4°C for 15 min and the supernate was collected. Protein concentration was determined by the Bradford method. Equal amounts of protein (20–40 μg) were loaded into SDS‐PAGE gels for separation. Protein was wet transferred to nitrocellulose membrane (Bio‐Rad, Mississauga, ON, Canada) and subsequently blocked for 1 h in 5% skimmed milk powder (w/v) dissolved in Tris‐buffered saline with Tween‐20 (TBST). Primary antibodies were incubated on the membrane overnight at 4°C. The next day the membrane was washed 3× for 5 min each, with TBST and incubated at room temperature with the appropriate secondary antibody conjugated to horseradish peroxidase (Santa Cruz, Mississauga, ON, Canada). The protein density was visualized using enhanced chemiluminescence (Bio‐Rad) on film or with an ImageStation 4000MM Pro (Carestream, Concord, ON, Canada). Long and short exposures were performed to isolate the linear range. Primary antibodies are detailed in Table 2.
Table 2.
List of antibodies
| Target | Manufacturer | Product ID | Lot no. | Primary dilution | Exposure (min) |
|---|---|---|---|---|---|
| PGC‐1α | Millipore | AB3242 | 2691399 | 1:1000 | 3 |
| LC3‐I/II | Cell Signalling | 4108 | 3 | 1:500 | 2 |
| p62 | Sigma | P0067 | 015M4877V | 1:3000 | 3 |
| BNIP3 | Dr L. A. Kirshenbaum | Gift | n/a | 1:1000 | 2 |
| NIX | Santa Cruz | sc‐166332 | D0114 | 1:200 | 2 |
| TFEB | Bethyl | A303‐673A | n/a | 1:4000 | 5 |
| LAMP‐1 | Abcam | ab24170 | GR3183900‐1 | 1:1000 | 2 |
| LAMP‐2 | Abcam | ab13524 | GR770‐14 | 1:1000 | 3 |
| Cathepsin D | Santa Cruz | sc‐6486 | J1111 | 1:1000 | 3 |
| Parkin | Cell Signalling | 4211 | 4 | 1:1000 | 2 |
| FoxO3 | Cell Signalling | 2497 | 6 | 1:1000 | 2 |
| p53 | Dr S. Benchimol | n/a | n/a | 1:50 | 5 |
| Beclin 1 | Cell Signalling | 3738 | 3 | 1:1000 | 2 |
| Aciculin | (Belkin et al. 1994) | Gift | n/a | 1:2000 | 1 |
| Optineurin | Santa Cruz | sc‐166576 | L2915 | 1:1000 | 3 |
| VDAC | Abcam | ab14734 | GR243577‐6 | 1:2000 | 1 |
Autophagy and mitophagy flux calculation
To determine the relative degree of autophagosome turnover or mitochondrial‐targeted turnover (mitophagy), colchicine and vehicle treatment conditions were employed. Western blotting of LC3‐II and p62 were performed in whole muscle extracts or IMF mitochondrial subfractions (isolated as described above) with all conditions represented on one SDS‐PAGE gel. Protein abundance was quantified from blots using Image J and values were corrected for corresponding loading controls (aciculin for whole muscle extracts, VDAC for mitochondria). The mean CON values were subtracted from the COL values of corresponding conditions (e.g. young CON COLC – mean young CON VEH) to yield autophagosome flux values.
High resolution respirometry
Half of the EDL muscle was used for high resolution respirometry (Oroboros O2k, Austria). Briefly, the instrument chambers were calibrated prior to sample isolation. The muscle sample was excised and immediately placed in ice‐cold BIOPS buffer (10 mm Ca‐EGTA, 0.1 μm free calcium, 20 mm imidazole, 20 mm taurine, 50 mm K‐MES, 0.5 mm DTT, 6.56 mm MgCl2, 5.77 mm ATP, 15 mm phosphocreatine, pH 7.1) followed by gentle mechanical separation of small muscle fibre bundles using fine forceps under a dissection microscope while on ice. Fibre bundles were incubated at 4°C for 30 min in saponin (50 μg mL–1) with gentle rocking. Fibre bundles were washed and weighed then added to the oxygen chamber in Miro5 buffer (110 mm sucrose, 60 mm potassium lactobionate, 0.5 mm EGTA, 3 mm MgCl2, 20 mm taurine, 10 mm KH2PO4, 20 mm HEPES, pH 7.1 at 30°C, and 0.1% BSA). The chambers were hyperoxygenated with 100% O2 followed by stabilization for a background reading. Glutamate (10 mm) and malate (2 mm) were added followed by succinate (10 mm) then ADP (2.5 mm) to assess basal and maximal respiration. Oxygen flux values were corrected for background readings and fibre mass. Respiratory control ratios were assessed to ensure quality of the fibres and mitochondrial respiration.
COX activity
As previously described (Chabi et al. 2008; Ljubicic et al. 2009; O'Leary et al. 2013), muscle lysates from young and aged TA were generated and incubated in the presence of fully reduced cytochrome c (Sigma). Equal amounts of sample were incubated with the reduced cytochrome c solution and the rate of oxidation over time was observed as the change in absorbance at 550 nm using a plate reader (Synergy HT, Bio‐tek, Thorold, ON, Canada).
Electron microscopy
Cubes (2 mm2) from the control and CCA EDL muscles were placed in a 2% glutaraldehyde solution in 0.1 mm sodium cacodylate, pH 7.3. Samples were embedded, stained and cut at the Advanced Imageing Centre at The Hospital for Sick Children (Toronto, ON, Canada) using standard procedures. About 35–40 images for each condition (n = 2) were captured on a FEI Tecnai 20 transmission electron microscope.
Histochemistry
The remainder of the EDL was placed in Optimal Cutting Temperature compound (OCT) and frozen in isopentane cooled on liquid nitrogen. When completely frozen, the samples were stored in the ‐80°C for future use. Samples were thawed to –20°C and mounted on a chuck in a cryostat (Cryotome SME; ThermoFisher, Mississauga, ON, Canada) with OCT. Samples were oriented for cross‐sectional cuts, cut in quadruple 10 μm thick sections, and transferred to clean microscope slides. All experimental conditions were represented on each microscope slide. Slices were then stained for cytochrome oxidase (COX) or succinate dehydrogenase (SDH) and dried as previously described (Menzies et al. 2013; Vainshtein et al. 2015a). Samples were covered with a thin glass coverslip and mounting media. Cross‐sections were imaged at 10x magnification using a Nikon Eclipse 90i upright microscope.
Statistics
Statistics were assessed using GraphPad Prism 6 software. A Student's t test or two‐way ANOVA with Tukey post hoc test were used where applicable. Due to unequal group sizes, repeated measures assessment or a three‐way ANOVA was unable to be performed. For Figs 4 and 6, separate two‐way ANOVAs were performed for (1) vehicle‐treated young and aged with CCA and (2) colchicine‐treated young and aged with CCA.
Figure 4. Mitophagy receptor expression with CCA and ageing.
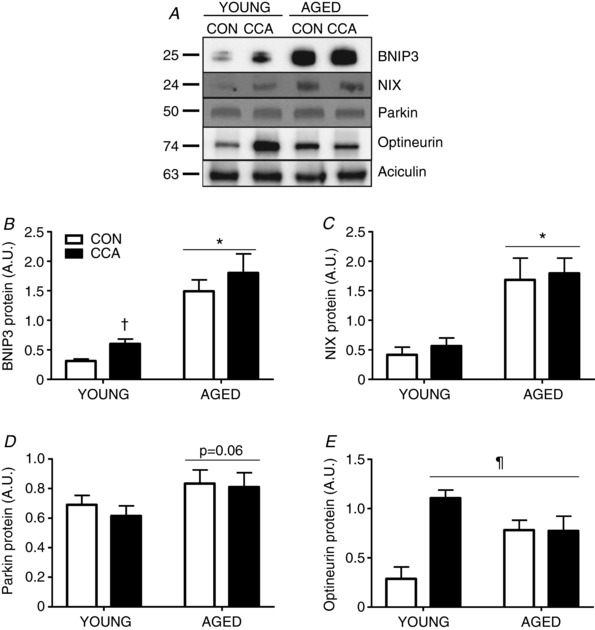
Protein expression of four known mitochondrial receptors for mitophagy were assessed through Western blotting in whole muscle protein extracts of vehicle‐treated animals (n = 4–6). A – E, representative blots are provided. Data were normalized to aciculin. Data are presented as means ± SEM. * P < 0.05 main effect of age; † P < 0.05 main effect of CCA; ¶ P < 0.05 vs. young CON. CON, control; CCA, chronic contractile activity.
Figure 6. Lysosomal markers with ageing and CCA.
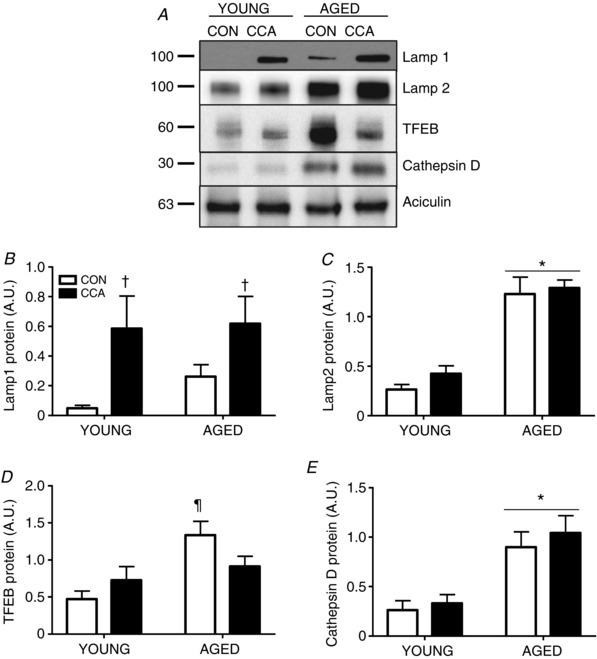
Lysosomes are the terminal step in autophagy/mitophagy. Assessment of protein indicators to lysosomal function were assessed including LAMP‐1 (n = 5–6, A and B), LAMP‐2 (n = 5–6, A and C), TFEB (n = 6, A and D) and Cathepsin D (n = 6, A and E) in vehicle‐treated whole muscle extracts. Protein expression was normalized to aciculin. Data are presented as means ± SEM. * P < 0.05 main effect of age; † P < 0.05 main effect of CCA; ¶ P < 0.05 age CON vs. young CON. CON, control; CCA, chronic contractile activity; TFEB, transcription factor EB, LAMP, lysosomal‐associated membrane protein.
Results
Effect of ageing and CCA on mitochondrial content
Aged animals were significantly larger than the young group and contained a larger fat mass (P < 0.05; Table 1), although no changes in food intake were noted. Additionally, aged animals had larger hearts, indicative of modest hypertrophy (P < 0.05; Table 1). Examination of muscle mass (TA and EDL) revealed that aged muscle was significantly smaller compared to muscle from the young animals (P < 0.05, Table 1). No change in muscle mass was noted following CCA or with colchicine injections in either age group. To confirm the induction of mitochondrial biogenesis in the vehicle‐treated age groups, COX enzyme activity was assessed as a representative measure of mitochondrial content. In young animals, CCA induced a 1.7‐fold increase (P < 0.05) in COX activity versus the contralateral muscle (Fig. 1 A). Furthermore, post hoc analysis revealed that COX activity in young animals following CCA was significantly higher than in aged CON and CCA muscle, by 2‐fold and 1.5‐fold, respectively (P < 0.05, Fig. 1 A). PGC‐1α protein expression was enhanced in young VEH‐treated muscle following CCA by 1.6‐fold (P < 0.05, Fig. 1 B). In contrast, CCA did not produce a significant enhancement of this key transcriptional coactivator in aged VEH‐treated muscle (Fig. 1 B).
Figure 1. Mitochondrial content with CCA.
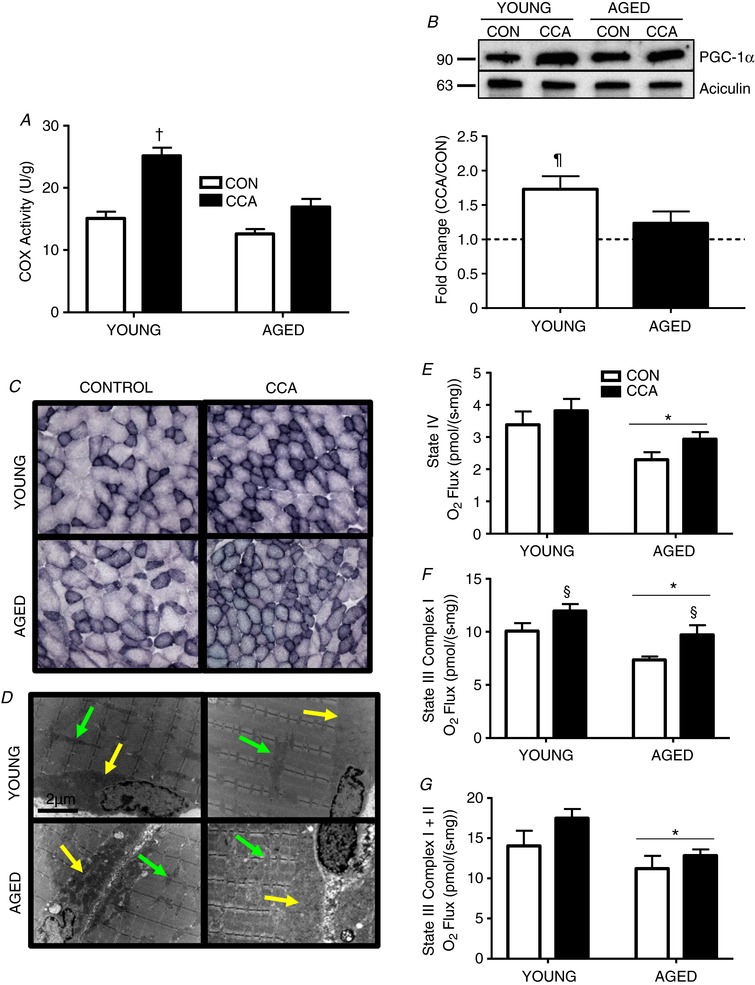
A, COX activity in vehicle‐treated young and aged muscle following CCA (n = 6–8). B, PGC‐1α protein expression expressed as fold change of CCA over CON (n = 5–6). C, SDH staining was performed on 10 μm sections. A representative image based on the assessment of two animals per condition is shown at 10× magnification. D, representative electron micrographs of mitochondria based on the assessment of two animals per condition (young and aged VEH‐treated muscle) are shown. Yellow arrows denote SS mitochondria and green arrows indicate IMF mitochondria. E–G, respiration was measured on intact saponin‐ permeabilized muscle fibre bundles for basal (E) and maximal (F and G), respiration states (n = 5–8). Data are presented as means ± SEM. † P < 0.05 CCA vs. all other conditions; ¶ P < 0.05 CCA vs. CON; * P < 0.05 main effect of age; § P < 0.05 main effect of CCA. CON, control; CCA, chronic contractile activity, COX; cytochrome oxidase; SDH, succinate dehydrogenase.
In support of these biochemical findings, qualitative histochemical staining for SDH in VEH‐treated muscle revealed similar observations as reflected by darker staining of fibres in young CCA muscle. A darker shade of SDH staining was also visible in aged muscle subjected to CCA compared to the respective control. It is interesting to note that the aged muscle contained many fibres with much more variable cross‐sectional area (Fig. 1 C). Additionally, qualitative electron micrographs of VEH‐treated muscle demonstrated enhanced thickness of the subsarcolemmal (SS) layer (yellow arrow) and the intermyofibrillar (IMF) density (green arrow) following CCA in young animals (Fig. 1 D). With ageing, the SS layer became notably thinner and less dense; however, CCA produced a modest enrichment of this SS layer (yellow arrow) in aged muscle, consistent with SS mitochondrial biogenesis. These results are similar to our previous findings (Iqbal et al. 2013).
Using permeabilized muscle fibres from VEH‐treated animals, basal State IV respiration was decreased with age (main effect, P < 0.05, Fig. 1 E). No change was observed with CCA for basal respiration in either age cohort (Fig. 1 E). Active, Complex I‐stimulated State III respiration was also diminished with age (main effect, P < 0.05, Fig. 1 F). The same effect was observed for Complex I + II‐stimulated State III respiration (Fig. 1 G). CCA also induced elevations (P < 0.05) in State III Complex I‐stimulated respiration in both age groups (Fig. 1 F). When these respiration data were normalized to COX activity, no significant differences among the conditions were noted (data not shown), indicating that the effects of age and CCA are reflective of changes in mitochondrial content per gram of muscle, rather than a result of alterations in mitochondrial composition.
Alterations in upstream autophagy regulators
Ageing resulted in a significant increase in forkhead box O3 (FoxO3) level in skeletal muscle. However, FoxO3 remained unaffected by CCA, regardless of age (Fig. 2 A and B). A main effect of CCA was observed on Beclin 1 protein expression, which was particularly evident from the 2.1‐fold increase observed in young muscle (P < 0.05; Fig. 2 A and C). Ageing also produced a large induction of Beclin 1 (P < 0.05) compared to young muscle in both CON and CCA conditions. p53 was undetectable in whole muscle extracts of young animals (data not shown), but was significantly enriched in aged muscle. CCA led to a marked 1.7‐fold increase (P < 0.05) in aged muscle (Fig. 2 D).
Figure 2. Upstream autophagy markers.
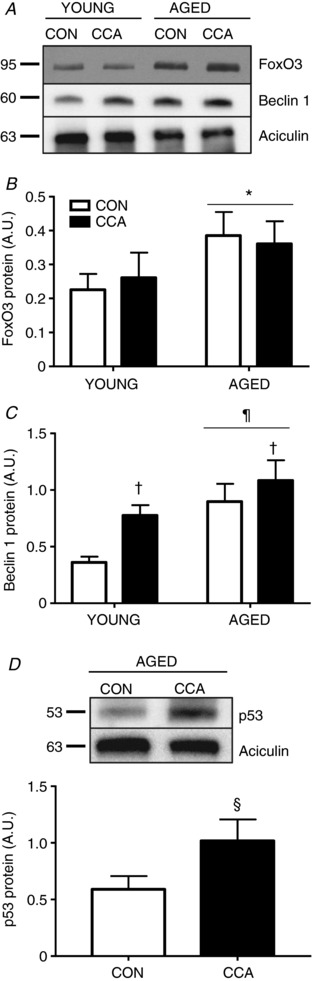
A, whole muscle protein extracts were assessed in vehicle‐treated muscle for FoxO3 (n = 7–8, A and B), Beclin 1 (n = 6, A and C), and p53 (n = 5, D). Representative blots are provided. Protein expression was normalized to aciculin. Data are presented as means ± SEM. * P < 0.05 main effect of age; † P < 0.05 main effect of CCA. ¶ P < 0.05 vs. young CON. § P < 0.05 vs. age CON. CON, control; CCA, chronic contractile activity.
Autophagosome turnover in young and aged muscle
No change in microtubule‐associated proteins 1A/1B light chain 3‐II (LC3‐II) protein expression was observed following CCA in young muscle (Fig. 3 A and B). Aged muscle displayed greater LC3‐II levels (main effect, P < 0.05) than young muscle (Fig. 3 A and B). Following CCA, no change in LC3‐II levels were noted in either young or aged muscle. As expected, colchicine treatment significantly enhanced LC3‐II accumulation in both young and aged muscle, ranging from 1.9‐ to 2.5‐fold (main effect, P < 0.05; Fig. 3 A and B) compared to respective vehicle‐treated conditions. With age, there was a greater accumulation of LC3‐II with autophagy inhibition compared to young muscle with colchicine treatment (P < 0.05). Interestingly, calculation of LC3‐II autophagosome flux (COL − VEH), revealed no alterations in young muscle after CCA (Fig. 3 C). However, ageing muscle presented with increased autophagic flux (P < 0.05), which also remained unchanged following CCA (Fig. 3 C).
Figure 3. Autophagy flux in young and aged muscle with CCA.
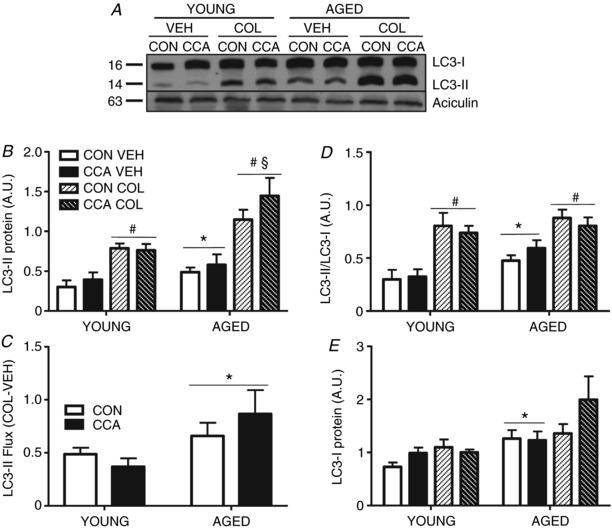
LC3‐II protein expression was measured in whole muscle samples to determine autophagic flux (n = 7) A–C, flux was calculated by subtracting VEH LC3‐II ratios from COL LC3‐II ratios (COL‐VEH; ratios were normalized to aciculin), for CON and CCA conditions in young and aged groups (n = 7). C, the LC3‐II/LC3‐I ratio was also assessed to compare and contrast against previous publications (n = 6–7). D, expression of the precursor LC3‐I was assessed under all experimental conditions (n = 6–7). E, representative blots are shown. Data are presented as means ± SEM. * P < 0.05, main effect of age; # P < 0.05, main effect of COL. § P < 0.05, aged COL vs. young COL, main effect. CON, control; CCA, chronic contractile activity; COL, colchicine; VEH, vehicle.
In agreement with the autophagy flux calculations, no difference was detected in the LC3‐II/LC3‐I ratio after CCA compared to CON in young muscle (Fig. 3 D). A 1.6‐fold elevation in the LC3‐II/LC3‐I ratio was detected in aged muscle basally compared to young resting muscle (P < 0.05). This ratio remained unchanged in aged muscle in response to CCA. In agreement with LC3‐II protein expression, we observed a predictable increase in the LC3‐II/LC3‐I ratio (main effect, P < 0.05) under colchicine‐treated conditions in both young and aged muscle, regardless of CCA. Neither CCA nor colchicine had an effect on LC3‐I levels in either age group. However, there was a main effect of ageing to increase the expression of LC3‐I (P < 0.05; Fig. 3 E).
Mitophagy markers with ageing and CCA
In young muscle, CCA increased BCL2/adenovirus E1B 19 kDa protein‐interacting protein 3 (BNIP3) expression by 1.9‐fold (P < 0.05, Fig. 4 A and B). Ageing produced a marked elevation in BNIP3 protein expression by 4.8‐fold (P < 0.05) and this remained unchanged following CCA. BNIP3‐like (BNIP3L/NIX) protein levels were not altered after CCA in young muscle (Fig. 4 A and C). Similar to BNIP3, a significant 4.0‐fold accumulation of NIX was noted in aged, resting muscle, and these high levels persisted following CCA (Fig. 4 A and C). The expression of Parkin in whole muscle extracts exhibited a trend towards an increase in ageing muscle (P = 0.06) and remained unaffected by CCA in both age groups (Fig. 4 D). Post hoc analysis of optineurin revealed a dramatic enhancement by 3.9‐fold (P < 0.05) with CCA in young muscle compared to its respective CON (Fig. 4 A and E). Optineurin levels were elevated basally in aged CON muscle by 2.7‐fold (P < 0.05), compared to young CON muscle. Optineurin levels were unresponsive to CCA in the muscle of ageing animals, in contrast to the large CCA‐induced enhancement found in young muscle (Fig. 4 A and E). Both age and CCA significantly increased the levels of optineurin above that found in young CON muscle (P < 0.05).
Mitophagy in young and aged muscle with CCA
Localization of LC3‐II to IMF mitochondria was not changed following CCA in young muscle (Fig. 5 A and B). Similarly, no change in LC3‐II positive IMF mitochondria was noted with ageing, irrespective of CCA when compared to young (Fig. 5 A and B). Colchicine‐mediated autophagy inhibition resulted in significant accumulations of LC3‐II on both young and aged mitochondria compared to vehicle, ranging from 1.4‐ to 3.6‐fold (main effect, P < 0.05; Fig. 5 B). IMF mitochondria from aged muscle accumulated more LC3‐II following colchicine treatment (main effect, P < 0.05) when compared to the young colchicine‐treated group. Notably, CCA caused a significant decrease in colchicine‐induced LC3‐II accumulation in both young and aged IMF mitochondria by ∼20% (P < 0.05; Fig. 5 B). Calculation of LC3‐II flux revealed a 1.6‐fold enhancement (P < 0.05) of basal mitophagy in aged, compared to young muscle (Fig. 5C). Interestingly, CCA produced decreases (P < 0.05) in LC3‐II IMF flux compared to CON muscle by 40% and 24% in young and aged muscle, respectively (Fig. 5 C).
Figure 5. IMF mitophagy flux.
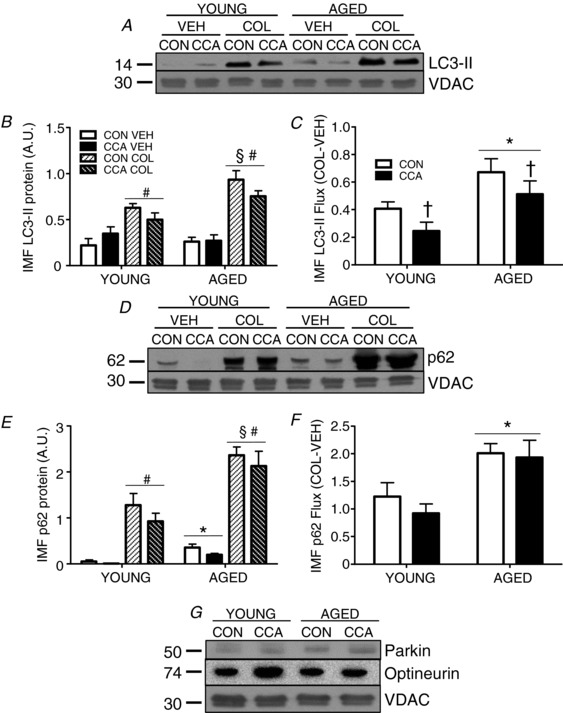
Mitochondria were isolated from the TA for all conditions. Localization of LC3‐II (n = 6–7, A and B) and p62 (n = 4–5; D and E) were assessed. Flux was calculated for both LC3‐II (n = 6–7, C) and p62 (n = 4–5, F) and calculated as described in Fig. 4. Protein density was normalized to the mitochondrial marker VDAC. Translocation of receptors to the mitochondria were also examined (n = 5, G). Data are presented as means ± SEM. * P < 0.05 main effect of age; † P < 0.05 main effect of CCA; # P <0.05 main effect of COL; § P < 0.05 aged COL vs. young COL, main effect. CON, control; CCA, chronic contractile activity; COL, colchicine; VEH, vehicle; IMF, intermyofibrillar; VDAC, voltage‐dependent anion channel.
p62 localization to IMF mitochondria was very low in muscle of young animals, and accumulated to significantly higher levels on mitochondria from aged muscle (P < 0.05, Fig. 5 D and E). CCA elicited 40–90% reductions in p62 localized to IMF mitochondria in young and aged muscle in the absence of colchicine. Colchicine treatment resulted in marked increases in p62 of between 6.6‐ and ∼200‐fold in both age groups (P < 0.05; Fig. 5 E). CCA had no significant effect on p62 mitophagy flux in young muscle (Fig. 5 F). With ageing, a 1.6‐fold elevation (P < 0.05) in IMF p62 flux was observed, and this remained unchanged following CCA. The localization of Parkin and optineurin to IMF mitochondria was unaffected by either CCA, or with age (Fig. 5 G).
Lysosomal alterations with ageing and CCA
Divergent responses were observed for lysosomal‐associated membrane proteins ‐1 and 2 (LAMP‐1/2), two abundant lysosomal membrane proteins. LAMP‐1 protein was induced in response to CCA in both young and aged muscle (main effect, P < 0.05; Fig. 6 A and B). No significant difference in LAMP‐1 was observed in aged, compared to young muscle (Fig. 6 B). In contrast, LAMP‐2 was not significantly altered by CCA in either age group, but exhibited an enhanced 4.7‐fold accumulation in aged muscle (P < 0.05; Fig. 6 A and C). In this study, CCA produced no significant change in TFEB content in young muscle. However, ageing alone led to an exaggerated 2.8‐fold (P < 0.05) increase in TFEB protein in comparison to young CON (Fig. 6 A and D). Interestingly, with the addition of CCA in the aged group, this increase was no longer present (Fig. 6 D). Cathepsin D expression was not altered by CCA in either young or aged muscle, but ageing muscle displayed a marked ∼3‐fold increase (P < 0.05), compared to young muscle (Fig. 6 A and E).
Discussion
With advancing age, there is a loss of muscle mass and performance. Identification of the potential cellular and molecular regulators that underlie this maladaptive phenomenon is critical as the number of aged individuals in the population continues to increase. Understanding the mechanisms that contribute to sarcopenia would be insightful to tailor beneficial strategies, such as exercise, to help preserve well‐being into the advanced years.
Mitochondria are essential organelles that have been documented to exhibit dysfunctional properties in aged muscle (Carter et al. 2015). These organelles have been associated with the maladaptive features of sarcopenia and remain under intense investigation in the literature. However, debate and controversy persist surrounding the connection between mitochondria and aging muscle. It has been documented that the generation and maintenance of mitochondria (biogenesis) in muscle with advancing age is hampered (Ljubicic & Hood, 2009; Ljubicic et al. 2009). Additionally, following exercise training protocols, aged muscle appears to harbour a lower capacity for the generation of new, healthy organelles than younger counterparts (Hood et al. 2016). Although this remains controversial, recent work has indicated that aged muscle can increase the transcription of PGC‐1α in response to contractile activity (Carter et al. 2018), as well as enhance its translational capacity in response to a training regime (Robinson et al. 2017). Nonetheless, little research has focused on the selective removal of organelles through mitophagy, a process also critical to maintaining organelle and muscle integrity. Furthermore, since mitophagy is highly dynamic, it is important to capture the turnover or ‘flux’ of this process, rather than relying on snapshot protein measurements of pathway components, or of the LC3‐II/LC3‐I ratio (Mizushima & Yoshimori, 2007; Castets et al. 2016; Klionsky, 2016; Yoshii & Mizushima, 2017). Thus, this study had two central aims: (1) to directly determine how autophagosome/mitophagy flux is changed in aged muscle, and (2) to determine how autophagosome/mitophagy flux adapts in response to a model of chronic exercise in both young and aged animals.
To gain insight into these aims, we utilized an established rodent model of ageing (Lushaj et al. 2008; Ballak et al. 2014), the Fisher 344 Brown Norway Hybrid rat and compared young (6 months) to aged (35‐36 months) animals. The aged cohort of this strain displayed hallmark features of aged muscle, including reduced muscle mass, lower mitochondrial content, decreased respiration and the presence of lipofuscin. Additionally, previous studies utilizing this model have characterized selected dysfunctional properties of mitochondria, including increased ROS production and greater apoptotic susceptibility (Chabi et al. 2008; Ljubicic et al. 2009).
Autophagy occurs at low levels basally, and is known to be upregulated with a variety of stimuli, including acute exercise (He et al. 2012; Vainshtein et al. 2015b). In order to capture the degree of autophagosome turnover through autophagy and mitophagy in young and aged muscle, we employed the autophagy inhibitor colchicine. Colchicine disrupts microtubules and impairs the ability of autophagosomes to travel to the lysosome for fusion and degradation of their sequestered contents (Amenta et al. 1977; Ju et al. 2010). Our preliminary data using electron microscopy indicated that autophagy inhibition through colchicine treatment produces an increased level of vacuolar inclusions within both young and aged muscle, as expected, but no disruption in sarcomere structure which might limit muscle contractility (H. N. Carter, Y. Kim, A. T. Erlich, D. Zarrin‐khat and D. A. Hood, unpublished observations). This drug has previously been used successfully for the measurement of autophagy and mitophagy flux in skeletal muscle (Ju et al. 2010, 2016; Mofarrahi et al. 2013; Vainshtein et al. 2015a, b ; Baehr et al. 2016). Thus, we compared the difference in the levels of LC3‐II between colchicine‐ and vehicle‐treated tissues to provide us with an approximation of the amount of autophagosomes with cargo, such as mitochondria, destined for lysosomal degradation. In the case of mitophagy, the isolation of mitochondria and subsequent Western blotting for LC3‐II reveals an approximation of the degree of organelles targeted for selective sequestration and recycling. Pilot work in Sprague‐Dawley rats injected with colchicine over 3 days, followed by the assessment of LC3‐II accumulation, revealed the success of the method. We observed significant increases in the amount of LC3‐II in both whole muscle and isolated mitochondria from the colchicine‐injected rats, compared to vehicle‐treated animals. Overall, these observations confirm that colchicine was effective in blocking the autophagy pathway in rat skeletal muscle.
We employed chronic contractile activity (CCA) as a model of exercise training to assess the response of autophagy/mitophagy. We have previously used this model with success to induce mitochondrial biogenesis in young and aged muscle (Ljubicic et al. 2009). Indeed, in our current study we observed a robust induction of mitochondria in young muscle following nine consecutive days of CCA, as demonstrated by elevated COX activity, enhanced respiration, darker SDH and COX histochemical staining and expansion of the SS and IMF populations observed through qualitative electron microscopy. Additionally, a significant induction of PGC‐1α was present in young muscle following CCA. Together, these observations confirm the high degree of malleability in young skeletal muscle, and indicate that the CCA protocol was effective at inducing mitochondrial biogenesis.
In agreement with our previous observations (Ljubicic et al. 2009), CCA in aged animals also induced mitochondrial biogenesis, but the degree of adaptation was reduced compared to that observed in young muscle. This was evident from attenuated increases in the key mitochondrial transcriptional coactivator PGC‐1α, along with reduced increases in COX activity, in aged compared to young muscle. Additionally, qualitative histochemical staining and electron micrography revealed increases in mitochondria following CCA in aged muscle, but to a lesser degree than in their younger counterparts. This is particularly interesting since the same absolute workload is applied to both age cohorts, suggesting that the responsiveness of aged muscle has diminished kinetics for mitochondrial biogenesis (Carter et al. 2015; Hood et al. 2016). While these observations appear to indicate that the capacity for exercise‐induced biogenesis is reduced, this remains debatable. For instance, a recent study in aged human muscle found that chronic exercise training is capable of inducing an enhanced abundance of mitochondrial proteins through increased protein synthesis (Robinson et al. 2017), suggesting that the capacity for the translation of mitochondrial proteins is not impaired with age. Further research on the simultaneous investigation of multiple steps within the gene expression pathway (e.g. transcription, translation, post‐translational trafficking) will be useful in determining whether exercise can serve to accelerate the pathways that may limit the mitochondrial adaptations in aged muscle.
To assess autophagy, we used LC3‐II as a key identifier of autophagosomes. We assessed the levels of LC3‐II under all conditions in whole muscle extracts with the intent of procuring an estimate of the amount of autophagosome flux occurring in young and aged muscle, with or without CCA. Treatment with colchicine greatly elevated LC3‐II levels in both young and aged cohorts, confirming that autophagy was successfully blocked. The benefit of performing flux calculations is to avoid the misinterpretation that could occur simply by assessing LC3‐II levels in untreated tissues. Greater LC3‐II levels could occur via two possible mechanisms, either by greater processing of LC3‐I to LC3‐II, or by the impaired degradation of autophagosomes which harbour LC3‐II, leading to enhanced levels of this marker (Klionsky, 2016). Thus, including a condition where autophagy was inhibited allowed us to gain an appreciation for the amount of autophagosomes destined for degradation.
Contrary to our hypothesis, LC3‐II autophagy flux in aged muscle was greater than in young muscle. There were no changes in LC3‐II autophagy flux brought about by CCA. These data were also reflected in the evaluation of p62 flux which show the same pattern (data not shown). If only the LC3‐II/I ratio had been evaluated, then the increase in this ratio in aged muscle could potentially have been interpreted in two ways: (1) as greater autophagy flux (i.e. more LC3‐II, more autophagosome formation, more drive for autophagy), or (2) as an impairment in degradation, and the subsequent accumulation of autophagosomes. Thus, by having a flux measurement this study we gained a clearer picture of the autophagy events occurring in aged muscle. Previous reports in young animal models with exercise training have concluded that autophagy flux is increased following training (Lira et al. 2013; Ju et al. 2016), yet our data, using direct flux measurements, or the LC3‐II/I ratio, reveal no change under these experimental conditions. The discrepancies in these results may have to do with the differences in the training protocols and animal models used and the muscles examined, and are likely to be influenced by the choice of pathway marker employed as a surrogate for autophagy flux measures.
The literature has also documented that specific muscle fibre types (e.g. fast vs. slow) exhibit differences in autophagy/mitophagy turnover. Some studies have concluded that oxidative fibres have higher rates of flux (Lira et al. 2013), while others have noted greater rates in predominantly glycolytic muscle (Mofarrahi et al. 2013; Paré et al. 2017). Fibre type changes in response to our short protocol of CCA (3 h day−1, 9 days) are unlikely to confound the results of this study, since a greater volume and time of stimulation is required to produce a shift in myosin heavy chain expression (Putman et al. 2001), and our previous work has documented enhanced fatigue resistance, but no change in half‐relaxation time or time to peak tension in young or aged skeletal muscle following this CCA protocol (Ljubicic et al. 2009).
Beyond bulk autophagy flux, we have a profound interest in the selective autophagy pathway directed towards dysfunctional mitochondria, termed mitophagy. Initially, we assessed the levels of receptors known to be involved in the targeted removal of these organelles in young and aged muscle. BNIP3, and its related family member NIX, are located in the outer mitochondrial membrane and each harbours a LC3‐interacting region (LIR). In aged muscle, these receptors were significantly upregulated. Parkin, an E3 ubiquitin ligase, is recruited to depolarized mitochondria by the kinase PTEN‐induced putative kinase 1 (PINK1) (Matsuda et al. 2010; Koyano et al. 2014; Matsuda, 2016). Upon recruitment, Parkin will ubiquitinate outer mitochondrial membrane proteins, such as VDAC (Sun et al. 2012) and Mfn2 (Gegg et al. 2010; Chen & Dorn, 2013), to identify the organelle for degradation. Similar to previous results, Parkin exhibited a trend for upregulation with ageing while optineurin, a receptor which is recruited to ubiquitin‐tagged mitochondria subsequent to phosphorylation (Wong & Holzbaur, 2015; Richter et al. 2016), remained unchanged with age. Despite this, the ratio of enhanced levels of these receptors with ageing suggests that the mitochondrial selection for mitophagy is high in aged muscle.
To evaluate mitophagy flux directly, mitochondria were isolated from the TA muscle and extracts were used to assess the organelle‐specific localization of mitophagy markers. In agreement with our whole muscle measures, LC3‐II localization was enriched in colchicine‐treated samples of both young and aged muscle. Flux analysis revealed that, in contrast to expectations, aged muscle exhibited greater LC3‐II mitophagy flux than young muscle. These findings were supported by our measurements of enhanced p62 mitophagy flux and localization on mitochondria, as well as the elevated expression of mitophagy receptors in mitochondria of aged muscle, as discussed above. Therefore, the enhanced rate of mitochondrial removal via this mitophagy pathway could be a contributing factor in the reduction in organelle content which can be observed in ageing muscle. On the other hand, the discrepancies evident in the literature on mitochondrial function (e.g. respiration) with age may depend, in large measure, on whether mitophagy is sufficiently upregulated to selectively remove the dysfunctional organelles, resulting in the maintenance of only a healthy pool of organelles, under the conditions of study.
Of great interest within our results was the observation that the adaptive response to CCA resulted in a significant reduction in mitophagy flux. It is known that under a variety of conditions in which respiratory dysfunction exists, exercise training or chronic contractile activity have led to an improvement in mitochondrial function, leading to reduced ROS signalling and AMPK activation (Taivassalo et al. 2001; Adhihetty et al. 2009; Ljubicic et al. 2009; Carter & Hood, 2012; Conley et al. 2013; Menzies et al. 2013). These are well‐known triggers for the activation of mitophagy. Thus, the adaptation to a reduced mitophagy flux is likely to be due to an improvement in the quality of the organelles brought about by CCA, and the attenuated signalling that accompanies this adaptation. Despite this decrease in mitophagy flux, CCA did elicit an increase in the expression of BNIP3 and optineurin receptors, exclusively in young muscle. In aged muscle, the same response to CCA was not observed, and in the case of optineurin, aged muscle was remarkably unresponsive. Previous work examining BNIP3 with exercise training has also observed increases in BNIP3 in whole muscle extracts (Lira et al. 2013; Ju et al. 2016). This increase in BNIP3 has been interpreted to mean that mitophagy must be increased as well. Our observations contradict previous conclusions which suggest that mitochondrial removal is enhanced following exercise training (Lira et al. 2013; Ju et al. 2016). However, in these prior cases only indirect measures of mitophagy were used to derive this conclusion.
Transcriptional regulation of autophagy components is necessary for changes in autophagy flux (Füllgrabe et al. 2016). Thus, we assessed the expression of the well‐known transcriptional regulators, FoxO3 (Mammucari et al. 2007) and p53 (Maiuri et al. 2010; Wang et al. 2013). In young muscle no CCA‐induced alterations were detectable in either protein. However, each of these proteins was significantly upregulated with age, in agreement with other studies (Tamilselvan et al. 2007; Ziaaldini et al. 2015; Wagatsuma et al. 2016). Interestingly, p53 was further enhanced with CCA in old muscle. Coupled with our observations of enhanced flux in aged muscle, the increase in these proteins may suggest enhanced transcription for autophagy genes in the ageing milieu. p53 has also been demonstrated to exert a role in mitochondrial biogenesis with exercise (Saleem et al. 2009; Saleem & Hood, 2013), thus the enhanced expression with ageing and CCA may also contribute to the production of new organelles with exercise. However, FoxO3 and p53 are subject to post‐translational modifications as well as changes in cellular distribution (e.g. nuclear/cytosolic shuttling) and further work investigating the cellular location and phosphorylation status of these transcription factors is required to fully understand their role in autophagy/mitophagy with exercise and ageing.
Further along the upstream autophagy pathway, numerous inputs converge upon Beclin 1, an essential regulator of the core autophagy machinery. In aged muscle, greater Beclin 1 was observed, similar to previous observations (Wohlgemuth et al. 2010; O'Leary et al. 2013; Sakuma et al. 2016). Depending on which protein is partnered with Beclin 1, autophagy can either be activated or inhibited. For instance, Bcl‐2 is negative regulator of autophagy when associated with Beclin 1 (Salminen et al. 2013). Other reports have noted that Bcl‐2 declines in ageing muscle (Ziaaldini et al. 2015), further supporting the conclusion autophagy is enhanced in aged muscle. We also observed an increase in Beclin 1 protein expression in young muscle exposed to CCA. Previous reports have identified an increase in Beclin 1 following both acute and chronic exercise (He et al. 2012; Ju et al. 2016), but some opposing results have also been documented (McMillan et al. 2015; Mejías‐Peña et al. 2016; Kim & Hood, 2017). He and colleagues (2012) observed a decreased association of Beclin 1 with Bcl‐2 within 30 min of acute exercise, despite a rise in total protein levels of Beclin 1. Thus, further examination of Beclin 1 interactions following training would provide insight on the significance of enhanced Beclin 1 expression following CCA.
The terminal step of the autophagy/mitophagy pathway is degradation of cargo within the low pH environment of the lysosome. We examined markers of the lysosome in young and aged conditions, and also with CCA. In muscle of young animals, we have previously observed that CCA leads to an early adaptive increase in lysosomal markers, indicative of organelle biogenesis prior to mitochondrial adaptations (Kim & Hood, 2017). In the current study, we observed a CCA‐induced increase in LAMP‐1, supporting this increase in the capacity of muscle with respect to the terminal step of autophagy. In contrast, we have previously reported that aged muscle exhibits morphological evidence of lysosomal impairment, as evident from observations of lipofuscin in electron micrographs (O'Leary et al. 2013). These lipofuscin granules were also evident in muscle from aged, but not young, animals in the current study (H. N. Carter, Y. Kim, A. T. Erlich, D. Zarrin‐khat and D. A. Hood, unpublished observations). This was coincident with enhanced expression of lysosomal protein markers. These two observations may suggest that aged skeletal muscle contains an accumulation of defective lysosomes, congruent with the lysosomal theory of ageing (Wiederanders & Oelke, 1984; Brunk & Terman, 2002). The increase in autophagy and mitophagy flux with ageing, measured up to the point of lysosomal cargo delivery, suggests that the lysosomes are overburdened and may become unable to process the incoming cargo, resulting in the formation of lipofuscin granules. Interestingly, CCA significantly lowered the expression of the master regulator of lysosomal biogenesis, TFEB, in aged muscle, suggesting that CCA promotes a corrective phenotype to both mitochondria and lysosomes in aged muscle. Future examination of TFEB cellular localization (Erlich et al. 2018), as well as its phosphorylation status, along with direct assessments of lysosomal function and degradation capacity, will be required to complete the assessment of autophagy and mitophagy flux with exercise in ageing muscle.
Additional information
Competing interests
None declared.
Author contributions
D.A.H. and H.N.C. conceived the experiment design, analysed the data and wrote the manuscript. H.N.C., Y.T., A.T.E. and D.Z. performed the experiments.
Funding
D. A. Hood holds a Canada Research Chair in Cell Physiology. H. N. Carter was supported by an NSERC Graduate scholarship. This work was supported by funding from Canadian Institutes of Health Research to D.A.H.
Acknowledgements
The authors wish to acknowledge Doug Holmyard for his assistance with electron micrographs and Drs L. A. Kirshenbaum and S. Benchimol for their generous gifts of BNIP3 and p53 antibodies, respectively.
Edited by: Scott Powers & Karyn Hamilton
This is an Editor's Choice article from the 15 August 2018 issue.
Linked articles This article is highlighted by a Perspective by Drake. To read this Perspective, visit https://doi.org/10.1113/JP276462. This article is also highlighted by a Journal Club article by Sanchez. To read the Journal Club, visit https://doi.org/10.1113/JP276580.
References
- Adhihetty PJ, Ljubicic V & Hood DA (2007). Effect of chronic contractile activity on SS and IMF mitochondrial apoptotic susceptibility in skeletal muscle. Am J Physiol Endocrinol Metab 292, E748–E755. [DOI] [PubMed] [Google Scholar]
- Adhihetty PJ, Uguccioni G, Leick L, Hidalgo J, Pilegaard H & Hood DA (2009). The role of PGC‐1alpha on mitochondrial function and apoptotic susceptibility in muscle. Am J Physiol Cell Physiol 297, C217–C125. [DOI] [PubMed] [Google Scholar]
- Alway SE, Mohamed JS & Myers MJ (2017). Mitochondria initiate and regulate sarcopenia. Exerc Sport Sci Rev 45, 58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amenta JS, Sargus MJ & Baccino FM (1977). Effect of microtubular or translational inhibitors on general cell protein degradation. Evidence for a dual catabolic pathway. Biochem J 168, 223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baehr LM, West DWD, Marcotte G, Marshall AG, De Sousa LG, Baar K & Bodine SC (2016). Age‐related deficits in skeletal muscle recovery following disuse are associated with neuromuscular junction instability and ER stress, not impaired protein synthesis. Aging (Albany NY) 8, 127–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballak SB, Degens H, de Haan A & Jaspers RT (2014). Aging related changes in determinants of muscle force generating capacity: a comparison of muscle aging in men and male rodents. Ageing Res Rev 14, 43–55. [DOI] [PubMed] [Google Scholar]
- Belkin AM, Klimanskaya I V, Lukashev ME, Lilley K, Critchley DR & Koteliansky VE (1994). A novel phosphoglucomutase‐related protein is concentrated in adherens junctions of muscle and nonmuscle cells. J Cell Sci 107, 159–173. [DOI] [PubMed] [Google Scholar]
- Brunk UT & Terman A (2002). The mitochondrial‐lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. Eur J Biochem 269, 1996–2002. [DOI] [PubMed] [Google Scholar]
- Carnio S, LoVerso F, Baraibar MA, Longa E, Khan MM, Maffei M, Reischl M, Canepari M, Loefler S, Kern H, Blaauw B, Friguet B, Bottinelli R, Rudolf R & Sandri M (2014). Autophagy impairment in muscle induces neuromuscular junction degeneration and precocious aging. Cell Rep 8, 1509–1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter HN, Chen CCW & Hood DA (2015). Mitochondria, muscle health and exercise with advancing age. Physiology 30, 208–223. [DOI] [PubMed] [Google Scholar]
- Carter HN & Hood DA (2012). Contractile activity‐induced mitochondrial biogenesis and mTORC1. Am J Physiol Cell Physiol 303, C540–C547. [DOI] [PubMed] [Google Scholar]
- Carter HN, Pauly M, Tryon LD & Hood DA (2018). Effect of contractile activity on PGC‐1α transcription in young and aged skeletal muscle. J Appl Physiol 124, 1605–1615. [DOI] [PubMed] [Google Scholar]
- Castets P, Frank S, Sinnreich M & Rüegg MA (2016). “Get the balance right”: Pathological significance of autophagy perturbation in neuromuscular disorders. J Neuromuscul Dis 3, 127–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabi B, Ljubicic V, Menzies KJ, Huang JH, Saleem A & Hood DA (2008). Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 7, 2–12. [DOI] [PubMed] [Google Scholar]
- Chen Y & Dorn GW (2013). PINK1‐phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching JK, Ju JS, Pittman SK, Margeta M & Weihl CC (2013). Increased autophagy accelerates colchicine‐induced muscle toxicity. Autophagy 9, 2115–2125. [DOI] [PubMed] [Google Scholar]
- Conley KE, Jubrias SA, Cress ME & Esselman PC (2013). Elevated energy coupling and aerobic capacity improves exercise performance in endurance‐trained elderly subjects. Exp Physiol 98, 899–907. [DOI] [PubMed] [Google Scholar]
- Conley KE, Jubrias SA & Esselman PC (2000). Oxidative capacity and ageing in human muscle. J Physiol 526, 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlich AT, Brownlee DM, Beyfuss K & Hood DA (2018). Exercise induces TFEB expression and activity in skeletal muscle in a PGC‐1α‐dependent manner. Am J Physiol Cell Physiol 314, C62–C72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füllgrabe J, Ghislat G, Cho D‐H & Rubinsztein DC (2016). Transcriptional regulation of mammalian autophagy at a glance. J Cell Sci 129, 3059–3066. [DOI] [PubMed] [Google Scholar]
- Gegg ME, Cooper JM, Chau K‐Y, Rojo M, Schapira AHV & Taanman J‐W (2010). Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin‐dependent manner upon induction of mitophagy. Hum Mol Genet 19, 4861–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouspillou G, Sgarioto N, Kapchinsky S, Purves‐Smith F, Norris B, Pion CH, Barbat‐Artigas S, Lemieux F, Taivassalo T, Morais JA, Aubertin‐Leheudre M & Hepple RT (2014). Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J 28, 1621–1633. [DOI] [PubMed] [Google Scholar]
- Grumati P, Coletto L, Schiavinato A, Castagnaro S, Bertaggia E, Sandri M & Bonaldo P (2011). Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI‐deficient muscles. Autophagy 7, 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, Korsmeyer S, Packer M, May HI, Hill JA, Virgin HW, Gilpin C, Xiao G, Bassel‐Duby R, Scherer PE, Levine B (2012). Exercise‐induced BCL2‐regulated autophagy is required for muscle glucose homeostasis. Nature 481, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepple RT (2014). Mitochondrial involvement and impact in aging skeletal muscle. Front Aging Neurosci 6, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloszy JO (1967). Biochemical adaptations in muscle. Effects of exercise on mitochondrial oxygen uptake and respiratory enzyme activity in skeletal muscle. J Biol Chem 242, 2278–2282. [PubMed] [Google Scholar]
- Hood DA, Tryon LD, Carter HN, Kim Y & Chen CCW (2016). Unravelling the mechanisms regulating muscle mitochondrial biogenesis. Biochem J 473, 2295–2314. [DOI] [PubMed] [Google Scholar]
- Hoppeler H, Howald H, Conley K, Lindstedt SL, Claassen H, Vock P & Weibel ER (1985). Endurance training in humans: aerobic capacity and structure of skeletal muscle. J Appl Physiol 59, 320–327. [DOI] [PubMed] [Google Scholar]
- Iqbal S, Ostojic O, Singh K, Joseph A‐M & Hood DA (2013). Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve 48, 963–970. [DOI] [PubMed] [Google Scholar]
- Jarvis JC & Salmons S (1991). A family of neuromuscular stimulators with optical transcutaneous control. J Med Eng Technol 15, 53–57. [DOI] [PubMed] [Google Scholar]
- Joseph A‐M, Adhihetty PJ, Buford TW, Wohlgemuth SE, Lees HA, Nguyen LM‐D, Aranda JM, Sandesara BD, Pahor M, Manini TM, Marzetti E & Leeuwenburgh C (2012). The impact of aging on mitochondrial function and biogenesis pathways in skeletal muscle of sedentary high‐ and low‐functioning elderly individuals. Aging Cell 11, 801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju J‐S, Jeon S‐I, Park J‐Y, Lee J‐Y, Lee S‐C, Cho K‐J & Jeong J‐M (2016). Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise‐trained condition. J Physiol Sci 66, 417–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju J‐S, Varadhachary AS, Miller SE & Weihl CC (2010). Quantitation of “autophagic flux” in mature skeletal muscle. Autophagy 6, 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent‐Braun JA & Ng A V (2000). Skeletal muscle oxidative capacity in young and older women and men. J Appl Physiol 89, 1072–1078. [DOI] [PubMed] [Google Scholar]
- Kim Y & Hood DA (2017). Regulation of the autophagy system during chronic contractile activity‐induced muscle adaptations. Physiol Rep 5, e13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klionsky DJ (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe J‐F, Saeki Y, Tanaka K & Matsuda N (2014). Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510, 162–166. [DOI] [PubMed] [Google Scholar]
- Kroemer G, López‐Otín C, Yuan J, Kroemer G, Kroemer G & Mandell M (2015). Autophagy: a druggable process that is deregulated in aging and human disease. J Clin Invest 125, 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M & Yan Z (2017). Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise‐induced mitophagy. Nat Commun 8, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leduc‐Gaudet J‐P, Picard M, Pelletier FS‐J, Sgarioto N, Auger M‐J, Vallée J, Robitaille R, St‐Pierre DH & Gouspillou G (2015). Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 6, 17923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon AS (2017). Attenuation of adverse effects of aging on skeletal muscle by regular exercise and nutritional support. Am J Lifestyle Med 11, 4–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, Hoehn KL & Yan Z (2013). Autophagy is required for exercise training‐induced skeletal muscle adaptation and improvement of physical performance. FASEB J 27, 4184–4193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljubicic V & Hood DA (2009). Diminished contraction‐induced intracellular signaling towards mitochondrial biogenesis in aged skeletal muscle. Aging Cell 8, 394–404. [DOI] [PubMed] [Google Scholar]
- Ljubicic V, Joseph A‐M, Adhihetty PJ, Huang JH, Saleem A, Uguccioni G & Hood DA (2009). Molecular basis for an attenuated mitochondrial adaptive plasticity in aged skeletal muscle. Aging (Albany NY) 1, 818–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lushaj EB, Johnson JK, McKenzie D & Aiken JM (2008). Sarcopenia accelerates at advanced ages in Fisher 344×Brown Norway rats. J Gerontol A Biol Sci Med Sci 63, 921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan EM, Paré M‐F, Baechler BL, Graham DA, Rush JWE & Quadrilatero J (2015). Autophagic signaling and proteolytic enzyme activity in cardiac and skeletal muscle of spontaneously hypertensive rats following chronic aerobic exercise. PLoS One 10, e0119382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Galluzzi L, Morselli E, Kepp O, Malik SA & Kroemer G (2010). Autophagy regulation by p53. Curr Opin Cell Biol 22, 181–185. [DOI] [PubMed] [Google Scholar]
- Mammucari C, Milan G, Romanello V, Masiero E, Rudolf R, Del Piccolo P, Burden SJ, Di Lisi R, Sandri C, Zhao J, Goldberg AL, Schiaffino S & Sandri M (2007). FoxO3 controls autophagy in skeletal muscle in vivo. Cell Metab 6, 458–471. [DOI] [PubMed] [Google Scholar]
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S & Sandri M (2009). Autophagy is required to maintain muscle mass. Cell Metab 10, 507–515. [DOI] [PubMed] [Google Scholar]
- Matsuda N (2016). Phospho‐ubiquitin: upending the PINK–Parkin–ubiquitin cascade. J Biochem 159, 379–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou Y, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N & Tanaka K (2010). PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189, 211–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne CN, Mokrusch T, Jarvis JC, Gilroy SJ & Salmons S (1993). Stimulation‐induced expression of slow muscle myosin in a fast muscle of the rat. Evidence of an unrestricted adaptive capacity. FEBS Lett 327, 297–300. [DOI] [PubMed] [Google Scholar]
- Mejías‐Peña Y, Rodriguez‐Miguelez P, Fernandez‐Gonzalo R, Martínez‐Flórez S, Almar M, de Paz JA, Cuevas MJ & González‐Gallego J (2016). Effects of aerobic training on markers of autophagy in the elderly. Age (Omaha) 38, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melov S, Shoffner JM, Kaufman A & Wallace DC (1995). Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Res 23, 4938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies KJ, Singh K, Saleem A & Hood DA (2013). Sirtuin 1‐mediated effects of exercise and resveratrol on mitochondrial biogenesis. J Biol Chem 288, 6968–6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BF, Robinson MM, Bruss MD, Hellerstein M & Hamilton KL (2012). A comprehensive assessment of mitochondrial protein synthesis and cellular proliferation with age and caloric restriction. Aging Cell 11, 150–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N & Yoshimori T (2007). How to interpret LC3 immunoblotting. Autophagy 3, 542–545. [DOI] [PubMed] [Google Scholar]
- Mofarrahi M, Guo Y, Haspel JA, Choi AM, Davis EC, Gouspillou G, Hepple RT, Godin R, Burelle Y & Hussain SN (2013). Autophagic flux and oxidative capacity of skeletal muscles during acute starvation. Autophagy 9, 1604–1620. [DOI] [PubMed] [Google Scholar]
- O'Leary MF, Vainshtein A, Iqbal S, Ostojic O & Hood DA (2013). Adaptive plasticity of autophagic proteins to denervation in aging skeletal muscle. Am J Physiol Cell Physiol 304, C422–C430. [DOI] [PubMed] [Google Scholar]
- Paré MF, Baechler BL, Fajardo VA, Earl E, Wong E, Campbell TL, Tupling AR & Quadrilatero J (2017). Effect of acute and chronic autophagy deficiency on skeletal muscle apoptotic signaling, morphology, and function. Biochim Biophys Acta ‐ Mol Cell Res 1864, 708–718. [DOI] [PubMed] [Google Scholar]
- Picard M, Ritchie D, Thomas MM, Wright KJ & Hepple RT (2011). Alterations in intrinsic mitochondrial function with aging are fiber type‐specific and do not explain differential atrophy between muscles. Aging Cell 10, 1047–1055. [DOI] [PubMed] [Google Scholar]
- Putman CT, Sultan KR, Wassmer T, Bamford JA, Skorjanc D & Pette D (2001). Fiber‐type transitions and satellite cell activation in low‐frequency‐stimulated muscles of young and aging rats. J Gerontol A Biol Sci Med Sci 56, B510‐9. [DOI] [PubMed] [Google Scholar]
- Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, Youle RJ & Dikic I (2016). Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci 113, 4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MM, Dasari S, Konopka AR, Johnson ML, Manjunatha S, Esponda RR, Carter RE, Lanza IR & Nair KS (2017). Enhanced protein translation underlies improved metabolic and physical adaptations to different exercise training modes in young and old humans. Cell Metab 25, 581–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanello V & Sandri M (2016). Mitochondrial quality control and muscle mass maintenance. Front Physiol 6, 422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooyackers OE, Adey DB, Ades PA & Nair KS (1996). Effect of age on in vivo rates of mitochondrial protein synthesis in human skeletal muscle. Proc Natl Acad Sci U S A 93, 15364–15369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Mariño G & Kroemer G (2011). Autophagy and aging. Cell 146, 682–695. [DOI] [PubMed] [Google Scholar]
- Rygiel KA, Picard M & Turnbull DM (2016). The ageing neuromuscular system and sarcopenia: a mitochondrial perspective. J Physiol 594, 4499–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma K, Aoi W & Yamaguchi A (2017). Molecular mechanism of sarcopenia and cachexia: recent research advances. Pflügers Arch ‐ Eur J Physiol 469, 573–591. [DOI] [PubMed] [Google Scholar]
- Sakuma K, Kinoshita M, Ito Y, Aizawa M, Aoi W & Yamaguchi A (2016). p62/SQSTM1 but not LC3 is accumulated in sarcopenic muscle of mice. J Cachexia Sarcopenia Muscle 7, 204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem A, Adhihetty PJ & Hood DA (2009). Role of p53 in mitochondrial biogenesis and apoptosis in skeletal muscle. Physiol Genomics 37, 58–66. [DOI] [PubMed] [Google Scholar]
- Saleem A & Hood DA (2013). Acute exercise induces tumour suppressor protein p53 translocation to the mitochondria and promotes a p53‐Tfam‐mitochondrial DNA complex in skeletal muscle. J Physiol 591, 3625–3636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A, Kaarniranta K & Kauppinen A (2013). Beclin 1 interactome controls the crosstalk between apoptosis, autophagy and inflammasome activation: Impact on the aging process. Ageing Res Rev 12, 520–534. [DOI] [PubMed] [Google Scholar]
- Salmons S & Jarvis JC (1991). Simple optical switch for implantable devices. Med Biol Eng Comput 29, 554–556. [DOI] [PubMed] [Google Scholar]
- Sebastián D, Sorianello E, Segalés J, Irazoki A, Ruiz‐Bonilla V, Sala D, Planet E, Berenguer‐Llergo A, Muñoz JP, Sánchez‐Feutrie M, Plana N, Hernández‐Álvarez MI, Serrano AL, Palacín M & Zorzano A (2016). Mfn2 deficiency links age‐related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J 35, e201593084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St‐Jean‐Pelletier F, Pion CH, Leduc‐Gaudet J‐P, Sgarioto N, Zovilé I, Barbat‐Artigas S, Reynaud O, Alkaterji F, Lemieux FC, Grenon A, Gaudreau P, Hepple RT, Chevalier S, Belanger M, Morais JA, Aubertin‐Leheudre M & Gouspillou G (2017). The impact of ageing, physical activity, and pre‐frailty on skeletal muscle phenotype, mitochondrial content, and intramyocellular lipids in men. J Cachexia Sarcopenia Muscle 8, 213–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Vashisht AA, Tchieu J, Wohlschlegel JA & Dreier L (2012). Voltage‐dependent anion channels (VDACs) recruit parkin to defective mitochondria to promote mitochondrial autophagy. J Biol Chem 287, 40652–40660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taivassalo T, Shoubridge EA, Chen J, Kennaway NG, DiMauro S, Arnold DL & Haller RG (2001). Aerobic conditioning in patients with mitochondrial myopathies: physiological, biochemical, and genetic effects. Ann Neurol 50, 133–141. [DOI] [PubMed] [Google Scholar]
- Tamilselvan J, Jayaraman G, Sivarajan K & Panneerselvam C (2007). Age‐dependent upregulation of p53 and cytochrome c release and susceptibility to apoptosis in skeletal muscle fiber of aged rats: role of carnitine and lipoic acid. Free Radic Biol Med 43, 1656–1669. [DOI] [PubMed] [Google Scholar]
- Terman A, Gustafsson B & Brunk UT (2006). Mitochondrial damage and intralysosomal degradation in cellular aging. Mol Aspects Med 27, 471–482. [DOI] [PubMed] [Google Scholar]
- Terman A, Kurz T, Navratil M, Arriaga EA & Brunk UT (2010). Mitochondrial turnover and aging of long‐lived postmitotic cells: the mitochondrial‐lysosomal axis theory of aging. Antioxid Redox Signal 12, 503–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainshtein A, Desjardins EM, Armani A, Sandri M & Hood DA (2015a). PGC‐1α modulates denervation‐induced mitophagy in skeletal muscle. Skelet Muscle 5, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vainshtein A, Tryon LD, Pauly M & Hood DA (2015b). Role of PGC‐1α during acute exercise‐induced autophagy and mitophagy in skeletal muscle. Am J Physiol Cell Physiol 308, C710–C719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagatsuma A, Shiozuka M, Takayama Y, Hoshino T, Mabuchi K & Matsuda R (2016). Effects of ageing on expression of the muscle‐specific E3 ubiquitin ligases and Akt‐dependent regulation of Foxo transcription factors in skeletal muscle. Mol Cell Biochem 412, 59–72. [DOI] [PubMed] [Google Scholar]
- Wang EY, Gang H, Aviv Y, Dhingra R, Margulets V & Kirshenbaum LA (2013). p53 mediates autophagy and cell death by a mechanism contingent on Bnip3. Hypertension 62, 70–77. [DOI] [PubMed] [Google Scholar]
- Wiederanders B & Oelke B (1984). Accumulation of inactive cathepsin D in old rats. Mech Ageing Dev 24, 265–271. [DOI] [PubMed] [Google Scholar]
- Wohlgemuth SE, Seo AY, Marzetti E, Lees HA & Leeuwenburgh C (2010). Skeletal muscle autophagy and apoptosis during aging: effects of calorie restriction and life‐long exercise. Exp Gerontol 45, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YC & Holzbaur ELF (2015). Temporal dynamics of PARK2/parkin and OPTN/optineurin recruitment during the mitophagy of damaged mitochondria. Autophagy 11, 422–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR & Mizushima N (2017). Monitoring and measuring autophagy. Int J Mol Sci 18, 1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziaaldini MM, Koltai E, Csende Z, Goto S, Boldogh I, Taylor AW & Radak Z (2015). Exercise training increases anabolic and attenuates catabolic and apoptotic processes in aged skeletal muscle of male rats. Exp Gerontol 67, 9–14. [DOI] [PubMed] [Google Scholar]