

Abstract
Key points
Prolonged exposure to vascular endothelial growth factor A (VEGF‐A) inhibits agonist‐mediated endothelial cell Ca2+ release and subsequent activation of intermediate conductance Ca2+‐activated K+ (IKCa) channels, which underpins vasodilatation as a result of endothelium‐dependent hyperpolarization (EDH) in mouse resistance arteries.
Signalling via mitogen‐activated protein/extracellular signal‐regulated kinase kinase (MEK) downstream of VEGF‐A was required to attenuate endothelial cell Ca2+ responses and the EDH‐vasodilatation mediated by IKCa activation.
VEGF‐A exposure did not modify vasodilatation as a result of the direct activation of IKCa channels, nor the pattern of expression of inositol 1,4,5‐trisphosphate receptor 1 within endothelial cells of resistance arteries.
These results indicate a novel role for VEGF‐A in resistance arteries and suggest a new avenue for investigation into the role of VEGF‐A in cardiovascular diseases.
Abstract
Vascular endothelial growth factor A (VEGF‐A) is a potent permeability and angiogenic factor that is also associated with the remodelling of the microvasculature. Elevated VEGF‐A levels are linked to a significant increase in the risk of cardiovascular dysfunction, although it is unclear how VEGF‐A has a detrimental, disease‐related effect. Small resistance arteries are central determinants of peripheral resistance and endothelium‐dependent hyperpolarization (EDH) is the predominant mechanism by which these arteries vasodilate. Using isolated, pressurized resistance arteries, we demonstrate that VEGF‐A acts via VEGF receptor‐2 (R2) to inhibit both endothelial cell (EC) Ca2+ release and the associated EDH vasodilatation mediated by intermediate conductance Ca2+‐activated K+ (IKCa) channels. Importantly, VEGF‐A had no direct effect against IKCa channels. Instead, the inhibition was crucially reliant on the downstream activation of the mitogen‐activated protein/extracellular signal‐regulated kinase kinase 1/2 (MEK1/2). The distribution of EC inositol 1,4,5‐trisphosphate (IP3) receptor‐1 (R1) was not affected by exposure to VEGF‐A and we propose an inhibition of IP3R1 through the MEK pathway, probably via ERK1/2. Inhibition of EC Ca2+ via VEGFR2 has profound implications for EDH‐mediated dilatation of resistance arteries and could provide a mechanism by which elevated VEGF‐A contributes towards cardiovascular dysfunction.
Keywords: VEGF‐A, endothelium‐derived hyperpolarizing factor, MEK, endothelial cell calcium, vasodilation
Key points
Prolonged exposure to vascular endothelial growth factor A (VEGF‐A) inhibits agonist‐mediated endothelial cell Ca2+ release and subsequent activation of intermediate conductance Ca2+‐activated K+ (IKCa) channels, which underpins vasodilatation as a result of endothelium‐dependent hyperpolarization (EDH) in mouse resistance arteries.
Signalling via mitogen‐activated protein/extracellular signal‐regulated kinase kinase (MEK) downstream of VEGF‐A was required to attenuate endothelial cell Ca2+ responses and the EDH‐vasodilatation mediated by IKCa activation.
VEGF‐A exposure did not modify vasodilatation as a result of the direct activation of IKCa channels, nor the pattern of expression of inositol 1,4,5‐trisphosphate receptor 1 within endothelial cells of resistance arteries.
These results indicate a novel role for VEGF‐A in resistance arteries and suggest a new avenue for investigation into the role of VEGF‐A in cardiovascular diseases.
Introduction
Vascular endothelial growth factor A (VEGF‐A) is a pro‐survival, angiogenic and permeability factor acting on endothelial cells (ECs) (Simons, Gordon and Claesson‐welsh, 2016). Although there are two receptors for VEGF‐A, its effects are mediated primarily through VEGF receptor‐2 (R2), whereas VEGF receptor‐1 (R1) merely acts as a decoy receptor in ECs, at least in the context of angiogenesis (Fong et al. 1995). On binding to VEGF‐A, VEGFR2 dimerizes and activates a number of pathways such as mitogen‐activated protein (MAP)/extracellular signal‐regulated kinase (ERK) kinase 1/2 (MEK1/2)‐ERK1/2, phosphatidylinositol‐4,5‐bisphosphate 3‐kinase‐Akt and MKK3/4/6‐p38 MAP kinase to initiate a range of cellular effects, including DNA synthesis, cell proliferation and an increase in permeability (Simons, Gordon and Claesson‐welsh, 2016). However, VEGFR2 dimerization appears only to explain part of the activation signal. In some cases, amplification of the signal occurs through the recruitment and binding of VEGF‐A to a co‐receptor, neuropilin‐1, enabling full activation of downstream pathways (Soker et al. 1998; Kawamura et al. 2008).
Although VEGF‐A is considered to stimulate the generation of nitric oxide (NO) from ECs leading to arterial vasodilatation (Ku et al. 1993; Itoh et al. 2002), the link between this signal and VEGFR is not clear. In human umbilical vein ECs (HUVECs), VEGF‐A increases EC intracellular Ca2+ ([Ca2+]i) through the activation of phospholipase Cγ and the subsequent production of inositol 1,4,5‐trisphosphate (IP3) (Favia et al. 2014). By contrast, in human microvasculature ECs (HMVECs), the increase in [Ca2+]i appears not to rely on IP3Rs but, instead, on Ca2+ influx through a receptor‐operated transient receptor potential channel (Cheng et al. 2006; Hamdollah Zadeh et al. 2008). An increase in EC Ca2+ will potentially activate NO synthase. However, to date, there has been no study linking a VEGF‐A evoked rise in EC Ca2+ and the release of NO in intact pressurized arteries.
In general, elevation of EC [Ca2+]i activates at least two distinct EC‐dependent vasodilator responses: one via NO release and the other as a result of endothelium‐derived hyperpolarization (EDH) with the EDH response predominating in smaller resistance arteries (Garland and Dora, 2017). In the latter, an increase in EC [Ca2+]i stimulates the opening of the small and intermediate conductance Ca2+‐activated K+ channels (SKCa, IKCa), which are localized to the endothelium. This results in an efflux of K+, causing hyperpolarization of the ECs. The hyperpolarizing current then spreads from the ECs to the neighbouring vascular smooth muscle cells (VSMC) via myoendothelial gap junctions to reduce the open probability of voltage‐dependent Ca2+ channels, causing vasodilatation (Edwards et al. 1998; Dora et al. 2008). EC‐dependent vasodilatation helps the maintenance of blood flow and ensures sufficient oxygen and nutrients are delivered to tissues. Reduced endothelium‐dependent vasodilatation occurs in a number of conditions, including diabetes, cancer and peripheral arterial disease (Lim et al. 2004; Stehr et al. 2010; Goel and Mercurio, 2013).
In the present study, we provide evidence to suggest that prolonged exposure to VEGF‐A and activation of the MEK1/2 pathway lead to a reduction in EC‐dependent vasodilatation in mouse isolated resistance arteries and that this reduction reflects a change in EC Ca2+ handling and reduced KCa activity.
Methods
Ethical approval
Ethical approval was obtained and all animal procedures were carried out in accordance with the UK Home Office Animals (Scientific Procedures) Act 1986. Experiments were carried out according to the guidelines laid down by the institution's animal welfare committee and regulations described in the Journal of Physiology editorial (Grundy, 2015).
Animals and tissue preparation
Wild‐type male C57BL/6J mice (Charles River, Kent, UK) between the ages of 11 and 16 weeks were used in the present study. Animals were housed in individually ventilated cages under a 12:12 h light/dark cycle and at 20–22°C. Standard chow and water were available ad libitum. Mice were killed in accordance with schedule 1 of the Animals (Scientific Procedures) Act by increasing the concentration of CO2 followed by cervical dislocation as confirmation of death. The mesentery was collected in cold MOPS buffer containing (in mm): 142.5 NaCl, 4.7 KCl, 1 CaCl2, 1.17 MgSO4, 3.0 MOPS, 1.2 NaH2PO4 (H2O), 11.0 glucose, 2.0 pyruvate and 0.02 EDTA at pH 7.40 ± 0.02.
Artery isolation and pressure myography
Third‐order mesenteric resistance arteries were dissected free of adhering adipocytes and connective tissue. Arteries were cannulated onto glass micropipettes (external diameter 100–120 μm), secured with 11‐0 sutures in a 2.0 mL temperature‐regulated chamber. Arteries were warmed to 37°C, gradually pressurized to a physiological pressure of 60 mmHg and allowed to equilibrate for 30 min (Beleznai et al. 2011). Agents were applied to the bath, except when VEGF‐A was perfused into the lumen. ZM323881 or UO126 were added to the bath 20 min prior to perfusion of either VEGF‐A and ZM323881 (VEGFR2 tyrosine kinase inhibitor) or VEGF‐A and UO126 (MEK1/2 inhibitor), respectively, into the lumen of artery at 1 μL min–1 using a beehive syringe pump. In preliminary experiments, 250 nm carboxyfluorescein was pumped together with VEGF‐A to establish the delay in delivering VEGF‐A into the lumen of arteries (15 min at 1 μL min–1). This time was added to the overall pumping time, meaning that t = 0 min was the time at which VEGF‐A reached the lumen. The luminal flow was stopped immediately prior to obtaining concentration–response curves.
Because the level of VEGF‐A ranges from undetectable to 0.1 nm under physiological conditions (Larsson, Sköldenberg and Eericson, 2002), rising to several nanomolar in pathological situations (Baker et al. 1995; Kaess et al. 2016), we compared responses to 1 pm, 0.1 nm and 1 nm VEGF‐A. Although VEGFR2 is phosphorylated within 5 min after VEGF‐A application, activation of downstream pathways varies from 10 to 60 min (Jang et al. 2017). Therefore, arteries were perfused for 60 min.
Diameter measurements were recorded on the stage of an inverted microscope (IX 70; Olympus, Tokyo, Japan) attached to a confocal scanning unit (FV500; Olympus). Artery viability was assessed with contraction to 1–3 μm phenylephrine (PE) followed by endothelium‐dependent vasodilatation to 1–10 μm SLIGRL, a PAR2 activating peptide (McGuire et al. 2002; Dora et al. 2003; Beleznai et al. 2011). Only arteries with dilatation >90% to 3 μm SLIGRL, reflecting undamaged endothelium, were used for the subsequent experiments. The outer diameter of the artery was measured using MetaMorph, version 7.7.4.0 (Molecular Devices, Sunnyvale, CA, USA). % Dilatation = (D – D constricted)/(D max – D constricted) × 100.
EC [Ca2+]i measurement in pressurized arteries
EC Ca2+ investigations were conducted on third‐order mesenteric resistance arteries from transgenic mice on a C57Bl/6J background expressing the genetically encoded Ca2+ indicator, GCaMP2, under the control of a connexin40 promoter [Tg(RP24‐25504‐GCaMP2)1 Mik mice] (Bagher, Davis and Segal, 2011). The mesenteric artery was isolated and pressurized as described above. Ca2+ measurements were taken from ECs at the bottom of the pressurized artery and images were obtained using a 40× water immersion objective (40×/1.15 NA objective, working distance = 0.25 mm; Olympus; excitation 488 nm, emission >505 nm) and acquired using Olympus FluoView 1200 software (FV10‐ASW) at ∼3 Hz with a gallium arsenide phosphide detector.
Ca2+ data were analysed using subcellular regions of interest as described previously (Garland et al. 2017), with each event categorized when viewing the original movie file during manual frame‐by‐frame playback. If a response radiated from a single point and was also synchronized and rapidly decayed within 20 μm2, this was termed local; Ca2+ waves were defined as asynchronous events that visibly propagated at least 20 μm along the length of an EC. All cells within a field of view (7–11 cells) were manually and individually analysed for Ca2+ event frequency. Results are presented as either frequency (events min–1) or F/F 0 traces, with the latter calculated by dividing the fluorescence intensity (F) by an average baseline fluorescence intensity (F 0). The number of local or propagating events were expressed as a percentage of total events.
Immunohistochemistry
Pressurized arteries were fixed with 2% paraformaldehyde for 10 min, blocked and permeabilized with 1% BSA in PBS‐Tween 20 (0.1%) for 1 h in luminal and abluminal space. Arteries were labelled with 2 μg mL–1 rabbit anti‐IP3R1 (PA1‐901; ThermoFisher Scientific, Waltham, MA, USA) and 2 μg mL–1 mouse anti‐ZO‐1 (339‐100; ThermoFisher Scientific) antibodies in 1% BSA PBS‐Tween 20 overnight at 4°C. This was followed by 2 h of incubation with 2 μg mL–1 goat anti‐rabbit 488 and anti‐mouse 405 secondary antibodies in 1% BSA PBS‐Tween 20. The nuclei and elastic lamina were labelled with 15 μm propidium iodide and 1 μm Alexa Fluor 633 hydrazide, respectively, for 20 min. Arteries were excited at 405, 488, 546 and 633 nm. The emitted fluorescence was collected from cells through the bottom wall of the artery through a water immersion objective (40×) and acquired using Olympus FluoView 1000 software (FV10‐ASW). Z‐stacks through the wall of the artery were obtained in 0.25 μm increments and reconstructed using Imaris, version 5.5 (Bitplane, Concord, MA, USA).
EC tube isolation
EC tubes were isolated as described previously (Socha et al. 2011; Socha and Segal, 2013; Garland et al. 2017), with modifications made for isolation from mouse mesenteric arteries. Arteries were dissected in cold dissection buffer containing (in mm): 137.0 NaCl, 5.6 KCl, 1.0 MgCl2, 10.0 Hepes, 10.0 glucose, 0.01 sodium nitroprusside and 0.1% BSA. Second‐ and third‐order mesenteric arteries were dissected free of surrounding tissue. One end of the artery was cannulated onto a glass micropipette (external diameter 100–120 μm) and the lumen flushed with cold dissection buffer to remove blood. Arteries were cut into 2–3 mm segments and transferred to a clear 1.5 mL microcentrifuge tube containing 1 mL of cold dissection buffer. Artery segments were washed twice with enzyme free dissociation buffer containing (in mm): 137.0 NaCl, 5.6 KCl, 1.0 MgCl2, 10.0 Hepes, 10.0 glucose, 2.0 CaCl2 and 0.1% BSA at 37°C. Segments were then incubated in dissociation buffer supplemented with 0.62 mg mL–1 papain, 1.0 mg mL–1 dithioerythritol and 1.5 mg mL–1 collagenase for 20–25 min at 37°C. Enzymatic digestion was terminated by aspiration of enzyme containing buffer and replaced with enzyme free dissociation buffer in a round culture dish for trituration. EC tubes were dissociated from surrounding vascular smooth muscle cells by gentle trituration using a glass micropipette (inner diameter 80–110 μm). A nanolitre injector (Nanoliter 2010) coupled with a Micro4 controller (World Precision Instruments, Sarasota, FL, USA) was mounted on an upright Olympus BX51WI microscope to allow for real‐time visualization of the trituration process.
RNA extraction and quantitative RT‐PCR
Arteries and EC tubes were isolated as described above. Four segments of second‐ and third‐order artery branches from one animal were pooled for each n value. Arteries were homogenized and RNA was extracted using the RNeasy plus Mini Kit (Qiagen, Valancia, CA, USA). For EC tubes, three >1 mm long EC tubes were pooled from one animal for each n value. Isolated EC tubes were transferred using a new glass micropipette into a clean round culture dish containing enzyme free dissociation buffer, and repeated three times to reduce VSMC contamination. RNA was then extracted from EC tubes using Cells‐to‐CT 1‐Step TaqMan Kit lysis buffer (ThermoFisher Scientific) in accordance with the manufacturer's instructions.
Quantitative RT‐PCR was carried out using the Cells‐to‐CT 1‐Step TaqMan Kit. Pre‐validated, gene specific FAM‐conjugated Taqman probes were purchased from ThermoFisher Scientific (Table 1). Reverse transcription was performed at 50°C for 20 min, followed by heat activation of Taq polymerase at 95°C for 30 s. PCR thermocycling conditions comprised 95°C for 15 s, then 60°C for 1 min, for 40 cycles, using the 7500 Fast Real‐Time PCR system (ThermoFisher Scientific). All samples were run in duplicates along with a no template negative control for each gene. Two housekeeping genes (Actb and β2M) were used to normalize gene expression. The relative expression was calculated using the ∆C t method [∆C t = C t (target gene) – C t (housekeeping gene)]. Success of VSMC removal was assessed by the expression of VSMC marker Acta2 (α‐SMA). C t > 37 was considered not to be detected.
Table 1.
Gene specific FAM‐conjugated Taqman primers
| Gene | Protein | Assay ID |
|---|---|---|
| Pecam‐1 | Platelet endothelial cell adhesion molecule‐1 (PECAM‐1) | Mm0124576_m1 |
| Acta2 | α‐Smooth muscle actin (α‐SMA) | Mm01204962_gh |
| β2Μ | β2 microglobulin (β2Μ) | Mm00437762_m1 |
| Actb | β‐actin | Mm04394036_g1 |
| Flt1 | Vascular endothelial growth factor receptor 1 (VEGFR1) | Mm00438980_m1 |
| Flk1 | Vascular endothelial growth factor receptor 2 (VEGFR2) | Mm01222435_m1 |
| Nrp1 | Neuropilin‐1 (NRP‐1) | Mm00435379_m1 |
| F2rl1 | Protease‐activated receptor 2 (PAR2) | Mm00433160_m1 |
| Itpr1 | Inositol trisphosphate receptor 1 (IP3R1) | Mm00439907_m1 |
| Itpr2 | Inositol trisphosphate receptor 2 (IP3R2) | Mm00444937_m1 |
| Itpr3 | Inositol trisphosphate receptor 3 (IP3R3) | Mm01306070_m1 |
Drugs
All other drugs were purchased from Sigma (St Louis, MO, USA), except mouse VEGF‐A164 (#493‐MV‐005) (R&D Systems, Minneapolis, MN, USA); diethylamin NONOate (#82100; DEA NONOate), ZM323881 (#2475) and UO126 (#1144) (Tocris Bioscience, St Louis, MO, USA); Apamin (#L8407) (Latoxan, Portes‐lès‐Valence, France); and SLIGRL (Auspep, Tullamarine, VIC, Australia). Stock solutions of ZM323881, UO126 and TRAM‐34 were prepared in DMSO and DEA NONOate in 10 mm NaOH. All other stock solutions were prepared in purified water.
Statistical analysis
All data are summarized as the mean ± SEM of n arteries (one per animal). The Ca2+ events data represent the analysis of at least seven cells per artery and values were averaged to provide one n value. Only active cells were used in the calculation of frequency. Statistical comparisons were made in Prism, version 7 (GraphPad Software Inc., San Diego, CA, USA) using an unpaired Student's t test, as well as one‐ or two‐way ANOVA with Bonferonni's post hoc test as appropriate. P < 0.05 was considered statistically significant.
Results
VEGF receptors are present in mouse resistance artery endothelial cells
RNA was extracted from either freshly isolated mesenteric arteries or from ECs alone isolated in the form of intact EC tubes. Pecam‐1 and Acta2 were used as EC and VSMC markers, respectively. Pecam‐1 expression was evident in both intact arteries and EC tubes, whereas the Acta2 signal was effectively absent from EC tubes, indicating that the EC samples were free from SMCs (Fig. 1 A). Flt1, Flk1 and their co‐receptor Nrp1 were each expressed in the EC tubes (Fig. 1 B). Despite the presence of VEGF receptors and functional endothelium, luminal perfusion of 0.1 or 1 nm VEGF‐A did not result in vasodilatation of arteries constricted with the α1‐adrenoceptor agonist PE (Fig. 1 C).
Figure 1. VEGF‐A receptor expression in mouse mesenteric resistance arteries.
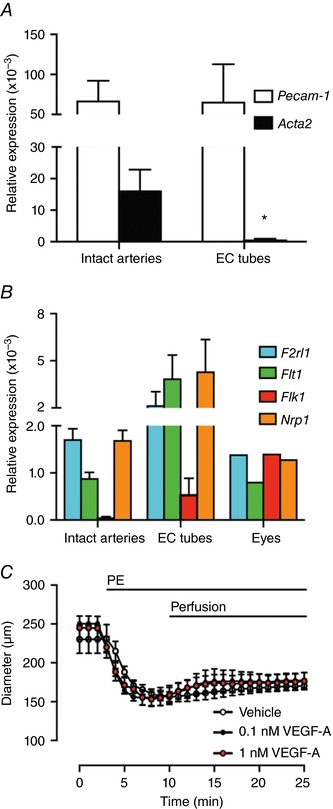
A, Pecam‐1 and Acta2 expression in intact arteries and isolated EC tubes. B, Flt1, Flk1 and Nrp1 expression (n = 4 animals). Eyes from a single animal were used as a positive control for the VEGF‐A receptors. C, arteries were pre‐constricted with PE. When pumped into the lumen of arteries, neither vehicle, nor VEGF‐A dilated the artery within 15 min (n = 3–4). * P < 0.05 compared to intact arteries.
VEGF‐A inhibits EDH‐mediated vasodilatation to SLIGRL
We investigated whether prolonged luminal exposure to 1 nm VEGF‐A, a concentration known to effectively activate VEGFR2 in rat arteries (Itoh et al. 2002; Egholm et al. 2016), might alter endothelium‐dependent vasodilatation to other agonists in small resistance arteries. SLIGRL readily dilates mouse resistance arteries via the EDH‐pathway and, to a lesser extent, NO (McGuire et al. 2002; Beleznai et al. 2011). Perfusion of 1 nm VEGF‐A for 60 min right‐shifted the concentration response curve to SLIGRL by >2.5‐fold (log EC50 –5.37 ± 0.02, n = 12) compared to vehicle (log EC50 –5.79 ± 0.01, n = 8), an effect that was not evident when arteries were perfused with 1 pm or 0.1 nm VEGF‐A (Fig. 2 A). The maximum dilatation to SLIGRL was not affected by 1 nm VEGF‐A (E Max 99.9 ± 0.1%, n = 12; vehicle: 99.6 ± 0.4%, n = 8).
Figure 2. VEGF‐A inhibits EDH‐mediated vasodilatation evoked by SLIGRL.
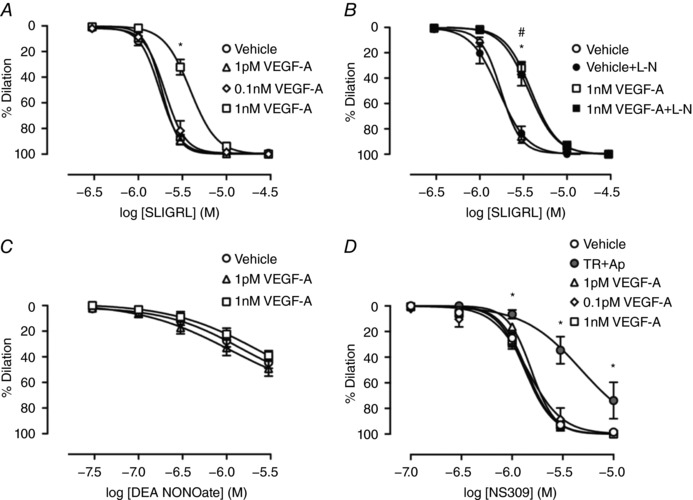
Arteries were perfused with either vehicle or VEGF‐A for 60 min. A, vasodilatation to SLIGRL (n = 7–8). B, vasodilatation to SLIGRL in the presence of l‐NAME (L‐N) (n = 5–8). C, vasodilatation to DEA NONOate (n = 7–9). D, vasodilatation to NS309 (n = 3–12). TR, TRAM‐34; Ap, apamin. * P < 0.05 compared to vehicle. # P < 0.05 compared to vehicle + L‐N.
To establish whether VEGF‐A affected the NO and/or EDH pathway, the NO synthase inhibitor l‐NAME was used. l‐NAME had no effect on SLIGRL‐mediated dilatation either when added alone or in the additional presence of VEGF‐A (Fig. 2 B), showing that inhibition of the NO pathway is not responsible for the inhibition of SLIGRL‐mediated vasodilatation by 1 nm VEGF‐A. Direct relaxation of VSMCs using the NO donor, DEA NONOate, was also unaltered by VEGF‐A (Fig. 2 C).
Because EC IKCa and SKCa channel activation underlies EDH‐evoked vasodilatation, their role was explored using NS309, a positive modulator of both channels. We tested the selectivity of NS309 to mediate vasodilatation via IKCa and SKCa channels using TRAM‐34 and apamin (Fig. 2 D). At 1 μm, NS309‐mediated vasodilatation was solely dependent on IKCa and SKCa activation. Approximately 70% of the vasodilatation to 3 μm NS309 was inhibited by IKCa and SKCa blockade, whereas a significant level of vasodilatation appeared to be independent of KCa channel activity at 10 μm (Fig. 2 D). Vasodilatation to NS309 was not altered by VEGF‐A at 1 and 3 μm NS309 (Fig. 2 D), suggesting the target may be upstream of EC KCa channels.
The effect of inhibition of IKCa and/or SKCa channels was then assessed against EDH using 1 μm TRAM‐34 and 100 nm apamin, respectively. Blocking IKCa with 1 μm TRAM‐34 reduced the EDH component of SLIGRL‐mediated vasodilatation to a level similar to VEGF‐A perfusion alone (Fig. 3 A). TRAM‐34 also attenuated SLIGRL‐mediated vasodilatation following VEGF‐A perfusion, although the effect was noticeably smaller compared to the vehicle perfused artery. The inhibitory effect against SLIGRL‐mediated vasodilatation was greater with the combined treatment of VEGF‐A perfusion and TRAM‐34 than with VEGF‐A alone. By contrast, SKCa inhibition with apamin did not affect SLIGRL‐mediated dilatation in the vehicle control group but markedly inhibited vasodilatation in VEGF‐A treated arteries, and more effectively than with VEGF‐A and TRAM‐34 (Fig. 3 B). Thus, when SKCa channels were blocked and EDH‐vasodilatation to SLIGRL depends on IKCa activation, VEGF‐A had a pronounced inhibitory effect. When both IKCa and SKCa were inhibited, vasodilatation to SLIGRL was significantly reduced compared to inhibition of either IKCa or SKCa channels alone, consistent with vasodilatation occurring via EDH. Under these conditions, the ability of VEGF‐A to inhibit SLIGRL‐mediated vasodilatation was lost (Fig. 3 C and D).
Figure 3. VEGF‐A inhibits IKCa and SKCa‐dependent EDH vasodilatation.
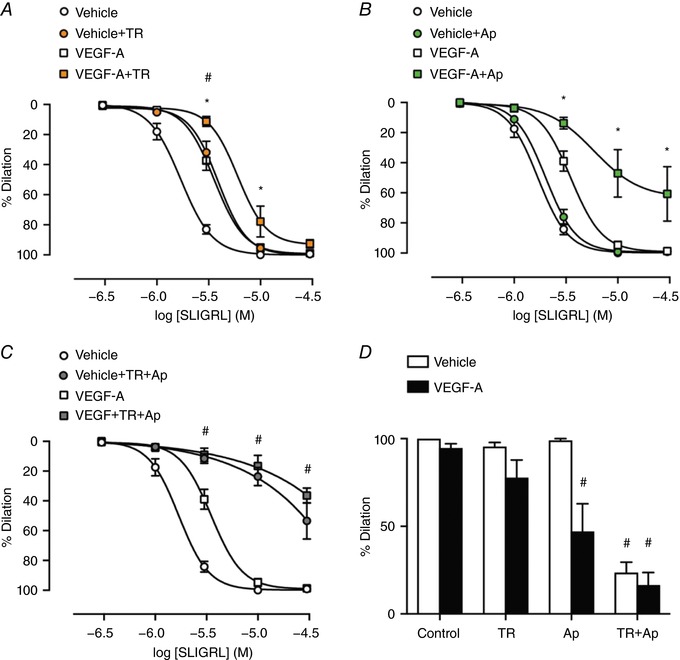
Arteries were perfused with either vehicle or 1 nm VEGF‐Afor 60 min. All experiments were performed in the presence of l‐NAME. Vehicle and VEGF‐A are from Fig. 2. A, vasodilatation to SLIGRL with the addition of TRAM‐34 (TR) (n = 5–13). B, vasodilatation with the addition of apamin (Ap) (n = 3–13). C, SLIGRL‐mediated vasodilatation in the presence of TRAM‐34 and apamin (n = 3–13). D, vasodilatation to 10 μm SLIGRL in the presence of IKCa and SKCa inhibitors. * P < 0.05 compared to VEGF‐A. # P < 0.05 compared to vehicle + TR.
EC Ca2+ signalling was inhibited by VEGF‐A exposure
Consistent with our previous studies, SLIGRL increased whole cell EC Ca2+ in pressurized murine mesenteric arteries associated with propagating waves (Fig. 4 A). In vehicle‐perfused control arteries, EC Ca2+ events developed locally then propagated along the cell (Ca2+ waves) (Fig. 4 A). By contrast, luminal perfusion with 1 nm VEGF‐A prevented local Ca2+ from evolving into propagating events (Figs 4 and 5 A and D). By examining each Ca2+ event separately, we observed the wave events propagating at least 20 μm from where the Ca2+ originated in the vehicle perfused arteries, although this propagation was absent in the VEGF‐A treated arteries in simulated with 3 μm SLIGRL (Fig. 5 A).
Figure 4. VEGF‐A pre‐exposure inhibits propagation of EC Ca2+ .
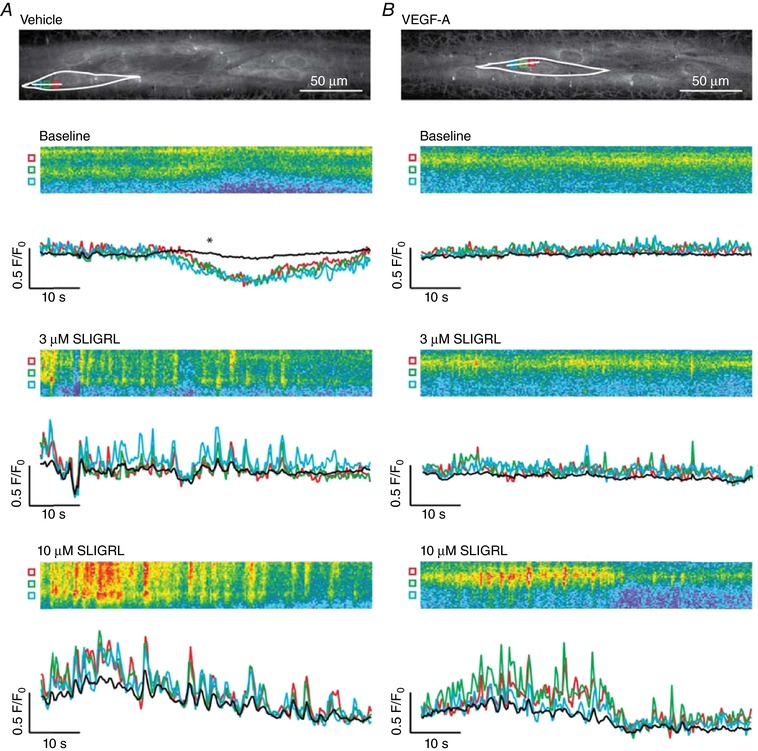
Pressurized arteries were luminally perfused with vehicle or 1 nm VEGF‐A for 60 min. Baseline Ca2+ was recorded for 1 min, SLIGRL was then added and remained in the bath for the duration of the recording. A, following perfusion of vehicle, 3 and 10 μm SLIGRL stimulated a clear increase in Ca2+ waves along the length of the cell. B, following perfusion of VEGF‐A, 3 μm SLIGRL stimulated less frequent, localized Ca2+ events, whereas 10 μm SLIGRL increased Ca2+ event frequency and Ca2+ waves propagated along the cell. Data are shown as line scans (corresponding to white lines in A and B) with fluorescence traces referring to subcellular regions of interest (coloured boxes in A and B). Black trace refers to whole‐cell recording. *Dip in intensity as a result of a movement artefact. Representative of five or six independent experiments.
Figure 5. VEGF‐A inhibits SLIGRL‐mediated Ca2+ events and wave propagation.
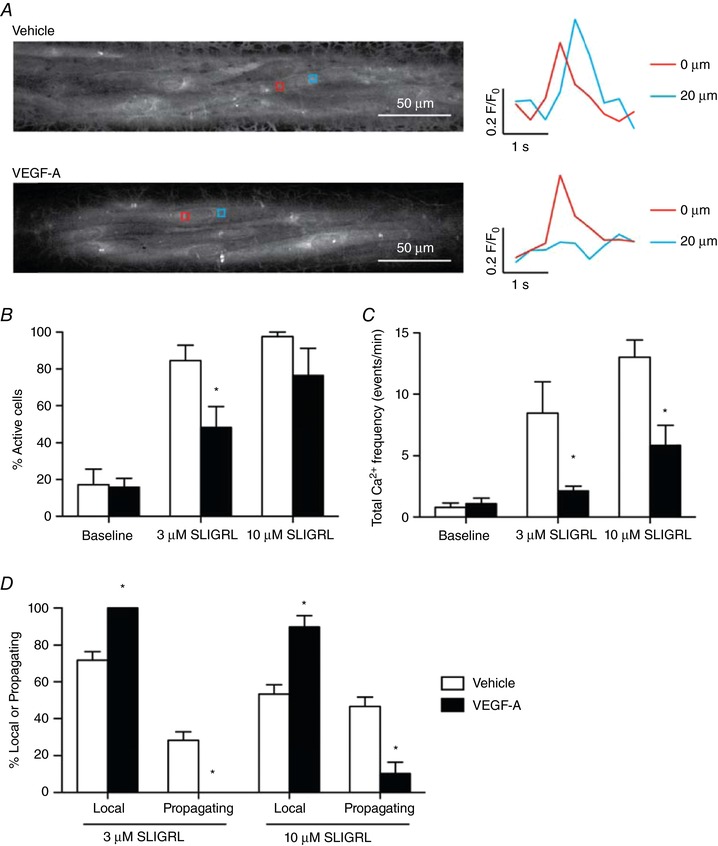
A, arteries were stimulated with 3 μm SLIGRL. Regions of interests were positioned 20 μm apart in active ECs: upper: vehicle perfused with trace to right showing a propagating wave; lower: 1 nm VEGF‐A perfused with a local event shown to the right. Representative of multiple cells within five or six independent experiments. B–D, summary of Ca2+ events. Following luminal perfusion with 1 nm VEGF‐A for 60 min, fewer ECs respond to 3 μm SLIGRL (B) and, of the active cells, there were fewer Ca2+ events (C). D, VEGF‐A treated arteries produced fewer propagating Ca2+ events compared to the vehicle‐treated arteries. n = 5–6; * P < 0.05 compared to vehicle.
Overall, VEGF‐A had a marked effect on the profile of Ca2+ events evoked by SLIGRL, reducing the number of active cells, the frequency of Ca2+ events and Ca2+ waves (Fig. 5).
VEGFR2‐MEK1/2 signalling is necessary for inhibition of both SLIGRL‐mediated vasodilatation and EC Ca2+ activity
To identify the mechanism responsible for attenuating EDH‐mediated vasodilatation, either 10 nm ZM323881, a selective VEGFR2 tyrosine kinase inhibitor (Whittles et al. 2002), or 10 μm UO126 (Boscolo, Mulliken and Bischoff, 2011), a selective blocker of MEK1/2, were used. The concentrations of ZM323881 and UO126 used have been demonstrated to selectively inhibit VEGFR2 and MEK1/2, respectively (Whittles et al. 2002; Tran and Neary, 2006; Boscolo, Mulliken and Bischoff, 2011). In each case, the inhibitory effects of VEGF‐A on EC Ca2+ handling and vasodilatation were prevented (Fig. 6), consistent with a central role for ERK1/2 downstream of VEGFR2 in the regulation of EC Ca2+ and the subsequent vasodilatation in response to SLIGRL.
Figure 6. VEGF‐A inhibits SLIGRL‐mediated EC Ca2+ events and vasodilatation via ERK1/2 signalling.
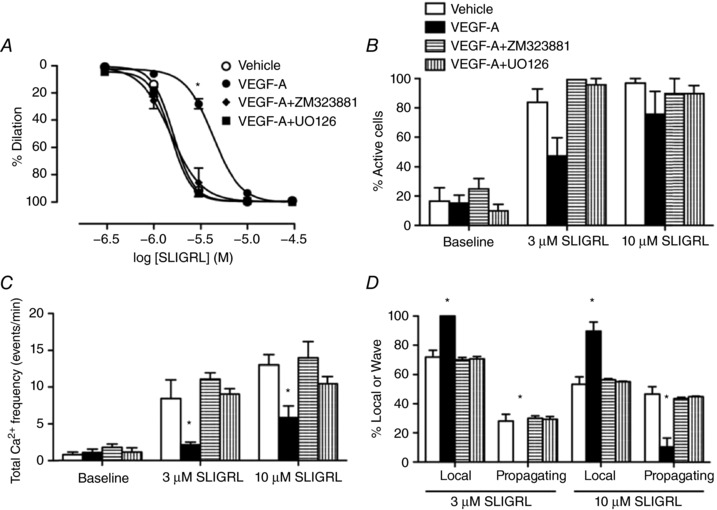
Vehicle and VEGF‐A are from Fig. 5. Arteries were perfused with either vehicle or 1 nm VEGF‐A with and without inhibitors for 60 min. A, ZM323881 and UO126 prevented VEGF‐A induced inhibition of SLIGRL‐mediated vasodilatation (n = 3–12). B–D, quantification of EC Ca2+ activities in arteries treated with ZM323881 or UO126 (n = 3–6). * P < 0.05 compared to vehicle.
IP3R1 distribution in ECs
The expression of Itpr1‐3 was found in freshly isolated intact arteries and EC tubes (Fig. 7 A). IP3R1 within ECs appeared as punctate strings forming a net‐like arrangement across the cells and along the cell borders. Clusters of IP3R1 were sometimes observed. There was no obvious change in IP3R1 distribution following 60 min perfusion with vehicle or 1 nm VEGF‐A (Fig. 7 B). ZO‐1 was used to distinguish the EC cell border and junctional integrity, and there was no change in ZO‐1 distribution following VEGF‐A perfusion.
Figure 7. Distribution of IP3R1 in ECs is not modified by VEGF‐A.
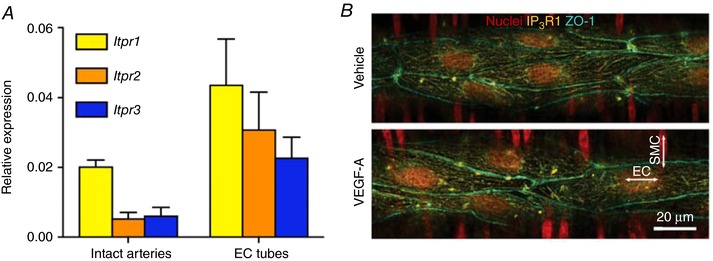
A, Itpr1‐3 expression in intact arteries and EC tubes (n = 4). B, pressurized arteries were perfused with either vehicle or 1 nm VEGF‐A for 60 min. Immunolabelling showed the IP3R1 distribution in ECs (horizontal nuclei). The distribution of ZO‐1 appears to be unaltered by VEGF‐A in arteries. Representative of three arteries per treatment.
Discussion
The present study provides evidence that prolonged stimulation with VEGF‐A, through the activation of VEGFR2 and the MEK1/2 pathway, inhibits EC Ca2+ mobilization and agonist‐evoked vasodilatation in mouse mesenteric resistance arteries. The altered Ca2+ handling prevented the effective activation of EC KCa channels, in particular IKCa, reducing the subsequent EDH‐mediated vasodilatation.
The majority of studies on the arterial action of VEGF‐A have focussed primarily on its acute role as a vasodilator. Longer‐term effects of VEGF‐A have not been reported, despite the fact that VEGF‐A is elevated chronically in numerous pathological conditions, including peripheral artery disease, diabetes associated complications and pre‐eclampsia (Baker et al. 1995; Stehr et al. 2010). Furthermore, high serum VEGF‐A has been linked to an increased likelihood of detrimental cardiovascular events (Kaess et al. 2016). Most studies rely on an antibody based enzyme linked immunosorbent assay to detect VEGF‐A in the blood. However, both the type of sample (serum/plasma) and the circulating levels of soluble VEGFR1 and VEGFR2 affect VEGF‐A binding to antibodies and, as such, reduce our ability to accurately determine the concentration of circulating VEGF‐A. Thus, the reported normal level for VEGF‐A in the blood ranges from undetectable to ∼100 pm, with values that can increase into the nanomolar range in cancer and pre‐eclampsia patients (Baker et al. 1995; Takahashi et al. 1995; Larsson, Sköldenberg and Eericson, 2002). In the present study, we used 1 nm VEGF‐A, which is known to effectively activate VEGFR2 and cause vasodilatation in a number of different vascular beds (Itoh et al. 2002; Egholm et al. 2016).
Using a previously established method to isolate fresh ECs rapidly from mesenteric arteries (Socha et al. 2011), we detected both VEGFR and NRP‐1 expression, confirming the presence of VEGF receptors in the ECs of resistance arteries. However, despite the presence of a functional endothelium and VEGFRs, we were unable to evoke vasodilatation with VEGF‐A in mouse mesenteric arteries. The VEGF‐A level rises gradually with perfusion, with the concentration peaking ∼5 min following the arrival of VEGF‐A in the lumen. Vasodilatation was not observed at any point, suggesting that neither low, nor high concentrations of VEGF‐A led to vasodilatation in mouse mesenteric arteries. In other vascular beds, VEGF‐A evoked vasodilatation is slow, often requiring 10–15 min before an effect is observed, which appears to be NO‐dependent. Because we found that exogenous NO only evoked moderate vasodilatation of mouse mesenteric arteries, the lack of vasodilatation to VEGF‐A may simply reflect a lack of responsivness in this artery. Although it has been reported that VEGF‐A can cause vasoconstriction once NOS is blocked (Egholm et al. 2016), we did not observe this phenomenon.
Despite a low arterial sensitivity to NO, SLIGRL evokes pronounced EC‐dependent vasodilatation via EDH (McGuire et al. 2002). This vasodilatation was inhibited after 60 min of exposure to VEGF‐A, showing the growth factor can negatively and significantly attenuate EDH‐vasodilatation. The inhibitory action of VEGF‐A did not increase further during repeated dose response curves ∼1 h later and in the presence of l‐NAME. This ability of VEGF‐A to reduce vasodilatation does not appear to be restricted to SLIGRL, and other studies have observed reduced vasodilatation to bradykinin using human dermal resistance arteries (Svedas et al. 2003). Supporting an inhibitory action against vasodilatation as a general mechanism and that high‐dose VEGF‐A infusion can cause a sustained increase in systemic vascular resistance (Yang et al. 1998).
SLIGRL‐evoked vasodilatation in mesenteric arteries from wild‐type mice relies on EDH, which is mediated by K+ efflux through EC IKCa and SKCa channels (Dora et al. 2003; Beleznai et al. 2011). However, VEGF‐A did not alter vasodilatation to the KCa channel opener NS309, a positive modulator of the channels, which mimics EDH‐vasodilatation. Because NS309 activates both SKCa and IKCa channels, this raised the possibility VEGF‐A modifies the input of only one type of channel. EC SKCa inhibition with apamin had a profound effect on SLIGRL‐mediated vasodilatation, although only following exposure to VEGF‐A, suggesting the growth factor targets IKCa to reduce vasodilatation. Interestingly, TRAM‐34 right‐shifted the SLIGRL (control) vasodilatation curve, indicating a predominant role for these channels in EDH‐vasodilatation, and contrasting with the absence of a similar shift when SKCa channels were blocked with apamin. Following VEGF‐A perfusion, there remained a small EDH component sensitive to TRAM‐34 inhibition. This suggests that VEGF‐A did not completely abolish the IKCa response. EDH‐vasodilatation was abolished by combined blockade of IKCa and SKCa and, not surprisingly, there was no inhibitory effect with VEGF‐A, highlighting EDH as the target for VEGF‐A mediated attenuation.
Boeldt et al. (2017) previously reported that VEGF‐A can affect connexion 43, which can inhibit the spread of hyperpolarizing current to mediate vasodilatation. However, the fact that NS309‐mediated vasodilatation was not modified at all by VEGF‐A suggests that current propagation and the EC KCa channels may not be the primary target for VEGF‐A attenuation. This raised the possibility that the inhibitory target was downstream of PAR2 receptor activation but upstream of KCa channels. SLIGRL acts through PAR2 to induce Ca2+ release via IP3Rs, with these Ca2+ events evolving into propagating waves spreading across the ECs. However, following exposure to VEGF‐A, the number of cells responding to SLIGRL was reduced and the underlying Ca2+ events became localized to smaller, more restricted sites. The restriction of Ca2+ release after exposure to VEGF‐A translated to a reduced global increase of Ca2+, which would be predicted to compromise the effective opening of EC KCa channels and thus reduce EDH‐vasodilatation. Although both IKCa and the SKCa channels are probably affected by the reduction in Ca2+ release, the reduction appeared to be particularly pronounced with IKCa input to EDH because, once SKCa channels were inhibited by apamin, the ability of SLIGRL to fully dilate arteries was dramatically reduced by prolonged exposure to VEGF‐A. The greater impact of IKCa blockade may reflect that these channels are localized in the fine myoendothelial projections, where ECs make contact with the VSMCs, whereas the SKCa channels are more uniformly distributed across the membrane (Dora et al. 2008). In HUVECs, other studies have reported that 250 pm VEGF‐A reduced the ATP‐mediated increase in frequency of Ca2+ bursts and [Ca2+]i (Boeldt et al. 2017). Therefore, it appears the effects of VEGF‐A are not limited to PAR2‐receptor mediated responses.
MEK1/2 signalling (and subsequent activation of ERK1/2) is a major signalling pathway downstream of VEGFR2 activation and it appears to be essential for VEGF‐A to exert an inhibitory effect on EC Ca2+ release. This suggests that ERK1/2, a serine/threonine kinase, may have a direct role in the regulation of EC Ca2+. Other studies suggest that ERK phosphorylates S436 of IP3R1 to reduce Ca2+ release and the frequency of Ca2+oscillations (Bai et al. 2006). This ERK1/2 phosphorylation site is absent in both IP3R2 and IP3R3 (Bai et al. 2006; Yang et al. 2006), which supports the possibility that SLIGRL‐mediated Ca2+ release and propagation are largely dependent on IP3R1 in the present study. When we examined the localization and distribution of IP3R1, we did not observe any obvious changes, and so alterated Ca2+ signalling was probably not caused by a redistribution of IP3R1. However, once appropriate phospho‐specific antibodies to IP3R1 become available for immunolabelling, it will be interesting to establish whether the IP3R1 is phosphorylated during exposure to VEGF‐A in pressurized artery ECs, as well as on which residues this occurs.
Our understanding of VEGF‐A signalling in the resistance vasculature is far from complete. The majority of data available have focussed on capillaries and the ability of VEGF‐A to evoke arterial vasodilatation. As a result of its angiogenic and pro‐survival properties in ECs, VEGF‐A has been trialled as a mean to stimulate therapeutic re‐vascularization. Although early studies that suggest VEGF‐A improves myocardial outcome in patients, the benefits of this approach were far less clear in later, more stringent trials (Losordo et al. 2002; Stewart et al. 2009). Although it has been suggested that VEGF‐A may reduce NO release evoked by other EC‐dependent vasodilators in vitro (Boeldt et al. 2017), the present study provides the first functional evidence to suggest that long‐term exposure to VEGF‐A prevents effective vasodilatation and that it does this by disrupting EDH‐vasodilatation. As such, VEGF‐A has the potential to increase peripheral resistance and compromise tissue blood flow, which may counteract any potential benefit of therapeutic angiogenesis.
Additional information
Competing interests
The authors declare that they have no competing interests.
Author contributions
XY conceived and designed the work and drafted the manuscript. XY and TB acquired, analysed and interpreted the data. PB, CJG and KAD helped design and interpret experiments and edited the manuscript. All authors approved the final version of the manuscript submitted for publication and are accountable for all aspects of the work. All persons designated as authors qualify for authorship and all those who qualify are listed.
Funding
This work was supported by the British Heart Foundation (PG/14/58/30998; FS/13/16/30199) and the Oxford BHF Centre of Research Excellence (RE/13/1/30181). KAD is a BHF‐funded Senior Basic Science Research Fellow.
Biography
Xi Ye received his PhD in Ophthalmology from the University of Bristol, UK, in 2015. Subsequently, he took up a postdoctoral position with Professor Kim Dora and Professor Chris Garland at the University of Oxford, UK, where he developed his interests in vasomotor regulation and its contributions towards health and disease. He is hoping to use both molecular and physiological approaches to investigate the pathogenesis and treatments for cardiovascular disease.

Edited by: Kim Barrett and Livia Hool
Contributor Information
Christopher J. Garland, Email: christopher.garland@pharm.ox.ac.uk
Kim A. Dora, Email: kim.dora@pharm.ox.ac.uk.
References
- Bagher, P. , Davis, M. and Segal, S. (2011). Visualizing calcium responses to acetylcholine convection along endothelium of arterolar networks in Cx40BAC‐GCaMP2 transgenic mice. Am J Physiol Heart Circ Physiol 301, H794–H802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai, G.‐R. , Yang, L.‐H. , Huang, X.‐Y. and Sun, F.‐Z. (2006). Inositol 1,4,5‐trisphosphate receptor type 1 phosphorylation and regulation by extracellular signal‐regulated kinase. Biochem Biophys Res Commun 348, 1319–1327. [DOI] [PubMed] [Google Scholar]
- Baker, P. , Krasnow, J. , Roberts, J. and Yeo, K. (1995). Elevated serum levels of vascular endothelial growth factor in patients with preeclampsia. Obstet Gynecol 86, 815–21. [DOI] [PubMed] [Google Scholar]
- Beleznai, T. , Takano, H. , Hamill, C. , Yarova, P. , Douglas, G. , Channon, K. and Dora, K. (2011). Enhanced K+‐channel‐mediated endothelium‐dependent local and conducted dilation of small mesenteric arteries from ApoE –/– mice. Cardiovasc Res 92, 199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeldt, D. S. , Krupp, J. , Yi, F. X. , Khurshid, N. , Shah, D. M. and Bird, I. M. (2017). Positive versus negative effects of VEGF165 on Ca2+ signaling and NO production in human endothelial cells. Am J Physiol Heart Circ Physiol 312, H173–H181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boscolo, E. , Mulliken, J. B. and Bischoff, J. (2011). VEGFR‐1 mediates endothelial differentiation and formation of blood vessels in a murine model of infantile hemangioma. Am J Pathol 179, 2266–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, H. , James, A. , Foster, R. , Hancox, J. and Bates, D. (2006). VEGF activates receptor‐operated cation channels in human microvascular endothelial cells. Arterioscler Thromb Vasc Biol 26, 1768–76. [DOI] [PubMed] [Google Scholar]
- Dora, K. A. , Gallagher, N. T. , Mcneish, A. and Garland, C. J. (2008). Modulation of endothelial cell KCa3.1 channels during endothelium‐derived hyperpolarizing factor signaling in mesenteric resistance arteries. Circ Res 102, 1247–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dora, K. , Sandow, S. , Gallagher, N. , Takano, H. , Rummery, N. , Hill, C. and Garland, C. (2003). Myoendothelial gap junctions may provide the pathway for EDHF in mouse mesenteric artery. J Vasc Res 40, 480–90. [DOI] [PubMed] [Google Scholar]
- Edwards, G. , Dora, K. , Gardener, M. , Garland, C. and Weston, A. (1998). K+ is an endothelium‐derived hyperpolarizing factor in rat arteries. Nature 396, 269–272. [DOI] [PubMed] [Google Scholar]
- Egholm, C. , Khammy, M. M. , Dalsgaard, T. , Mazur, A. , Tritsaris, K. , Hansen, A. J. , Aalkjaer, C. and Dissing, X. S. (2016). GLP‐1 inhibits VEGFA‐mediated signaling in isolated human endothelial cells and VEGFA‐induced dilation of rat mesenteric arteries. Am J Physiol Heart Circ Physiol 1311, H1214–H1224. [DOI] [PubMed] [Google Scholar]
- Favia, A. , Desideri, M. , Gambara, G. , D'Alessio, A. , Ruas, M. , Esposito, B. , Del Bufalo, D. , Parrington, J. , Ziparo, E. , Palombi, F. , Galione, A. and Flippini, A. (2014). VEGF‐induced neoangiogenesis is mediated by NAADP. Proc Natl Acad Sci U S A 111, E4706–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong, G. , Rossant, J. , Gertsenstein, M. and Breitman, M. (1995). Role of the Flt‐1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376, 66–70. [DOI] [PubMed] [Google Scholar]
- Garland, C. J. , Bagher, P. , Powell, C. , Ye, X. , Lemmey, H. A. L. , Borysova, L. and Dora, K. A. (2017). Voltage‐dependent Ca2+ entry into smooth muscle during contraction promotes endothelium‐mediated feedback vasodilation in arterioles. Sci Signal 10, eaal3806. [DOI] [PubMed] [Google Scholar]
- Garland, C. J. and Dora, K. A. (2017). EDH: endothelium‐dependent hyperpolarization and microvascular signalling. Acta Physiol (Oxf) 219, 152–161. [DOI] [PubMed] [Google Scholar]
- Goel, H. L. and Mercurio, A. M. (2013). VEGF targets the tumour cell. Nat Rev Cancer 13, 871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy, D. (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdollah Zadeh, M. , Glass, C. A. , Magnussen, A. , Hancox, J. and Bates, D. O. (2008). VEGF‐mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation 15, 605–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, S. , Brawley, L. , Wheeler, T. , Anthony, F. W. , Poston, L. and Hanson, M. A. (2002). Vasodilation to vascular endothelial growth factor in the uterine artery of the pregnant rat is blunted by low dietary protein intake. Pediatr Res 51, 485–491. [DOI] [PubMed] [Google Scholar]
- Jang, K. , Kim, M. , Gilbert, C. A. , Simpkins, F. , Ince, T. A. and Slingerland, J. M. (2017). VEGFA activates an epigenetic pathway upregulating ovarian cancer‐initiating cells, EMBO Mol Med, 9, 304–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaess, B. M. , Preis, S. R. , Beiser, A. , Sawyer, D. B. , Chen, T. C. , Seshadri, S. and Vasan, R. S. (2016). Circulating vascular endothelial growth factor and the risk of cardiovascular events. Heart 102, 1898–1901. [DOI] [PubMed] [Google Scholar]
- Kawamura, H. , Li, X. , Harper, S. J. , Bates, D. O. and Claesson‐Welsh, L. (2008). Vascular endothelial growth factor (VEGF)‐A165b is a weak in vitro agonist for VEGF receptor‐2 due to lack of coreceptor binding and deficient regulation of kinase activity. Cancer Res 68, 4683–4692. [DOI] [PubMed] [Google Scholar]
- Ku, D. D. , Zaleski, J. K. , Liu, S. and Brock, T. A. (1993). Vascular endothelial growth factor induces EDRF‐dependent relaxation in coronary arteries. Am J Physiol Heart Circ Physiol 265, H586–H592. [DOI] [PubMed] [Google Scholar]
- Larsson, A. , Sköldenberg, E. and Eericson, H. (2002). Serum and plasma levels of FGF‐2 and VEGF in healthy blood donors. Angiogenesis 5, 107–110. [DOI] [PubMed] [Google Scholar]
- Lim, H. , Blann, A. , Chong, A. , Freestone, B. and Lip, G. (2004). Plasma vascular endothelial growth factor, angiopoietin‐1, and angiopoietin‐2 in diabetes: implications for cardiovascular risk and effects of multifactorial intervention. Diabetes Care 27, 2918–2924. [DOI] [PubMed] [Google Scholar]
- Losordo, D. W. , Vale, P. R. , Hendel, R. C. , Milliken, C. E. , Fortuin, F. D. , Cummings, N. , Schatz, R. A. , Asahara, T. , Isner, J. M. and Kuntz, R. E. (2002). Clinical investigation and reports trial of myocardial vascular endothelial growth factor 2 gene transfer by catheter delivery in patients with chronic myocardial ischemia. Circulation 105, 2012–2018. [DOI] [PubMed] [Google Scholar]
- McGuire, J. J. , Hollenberg, M. D. , Andrade‐Gordon, P. and Triggle, C. R. (2002). Multiple mechanisms of vascular smooth muscle relaxation by the activation of proteinase‐activated receptor 2 in mouse mesenteric arterioles. Br J Pharmacol 135, 155–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons, M. , Gordon, E. and Claesson‐welsh, L. (2016). Mechanisms and regulation of endothelial VEGF receptor signalling. Nat Rev Mol Cell Biol 17, 611–625. [DOI] [PubMed] [Google Scholar]
- Socha, M. , Hakim, C. , Jackson, W. and Segal, S. (2011). Temperatur effects on morphological integrity and Ca2+ signaling in freshly isolated murine feed artery endothelial cell tubes. Am J Physiol Heart Circ Physiol 301, H773–H783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Socha, M. J. and Segal, S. S. (2013). Isolation of microvascular endothelial tubes from mouse resistance arteries. J Vis Exp 25, e50759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soker, S. , Takashima, S. , Miao, H. Q. , Neufeld, G. and Klagsbrun, M. (1998). Neuropilin‐1 is expressed by endothelial and tumor cells as an isoform‐specific receptor for vascular endothelial growth factor. Cell 92, 735–745. [DOI] [PubMed] [Google Scholar]
- Stehr, A. , Töpel, I. , Müller, S. , Unverdorben, K. , Geissler, E. K. , Kasprzak, P. M. , Schlitt, H. J. and Steinbauer, M. (2010). VEGF: a surrogate marker for peripheral vascular disease. Eur J Endovasc Surg 39, 330–332. [DOI] [PubMed] [Google Scholar]
- Stewart, D. J. , Kutryk, M. J. B. , Fitchett, D. , Freeman, M. , Camack, N. , Su, Y. , Della Siega, A. , Bilodeau, L. , Burton, J. R. and Proulx, G. (2009). VEGF gene therapy fails to improve perfusion of ischemic myocardium in patients with advanced coronary disease: results of the NORTHERN trial. Mol Ther 17, 1109–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svedas, E. , Islam, K. B. , Nisell, H. and Kublickiene, K. R. (2003). Vascular endothelial growth factor induced functional and morphologic signs of endothelial dysfunction in isolated arteries from normal pregnant women. Am J Obstet Gynecol 188, 168–176. [DOI] [PubMed] [Google Scholar]
- Takahashi, Y. , Kitadai, Y. , Bucana, C. D. , Cleary, K. R. and Ellis, L. M. (1995). Expression of vascular endothelial growth factor and its receptor, KDR, correlates with vascularity, metastasis, and proliferation of human colon cancer. Cancer Res 55, 3964–3968. [PubMed] [Google Scholar]
- Tran, M. D. and Neary, J. T. (2006). Purinergic signaling induces thrombospondin‐1 expression in astrocytes. Proc Natl Acad Sci U S A 103, 9321–9326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittles, C. E. , Pocock, T. M. , Wedge, S. R. , Kendrew, J. , Hennequin, L. F. , Harper, S. J. and Bates, D. O. (2002). ZM323881, a novel inhibitor of vascular endothelial growth factor‐receptor‐2 tyrosine kinase activity. Microcirculation 9, 513–522. [DOI] [PubMed] [Google Scholar]
- Yang, L. , Bai, G. , Huang, X. and Sun, F. (2006). ERK binds, phosphorylates InsP3 type 1 receptor and regulates intracellular calcium dynamics in DT40 cells. Biochem Biophys Res Commun 349, 1339–1344. [DOI] [PubMed] [Google Scholar]
- Yang, R. , Bunting, S. , Ko, A. , Leyt, B. , Modi, N. , Zioncheck, T. , Ferrara, N. and Jin, H. (1998). Substantially attenuated hemodynamic responses to Escherichia coli‐ derived vascular endothelial growth factor given by intravenous infusion compared with bolus injection. J Pharmacol Exp Ther 284, 103–110. [PubMed] [Google Scholar]