

Abstract
Key points
Membrane rafts (MRs)–redox signalling pathway is activated in response to transforming growth factor‐β1 (TGF‐β1) stimulation in renal tubular cells.
This pathway contributes to TGF‐1β‐induced epithelial–mesenchymal transition (EMT) in renal tubular cells.
The the MRs–redox signalling pathway is activated in renal tubular cells isolated from angiotensin II (AngII)‐induced hypertensive rats.
Inhibition of this pathway attenuated renal inflammation and fibrosis in AngII‐induced hypertension.
Abstract
The membrane rafts (MRs)–redox pathway is characterized by NADPH oxidase subunit clustering and activation through lysosome fusion, V‐type proton ATPase subunit E2 (encoded by the Atp6v1e2 gene) translocation and sphingomyelin phosphodiesterase 1 (SMPD1, encoded by the SMPD1 gene) activation. In the present study, we hypothesized that the MRs–redox‐derived reactive oxygen species (ROS) are involved in renal inflammation and fibrosis by promoting renal tubular epithelial–mesenchymal transition (EMT). Results show that transforming growth factor‐β1 (TGF‐β1) acutely induced MR formation and ROS production in NRK‐52E cells, a rat renal tubular cell line. In addition, transfection of Atp6v1e2 small hairpin RNAs (shRNA) and SMPD1 shRNA attenuated TGF‐β1‐induced changes in EMT markers, including E‐cadherin, α‐smooth muscle actin (α‐SMA) and fibroblast‐specific protein‐1 (FSP‐1) in NRK‐52E cells. Moreover, Erk1/2 activation may be a downstream regulator of the MRs–redox‐derived ROS, because both shRNAs significantly inhibited TGF‐β1‐induced Erk1/2 phosphorylation. Further in vivo study shows that the renal tubular the MRs–redox signalling pathway was activated in angiotensin II (AngII)‐induced hypertension, as indicated by the increased NADPH oxidase subunit Nox4 fraction in the MR domain, SMPD1 activation and increased ROS content in isolated renal tubular cells. Finally, renal transfection of Atp6v1e2 shRNA and SMPD1 shRNA significantly prevented renal fibrosis and inflammation, as indicated by the decrease of α‐SMA, fibronectin, collagen I, monocyte chemoattractant protein‐1 (MCP‐1), intercellular cell adhesion molecule‐1 (ICAM‐1) and tumour necrosis factor‐α (TNF‐α) in kidneys from AngII‐infused rats. It was concluded that the the MRs–redox signalling pathway is involved in TGF‐β1‐induced renal tubular EMT and renal inflammation/fibrosis in AngII‐induced hypertension.
Keywords: membrane rafts, epithelial‐mesenchymal transition, renal fibrosis, angiotensin II
Key points
Membrane rafts (MRs)–redox signalling pathway is activated in response to transforming growth factor‐β1 (TGF‐β1) stimulation in renal tubular cells.
This pathway contributes to TGF‐1β‐induced epithelial–mesenchymal transition (EMT) in renal tubular cells.
The the MRs–redox signalling pathway is activated in renal tubular cells isolated from angiotensin II (AngII)‐induced hypertensive rats.
Inhibition of this pathway attenuated renal inflammation and fibrosis in AngII‐induced hypertension.
Introduction
Tubulointerstitial fibrosis is the common final pathway of diverse forms of chronic renal disease that contributes to both organ insufficiency and failure (Mao et al. 2008). Transforming growth factor‐β1 (TGF‐β1) plays a pivotal role in the progression of renal fibrosis, by promoting renal tubular epithelial–mesenchymal transition (EMT), resulting in increased synthesis and accumulation of extracellular matrix (ECM) proteins, and the inhibition of their degradation (Qiao et al. 2015). During EMT, tubular epithelial cells are transformed into myofibroblasts through a stepwise process, including loss of cell–cell adhesion and E‐cadherin expression, de novo α‐smooth muscle actin (α‐SMA) expression, actin reorganization, tubular basement membrane disruption, cell migration, and fibroblast invasion with production of profibrotic molecules such as collagen types I and III and fibronectin (Bedi et al. 2008).
It has been well documented that membrane rafts (MRs) are enriched in cholesterol with saturated acyl chains such as sphingolipids and glycosphingolipids, and that raft formation is driven by lipids and protein interactions (Lang, 2007; Vetrivel & Thinakaran, 2010; Xu et al. 2012). Lysosome was trafficked to the plasma membrane and fused with plasma membrane in response to Fas ligand (FasL), tumour necrosis factor‐related apoptosis‐inducing ligand (TRAIL) and angiotensin II (AngII) stimulation in coronary arterial endothelial cells and in mesenteric endothelial cells. During lysosome fusion, sphingomyelin phosphodiesterase 1 (SMPD1, encoded by the SMPD1 gene), also known as lysosomal acid sphingomyelinase (ASMase), is translocated to the plasma membrane and activated there (Han et al. 2011, 2012, 2016; Li et al. 2013). The product of SMPD1, ceramide, then triggers clustering of MRs and the subunits of NADPH oxidase, leading to activation of this enzyme (Han et al. 2011, 2012; Li et al. 2013). Notably, it was found that translocated lysosomal V‐type proton ATPase subunit E2 (encoded by the Atp6v1e2 gene) critically contributes to the formation of a local acid microenvironment to facilitate activation of SMPD1 and consequent MR aggregation (Xu et al. 2012). This signalling pathway represents the initial pathological change of endothelial cells and may finally lead to coronary atherosclerosis and hypertension (Zhang et al. 2006; Han et al. 2012). Callera et al. (2011) reported that c‐Src activation via trafficking to the MR domain and phosphorylation mediates the pro‐inflammatory signalling in response to aldosterone stimulation in vascular smooth muscle cells. However, it remains largely unknown whether MRs are activated and play a role in vivo.
It has been reported that NADPH oxidase plays a critical role in EMT in response to various stimulations including TGF‐β1, AngII, glucose and advanced oxidation protein products in various cell types including renal tubular cells, peritoneal mesothelial cells, intestinal epithelial cells and melanoma cells (Rhyu et al. 2005; Chang et al. 2011; Liu et al. 2012; He et al. 2015; Das et al. 2016; Xu et al. 2017). However, the mechanisms underlying how NADPH oxidase is activated in response to these stimulations remain largely unknown. In the present study, we hypothesized that the MRs–redox signalling pathway contributes to TGF‐β1‐induced EMT in renal tubular cells, and inflammation/fibrosis in AngII‐induced hypertension. Predesigned Atp6v1e2 small hairpin RNAs (shRNA) and SMPD1 shRNA were delivered to renal tubular cells to evaluate the involvement of the MRs–redox signalling pathway in TGF‐β1‐induced EMT. Next, both shRNAs were transfected into kidney to evaluate whether the MRs–redox signalling pathway is involved in inflammation and fibrosis in AngII‐induced hypertension. Our results show that the MRs–redox signalling pathway plays a critical role in TGF‐β1‐induced renal tubular EMT in vitro, and is involved in renal inflammation/fibrosis in AngII‐induced hypertension in vivo.
Methods
Ethical approval
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health (NIH Publication No. 85‐23, revised 1996), was approved by the Ethics Review Board of Ruijin Hospital, and conforms to the principles and regulations described by Grundy (2015).
Cell culture and animal treatment
NRK‐52E cells, a rat renal tubular cell line, were purchased from the cell bank of Shanghai Institutes of Biological Sciences, and maintained in DMEM/Ham's F12 (DMEM/F12) medium, supplemented with 10% fetal calf serum (FCS), glutamine (2 mM), penicillin (100 IU/ml) and streptomycin (100 μg/ml). Cells were cultured at 37°C in a humidified atmosphere of 5% CO2 in air (Han et al. 2010, 2013). For EMT experiments, cells were treated with 10 ng/ml TGF‐β1 for 48 h.
Male Sprague‐Dawley rats (∼ 280 g) were purchased from the Shanghai Experimental Animal Center, China. Four groups of animals were included: vehicle infusion + control plasmids (control), AngII infusion + control plasmids (AngII), AngII infusion + Atp6v1e2 shRNA plasmids (AngII + Atp6v1e2 shRNA), and AngII infusion + SMPD1 shRNA plasmids (AngII + SMPD1 shRNA). Rats were anaesthetized with isoflurane (2%) throughout the surgery. AngII (Sigma‐Aldrich, St Louis, MO, USA; 200 ng/kg/min) was infused for 2 weeks using ALZET mini‐osmotic pumps (model 2002) implanted intraperitoneally during the surgery above. Systolic blood pressure was measured weekly by a tail‐cuff method.
Transfection of DNA into cells and kidney
Predesigned rat Atp6v1e2 shRNA (SKU, TG707501; shRNA sequence, ATAGACGCCAAGGCAGAGGAAGAGTTCAA) and rat SMPD1 shRNA (SKU, TF700757; shRNA sequence, TCTGGCAACAGTCTCGACAAGATCAGCTA) were from Origene (Rockville, MD, USA). Plasmid transfection into NRK‐52E cells was performed using TurboFection 8.0 (Origene) as per the manufacturer's instructions. Transfection of shRNA into rat kidneys was performed as we have described previously (Yi et al. 2009; Zhu et al. 2011, 2012). In brief, rats were anesthetized with 2% isoflurane, the abdomen was opened and the kidney was exposed, and the renal artery and vein were temporarily blocked. Then, 50 μg of plasmids mixed with 8 μl of in vivo jetPEI (Polyplus Transfection, New York, NY, USA) in 10% glucose (600 μl) was injected into the kidneys via the left renal artery. After injection, an ultrasound transducer (Sonitron 2000, Rich‐Mar, Chattanooga, TN, USA) was applied directly onto the kidney with an output of 1 MHz at 10% power for a total of 60 s with 30 s intervals. Finally, the renal artery and vein were unblocked to recover renal blood flow: the total blockage time was less than 5 min. This technique has been shown to effectively deliver DNA into the renal cells without toxicity to the kidney (Newman & Bettinger, 2007).
Confocal analysis of MR clusters
MR staining was performed as previously described (Han et al. 2012). In brief, monosialotetrahexosylganglioside (GM1) enriched in MRs was stained by Alexa 488‐labelled cholera toxin B (Alexa 488‐CTXB 1 μg/ml, 2 h, Molecular Probes, Eugene, OR, USA). Staining was visualized through sequentially scanning on a confocal laser‐scanning apparatus (Zeiss LSM510), equipped with a 60× objective. The vast majority of resting cells displayed a homogeneous distribution of fluorescence throughout the membrane, and these cells were marked as negative; clustering was defined as one or several intense spots or patches in each cell; results are given as the percentage of cells showing one or more clusters after the indicated treatment.
Measurement of reactive oxygen species by dihydroethidium imaging
Dihydroethidium (DHE) was used to evaluate the amount of oxidant formation as described previously (Leung et al. 2016). Cells were incubated in Krebs solution containing 5 μmol/l DHE (Life Technologies, Inc., Rockville, MD) for 10 min at 37°C. Samples were examined with a confocal laser‐scanning apparatus (Zeiss LSM510) at an excitation/emission of 488/605 nm.
Immunofluorescence microscopy
Immunofluorescence staining was performed using cultured renal tubular cells on cover slips as described previously (Han et al. 2013). After fixation, the cells were incubated with goat anti‐E‐cadherin (Abcam, Cambridge, MA, USA; ab76055), mouse anti‐α‐smooth muscle actin (α‐SMA) (R&D Systems, Minneapolis, MN, USA; MAB1420) or goat anti‐fibroblast‐specific protein‐1 (FSP‐1; 1:50 dilution) (Santa Cruz Biotechnology, Santa Cruz, CA, USA) antibodies, respectively, at 4°C overnight. After washing, the slides were incubated with corresponding Alex‐555‐labelled secondary antibodies and then mounted and subjected to examinations using on a confocal laser‐scanning apparatus (Zeiss LSM510). These experiments were performed to observe the changes of EMT markers in renal tubular cells. Integrated optical intensity (IOD) was calculated by using image‐pro plus v6.0 software (Media Cybernetics, Silver Springs, MD, USA).
Measurement of renal blood flow and harvest of kidney
A factory‐calibrated flow probe (1RB series, Transonic Systems, Ithaca, NY, USA) was placed around the left renal artery and covered in conducting jelly, and renal blood flow was measured as described previously (Hamza & Kaufman, 2004). Rats were anaesthetized with pentobarbital (50 mg/kg i.p.) and quickly decapitated. The kidneys were cut longitudinally. Half of the kidney was fixed in 10% neutral buffered formalin and the other half was frozen in liquid nitrogen and stored at −80°C.
Histology and immunohistochemistry
Kidneys were fixed in 4% paraformaldehyde, embedded in paraffin and cut into 4 μm paraffin sections. Changes in renal morphology and fibrosis were evaluated using Masson trichrome staining. Immunohistochemistry was performed as described previously (Zhou et al. 2014). Slides were deparaffinized in xylene and hydrated in a graded ethanol series. All sections were then incubated with anti‐fibronectin antibody (Abcam, ab45688), anti‐α‐SMA antibody (R&D Systems, MAB1420) and horseradish peroxidase (HRP)‐labelled goat anti‐rabbit/mouse polyclonal antibody. The slices were then developed with 3,3‐diaminobenzidine (DAB) for colour reaction, and counterstained with haematoxylin.
Isolation and characterization of proximal tubular cells
Renal tubular epithelial cells were isolated as previously described (Brendler‐Schwaab & Herbold, 1997). In brief, rats were anaesthetized with isoflurane and kidneys were harvested after cardial perfusion with Hanks 199 medium (22340‐020, Gibco, Life Technologies) with 0.5% Collagenase Type I (Worthington Inc., Lakewood, NJ, USA). Kidneys were digested at 37°C in a bubble‐agitated collagenase I solution for 40 min, the resulting crude suspension was filtrated over gauze mesh of size 250 μm and subsequently centrifuged at approximately 500 g for 30 s. The resulting pellet was resuspended in 30 ml, and the suspension was carefully underlaid with 5 ml of 100% Percoll using a syringe. Subsequent centrifugation was performed at 17,200 g and 4°C for 15 min. The layer directly above the 100% Percoll cushion was collected for flotation assay, measurement of SMPD1 activity and reactive oxygen species (ROS) measurement with lucigenin. Finally, we evaluated the expression of the proximal tubular brush border enzyme, alkaline phosphatase, as described previously (Terryn et al. 2007). Results show that the mRNA level and protein level of both Atp6v1e2 and SMPD1 were significantly decreased when NRK‐52E cells were transfected with their respective shRNAs (data not shown). The effectiveness of both shRNAs was further confirmed in our in vivo study. Renal tubular cells were isolated in the four groups of rats at the end of treatments, and Atp6v1e2 and SMPD1 protein levels were then evaluated by western blot assay. Protein levels of Atp6v1e2 and SMPD1 were significantly lower in proximal tubular cells isolated from rat kidneys transfected with Atp6v1e2 and SMPD1 shRNAs compared with those from control shRNA (data not shown). These results suggest that both shRNAs work efficiently in vitro and in vivo.
Floatation of MR fractions
Detergent‐resistant membrane fraction flotation was used to evaluate MR clustering in kidney (Han et al. 2012). In brief, isolated renal tubular cells were layered onto a step gradient consisting of 40%, 30% and 5% OptiPrep Density Gradient medium, and the samples were then centrifuged at 32,000 rpm for 30 h at 4°C using a P40ST rotor. Fractions were collected from top to bottom. After addition of an equal volume of 30% trichloroacetic acid, the precipitated proteins were spun down by centrifugation at 13,000 rpm at 4°C for 15 min. The protein pellet was carefully washed with cold acetone and then used for immunoblot analysis.
Assay of SMPD1 activity
The activity of SMPD1 was measured as described previously (Yi et al. 2004). Briefly, isolated renal tubular cells (20 μg) were incubated with 0.02 μCi of N‐methyl‐[14C]‐sphingomyelin in 100 μl acidic reaction buffer containing 100 mmol/l sodium acetate, and 0.1% Triton X‐100, pH 5.0 at 37°C for 15 min. The reaction was terminated with 1.5 ml chloroform/methanol (2:1) and 0.2 ml double‐distilled water. After vortexing and centrifuging at 1000 g for 5 min to separate into two phases, the upper aqueous phase containing the N‐methyl‐[14C]‐sphingomyelin metabolite, 14C‐choline phosphate, was measured in a Beckman liquid scintillation counter. The rate of choline phosphate formation was calculated to represent enzyme activity.
Lucigenin measurement of NADPH oxidase activity in kidney tissue
Superoxide production in isolated renal tubular cells was measured with the use of lucigenin chemiluminescence as described previously (Cheng et al. 2016). In brief, 200 μg of tissue homogenate and 5 μmol/l lucigenin were added into each well of a 96‐well microplate and then incubated at 37°C for 10 min. Chemiluminescence was recorded by a chemiluminescence reader (BLR‐201, Aloka, Twinsburg, OH, USA).
Western blot
Culture of NRK‐52E cells, isolation of renal tubular cells, and western blotting, were performed as described previously (Li et al. 2007; Bao et al. 2010; Cheng et al. 2011). Briefly, after boiling for 5 min at 95°C in 5× loading buffer, cell or kidney protein was subjected to SDS‐PAGE, transferred onto a PVDF membrane and blocked by solution with dry milk. The membrane was probed with the following primary antibodies: SMPD1 (Abcam, ab83354), Atp6v1e2 (Novus Biological, Littleton, CO, USA, NBP2‐15519), E‐cadherin (Abcam, ab76055), fibronectin (Abcam, ab45688), α‐SMA (R&D Systems, MAB1420), FSP‐1 (Abcam, ab197896), collagen I (Abcam, ab21287), p‐Erk1/2 (Cell Signaling, Danvers, MA, USA, 9101), t‐Erk1/2 (Cell Signaling, 4696), aquaporin‐1 (Abcam, ab46182) N‐cadherin (Abcam, ab18203), aminopeptidase A (Abcam, ab36122), uromucoid (Abcam, ab217354), monocyte chemoattractant protein‐1 (MCP‐1, Abcam, ab25124), intercellular cell adhesion molecule‐1 (ICAM‐1, Abcam, ab171123), Nox4 (Abcam, ab109225) and tumour necrosis factor‐α (TNF‐α, Abcam, ab9755), overnight at 4°C followed by incubation with HRP‐labelled secondary antibody (1:5000, Santa Cruz Biotechnology); β‐actin was detected by using HRP‐labelled anti‐GADPH antibody (1:5000, Santa Cruz Biotechnology) as a loading control. The immunoreactive bands were detected by chemiluminescence methods and visualized on Kodak Omat X‐ray films. Densitometric analysis of the images obtained from X‐ray films was performed using the Image J software (NIH, Bethesda, MD, USA).
RNA extraction and quantitative real‐time PCR
Total RNA from kidney was extracted using TRIzol solution (Life Technologies, Inc., Rockville, MD, USA). cDNA was generated by using oligo (dT) primer and PrimeScript RT Reagent Kit (Takara, Shiga, Japan). Quantitative PCR (qPCR) was performed and analysed by kinetic real‐time PCR using the ViiA 7 Real‐Time PCR System (Applied Biosystems, Foster City, CA, USA) with SYBR Premix Ex Taq™ (Takara) for relative quantification of the indicated genes. The sequence of primers was as follows: Atp6v1e2, sense, 5’‐GAGGAAGGTGTACGTGTTCTATG‐3’, antisense, 5’‐AGAACCCAGAAGTCGTCAAAG‐3’; SMPD1, sense, 5’‐CAGTTCTTTGGCCACACTCA‐3’, antisense, 5’‐CGGCTCAGAGTTTCCTCATC‐3’; MCP‐1, sense, 5’‐TATGCAGGTCTCTGTCACGC‐3’, antisense, 5’‐AAGTGTTGAACCAGGATTCACA‐3’; ICAM‐1, sense, 5’‐CCTGGGTCATAATTGTTGGTG‐3’, antisense, 5’‐AGGAAGTCAGCCTTTCTTGG‐3’; TNF‐α, sense, 5’‐AAATGGGCTCCCTCTCATCAGTTC‐3’, antisense, 5’‐TCTGCTTGGTGGTTTGCTACGAC‐3’. Relative gene expressions were calculated in accordance with the ΔΔCt method. Relative mRNA levels were expressed based on the values of 2−ΔΔCt as described by Han et al. (2013).
Statistics
Data are presented as mean ± SEM. Significant differences between and within multiple groups were examined using one‐way or two‐way ANOVA for repeated measures, followed by Duncan's multiple‐range test. A Student's t‐test was used to detect significant differences between two groups. P < 0.05 was considered statistically significant.
Results
TGF‐β1 induced the MRs–redox signalling pathway in renal tubular cells
The effect of TGF‐β1 on MR clustering in renal tubular cells was first evaluated by staining with labelling of ganglioside GM1 coupled with a specific marker cholera toxin B (CTXB) subunit. Figure 1 A shows that MRs were diffusely distributed across the cell membrane under control conditions as indicated by the weak, diffused green fluorescence. Upon stimulation with TGF‐β1 at 10 ng/ml for 20 min, clustering of MRs occurred, as indicated by the formation of brighter patches of green fluorescence. Summarized data in Fig. 1 A show that only a small percentage of the resting cells exhibited these clusters (15.6%), 5 ng/ml TGF‐β1 significantly increased MR clustering, and 10 ng/ml TGF‐β1 reached the highest stimulatory effect on MR clustering in renal tubular cells.
Figure 1. TGF‐β1 induced MRs formation and ROS production in renal tubular cells.
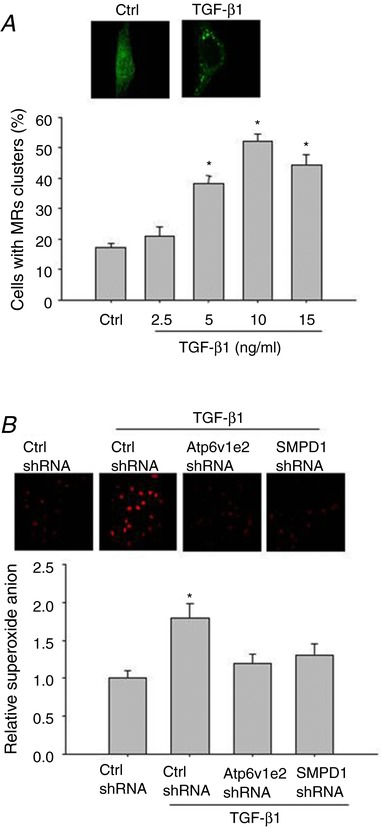
A, representative confocal fluorescence images (upper panel) and summarized data (lower panel) showing the effect of TGF‐β1 on MR formation. B, representative graphical representation (upper panel) and summarized data (lower panel) showing the effect of Atp6v1e2 shRNA and SMPD1 shRNA on TGF‐β1‐induced ROS production in DHE staining. Results are the means ± SEM of measurements from five separate preparations. * P < 0.05 vs. control group in A, * P < 0.05 vs. other groups in B. [Color figure can be viewed at http://wileyonlinelibrary.com]
We then evaluated whether inhibition of Atp6v1e2 and SMPD1 would attenuate TGF‐β1‐induced MR formation and ROS production given that previous studies show that both proteins are critical in MR formation and NADPH oxidase activation. Figure 1 B shows that the transfection of both shRNAs significantly inhibited TGF‐β1‐induced MR formation (Control, 15.6 ± 1.13%; TGF‐β1, 50.8 ± 4.18%; TGF‐β1 + Atp6v1e2 shRNA, 18.4 ± 3.4%; TGF‐β1 + SMPD1 shRNA, 20.5 ± 2.2%. P< 0.05 TGF‐β1 vs. other groups). Accordingly, TGF‐β1 significantly increased O2.− generation, and this effect was significantly inhibited by Atp6v1e2 shRNA and SMPD1 shRNA (Fig. 1 B). These results indicate that the MRs–redox signalling pathway was activated in response to TGF‐β1 stimulation in renal tubular cells.
Involvement of the MRs–redox signalling pathway in TGF‐β1‐induced EMT in renal tubular cells
It has been shown that NADPH oxidase‐derived ROS mediate TGF‐β1‐induced EMT in renal tubular cells, so we determined whether the MRs–redox signalling pathway is involved in TGF‐β1‐induced EMT. As shown in Fig. 2, TGF‐β1 significantly decreased the epithelial marker E‐cadherin, and increased mesenchymal markers including cytoskeletal protein α‐SMA and signal transduction protein FSP‐1, indicating that EMT occurred in response to TGF‐β1 stimulation. In cells pre‐treated with Atp6v1e2 shRNA and SMPD1 shRNA, TGF‐β1‐induced EMT was significantly inhibited, as indicated by the increase in epithelial marker E‐cadherin, and decrease in mesenchymal markers α‐SMA and FSP‐1 compared with the TGF‐β1‐treated group (Fig. 2). These results indicate that the MRs–redox signalling pathway contributes to TGF‐β1‐induced EMT.
Figure 2. Atp6v1e2 shRNA and SMPD1 shRNA inhibit TGF‐β1‐induced changes in E‐cad, α‐SMA and FSP‐1 as found by western blot assay and immunofluorescence microscopy assay.
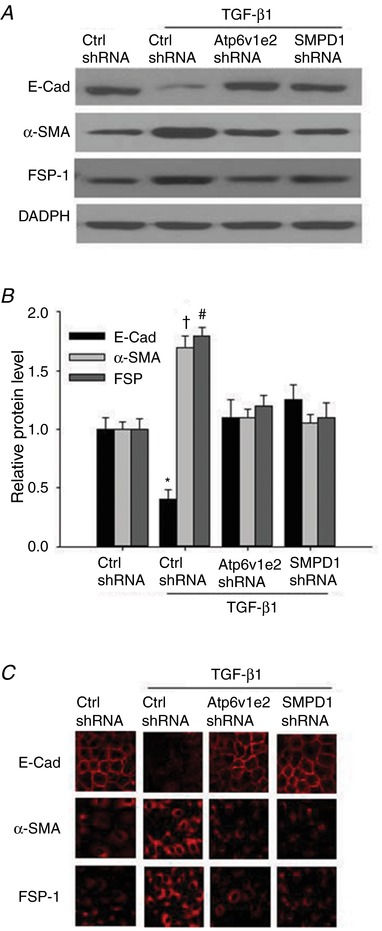
Representative western blot images (A) and summarized data (B) showing the inhibitory effect of Atp6v1e2 shRNA and SMPD1 shRNA on TGF‐β1‐induced decrease in the epithelial marker E‐cad, and increases in mesenchymal markers α‐SMA and FSP‐1. C, representative images showing the inhibitory effect of Atp6v1e2 shRNA and SMPD1 shRNA on TGF‐β1‐induced decreases in the epithelial marker E‐cadherin, and increases in mesenchymal markers α‐SMA and FSP‐1 by immunofluorescence microscopy assay. E‐Cad, E‐cadherin; α‐SMA, α‐smooth muscle actin; FSP‐1, fibroblast‐specific protein‐1. Results are the means ± SEM of measurements from five separate preparations. * P < 0.05, † P < 0.05 and # P < 0.05 vs. other groups for E‐cadherin, α‐SMA and FSP‐1, respectively. [Color figure can be viewed at http://wileyonlinelibrary.com]
Atp6v1e2 shRNA and SMPD1 shRNA attenuated TGF‐β1‐induced Erk1/2 phosphorylation in renal tubular cells
It has been shown that Erk1/2 phosphorylation is a downstream regulator of ROS and plays a critical role in TGF‐β1‐induced EMT. We thus determined whether MR‐derived ROS were involved in TGF‐β1‐induced Erk1/2 phosphorylation. As shown in Fig. 3, TGF‐β1 time‐dependently increased Erk1/2 phosphorylation. When the cells were treated with Atp6v1e2 shRNA and SMPD1 shRNA, the stimulatory effect of TGF‐β1 was almost fully lost (Fig. 3). These results suggest that the MRs–redox signalling pathway contributes to TGF‐β1‐induced EMT through Erk1/2 phosphorylation.
Figure 3. Atp6v1e2 shRNA and SMPD1 shRNA inhibited TGF‐β1‐induced Erk1/2 phosphorylation in renal tubular cells.
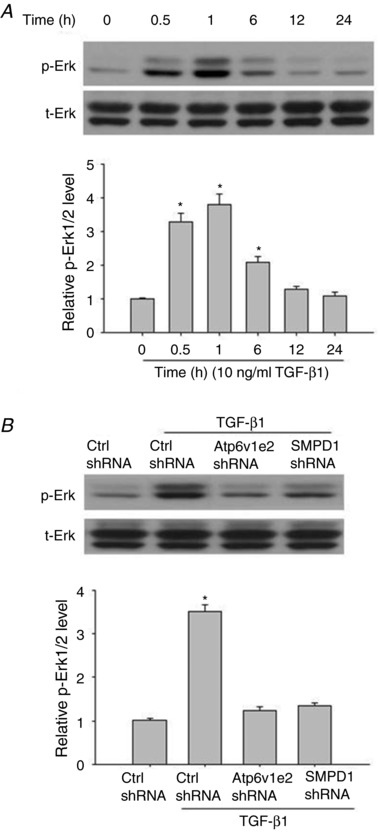
A, representative western blot images (upper panel) and summarized data (lower panel) showing the time‐dependent effect of TGF‐β1 on Erk1/2 phosphorylation. B, representative western blot images (upper panel) and summarized data (lower panel) showing the inhibitory effect of Atp6v1e2 shRNA and SMPD1 shRNA on TGF‐β1‐induced Erk1/2 phosphorylation. Results are the means ± SEM of measurements from five separate preparations. * P < 0.05 vs. control group in A; * P < 0.05 vs. other groups in B.
Inhibitory effect of simvastatin on TGF‐β1‐induced ROS production, EMT, and Erk1/2 phosphorylation in renal tubular cells
It has been reported that statins have a protective role in renal EMT and fibrosis, and that ROS are important in mediating the protective effect of statins. We then evaluated whether statins would inhibit TGF‐β1‐induced ROS production, EMT, and Erk1/2 phosphorylation in renal tubular cells. The results show that simvastatin significantly inhibited TGF‐β1‐induced ROS production (Control: 1.0 ± 0.07; TGF‐β1: 1.82 ± 0.13; TGF‐β1 + Sim: 1.15 ± 0.08). Furthermore, simvastatin significantly inhibited TGF‐β1‐induced EMT, and Erk1/2 phosphorylation in renal tubular cells (Fig. 4). These results suggest that the protective effect of statins involves decreasing ROS and subsequent Erk1/2 and EMT in renal tubular EMT.
Figure 4. Simvastatin attenuated TGF‐β1‐induced EMT and Erk1/2 phosphorylation in renal tubular cells.
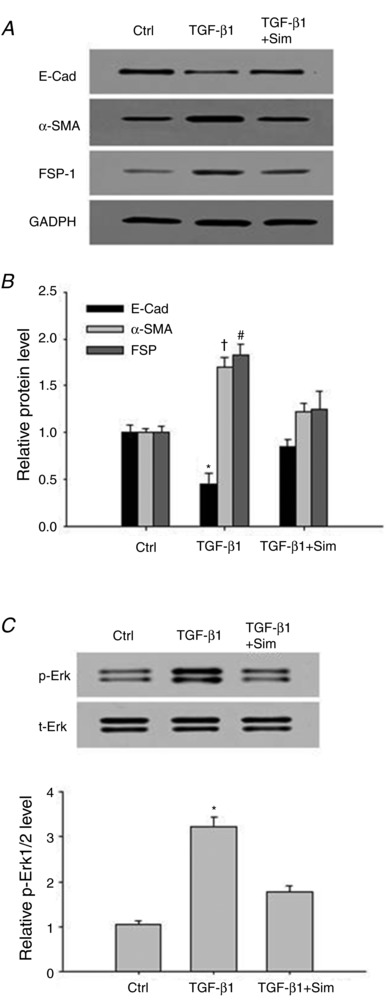
Representative western blot images (A) and summarized data (B) showing the inhibitory effect of simvastatin on TGF‐β1‐induced decrease in epithelial marker E‐cad, and increases in mesenchymal markers α‐SMA and FSP‐1. C, representative western blot images (upper panel) and summarized data (lower panel) showing the inhibitory effect of simvastatin on TGF‐β1‐induced Erk1/2 phosphorylation. E‐Cad, E‐cadherin; α‐SMA, α‐smooth muscle actin; FSP‐1, fibroblast‐specific protein‐1. Results are the means ± SEM of measurements from five separate preparations. * P < 0.05, † P < 0.05 and # P < 0.05 vs. other groups for E‐cadherin, α‐SMA and FSP‐1, respectively in B; * P < 0.05 vs. other groups in C.
Effects of Atp6v1e2 shRNA and SMPD1 shRNA on systolic blood pressure and renal blood flow in AngII‐infused rats
Figure 5 A shows that chronic AngII infusion caused a significant systolic blood pressure increase. There was no difference in AngII‐induced increases in systolic pressure between rats treated with AngII + control plasmids, AngII + Atp6v1e2 shRNA and AngII + SMPD1 shRNA (Fig. 5 A). As shown in Fig. 5 B, there was no difference in renal blood flow among the four animal groups at the end of the treatment. Similarly, there was no difference in body weight among the four animal groups at the end of the treatment (Control, 390.8 ± 13.3 g; AngII, 376.1 ± 9.8 g; AngII + Atp6v1e2, 371.34 ± 12.6 g; AngII + SMPD1 shRNA, 361.3 ± 19.5 g). These results suggest that the effect of Atp6v1e2 shRNA and SMPD1 shRNA was not through the alteration of blood pressure and renal blood flow.
Figure 5. Effect of Atp6v1e2 shRNA and SMPD1 shRNA on systolic blood pressure and renal blood flow in AngII‐infused rats.
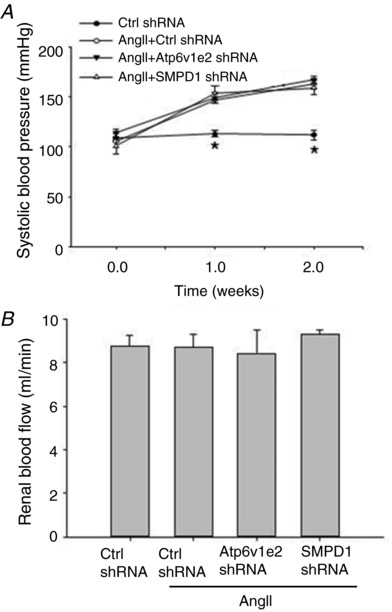
Summarized data showing systolic blood pressure (A) and renal blood flow (B) in control rats and AngII‐infused rats with or without Atp6v1e2 shRNA and SMPD1 shRNA treatments. n = 6–8 rats. * P < 0.05 vs. other groups in the same treatment.
Atp6v1e2 shRNA and SMPD1 shRNA attenuated α‐SMA expression and collagen deposition in AngII‐infused rats
Next, we investigated whether the MRs–redox signalling pathway is involved in renal fibrosis in AngII‐infused rats, as EMT plays a critical role in chronic kidney damage. As shown in Fig. 6 A, AngII‐infused rats displayed more collagen deposition as determined by Masson staining in the kidney interstitium compared to that in control rats. The renal fibrotic changes were further assessed by fibronectin and α‐SMA immunostaining for activated myofibroblasts, which are the major components of the intracellular matrix. As shown in Fig. 6 A, AngII infusion resulted in a substantial increase in activated fibroblasts in the interstitium and renal tubules, and these changes were significantly inhibited in AngII‐infused rat kidney transfected with Atp6v1e2 shRNA and SMPD1 shRNA.
Figure 6. Atp6v1e2 shRNA and SMPD1 shRNA prevent AngII‐infused rats from developing renal fibrosis.
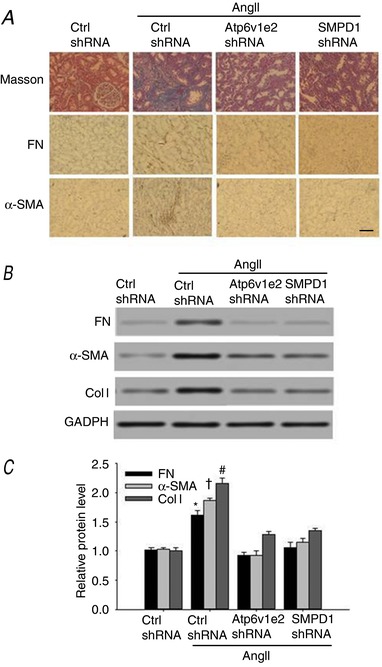
A, representative images of Mason's staining, fibronectin staining and α‐SMA staining at 400× (scale bar 50 μm) magnification in control and AngII‐infused rats with or without Atp6v1e2 shRNA and SMPD1 shRNA treatments. Representative western blot images (B) and summarized data (C) showing fibronectin, α‐SMA and collagen I content in control and AngII‐infused rats with or without Atp6v1e2 shRNA and SMPD1 shRNA treatments. FN, fibronectin; α‐SMA, α‐smooth muscle actin; Col I, collagen I. n = 6–8 rats. * P < 0.05, † P < 0.05 and # P < 0.05 vs. other groups for fibronectin, α‐SMA and collagen I, respectively. [Color figure can be viewed at http://wileyonlinelibrary.com]
The changes of fibronectin, α‐SMA and collagen deposition were further examined by western blot assay. As shown in Fig. 6 B and C, the expression of fibronectin, α‐SMA and collagen I was significantly increased in kidney from AngII‐infused rats. Treatment with Atp6v1e2 shRNA and SMPD1 shRNA significantly reduced the AngII‐induced deposition of collagen, as well as increased expression of fibronectin and α‐SMA (Fig. 6 B, C). These results suggest that the MRs–redox signalling pathway contributes to AngII‐induced renal fibrosis.
Atp6v1e2 shRNA and SMPD1 shRNA attenuated clustering of Nox4 in MR domains, SMPD1 activation and ROS production in renal tubular cells isolated from AngII‐infused rats
We then further examined the changes of the MRs–redox signalling pathway in renal tubular cells from AngII‐infused rats. Renal tubular cells were collected by Percoll density gradient centrifugation, and these isolated renal tubular cells were used for three‐layer flotation assays, SMPD1 activity measurement and ROS production. As shown in Fig. 7 A, expression of flotillin‐1, a specific MR marker, was primarily found in the fractions (fraction 2–4) between the 5% and 30% gradients. Nox4 was mainly located in the non‐MR domain in kidney from control rats, and it was mainly detected in the MR domain in kidney from AngII‐infused rats (Ctrl, 6.4 ± 0.7%; AngII, 34.1 ± 3.8%; AngII + Atp6v1e2 shRNA, 7.2 ± 1.4%; AngII+ SMPD1 shRNA, 8.7 ± 1.9%). SMPD1 activity and ROS production were also significantly increased in renal tubular cells from AngII‐infused rats (Fig. 7 B, C). As expected, the increased translocation of Nox4 in the MR domain, SMPD1 activity and ROS production were significantly attenuated in renal tubular cells from AngII‐infused rats treated with Atp6v1e2 shRNA and SMPD1 shRNA (Fig. 7 A–C). These results suggest that the MRs–redox signalling pathway was activated in kidney from AngII‐infused rats.
Figure 7. Atp6v1e2 shRNA and SMPD1 shRNA prevent Nox4 translocation to MR domains, SMPD1 activation and increase ROS production in tubular cells isolated from AngII‐infused rats.
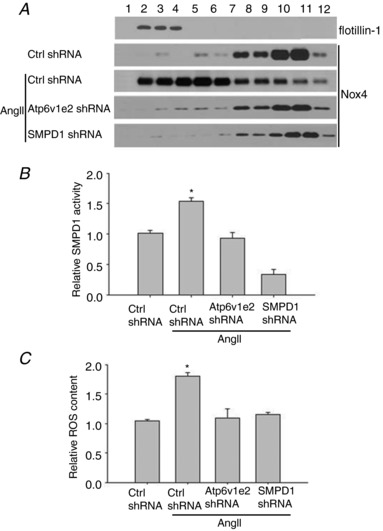
A, Atp6v1e2 shRNA and SMPD1 shRNA prevented translocation of Nox4 to the MR domain in renal tubular cells in AngII‐infused rats; numbers (1–12) on the top indicate membrane fractions isolated by gradient centrifugation from the top to bottom, fraction 1 was very light fractions without proteins, fractions 2–4 were light fractions designated as MRs based on the presence of the MR marker protein, flotillin‐1, the heavier fractions 5–12 were designated as non‐raft fractions which contained membrane and cytosolic proteins. Effect of Atp6v1e2 shRNA and SMPD1 shRNA on SMPD1 activity (B) and ROS production (C) in renal tubular cells in AngII‐infused rats. n = 6–8 rats. * P < 0.05 vs. other groups.
Atp6v1e2 shRNA and SMPD1 shRNA attenuated the increased renal expression of MCP‐1, TNF‐α and ICAM‐1 in AngII‐infused rats
It has been shown that inflammation plays a critical role in renal fibrosis, and we then evaluated whether inhibition of the MRs–redox signalling pathway would attenuate increased expression of inflammatory factors. As shown in Fig. 8, the mRNA and protein levels of MCP‐1, TNF‐α and ICAM‐1 were significantly increased in kidney from AngII‐infused rats compared with that from control rats. Notably, the increased expression of all three subunits was decreased to the control level in kidney from AngII‐infused rats treated with Atp6v1e2 shRNA and SMPD1 shRNA. These results indicate that the MRs–redox signalling pathway was involved in renal inflammation in AngII‐infused rats.
Figure 8. Inhibitory effect of Atp6v1e2 shRNA and SMPD1 shRNA on the increased expression of MCP‐1, ICAM‐1 and TNF‐α in kidney from AngII‐infused rats.
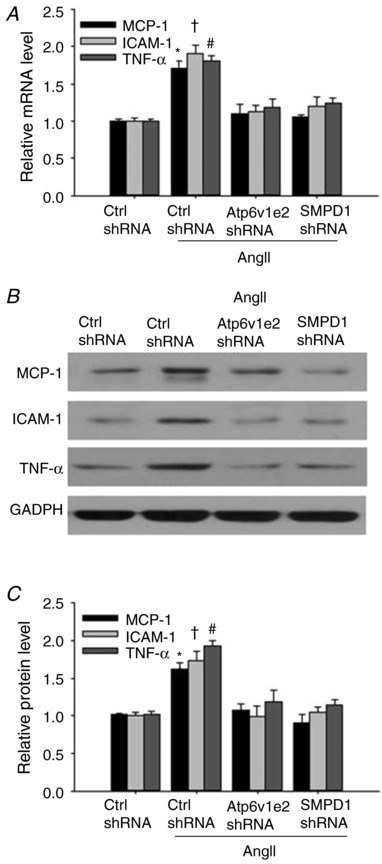
A, summarized data showing the effect of Atp6v1e2 shRNA and SMPD1 shRNA on MCP‐1, ICAM‐1 and TNF‐α mRNA level in kidney from AngII‐infused rats. Representative gel documents (B) and summarized data (C) showing the effect of Atp6v1e2 shRNA and SMPD1 shRNA on MCP‐1, ICAM‐1 and TNF‐α protein levels in kidney from AngII‐infused rats. n = 6–8 rats. * P < 0.05, † P < 0.05 and # P < 0.05 vs. other groups for MCP‐1, ICAM‐1 and TNF‐α, respectively.
Discussion
In the present study, we evaluated whether the MRs–redox signalling pathway is involved in TGF‐β1‐induced renal tubular EMT and renal fibrosis. We used Atp6v1e2 shRNA plasmid and SMPD1 shRNA plasmid to specifically decrease the expression of both enzymes. Our results show that both shRNA treatments prevented TGF‐β1‐induced EMT. Further in vivo study shows that the MRs–redox signalling pathway was activated in kidney from AngII‐infused rats as indicated by increased translocation of NADPH oxidase to the MR domain, increased SMPD1 activity and increased ROS production. Notably, these changes were significantly inhibited when Atp6v1e2 shRNA and SMPD1 shRNA were transfected to the kidney. Meanwhile, renal fibrosis and inflammation were also significantly inhibited in kidney from AngII‐infused rats. These results suggest that the MRs–redox signalling pathway was involved in TGFβ‐1‐induced renal tubular EMT and renal fibrosis/inflammation.
First, the present study shows that TGF‐β1 induced the formation of MR clusters on the cell membrane of renal tubular cells by using confocal microscopic analysis with CTX as a marker. CTX specifically binds to ganglioside GM1, which partitions into MRs, and so has been extensively used to evaluate MR clustering in endothelial cells in our previous studies (Han et al. 2012), as well as in other labs (Wang et al. 2013). It has been shown that NADPH oxidase‐derived ROS play a critical role in EMT in response to various stimulators such as TGF‐β1, AngII and glucose (Rhyu et al. 2005; Chang et al. 2011; Liu et al. 20122; He et al. 2015; Das et al. 2016; Xu et al. 2017), and that ROS mediate TGF‐β1‐induced renal tubular epithelial cells, hepatic stellate cells and pulmonary fibroblasts transdifferentiation (Rhyu et al. 2005; Bocchino et al. 2010; Ghatak et al. 2011). Consistent with these findings, the present study shows that increased ROS production via the MRs–redox signalling pathway was involved in TGF‐β1‐induced EMT in western blot and immunofluorescence assays. In contrast to previous studies showing that increased expression of NADPH oxidase subunits is responsible for these changes (Bondi et al. 2010), this finding provides a new mechanism in which TGF‐β1 activated NADPH oxidase through the MRs–redox signalling pathway without the change in NADPH oxidase expression. This finding is consistent with our previous studies showing that the MRs–redox signalling pathway is activated in response to FasL, TRAIL and AngII in coronary arterial endothelial cells and mesenteric endothelial cells (Zhang et al. 2006; Han et al. 2011, 2012; Li et al. 2013). Furthermore, we show that Erk1/2 was involved in MR‐mediated EMT in renal tubular cells, which is consistent with previous studies showing that Erk1/2 plays a critical role in renal tubular cells and in mammary gland epithelial cells (Rhyu et al. 2005; Bondi et al. 2010; Zhu et al. 2012). It has been reported that statins have a protective effect on renal fibrosis, and this effect may be caused by its inhibitory effect on renal EMT (Patel et al. 2006; Chade et al. 2008). Furthermore, it has been shown that statins inhibited TGF‐β1‐induced ROS production and Erk1/2 phosphorylation in renal tubular cells (Yoshikawa et al. 2010; Ishibashi et al. 2012). Consistent with these studies, the present study shows that simvastatin effectively inhibited GF‐β1‐induced ROS, Erk1/2 phosphorylation and EMT in renal tubular cells.
Furthermore, we used three‐layer flotation to evaluate the change of MR components by using proximal tubular cells isolated from kidney (Brendler‐Schwaab & Herbold, 1997; Yi et al. 2009). It was found that the MRs–redox signalling pathway was activated, as indicated by the increased fraction of Nox4 in the MR domain, increased SMPD1 activity and ROS production in renal tubular cells isolated from kidney in AngII‐infused rats. Importantly, these changes were significantly inhibited by Atp6v1e2 shRNA and SMPD1 shRNA, two critical enzymes in the MRs–redox signalling pathway. These in vivo findings are consistent with cell studies showing increased ROS production, increased Nox4 translocation and SMPD1 activation when the MRs–redox signalling pathway was activated in renal tubular cells in the present study, and in coronary endothelial cells and mesenteric endothelial cells in previous studies (Han et al. 2011, 2016). Of note, the present study extended our previous cell studies by showing that this MRs–redox signalling was activated and contributed to ROS production in AngII‐infused rats.
Consistent with previous studies showing that NADPH oxidase activation contributes to renal fibrosis, the present study demonstrates that inhibition of the MRs–redox signalling pathway with Atp6v1e2 shRNA and SMPD1 shRNA significantly attenuated renal fibrosis in AngII‐infused, rats as indicated by the decreased expression of fibronectin and collagen I in kidney from AngII‐infused rats. These findings are consistent with a previous study showing that the SMPD1 inhibitor amitriptyline improved renal function and renal fibrosis in a unilateral ureter obstruction (UUO) animal model (Achar et al. 2009). Furthermore, Cao et al. (2012) demonstrated that Atp6v1e2‐mediated pH change was involved in TGF‐β1‐induced EMT in rat renal tubular cells, which also plays a critical role in MR formation (Xu et al. 2012). Importantly, the present study shows that inhibition of the MRs–redox signalling pathway decreased the expression of EMT marker α‐SMA. It has been reported that tubulointerstitial fibrosis, a major feature of diabetic nephropathy, occurs due to the accumulation of interstitial ECM such as fibronectin, collagen I and collagen IV derived from activated fibroblasts/myofibroblasts in the kidney (Gilbert & Cooper, 1999; Zeisberg et al. 2001). Although the origin of myofibroblasts remains controversial, EMT is well recognized as an important source of myofibroblasts (Kalluri & Neilson, 2003). Myofibroblasts that express α‐SMA as the cell marker are a major source of ECM production. Indeed, it has been demonstrated that up to one‐third of all disease‐related fibroblasts can originate from tubular epithelia at the site of injury through EMT (Iwano et al. 2002), suggesting that EMT is one of the major mechanisms of tubulointerstitial fibrosis. These studies are consistent with our present study by showing that MRs–redox signalling contributes to TGFβ‐1‐mediated EMT in renal tubular cells and that this MRs–redox‐mediated EMT contributes to renal fibrosis in kidney from AngII‐infused rats.
It has been shown that the intracellular ROS in renal cells, such as tubular epithelial cells, mesangial cells and fibroblasts, were mainly derived from NADPH oxidases, which were up‐regulated in diabetic nephropathy (Li & Shah, 2003; Gill & Wilcox, 2006; Bedard & Krause, 2007). In particular, tubular cells are an important source of ROS in the kidney (Ricardo & Diamond, 1998). ROS mediate multiple cellular functions, including proliferation, apoptosis, migration and differentiation, and ROS at low concentrations can be used as signal transduction molecules (Liu et al. 2014). It has been reported that ROS can stimulate inflammatory effects in various organs including kidney (Turgut & Bolton, 2010; Sedeek et al. 2013). Consistent with these findings, the present study shows that inhibition of the tubular MRs–redox signalling pathway effectively inhibited the increase of MCP‐1, ICAM‐1 and TNF‐α in kidney from AngII‐infused rats. These studies suggest that decreased inflammation may contribute to the protective effect of MRs–redox inhibition on renal fibrosis (Jiang et al. 2007; Babelova et al. 2012; Lee et al. 2012; Sedeek et al. 2013).
In summary, the present study has demonstrated that the MRs–redox signalling pathway is involved in TGF‐β1‐induced EMT in renal tubular cells and contributes to chronic kidney damage in AngII‐induced hypertensive rats. Given the high oxidative status in chronic kidney diseases, this finding may provide a pharmacological target for the prevention and treatment of renal fibrosis.
Additional information
Competing interests
None declared.
Author contributions
W.Q.H., L.X., X.F.T., W.D.C., Y.J.W. and P.J.G. conceived and designed the experimental studies. W.Q.H., L.X. and Y.J.W. performed the animal studies and made the physiological measurements. W.Q.H., L.X. and W.D.C. performed the cell and molecular study. W.Q.H., L.X. and P.J.G. prepared the figures, tables and manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by the National Natural Science Foundation of China (81100184, 81230071, 81570221, 91539202, 81200203 and 81300089), Shanghai Nature Science Foundation (17ZR1423700), the Pujiang Program of the Shanghai Science and Technology Committee (14PJ1406400), the Shanghai Medical Bureau Fund (201540037), the Scientific Fund of Shanghai Jiao Tong University School of Medicine (14XJ10042), and the Scientific Research Foundation for the Returned Overseas Chinese Scholars of the State Education Ministry.
Biography
Wei‐Qing Han is an associate professor in Ruijin Hospital, Shanghai, China. His PhD research was focused on vascular biology at the Shanghai Institutes of Biological Sciences. His postdoc study was mainly on the membrane raft–lipid raft signalling pathway in coronary arterial endothelial dysfunction at the Virginia Commonwealth University. Currently, his research is focused on the role of membrane raft–redox signalling pathway in animal studies such as hypertension and diabetes. The main challenge has been to evaluate the involvement of membrane raft in human hypertension. These results may greatly improve our understanding of the role of reactive oxygen species in cardiovascular diseases.

Edited by: Kim Barrett & Dennis Brown
References
- Achar E, Maciel TT, Collares CF, Teixeira VP & Schor N. (2009). Amitriptyline attenuates interstitial inflammation and ameliorates the progression of renal fibrosis. Kidney Int 75, 596–604. [DOI] [PubMed] [Google Scholar]
- Babelova A, Avaniadi D, Jung O, Fork C, Beckmann J, Kosowski J, Weissmann N, Anilkumar N, Shah AM, Schaefer L, Schroder K & Brandes RP. (2012). Role of Nox4 in murine models of kidney disease. Free Radic Biol Med 53, 842–853. [DOI] [PubMed] [Google Scholar]
- Bao JX, Jin S, Zhang F, Wang ZC, Li N & Li PL. (2010). Activation of membrane NADPH oxidase associated with lysosome‐targeted acid sphingomyelinase in coronary endothelial cells. Antioxid Redox Signal 12, 703–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K & Krause KH. (2007). The NOX family of ROS‐generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87, 245–313. [DOI] [PubMed] [Google Scholar]
- Bedi S, Vidyasagar A & Djamali A. (2008). Epithelial‐to‐mesenchymal transition and chronic allograft tubulointerstitial fibrosis. Transplant Rev 22, 1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchino M, Agnese S, Fagone E, Svegliati S, Grieco D, Vancheri C, Gabrielli A, Sanduzzi A & Avvedimento EV. (2010). Reactive oxygen species are required for maintenance and differentiation of primary lung fibroblasts in idiopathic pulmonary fibrosis. PLoS One 5, e14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondi CD, Manickam N, Lee DY, Block K, Gorin Y, Abboud HE & Barnes JL. (2010). NAD(P)H oxidase mediates TGF‐beta1‐induced activation of kidney myofibroblasts. J Am Soc Nephrol 21, 93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendler‐Schwaab SY & Herbold BA. (1997). A new method for the enrichment of single renal proximal tubular cells and their first use in the comet assay. Mutat Res 393, 175–178. [DOI] [PubMed] [Google Scholar]
- Callera GE, Yogi A, Briones AM, Montezano AC, He Y, Tostes RC, Schiffrin EL & Touyz RM. (2011). Vascular proinflammatory responses by aldosterone are mediated via c‐Src trafficking to cholesterol‐rich microdomains: role of PDGFR. Cardiovasc Res 91, 720–731. [DOI] [PubMed] [Google Scholar]
- Cao X, Yang Q, Qin J, Zhao S, Li X, Fan J, Chen W, Zhou Y, Mao H & Yu X. (2012). V‐ATPase promotes transforming growth factor‐beta‐induced epithelial‐mesenchymal transition of rat proximal tubular epithelial cells. Am J Physiol Renal Physiol 302, F1121–1132. [DOI] [PubMed] [Google Scholar]
- Chade AR, Zhu XY, Grande JP, Krier JD, Lerman A & Lerman LO. (2008). Simvastatin abates development of renal fibrosis in experimental renovascular disease. J Hypertens 26, 1651–1660. [DOI] [PubMed] [Google Scholar]
- Chang J, Jiang Z, Zhang H, Zhu H, Zhou SF & Yu X. (2011). NADPH oxidase‐dependent formation of reactive oxygen species contributes to angiotensin II‐induced epithelial‐mesenchymal transition in rat peritoneal mesothelial cells. Int J Mol Med 28, 405–412. [DOI] [PubMed] [Google Scholar]
- Cheng PW, Lu PJ, Chen SR, Ho WY, Cheng WH, Hong LZ, Yeh TC, Sun GC, Wang LL, Hsiao M & Tseng CJ. (2011). Central nicotinic acetylcholine receptor involved in Ca2+‐calmodulin‐endothelial nitric oxide synthase pathway modulated hypotensive effects. Br J Pharmacol 163, 1203–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X, Zheng X, Song Y, Qu L, Tang J, Meng L & Wang Y. (2016). Apocynin attenuates renal fibrosis via inhibition of NOXs‐ROS‐ERK‐myofibroblast accumulation in UUO rats. Free Radic Res 50, 840–852. [DOI] [PubMed] [Google Scholar]
- Das SJ, Lovicu FJ & Collinson EJ. (2016). Nox4 plays a role in TGF‐β‐dependent lens epithelial to mesenchymal transition. Invest Ophthalmol Vis Sci 57, 3665–3673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghatak S, Biswas A, Dhali GK, Chowdhury A, Boyer JL & Santra A. (2011). Oxidative stress and hepatic stellate cell activation are key events in arsenic induced liver fibrosis in mice. Toxicol Appl Pharmacol 251, 59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert RE & Cooper ME. (1999). The tubulointerstitium in progressive diabetic kidney disease: more than an aftermath of glomerular injury? Kidney Int 56, 1627–1637. [DOI] [PubMed] [Google Scholar]
- Gill PS & Wilcox CS. (2006). NADPH oxidases in the kidney. Antioxid Redox Signal 8, 1597–1607. [DOI] [PubMed] [Google Scholar]
- Grundy D. (2015). Principles and standards for reporting animal experiments in The Journal of Physiology and Experimental Physiology. J Physiol 593, 2547–2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza SM & Kaufman S. (2004). Splenorenal reflex modulates renal blood flow in the rat. J Physiol 558, 277–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han WQ, Chen WD, Zhang K, Liu JJ, Wu YJ & Gao PJ. (2016). Ca2+‐regulated lysosome fusion mediates angiotensin II‐induced lipid raft clustering in mesenteric endothelial cells. Hypertens Res 39, 227–236. [DOI] [PubMed] [Google Scholar]
- Han WQ, Wong WT, Tian XY, Huang Y, Wu LY, Zhu DL & Gao PJ. (2010). Contributory role of endothelium and voltage‐gated potassium channels in apocynin‐induced vasorelaxations. J Hypertens 28, 2102–2110. [DOI] [PubMed] [Google Scholar]
- Han WQ, Xia M, Xu M, Boini KM, Ritter JK, Li NJ & Li PL. (2012). Lysosome fusion to the cell membrane is mediated by the dysferlin C2A domain in coronary arterial endothelial cells. J Cell Sci 125, 1225–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han WQ, Xia M, Zhang C, Zhang F, Xu M, Li NJ & Li PL. (2011). SNARE‐mediated rapid lysosome fusion in membrane raft clustering and dysfunction of bovine coronary arterial endothelium. Am J Physiol Heart Circ Physiol 301, H2028–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han WQ, Zhu Q, Hu J, Li PL, Zhang F & Li N. (2013). Hypoxia‐inducible factor prolyl‐hydroxylase‐2 mediates transforming growth factor β1‐induced epithelial‐mesenchymal transition in renal tubular cells. Biochim Biophys Acta 1833, 1454–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He T, Guan X, Wang S, Xiao T, Yang K, Xu X, Wang J & Zhao J. (2015). Resveratrol prevents high glucose‐induced epithelial‐mesenchymal transition in renal tubular epithelial cells by inhibiting NADPH oxidase/ROS/ERK pathway. Mol Cell Endocrinol 402, 13–20. [DOI] [PubMed] [Google Scholar]
- Ishibashi Y, Yamagishi S, Matsui T, Ohta K, Tanoue R, Takeuchi M, Ueda S, Nakamura K & Okuda S. (2012). Pravastatin inhibits advanced glycation end products (AGEs)‐induced proximal tubular cell apoptosis and injury by reducing receptor for AGEs (RAGE) level. Metabolism 61, 1067–1072. [DOI] [PubMed] [Google Scholar]
- Iwano M, Plieth D, Danoff TM, Xue C, Okada H & Neilson EG. (2002). Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clinical Invest 110, 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Wang XX, Scherzer P, Wilson P, Tallman J, Takahashi H, Li J, Iwahashi M, Sutherland E, Arend L & Levi M. (2007). Farnesoid X receptor modulates renal lipid metabolism, fibrosis, and diabetic nephropathy. Diabetes 56, 2485–2493. [DOI] [PubMed] [Google Scholar]
- Kalluri R & Neilson EG. (2003). Epithelial‐mesenchymal transition and its implications for fibrosis. J Clin Invest 112, 1776–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T. (2007). SNARE proteins and ‘membrane rafts’. J Physiol 585, 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee IT, Shih RH, Lin CC, Chen JT & Yang CM. (2012). Role of TLR4/NADPH oxidase/ROS‐activated p38 MAPK in VCAM‐1 expression induced by lipopolysaccharide in human renal mesangial cells. Cell Commun Signal 10, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung SB, Zhang H, Lau CW & Lin ZX. (2016). Attenuation of blood pressure in spontaneously hypertensive rats by acupuncture was associated with reduction oxidative stress and improvement from endothelial dysfunction. Chin Med 11, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JM & Shah AM. (2003). ROS generation by nonphagocytic NADPH oxidase: potential relevance in diabetic nephropathy. J Am Soc Nephrol 14, S221–226. [DOI] [PubMed] [Google Scholar]
- Li N, Yi F, Sundy CM, Chen L, Hilliker ML, Donley DK, Muldoon DB & Li PL. (2007). Expression and actions of HIF prolyl‐4‐hydroxylase in the rat kidneys. Am J Physiol Renal Physiol 292, F207–216. [DOI] [PubMed] [Google Scholar]
- Li X, Han WQ, Boini KM, Xia M, Zhang Y & Li PL. (2013). TRAIL death receptor 4 signaling via lysosome fusion and membrane raft clustering in coronary arterial endothelial cells: evidence from ASM knockout mice. J Mol Med (Berl) 91, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Gomez Garcia AM & Meyskens FL, Jr (2012). NADPH oxidase 1 overexpression enhances invasion via matrix metalloproteinase‐2 and epithelial‐mesenchymal transition in melanoma cells. J Invest Dermatol 132, 2033–2041. [DOI] [PubMed] [Google Scholar]
- Liu XX, Zhou HJ, Cai L, Zhang W, Ma JL, Tao XJ & Yu JN. (2014). NADPH oxidase‐dependent formation of reactive oxygen species contributes to transforming growth factor β1‐induced epithelial‐mesenchymal transition in rat peritoneal mesothelial cells, and the role of astragalus intervention. Chin J Integr Med 20, 667–674. [DOI] [PubMed] [Google Scholar]
- Mao H, Li Z, Zhou Y, Li Z, Zhuang S, An X, Zhang B, Chen W, Nie J, Wang Z, Borkan SC, Wang Y & Yu X. (2008). HSP72 attenuates renal tubular cell apoptosis and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol 295, F202–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman CM & Bettinger T. (2007). Gene therapy progress and prospects: ultrasound for gene transfer. Gene Therapy 14, 465–475. [DOI] [PubMed] [Google Scholar]
- Patel S, Mason RM, Suzuki J, Imaizumi A, Kamimura T & Zhang Z. (2006). Inhibitory effect of statins on renal epithelial‐to‐mesenchymal transition. Am J Nephrol 26, 381–387. [DOI] [PubMed] [Google Scholar]
- Qiao X, Wang L, Wang Y, Zhao N, Zhang R, Han W & Peng Z. (2015). Intermedin is upregulated and attenuates renal fibrosis by inhibition of oxidative stress in rats with unilateral ureteral obstruction. Nephrology (Carlton) 20, 820–831. [DOI] [PubMed] [Google Scholar]
- Rhyu DY, Yang Y, Ha H, Lee GT, Song JS, Uh ST & Lee HB. (2005). Role of reactive oxygen species in TGF‐beta1‐induced mitogen‐activated protein kinase activation and epithelial‐mesenchymal transition in renal tubular epithelial cells. J Am Soc Nephrol 16, 667–675. [DOI] [PubMed] [Google Scholar]
- Ricardo SD & Diamond JR. (1998). The role of macrophages and reactive oxygen species in experimental hydronephrosis. Semin Nephrol 18, 612–621. [PubMed] [Google Scholar]
- Sedeek M, Nasrallah R, Touyz RM & Hebert RL. (2013). NADPH oxidases, reactive oxygen species, and the kidney: friend and foe. J Am Soc Nephrol 24, 1512–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terryn S, Jouret F, Vandenabeele F, Smolders I, Moreels M, Devuyst O, Steels P & Van Kerkhove E. (2007). A primary culture of mouse proximal tubular cells, established on collagen‐coated membranes. Am J Physiol Renal Physiol 293, F476–485. [DOI] [PubMed] [Google Scholar]
- Turgut F & Bolton WK. (2010). Potential new therapeutic agents for diabetic kidney disease. Am J Kidney Dis 55, 928–940. [DOI] [PubMed] [Google Scholar]
- Vetrivel KS & Thinakaran G. (2010). Membrane rafts in Alzheimer's disease β‐amyloid production. Biochim Biophys Acta 1801, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang R, Bi J, Ampah KK, Zhang C, Li Z, Jiao Y, Wang X, Ba X & Zeng X. (2013). Lipid raft regulates the initial spreading of melanoma A375 cells by modulating beta1 integrin clustering. Int J Biochem Cell Biol 45, 1679–1689. [DOI] [PubMed] [Google Scholar]
- Xu M, Xia M, Li XX, Han WQ, Boini KM, Zhang F, Zhang Y, Ritter JK & Li PL. (2012). Requirement of translocated lysosomal V1 H+‐ATPase for activation of membrane acid sphingomyelinase and raft clustering in coronary endothelial cells. Mol Biol Cell 23, 1546–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Sun S, Xie F, Ma J, Tang J, He S & Bai L. (2017). Advanced oxidation protein products induce epithelial‐mesenchymal transition of intestinal epithelial cells via a PKC δ‐mediated, redox‐dependent signaling pathway. Antiox Redox Signal 27, 37–56. [DOI] [PubMed] [Google Scholar]
- Yi F, Xia M, Li N, Zhang C, Tang L & Li PL. (2009). Contribution of guanine nucleotide exchange factor Vav2 to hyperhomocysteinemic glomerulosclerosis in rats. Hypertension 53, 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi F, Zhang AY, Janscha JL, Li PL & Zou AP. (2004). Homocysteine activates NADH/NADPH oxidase through ceramide‐stimulated Rac GTPase activity in rat mesangial cells. Kidney Int 66, 1977–1987. [DOI] [PubMed] [Google Scholar]
- Yoshikawa M, Hishikawa K, Idei M & Fujita T. (2010). Trichostatin a prevents TGF‐β1‐induced apoptosis by inhibiting ERK activation in human renal tubular epithelial cells. Eur J Pharmacol 642, 28–36. [DOI] [PubMed] [Google Scholar]
- Zeisberg M, Bonner G, Maeshima Y, Colorado P, Muller GA, Strutz F & Kalluri R. (2001). Renal fibrosis: collagen composition and assembly regulates epithelial‐mesenchymal transdifferentiation. Am J Pathol 159, 1313–1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang AY, Yi F, Zhang G, Gulbins E & Li PL. (2006). Lipid raft clustering and redox signaling platform formation in coronary arterial endothelial cells. Hypertension 47, 74–80. [DOI] [PubMed] [Google Scholar]
- Zhou X, Zhang J, Xu C & Wang W. (2014). Curcumin ameliorates renal fibrosis by inhibiting local fibroblast proliferation and extracellular matrix deposition. J Pharmacol Sci 126, 344–350. [DOI] [PubMed] [Google Scholar]
- Zhu Q, Liu M, Han WQ, Li PL, Wang Z & Li N. (2012). Overexpression of HIF prolyl‐hydoxylase‐2 transgene in the renal medulla induced a salt sensitive hypertension. J Cell Mol Med 16, 2701–2707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Q, Wang Z, Xia M, Li PL, Van Tassell BW, Abbate A, Dhaduk R & Li N. (2011). Silencing of hypoxia‐inducible factor‐1α gene attenuated angiotensin II‐induced renal injury in Sprague‐Dawley rats. Hypertension 58, 657–664. [DOI] [PMC free article] [PubMed] [Google Scholar]