

Abstract
Copepods harbor diverse bacterial communities, which collectively carry out key biogeochemical transformations in the ocean. However, bulk copepod sampling averages over the variability in their associated bacterial communities, thereby limiting our understanding of the nature and specificity of copepod–bacteria associations. Here, we characterize the bacterial communities associated with nearly 200 individual Calanus finmarchicus copepods transitioning from active growth to diapause. We find that all individual copepods sampled share a small set of “core” operational taxonomic units (OTUs), a subset of which have also been found associated with other marine copepod species in different geographic locations. However, most OTUs are patchily distributed across individual copepods, thereby driving community differences across individuals. Among patchily distributed OTUs, we identified groups of OTUs correlated with common ecological drivers. For instance, a group of OTUs positively correlated with recent copepod feeding served to differentiate largely active growing copepods from those entering diapause. Together, our results underscore the power of individual-level sampling for understanding host–microbiome relationships.
Introduction
Calanoid copepods play important roles in marine ecosystems, serving both as lynchpins of aquatic food webs [1] and vehicles of vertical carbon transport [1, 2]. While typically no more than a millimeter in length, copepods also serve as nutrient-rich habitats for tens of thousands of bacteria [3–5], which are distinct from those “free-living” in ambient seawater [3, 6]. These copepod-associated bacterial communities initiate key chemical processes, including carbon degradation and nitrogen fixation [7–11]. Thus, they likely exert a fundamental control on biogeochemical transformations in the marine environment. However, what controls the composition of copepod-associated bacterial communities, including associations with their host, remains poorly understood.
Living copepods present a dynamic habitat whose properties may depend strongly on host physiology [5]. When copepods feed, they amass food particles in their guts, thus presenting a nutrient-rich (albeit often undersaturated in oxygen) environment compared to the surrounding seawater [8, 12]. Moreover, as copepods feed and defecate, they release nutrients into their surroundings, which bacteria can then exploit [5, 13]. Accordingly, bacteria are not uniformly distributed over the copepod surface, but instead, are often concentrated at the gut, mouth, and anus [3, 14]. The frequency with which copepods molt can also affect their associated bacterial communities by modulating the rate of community turnover [5]. Additionally, as copepods migrate vertically through the water column, their associated bacteria travel with them, further shaping the environment to which they are exposed [5, 15].
Altogether, copepod physiology is likely to have a profound influence on their associated bacterial flora. However, given that the physiological state of a copepod could vary drastically from individual to individual, bulk sampling of copepods could lead to physiological averaging that would mask these effects. Individual sampling of copepods also offers the opportunity to study co-occurrence patterns among bacterial taxa within communities, thereby allowing us to identify robust statistical associations between taxa driven by shared niche preferences or interspecific dependencies, for instance [16].
One of the most dramatic physiological transitions that copepods in the family Calanidae undergo is the transition from active growth to a diapausing (dormant) state. This strategy is employed by several temperate and polar species as a means of persisting during unfavorable periods, such as when predators are abundant or food is limited (reviewed by Baumgartner and Tarrant [17]). Each year, an enormous number of C. finmarchicus juveniles—tens of thousands per square meter in the North Atlantic—enter diapause [2, 18, 19]. This transition coincides with a number of physiological changes, including cessation of feeding [20, 21], delayed molting [22, 23], pH changes and accumulation of ammonium in hemolymph [24, 25], differential gene expression [26–28], and catabolism of lipids accumulated in an enlarged oil sac during the late juvenile stages [20, 29]. Importantly, copepods entering diapause migrate vertically from the surface to deep ocean basins, bringing with them vast amounts of carbon in the form of lipid stores that is eventually sequestered in the deep ocean via respiration [2, 20]. This process (the so-called “lipid pump”) may be a vital determinant of global carbon fluxes in the ocean [2]. The bacterial communities associated with diapausing copepods may contribute to this lipid pump, much like the microbe-mediated “biological pump” [30]. Nonetheless, to date, how copepod-associated bacterial communities change during a copepod’s transition to diapause has not been characterized.
The goals of this study were to (a) characterize variability in copepod-associated bacterial communities across many individual copepods, and (b) assess the potential drivers underlying this variability, including host physiology (via morphological proxies). To this end, we collected 189 individual copepods (Calanus finmarchicus stage C5 copepodites from Trondheimsfjord, Norway) on two separate dates during the early summer (6 June 2012 and 11 June 2012). At this time of year, C5 copepodites have typically just begun their descent from the surface ocean, and thus are in the early stages of the transition from active growth to a diapausing (dormant) state (Materials and methods; Supplementary Methods). On each of these dates, we sampled copepods inhabiting two depth strata (shallow, 0–50 m, and deep, 250–340 m below the surface), thereby allowing us to sample a physiological gradient that included actively growing and diapausing states (Fig. 1a). Accordingly, for each individual, we quantified morphological characteristics that mark the physiological transition between active growth and diapause. We also characterized the composition of the copepod-associated bacterial community via 16S rRNA amplicon sequencing.
Fig. 1.

Sampling of individual copepods from the North Atlantic. a Individual copepods were collected over two sampling dates at Tröllet Station within Trondheimsfjord, Norway (189 individuals in total; C. finmarchicus C5 copepodids). To capture individuals in a wide range of physiological states, copepods were collected from surface (0–50 m depths) and deep (250–350 m depths) seawater. At the surface, copepods were largely in an active state, while at depth, copepods were often in diapause (dormant). Among copepod individuals, physiological differences were manifested by changes in morphology, including differences in b prosome (body) volume, c oil sac fractional fullness, d the presence of food in the gut, e and overall bacterial load. Procedures for quantifying each of these characteristics are described in Materials and methods. In all plots, black bars indicate mean value
Results and discussion
Morphological differences within and between deep- and shallow-dwelling copepod populations
Copepods collected from shallow and deep seawater displayed the morphological hallmarks of active growth and diapause, respectively, although copepods from each collection depth exhibited substantial inter-individual variability (Fig. 1b–e). Consistent with previous studies [27, 28], deep-dwelling copepods had significantly larger body volumes (Fig. 1b, p = 6 × 10−19, two-sided Mann–Whitney U test) and fuller oil sacs (Fig. 1c, p = 5 × 10−16, two-sided Mann–Whitney U test) on average than their shallow-dwelling counterparts. Deep-dwelling copepods were also significantly less likely to have food in their guts (Fig. 1d, p = 4 × 10−8, Fisher’s exact test) and, on average, harbored fewer bacterial cells (Fig. 1e, p = 6 × 10−5, two-sided Mann–Whitney U test) than those from shallow seawater. However, even copepods sampled from the same depth varied substantially in these morphological characteristics, most likely due to temporal asynchronicity in their transition to diapause (Fig. 1b–e). Indeed, for each characteristic, the variation (standard deviation) within a single depth stratum was comparable to the difference in means between depth strata (Table S1). Thus, we hypothesized that variability in bacterial communities might be associated with this morphological variability between copepod individuals—potentially indicative of underlying physiological differences.
Significant compositional variability, but a common core across copepod-associated bacterial communities
Consistent with our hypothesis, individual copepods varied substantially in the bacterial communities they harbored. To characterize this variability, we focused on the 241 OTUs whose mean abundance across all copepod individuals was above a defined threshold (fmin = 2 × 10−4) (Materials and methods; Fig. 2; Fig. S1). This OTU subset accounts for 90% of all sequenced reads from copepod-associated bacterial communities, thus representing a substantial portion of the diversity.
Fig. 2.

Physiologically diverse copepods share a common “core”, but many OTUs are patchily distributed across copepod individuals. Compositional heat map of a subset of OTUs (mean abundance >2 × 10−4) present over nearly 200 copepod individuals. The left heat map shows the “core” microbiome (OTUs present in >90% of copepod-associated communities). The right heat map shows OTUs that are patchily distributed across copepods (present on <90% of copepods). In both heat maps, OTUs are clustered by their correlation (distance metric: log-transformed Pearson correlation as estimated with SparCC; clustered with Ward’s minimum variance method). Copepods are clustered by the overall similarity of their communities across core and patchily distributed components (distance metric: Euclidean distance between log-transformed relative abundances; clustered with Ward’s minimum variance method). The leftmost color-coded column indicates the depth at which an individual copepod was collected (blue, shallow; red, deep). The second from left color-coded column indicates the date on which the sample was collected (light gray, 6/6/2012; dark gray, 6/11/2012). The topmost lines indicate which OTUs fall into one of the five most commonly observed taxonomic families. OTU clusters from Fig. 4 (e.g., “Cluster 1”) are indicated. One particular bacterial taxon, seq1, is also indicated
Among this subset of OTUs, most were patchily distributed across individual copepods, with 207 out of 241 OTUs present on fewer than 90% of copepod individuals (Materials and methods). These OTUs were taxonomically diverse, with representatives from Gammaproteobacteria, Alphaproteobacteria, and Flavobacteria. Interestingly, a number of taxa that are consistently enriched in surveys of copepod-associated communities were common among these patchily distributed OTUs (e.g., Flavobacteriaceae, Oceanospirillaceae, and Pseudoalteromonadaceae; Fig. 2) [5, 31, 32]. Thus, these OTUs are not stably associated with the C. finmarchicus individuals collected in this study; indeed, there is significant copepod-to-copepod variability in their abundances that would have been masked by bulk copepod sampling.
Despite significant variability in the bacterial communities associated with individual copepods, we also identified a set of “core” OTUs, each of which was detected on more than 90% of the copepods sampled (Fig. 2). This core set comprised 34 OTUs (out of the 241 above our abundance threshold, Fig. S1; note that seq68, seq111, seq303, seq170, and seq10239 were recovered at low abundance in three blank samples, and thus, should be interpreted with caution, see Materials and methods). They represented a range of diverse bacterial phyla, including Actinobacteria, Bacteroidetes, and Proteobacteria. However, 25 of the 34 core OTUs were Proteobacteria, with 10 each being Gammaproteobacteria and Alphaproteobacteria. Of the gammaproteobacterial lineages, four were Moraxellaceae, a bacterial family whose specific associations with C. finmarchicus have been documented previously [31]. The other four gammaproteobacterial OTUs were Vibrionaceae (Fig. 2, Table S2). By 16S rRNA V4 sequence, three of the four sequences were most similar to Vibrio sp. AND4 (Fig. S3), a strain closely related to the V. harveyi group [33], although the length of the amplicon is insufficient for confident species-level classification. The remainder were representatives of Pseudomonadaceae and Halomonadaceae. Together, this core set of OTUs comprised a highly variable fraction of the community across individual copepods (range: 1.5–93%).
Generality of the core microbiome across geography and copepod species
Given that these core OTUs were ubiquitous members of the copepod microbiome in our samples (C. finmarchicus from Trondheimsfjord, Norway), we next probed the generality of their associations across copepod species and geographic locations. To do so, we performed a sequence-level comparison of the 16S rRNA V4 region between core OTUs observed in this study and those previously found associated with three different species of copepods (Acartia longiremis, Centropages hamatus, and Calanus finmarchicus) collected from the Gulf of Maine [31]. We evaluated location specificity by comparing OTUs associated with copepods of the same species (C. finmarchicus) from distinct geographic locations (Trondheimsfjord, Norway vs. Gulf of Maine). We evaluated species specificity by comparing our core OTUs to those found on two non-Calanus copepod species. Note that different primers were used to amplify 16S rRNA hypervariable regions in our study (515F, 806R; Materials and methods) compared to Moisander et al. (Bact 341F, Bact 785F; [34]), which may cause us to underestimate the number of shared OTUs. Additionally, the size of the amplicon (<300 base pairs) may lead us to underestimate diversity in the sample [35].
We found that 23 out of 34 core OTUs were location-specific, lacking close relatives on C. finmarchicus copepods from the Gulf of Maine. However, a subset of core OTUs was found at both geographic locations: six had an exact sequence match, and five had one mismatch, to OTUs observed on Gulf of Maine C. finmarchicus copepods (Table S2). Interestingly, of the 11 core OTUs with close sequence matches, four were from the family Moraxellaceae (genus Acinetobacter). Representatives of this family have previously been found associated with C. finmarchicus (among other copepod species), and are thought to exploit nutrient-rich patches, like copepods, in the environment [6, 32, 36].
We also observed that a subset of our core OTUs were not copepod species-specific, as their close relatives have been found to colonize non-Calanus copepod species. In particular, 8 out of 34 core OTUs had an exact sequence match in bacterial communities associated with at least one non-Calanus copepod species studied in [31] (Acartia longiremis or Centropages hamatus), including representatives from the genera Photobacterium, Halomonas, and Pelomonas (Table S2). Notably, overlapping geography cannot explain the occurrence of shared core OTUs, since non-Calanus copepod species from Moisander et al. were collected from a different geographic location (the Gulf of Maine) than those in our study. Furthermore, similar feeding patterns are unlikely to account for the overlap, since these copepod species span a wide range of feeding lifestyles, including herbivorous, omnivorous, and detritivorous lifestyles. Instead, this result suggests that different copepod species may exert some similar selective forces on their associated bacterial communities.
Copepod host physiology shapes copepod-associated bacterial communities
When individual copepods were clustered by the similarity (Spearman correlation) of their associated bacterial communities, they largely grouped by the vertical depth at which they were collected (Fig. 2). This suggested that differences in bacterial composition between shallow and deep seawater could account for variability in community composition between shallow- and deep-dwelling copepods, as copepods can be colonized by bacteria from the ambient seawater [15].
However, we found that the vertical depth at which a copepod was sampled was most likely an indirect predictor of the composition of its associated bacterial community. In particular, when controlling for copepod physiology via the morphological variables that we measured (body volume, oil sac fullness, the presence of food in the gut), copepod vertical collection depth only accounted for a small portion of the dissimilarity between their bacterial communities (Table S3, R2 = 0.026, p < 10−3, bivariate PERMANOVA with 1,000,000 permutations; significance of dispersion differences between depth groups: p < 2.2 × 10−16). Indeed, in a principal coordinates analysis, copepod-associated bacterial communities clustered distinctly from assemblages in the ambient seawater (Fig. 3). Furthermore, the bacterial communities from shallow-dwelling copepods were no more similar to shallow seawater than to deep seawater (and likewise for deep-dwelling copepods) (Fig. S4). Thus, it is unlikely that ambient seawater composition differentiates communities inhabiting deep- and shallow-dwelling copepods.
Fig. 3.
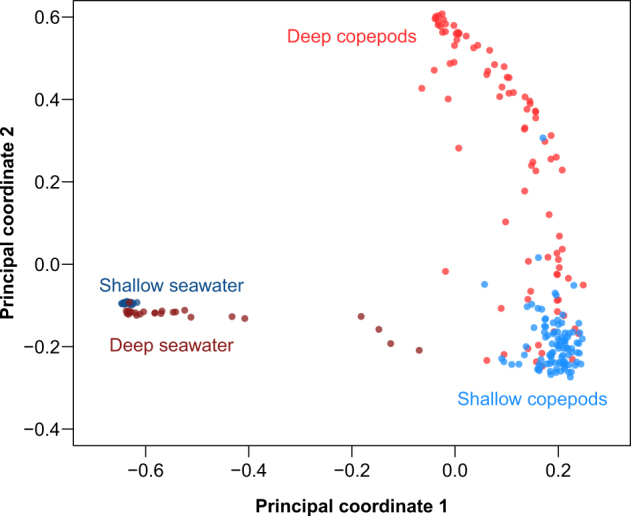
Copepod-associated bacterial communities are distinct from free-living bacterial communities in the ambient seawater. Biplot for a principal coordinates analysis (PCoA) of bacterial assemblages from individual copepods and the seawater from which they were collected
Instead, we hypothesized that differences in individual-specific morphological characteristics shape the bacterial niche, and thus are important drivers of bacterial community composition. In particular, the specific habitat provided by an individual copepod, including its feeding history, may shape local selective pressures [5, 12], thereby fostering significant inter-individual variability in copepod-associated bacterial community composition. Moreover, bacteria exist at much higher cell densities on each copepod individual than in the ambient seawater [5], suggesting that local interactions between bacterial taxa may also influence bacterial community composition.
We took a two-step approach to quantify statistical associations between individual copepod-level characteristics (including copepod host physiology and co-occurring bacterial taxa) and copepod-associated bacterial community composition. First, we characterized the network of correlations between individual OTUs, which allowed us to identify clusters of OTUs whose distribution across individual copepods was strongly positively correlated. Second, using a multivariate linear regression approach (Materials and methods; Supplementary Methods), we identified putative associations linking these clusters to each other and to the morphological characteristics that we quantified for individual copepods. Importantly, this multivariate regression approach allowed us to quantify the effect of each morphological characteristic independently, controlling for the effects of the other morphological characteristics.
Identifying clusters of highly correlated OTUs within the copepod microbiome
Overall, we identified seven clusters of OTUs, each of which contained OTUs with strong correlations across individual copepods. To identify this structure, we used SparCC to estimate the Pearson correlation of log-transformed OTU abundances across all individual copepods, focusing on the 241 OTUs whose mean abundance was above our pre-defined threshold (Fig. S1; Materials and methods). Of these OTUs, most (177 out of 241) were negligibly correlated with any of the other OTUs. However, the remaining OTUs (64 out of 241) grouped into seven clusters (Fig. 4; Table S4). Within clusters, pairs of OTUs were typically highly positively correlated, while OTUs from different clusters were often negatively correlated. The magnitudes of OTU–OTU correlations were consistent across sampling dates (correlation = 0.63; Fig. S5), suggesting that these associations are biologically reproducible. Notably, these clusters differed in their level of phylogenetic coherence: some clusters (e.g., Cluster 6; Fig. 4) only contained OTUs from a single taxonomic class (Gammaproteobacteria), while others (e.g., Cluster 2, Fig. 4) contained representatives from several disparate classes (Actinobacteria, Alphaprotebacteria, and others).
Fig. 4.
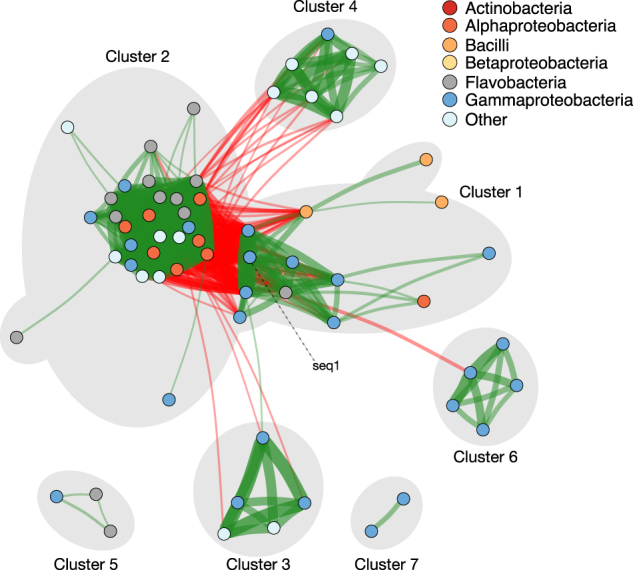
Across hundreds of copepod-associated communities, bacteria form a small number of highly positively correlated clusters. Nodes represent individual OTUs and are colored by taxonomy at the class level. Edges indicate positive (green) or negative (red) correlations between OTUs as approximated by SparCC. The magnitude of the correlation is proportional to the width of the edge. Nodes for which all inter-OTU correlations are negligible (|ρ| < 0.35) are not shown. Nodes are arranged according to a Fruchterman–Reingold force-directed graph drawing algorithm [47]. The positions of some nodes have been adjusted for disambiguation of cluster designations. Clusters of highly positively correlated OTUs are outlined in gray. One particular OTU, seq1, is indicated
Building on our identification of strongly correlated clusters of copepod-associated OTUs, we next sought to identify the ecological factors that may influence their distributions across individual copepods. The compositional nature of the bacterial community data precludes the analysis of cluster relative abundances directly [37]. Therefore, we applied a standard additive log-ratio transform, with which we quantified the log-transformed ratio of the abundance of each cluster relative to the abundance of the core microbiome for each copepod individual [37]; Materials and Methods; Supplementary Methods). We then used multivariate regression models to identify associations between each cluster’s log-ratio-transformed abundance and copepod-specific morphological characteristics, as well as the abundances of other clusters. Importantly, rather than identifying associations conditioned on a particular regression model, we used Bayesian model averaging to calculate the probability that an independent variable had a non-zero effect over all models [38].
Using this approach, we identified several significant associations, including those between bacterial clusters and copepod morphology, as well as those between pairs of bacterial clusters. These associations allowed us to identify possible ecological factors underlying fine-scale differentiation in copepod-associated bacterial communities at the level of individual copepod hosts.
Factors influencing bacterial cluster abundances within the copepod microbiome
We first identified a positive association between Cluster 2—a cluster that was enriched among shallow-dwelling copepods compared to their deep-dwelling counterparts (Fig. 2)—and the presence of food in the copepod gut (Fig. 5). In particular, when food was present in the gut, the abundance of Cluster 2 was high relative to the core microbiome, but was significantly lower among copepods where food was absent. Given that 10 out of the 29 OTUs in Cluster 2 were Flavobacteriacaeae, our findings support a growing body of evidence that Flavobacteriaceae (and perhaps other clades) are associated with the food a copepod ingests, rather than any intrinsic feature of a copepod [5, 31].
Fig. 5.
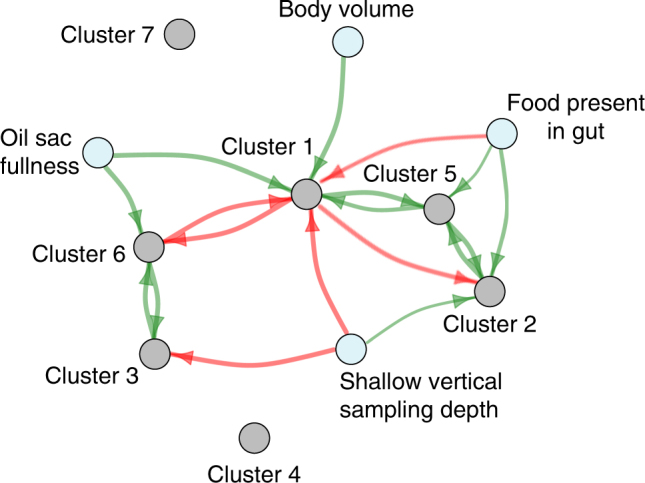
Cluster abundances are associated with host morphology, as well as the abundances of other clusters. Nodes represent clusters of positively correlated OTUs (gray) or copepod morphological characteristics (light blue). Directed edges indicate the association of a given independent variable on the summed abundance of all OTUs in a cluster, controlling for the effects of all other independent variables (green: positive association; red: negative association). The width of the edge is proportional to the probability that an independent variable has a non-zero effect on the cluster abundance, as determined by Bayesian model averaging. Edges for which the probability of a non-zero effect is less than 90% are not shown (Table S5)
Many mechanisms may account for the strong positive association between Cluster 2 abundance and food in the gut. However, one possible link is that Flavobacteriaceae are often highly abundant on diatoms [39] and other phytoplankton [40], which are typical sources of food for C. finmarchicus. Therefore, Flavobacteriaceae (and perhaps other Cluster 2 OTUs) may reach high abundance within the copepod microbiome by “hitchhiking” into the gut on an ingested food particle. Alternatively, Cluster 2 OTUs may typically reside at low levels in the copepod gut, but grow rapidly on the nutrients provided when copepods feed. Regardless of mechanism, the presence of food in the gut (through its effect on Cluster 2) is a significant differentiator of copepod microbiomes across vertical depths.
Among copepods collected at a single vertical depth, we also identified differences in their associated bacterial communities that could be linked to host-specific morphological variability. In particular, among deep-dwelling copepods, we identified two distinct subgroups based upon their associated bacterial communities (Fig. 2). One subgroup corresponded to those that harbored OTUs from Clusters 3 and 6 at high relative abundance, while the other subgroup was comparatively depleted in OTUs from these clusters. Interestingly, of the six OTUs classified as Oceanospirillaceae across all clusters, four were found in Cluster 6 (comprising 80% of Cluster 6). This suggests that the Oceanospirillaceae present in our study change in abundance as a correlated unit defined by Cluster 6.
Using our regression approach, we found that the abundance of Cluster 6 relative to the core microbiome was positively associated with the fullness of a copepod’s oil sac, a repository of nutrient-rich lipids amassed during the transition to diapause (Fig. 5). To our knowledge, the effect of oil sac fullness on the copepod microbiome has not been explored previously, and thus, the mechanisms of this association remain unclear. We hypothesize that oil sac fullness does not directly influence the copepod microbiome, since it is a closed organ. Instead, it may reflect the environmental conditions while the copepod was developing at the surface, including prey abundance and composition, temperature (which affects size and metabolic rate), and seasonality.
Finally, we identified one cluster, Cluster 1, a hub in the association network, which mediates differences in the diversity of bacterial communities harbored by shallow- and deep-dwelling copepods. Notably, this cluster was a defining feature of the communities associated with deep-dwelling copepods: on average, the relative abundance of Cluster 1 was significantly higher among deep-dwelling copepods (mean: 48%) than among shallow-dwelling copepods (mean: 2%) (p = 4 × 10−25, two-sided Mann–Whitney U test). While this cluster contained a diverse mixture of taxonomic classes (Fig. 4), it was dominated by a single OTU (seq1) that, alone, reached a maximum relative abundance of nearly 90% on some individual copepods (Fig. 2). Surprisingly, seq1’s closest taxonomic relatives are from the genus Marinimicrobium, a common genus of Gammaproteobacteria whose association with brine shrimp has been documented previously [41], but whose associations with copepods have not been demonstrated. Thus, the bacterial communities associated with deep-dwelling copepods are dominated by OTUs whose roles in the copepod microbiome have not, to our knowledge, been previously described.
Despite the high abundance of Cluster 1, its underlying drivers remain unclear. Using our multivariate regression approach, we found that the abundance of Cluster 1 relative to the core microbiome was positively associated with copepod body volume and oil sac fullness and was negatively associated with shallow sampling depth and the presence of food in the gut, all of which suggest that Cluster 1 may become more abundant as copepods transition to diapause. Cluster 1’s associations with Clusters 5 and 6 also indicate the possible effects of inter-cluster interactions.
Conclusion
Previous studies have demonstrated that individual copepods serve as discrete “patches” on which bacterial communities form. Here, we leverage the statistical power afforded by highly resolved analysis of individuals within copepod populations to determine how bacterial communities are structured on individual copepod patches. We demonstrate that copepod-associated OTUs can be categorized into three groups, based on their distribution across individual copepods.
The first group is a core set of 34 OTUs shared across nearly all individual copepods sampled. Among the core OTUs were several taxonomic groups previously found associated with marine copepods including representatives from the genus Vibrionaceae and Moraxellaceae. Members of this conserved core may respond to ecological selective pressures that exist across copepod individuals.
The second group, a subset of 64 OTUs that were patchily distributed across copepod individuals, form discrete clusters, each of which is correlated with a unique set of local ecological selective pressures, including copepod-specific physiology (e.g., recent feeding) and co-colonizing bacteria. Among these, we identified multiple clusters that differentiate communities colonizing actively growing vs. diapausing copepods. These include Cluster 2, a Flavobacteriaceae-enriched cluster whose abundance is positively correlated with the presence of food in the copepod gut.
The third group, the remaining 143 patchily distributed OTUs, consists of OTUs whose ecological drivers remain unknown, but whose abundances vary significantly across individual copepods. We hypothesize that OTUs within this group are transiently associated with copepods, and thus, may not respond to ecological selective pressures on copepods.
Given the underlying organization of the copepod microbiome, further work should be aimed at understanding how these groups influence global ecosystem processes. For instance, patchily distributed OTUs associated with the presence of food in the gut may most directly influence carbon remineralization. Ultimately, future work incorporating these factors will lead to a better understanding of copepod–bacterial interactions, and how these interactions affect the global marine ecosystem. More generally, given the prevalence of microscale “patch” structures in other ecosystems, we believe this work may inspire future studies of microbial communities at local scales [16].
Materials and methods
Sampling of individual copepods and seawater
Copepods were sampled at Tröllet Station in Trondheimsfjord, Norway (63°29′N, 10°18′E; 430 m water depth) at two different depth strata (shallow, 0–50 m, and deep, 250–350 m deep) on two separate dates during the early summer (6 June 2012 and 11 June 2012), at which time, some individuals within the population had already descended from the surface ocean and entered into diapause. The surrounding seawater was also sampled within each depth stratum on each of the sampling dates. Detailed sampling protocols are available in Supplementary Methods.
Quantification of morphological characteristics
For all individual copepods, we quantified the prosome (body) volume, oil sac volume measured as a faction of its apparent size-specific maximum (fractional fullness), and the presence or absence of food in the gut. The presence or absence of food in the gut was assessed in live copepods—individual copepods were examined with a stereomicroscope for the presence of colored material in the gut, which indicates undigested algal matter or detritus. While C. finmarchicus is primarily herbivorous, they have been observed to consume microzooplankton during periods of low phytoplankton abundances [42], which may not be visually apparent. The season during which we sampled (late spring) is generally one of high phytoplankton abundance in Norwegian waters, so we expected that the main diet of the sampled C. finmarchicus to be phytoplankton, which is visible in the gut. All other morphological characteristics were quantified with digital images as described in Supplementary Methods.
Quantifying bacterial abundance on copepods and in seawater
The total number of bacteria present per copepod was quantified via fluorescence microscopy as previously described [48] with modifications described in Supplementary Methods.
DNA extraction from individual copepod and seawater samples
Total genomic DNA was extracted using a modification of a previously described protocol [49]. Briefly, samples were bead-beaten in 2-mL tubes with a mixture of 0.1-mm diameter silica, 1.4-mm diameter zirconium, and 4-mm diameter silica beads (OPS Diagnostics PFMM 4000-100-28). After treatments with lysozyme and proteinase K, genomic DNA was extracted from samples in phenol:chloroform:isoamyl alcohol (25:24:1 v/v) solution. Subsequently, DNA was precipitated from samples overnight in isopropanol with GlycoBlue (1 µL; Life Technologies #AM9516) as a co-precipitant. Precipitated DNA was resuspended in water (molecular biology grade) and stored at −20 °C. All copepod and seawater samples were extracted in a random order to avoid bias, and blanks were processed simultaneously. Details of DNA extraction procedure are given in Supplementary Methods.
16S rRNA amplicon sequencing of copepod- and seawater-associated bacterial communities
Amplicon libraries (16S rRNA gene V4 hypervariable region) were prepared with modifications to a previously described protocol [52]. Details are described in Supplementary Methods. To ensure that seawater and copepod samples were comparable, seawater samples were diluted 100-fold in water (molecular biology grade) such that identical amplification schemes could be used for all samples.
After amplicon libraries were prepared, samples were multiplexed for sequencing. Copepod 18S rRNA amplicons were removed from this mixture via gel extraction (NucleoSpin® Gel and PCR Clean-up kit, Macherey-Nagel #740609) before sequencing. Multiplexed, gel extracted samples were sequenced on an Illumina MiSeq (paired-end, 250 + 250) at the BioMicro Center (Massachusetts Institute of Technology, Cambridge, MA). Reads were merged and quality filtered with custom scripts calling USEARCH [43], mothur [50], and SmileTrain (https://github.com/almlab/SmileTrain). Details of 16S rRNA sequencing analysis are given in Supplementary Methods.
Identifying operational taxonomic units via distribution-based clustering
Operational taxonomic units (OTUs) were identified using distribution-based clustering (DBC), an algorithm that uses both genetic distance and the distribution of sequences across samples to group reads into OTUs. This approach reduces the number of OTUs with redundant information and improves the power of many downstream analyses to describe biologically relevant trends [52].
A two-step process was used to identify OTUs. First, the DBC algorithm was applied to quality-filtered reads with a high abundance threshold (--k_fold 10) to remove reads that likely arose through PCR/sequencing errors. This step generated 15,060 OTUs across all seawater and copepod samples. Second, the DBC algorithm was applied to this set of OTUs (95% identity threshold, --k_fold 0). This step merged OTUs with representative sequences that were >95% identical and were similarly distributed over samples. Altogether, this resulted in 9642 OTUs across all seawater and copepod samples.
Taxonomic assignment of individual OTUs was performed with the RDP Classifier [51]. OTUs that were annotated as “Cryptomonadaceae”, “Chlorophyta”, “Bacillariophyta”, “Streptophyta”, or “Chloroplast” were removed prior to normalization, since these OTUs are likely non-bacterial in origin. After removal of these OTUs, 9487 OTUs remained for subsequent analyses.
Identifying “abundant” OTU subset
From the 9487 non-“Chloroplast” OTUs, we identified a subset of 241 OTUs that were used for subsequent analyses. First, for each of the two sampling dates (6 June 2012 and 11 June 2012), we identified the list of OTUs whose mean relative abundance across all copepods sampled on that date was greater than 2 × 10−4. These lists comprised roughly 300 OTUs for each date. Second, we found the intersection of these two OTU lists, identifying a set of 241 OTUs whose mean relative abundance across both sampling dates was above the threshold (Fig. S1). Together, these 241 OTUs corresponded to 89.9% of sequenced reads from all copepod samples.
Analysis of sequencing blank samples
Given that copepod-associated bacterial communities provide low DNA input, analysis of analogously processed blank samples is important to ensure that reagent contamination does not dominate sequencing signal. To this end, we sequenced seven blank samples distributed across three 96-well plates, one of which contained only samples from ambient seawater, while the remainder contained only copepod-associated community samples.
Overall, we find 1925 sequenced reads in total across seven blank samples. Of those, 1507 reads came from two blank samples, both of which were on the ambient seawater 96-well plate. Sequences in these blanks were consistent with bacteria that are marine in origin, suggesting that contamination of the blanks by seawater may have occurred. However, samples from seawater had high input DNA concentrations, as well as high library concentrations compared to the blanks, suggesting that this contamination likely has only a minor effect on those samples. Furthermore, this contamination is unlikely to affect sequencing results from copepod samples, since they were prepared independently on separate 96-well plates.
In the remaining five blank samples (with 418 reads total), we found rare and inconsistent representation of the 241 OTUs that we focused on in this study. Of the 241 OTUs, 190 OTUs did not have a sequence representative in any blank sample. Among the remainder of OTUs, 32 OTUs were observed in one blank (20 by only 1 sequenced read), 12 OTUs in two blanks, and 7 OTUs in three blanks. No OTUs were observed in all blanks, suggesting that they did not arise due to consistent reagent contamination. Since OTUs were not observed consistently in the blanks, we have chosen not to remove them from the analysis. Instead, we have indicated in the text which OTUs of interest may be affected by the results from the blanks.
Defining the “core” and “patchily distributed” microbiome
Here, we defined the “core” microbiome as OTUs that were present at non-zero relative abundance in at least 90% of copepod-associated bacterial communities. All OTUs that did not meet this criterion were considered to be patchily distributed across copepod individuals. Note that our goal was to identify core OTUs shared across individual copepods in a conservative manner that avoids false positives. Ideally, this would entail a definition of “core” OTUs as those present on 100% of copepods sampled. However, due to low sequencing coverage and/or low fractional abundances of OTUs, we expect core OTUs to be missed in some fraction of samples. Furthermore, we found empirically that the distribution of OTUs across copepods is multimodal, with OTUs occupying ≥90% of copepods creating a large, distinct peak in a histogram of the number of copepods on which an OTU is represented (Fig. S2).
Classification of Vibrio OTUs from the core microbiome
To compare Vibrionaceae OTUs to previously sequenced Vibrionaceae, we first amassed a collection of over 500 Vibrio genomes, from NCBI GenBank. Consensus 16S rRNA sequences were identified for each genome by: (1) identifying all copies of 16S rRNA for a given genome with barrnap (https://github.com/tseemann/barrnap) and (2) identifying one or more consensus sequences with USEARCH [43]. Using the SILVA Incremental Aligner (SINA), we performed a multiple sequence alignment of these consensus sequences (536 in total) and the Vibrio OTU sequences observed in this study [44]. We generated a phylogenetic tree from this alignment with FastTree [45], using Shewanella as an outgroup.
Comparison of core OTUs to Moisander et al. (2015) study
Sequenced reads (16S rRNA gene, V3–V4 hypervariable regions) from [31] were downloaded from the NCBI SRA database [31]. Reads were quality filtered as previously described and were truncated to only include the V4 hypervariable region. Reads were dereplicated with USEARCH [43] in order to identify OTUs at 100% identity. OTU sequences from [31] were compared to those observed in this study using UBLAST [43].
Using PERMANOVA to identify broad associations between metadata and bacterial communities
To assess sources of variation across copepod-associated bacterial communities, we carried out a permutational MANOVA (PERMANOVA) [46]. All analyses were performed with the adonis function from the R vegan package with a Euclidean distance metric and 106 permutations.
Identifying correlated clusters of OTUs
We applied SparCC with the default parameters (−i 20, −x 10, −t 0.1) to compositional abundance data for the 241 OTUs whose mean abundance surpassed the defined threshold (Fig. S1). This procedure generated a 241 × 241 matrix of pairwise correlations between all OTUs. We then identified clusters of OTUs where the within-cluster correlation was strongly positive using a walktrap community detection algorithm. Details of computation can be found in Supplementary Methods.
Multivariate regressions
We used multivariate linear regression to identify associations between the bacterial cluster abundances and numerous predictors, including copepod morphological characteristics and the abundances of other bacterial clusters. Briefly, we (a) transformed the bacterial cluster relative abundance data with an additive log-ratio transform and (b) used Bayesian model averaging to calculate the probability that a given predictor has a non-zero effect on the cluster abundance across all possible regression models formulated from combinations of explanatory variables. Details of the model formulation and implementation of the analysis can be found in Supplementary Methods.
Electronic supplementary material
Acknowledgements
This work was supported in part by National Science Foundation grants OCE-1132567 to MFB and AMT and OCE-1435993 to MFP. MSD was supported by the Department of Defense through the National Defense Science and Engineering Graduate (NDSEG) Fellowship program. AAA was supported by an EPA STAR Fellowship, NSF GRFP, and the WHOI Ocean Venture Fund. We thank Sarah Preheim for help with performing the distribution-based clustering, Michael Cutler for providing advice regarding 16S rRNA library preparation, Nadine Lysiak for assistance in the field, Morgan Rubanow for assistance with the morphometric analysis, Krista Longnecker for her advice regarding the bacterial abundance counts, Bjørn Henrik Hansen, Dag Altin, Trond Nordtug, and Anders Olsen for significant logistical support in Trondheim, Norway, and the captain and crew of the R/V Gunnerus.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
The online version of this article (10.1038/s41396-018-0182-1) contains supplementary material, which is available to authorized users.
References
- 1.Turner JT. The importance of small planktonic copepods and their roles in Pelagic marine food webs. Zool Stud. 2004;43:255–66. [Google Scholar]
- 2.Jónasdóttir SH, Visser AW, Richardson K, Heath MR. Seasonal copepod lipid pump promotes carbon sequestration in the deep North Atlantic. Proc Natl Acad Sci USA 2015;2015:12110. [DOI] [PMC free article] [PubMed]
- 3.Møller EF, Riemann L, Søndergaard M. Bacteria associated with copepods: abundance, activity and community composition. Aquat Microb Ecol. 2007;47:99–106. doi: 10.3354/ame047099. [DOI] [Google Scholar]
- 4.Sochard MR, Wilson DF, Austin B, Colwell RR. Bacteria associated with the surface and gut of marine copepods. Appl Environ Microbiol. 1979;37:750–9. doi: 10.1128/aem.37.4.750-759.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tang KW, Turk V, Grossart HP. Linkage between crustacean zooplankton and aquatic bacteria. Aquat Microb Ecol. 2010;61:261–77. doi: 10.3354/ame01424. [DOI] [Google Scholar]
- 6.De Corte D, Lekunberri I, Sintes E, Garcia J, Gonzales S, Herndl GJ. Linkage between copepods and bacteria in the North Atlantic Ocean. Aquat Microb Ecol. 2014;72:215–25. doi: 10.3354/ame01696. [DOI] [Google Scholar]
- 7.Scavotto RE, Dziallas C, Bentzon-Tilia M, Riemann L, Moisander PH. Nitrogen-fixing bacteria associated with copepods in coastal waters of the North Atlantic Ocean. Environ Microbiol. 2015;17:3754–65. doi: 10.1111/1462-2920.12777. [DOI] [PubMed] [Google Scholar]
- 8.Tang KW, Glud RN, Glud A, Rysgaard S, Nielsen TG. Copepod guts as biogeochemical hotspots in the sea: evidence from microelectrode profiling of Calanus spp. Limnol Oceanogr. 2011;56:666–72. doi: 10.4319/lo.2011.56.2.0666. [DOI] [Google Scholar]
- 9.Turner JT. Zooplankton fecal pellets, marine snow, phytodetritus and the ocean’s biological pump. Prog Oceanogr. 2015;130:205–48. doi: 10.1016/j.pocean.2014.08.005. [DOI] [Google Scholar]
- 10.Bickel SL, Tang KW. Carbon substrate usage by zooplankton-associated bacteria, phytoplankton-associated bacteria, and free-living bacteria under aerobic and anaerobic conditions. Mar Biol. 2014;161:2233–42. doi: 10.1007/s00227-014-2501-z. [DOI] [Google Scholar]
- 11.Bickel SL, Tang KW. Microbial decomposition of proteins and lipids in copepod versus rotifer carcasses. Mar Biol. 2010;157:1613–24. doi: 10.1007/s00227-010-1434-4. [DOI] [Google Scholar]
- 12.Tang KW. Copepods as microbial hotspots in the ocean: effects of host feeding activities on attached bacteria. Aquat Microb Ecol. 2005;38:31–40. doi: 10.3354/ame038031. [DOI] [Google Scholar]
- 13.Møller EF, Thor P, Nielsen TG. Production of DOC by Calanus finmarchicus, C. glacialis and C. hyperboreus through sloppy feeding and leakage from fecal pellets. Mar Ecol Prog Ser. 2003;262:185–91. doi: 10.3354/meps262185. [DOI] [Google Scholar]
- 14.Carman KR, Dobbs FC. Epibiotic microorganisms on copepods and other marine crustaceans. Microsc Res Tech. 1997;37:116–35. doi: 10.1002/(SICI)1097-0029(19970415)37:2<116::AID-JEMT2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 15.Grossart HP, Dziallas C, Leunert F, Tang KW. Bacteria dispersal by hitchhiking on zooplankton. Proc Natl Acad Sci USA. 2010;107:11959–64. doi: 10.1073/pnas.1000668107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cordero OX, Datta MS. Microbial interactions and community assembly at microscales. Curr Opin Microbiol. 2016;31:227–34. doi: 10.1016/j.mib.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baumgartner MF, Tarrant AM. The physiology and ecology of diapause in marine copepods. Annu Rev Mar Sci. 2017;9:387–411. doi: 10.1146/annurev-marine-010816-060505. [DOI] [PubMed] [Google Scholar]
- 18.Gislason A, Silva T. Abundance, composition, and development of zooplankton in the Subarctic Iceland Sea in 2006, 2007, and 2008. ICES J Mar Sci. 2012;69:1263–76. doi: 10.1093/icesjms/fss070. [DOI] [Google Scholar]
- 19.Heath M, Boyle P, Gislason A, Gurney W, Hay S, Head E, et al. Comparative ecology of over-wintering Calanus finmarchicus in the northern North Atlantic, and implications for life-cycle patterns. ICES J Mar Sci. 2004;61:698–708. doi: 10.1016/j.icesjms.2004.03.013. [DOI] [Google Scholar]
- 20.Hirche HJ. Overwintering of Calanus finmarchicus and Calanus helgolandicus. Mar Ecol Prog Ser. 1983;11:281–90. doi: 10.3354/meps011281. [DOI] [Google Scholar]
- 21.Hirche HJ. Spatial distribution of digestive enzyme activities of Calanus finmarchicus and C.hyperboreus in Fram Strait/Greenland Sea. J Plankton Res. 1989;11:431–43. doi: 10.1093/plankt/11.3.431. [DOI] [Google Scholar]
- 22.Miller CB, Cowles TJ, Wiebe PH, Copley NJ, Grigg H. Phenology in Calanus finmarchicus; hypotheses about control mechanisms. 1991. http://ir.library.oregonstate.edu/xmlui/handle/1957/17727 (accessed March 28, 2016).
- 23.Arashkevich EG, Tande KS, Pasternak AF, Ellertsen B. Seasonal moulting patterns and the generation cycle of Calanus finmarchicus in the NE Norwegian Sea, as inferred from gnathobase structures, and the size of gonads and oil sacs. Mar Biol. 2004;146:119–32. doi: 10.1007/s00227-004-1416-5. [DOI] [Google Scholar]
- 24.Sartoris FJ, Thomas DN, Cornils A, Schnack-Schiel SB. Buoyancy and diapause in Antarctic copepods: the role of ammonium accumulation. Limnol Oceanogr. 2010;55:1860–4. doi: 10.4319/lo.2010.55.5.1860. [DOI] [Google Scholar]
- 25.Schrunder S, Schnack-Schiel SB, Auel H, Sartoris FJ. Control of diapause by acidic pH and ammonium accumulation in the hemolymph of Antarctic copepods. PLoS ONE. 2013;8:e77498. doi: 10.1371/journal.pone.0077498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tarrant AM, Baumgartner MF, Hansen BH, Altin D, Nordtug T, Olsen AJ. Transcriptional profiling of reproductive development, lipid storage and molting throughout the last juvenile stage of the marine copepod Calanus finmarchicus. Front Zool. 2014;11:1. doi: 10.1186/s12983-014-0091-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tarrant AM, Baumgartner MF, Verslycke T. Differential gene expression in diapausing and active Calanus finmarchicus (Copepoda) Mar Ecol Prog Ser. 2008;355:193–207. doi: 10.3354/meps07207. [DOI] [Google Scholar]
- 28.Aruda AM, Baumgartner MF, Reitzel AM, Tarrant AM. Heat shock protein expression during stress and diapause in the marine copepod Calanus finmarchicus. J Insect Physiol. 2011;57:665–75. doi: 10.1016/j.jinsphys.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 29.Miller CB, Crain JA, Morgan CA. Oil storage variability in Calanus finmarchicus. ICES J Mar Sci J Cons. 2000;57:1786–99. doi: 10.1006/jmsc.2000.0975. [DOI] [Google Scholar]
- 30.Jiao N, Herndl GJ, Hansell DA, Benner R, Kattner G, Wilhelm SW, et al. Microbial production of recalcitrant dissolved organic matter: long-term carbon storage in the global ocean. Nat Rev Microbiol. 2010;8:593–9. doi: 10.1038/nrmicro2386. [DOI] [PubMed] [Google Scholar]
- 31.Moisander PH, Sexton AD, Daley MC. Stable associations masked by temporal variability in the marine copepod microbiome. PloS ONE. 2015;10:e0138967. doi: 10.1371/journal.pone.0138967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shoemaker KM, Moisander PH (2015). Microbial diversity associated with copepods in the North Atlantic subtropical gyre. FEMS Microbiol Ecol. 10.1093/femsec/fiv064. [DOI] [PubMed]
- 33.Urbanczyk H, Ogura Y, Hayashi T. Taxonomic revision of Harveyi clade bacteria (family Vibrionaceae) based on analysis of whole genome sequences. Int J Syst Evol Microbiol. 2013;63:2742–51. doi: 10.1099/ijs.0.051110-0. [DOI] [PubMed] [Google Scholar]
- 34.Klindworth A, Pruesse E, Schweer T, Peplies J, Quast C, Horn M, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41:e1. doi: 10.1093/nar/gks808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yarza P, Yilmaz P, Pruesse E, Glöckner FO, Ludwig W, Schleifer KH, et al. Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol. 2014;12:635–45. doi: 10.1038/nrmicro3330. [DOI] [PubMed] [Google Scholar]
- 36.Gerdts G, Brandt P, Kreisel K, Boersma M, Schoo KL, Wichels A. The microbiome of North Sea copepods. Helgol Mar Res. 2013;67:757–73. doi: 10.1007/s10152-013-0361-4. [DOI] [Google Scholar]
- 37.Aitchison J. The statistical analysis of compositional data. London, New York: Chapman and Hall; 1986. [Google Scholar]
- 38.Raftery AE. Bayesian model selection in social research. Sociol Methodol. 1995;25:111. doi: 10.2307/271063. [DOI] [Google Scholar]
- 39.Amin SA, Parker MS, Armbrust EV. Interactions between diatoms and bacteria. Microbiol Mol Biol Rev. 2012;76:667–84. doi: 10.1128/MMBR.00007-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchan A, LeCleir GR, Gulvik CA, González JM. Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat Rev Microbiol. 2014;12:686–98. doi: 10.1038/nrmicro3326. [DOI] [PubMed] [Google Scholar]
- 41.Riddle MR, Baxter BK, Avery BJ. Molecular identification of microorganisms associated with the brine shrimp Artemia franciscana. Aquat Biosyst. 2013;9:7. doi: 10.1186/2046-9063-9-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ohman MD, Runge JA. Sustained fecundity when phytoplankton resources are in short supply: omnivory by Calanus finmarchicus in the Gulf of St. Lawrence. Limnol Oceanogr. 1994;39:21–36. doi: 10.4319/lo.1994.39.1.0021. [DOI] [Google Scholar]
- 43.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinforma Oxf Engl. 2010;26:2460–1. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 44.Pruesse E, Peplies J, Glockner FO. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics. 2012;28:1823–9. doi: 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Price MN, Dehal PS, Arkin AP. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010;5:e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
- 47.Fruchterman TMJ, Reingold EM. Graph drawing by force-directed placement. Softw Pract Exp. 1991;21:1129–64. doi: 10.1002/spe.4380211102. [DOI] [Google Scholar]
- 48.Tang KW, Bickel SL, Dziallas C, Grossart HP. Microbial activities accompanying decomposition of cladoceran and copepod carcasses under different environmental conditions. Aquat Microb Ecol. 2009;57:89–100. doi: 10.3354/ame01331. [DOI] [Google Scholar]
- 49.Zhou J, Bruns MA, Tiedje JM. DNA recovery from soils of diverse composition. Appl Environ Microbiol. 1996;62:316–322. doi: 10.1128/aem.62.2.316-322.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009;75:7537–7541. [DOI] [PMC free article] [PubMed]
- 51.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naïve Bayesian Classifier for Rapid Assignment of rRNA Sequences into the New Bacterial Taxonomy. Appl Environ Microbiol 2007;73:5261–5267. [DOI] [PMC free article] [PubMed]
- 52.Preheim SP, Perrotta AR, Martin-Platero AM, Gupta A, Alm EJ. Distribution-based clustering: using ecology to refine the operational taxonomic unit. Appl Environ Microbiol 2013;79:6593–6603. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.