

Abstract
Cells sense myriad signals during G1, and a rapid response to prevent cell cycle entry is of crucial importance for proper development and adaptation. Cln3, the most upstream G1 cyclin in budding yeast, is an extremely short‐lived protein subject to ubiquitination and proteasomal degradation. On the other hand, nuclear accumulation of Cln3 depends on chaperones that are also important for its degradation. However, how these processes are intertwined to control G1‐cyclin fate is not well understood. Here, we show that Cln3 undergoes a challenging ubiquitination step required for both degradation and full activation. Segregase Cdc48/p97 prevents degradation of ubiquitinated Cln3, and concurrently stimulates its ER release and nuclear accumulation to trigger Start. Cdc48/p97 phosphorylation at conserved Cdk‐target sites is important for recruitment of specific cofactors and, in both yeast and mammalian cells, to attain proper G1‐cyclin levels and activity. Cdk‐dependent modulation of Cdc48 would subjugate G1 cyclins to fast and reversible state switching, thus arresting cells promptly in G1 at developmental or environmental checkpoints, but also resuming G1 progression immediately after proliferative signals reappear.
Keywords: Cdc48, cell cycle, Cln3, ER release, Start
Subject Categories: Cell Cycle; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Cdc48, also called p97 in metazoans, is a highly conserved chaperone of the AAA‐ATPases family involved in cellular functions as diverse as endoplasmic reticulum‐associated degradation (ERAD), transcriptional control, cell cycle progression, membrane fusion, autophagy, and DNA repair (Jentsch & Rumpf, 2007; van den Boom & Meyer, 2018). Cdc48 forms a homohexameric complex, and each subunit contains two AAA domains that hydrolyze ATP inducing conformational changes in the whole complex (DeLaBarre & Brunger, 2005), and play essential functions in peptide unfolding by translocation (Bodnar & Rapoport, 2017). By means of multiple associated factors (Dargemont & Ossareh‐Nazari, 2012), Cdc48 binds ubiquitinated proteins and segregates them from protein complexes, cell membranes, and chromatin, for either proteasomal degradation, recycling, or functional release (Elsasser & Finley, 2005; Jentsch & Rumpf, 2007; van den Boom & Meyer, 2018). Regarding cell cycle progression, Cdc48 has been unequivocally shown to be involved in degradation of replication licensing factor Cdt1 in S phase (Franz et al, 2011; Raman et al, 2011), helicase dissociation after DNA replication (Maric et al, 2014), and, at the end of mitosis, in spindle disassembly (Cao et al, 2003) and chromatin decondensation (Mérai et al, 2014). However, although there is evidence to suggest a role of Cdc48 in cell cycle entry, its relevance under normal proliferation conditions is controversial (Archambault et al, 2004; Hsieh & Chen, 2011).
Irreversible commitment to cell cycle entry in budding yeast takes place at Start, a late G1 event (Hartwell et al, 1974). Cyclin Cln3 acts as the most upstream activator of Start, where a transcriptional wave driven by SBF (Swi6‑Swi4) and MBF (Mbp1‑Swi6) induces the expression of ca. 200 genes to trigger cell cycle entry (Ferrezuelo et al, 2010; Eser et al, 2011). Cyclin Cln3 forms a complex with Cdc28, the cell cycle Cdk in budding yeast, which phosphorylates the inhibitor Whi5 to activate G1/S transcription (Bertoli et al, 2013). Two other G1 cyclins, Cln1 and Cln2, are under G1/S transcriptional control and create a positive feedback loop that provides Start with robustness and irreversibility (Skotheim et al, 2008; Charvin et al, 2010). Remarkably, the core of the Start network is exquisitely conserved at the onset of the mammalian cell cycle, where Cdk4,6‑cyclin D complexes phosphorylate RB and activate E2F‑DP transcription factors in a positive feedback loop involving Cdk2‐cyclin E (Johnson & Skotheim, 2013).
Cell size homeostasis in budding yeast is a hallmark of proper coordination between growth and cell cycle entry machineries, which is mainly exerted through the Start network (Jorgensen & Tyers, 2004). Whi5, a key Start inhibitor synthesized during the previous cycle, is diluted by growth in G1 and has been proposed as a key factor for setting the critical size (Schmoller et al, 2015). On the other hand, as they are intrinsically unstable, G1 cyclins are thought to transmit growth rate information for adapting cell size to environmental conditions. Cln3 cyclin is kept at low and nearly constant levels throughout G1, but is retained at the ER to prevent premature nuclear accumulation and unscheduled execution of Start (Aldea et al, 2007). In late G1, Hsp70‐Hsp40 chaperones participate in releasing Cln3 from the ER, thus favoring accumulation of the Cdc28‐Cln3 complex in the nucleus to activate the G1/S regulon (Vergés et al, 2007). In mammalian cells, cyclin D1 depends on Hsp70 chaperone activity to form trimeric complexes with Cdk4 and NLS‐containing KIP proteins (p21, p27, p57) that drive their nuclear accumulation (Diehl et al, 2003). Ydj1, the same Hsp40 chaperone involved in releasing Cln3 from the ER, has also been shown to play an important role in the degradation mechanisms of this G1 cyclin (Yaglom et al, 1996). These conflicting effects suggest the existence of unknown mechanisms specifying the molecular fate of G1 cyclins at the earliest steps of cell cycle entry (Fig 1A).
Figure 1. Cdc48 regulates cell cycle entry and the critical size in budding yeast.
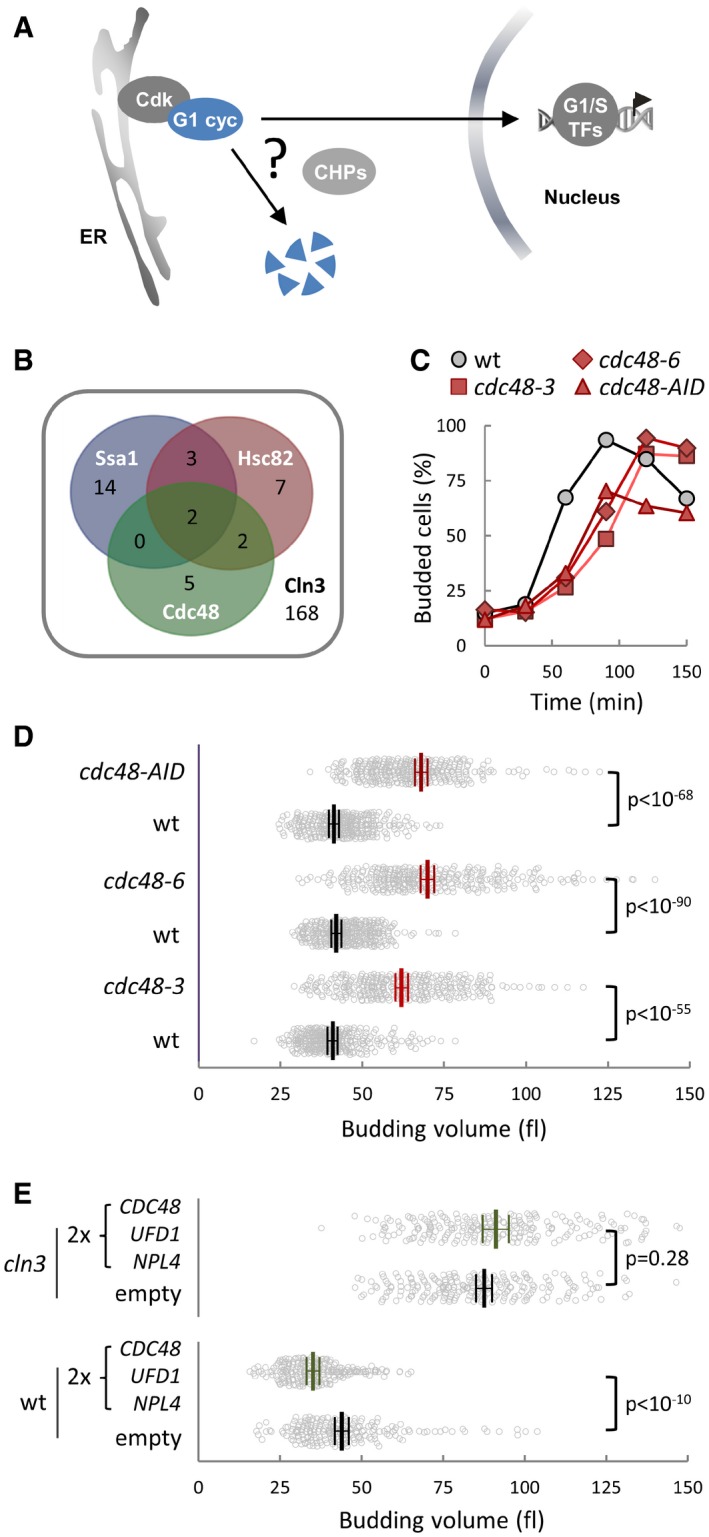
- Ssa1‐Ydj1 chaperones (CHPs) are important for nuclear accumulation of G1‐cyclin Cln3 but also for its proteasomal destruction. However, how these opposing fates are regulated is unknown.
- Venn diagram of physical interactors of Ssa1, Hsc82, and Cdc48 that display genetic or physical interactions with Cln3.
- Budding frequencies of wild‐type (wt), cdc48‐3, cdc48‐6, and cdc48‐AID newborn daughter cells during growth at the restrictive temperature (37°C) in the presence of auxin to induce degradation of Cdc48‐AID.
- Individual volumes at budding of cells as in (C). Mean values (N > 300, thick lines), confidence limits (α = 0.05, thin lines) for the mean and P‐values obtained from t‐tests are also shown.
- Individual volumes at budding of wild‐type (wt) and cln3 cells transformed with a centromeric vector empty (ctrl) or carrying the CDC48, UFD1, and NPL4 genes. Mean values (N > 200), confidence limits (α = 0.05, thin lines) for the mean and P‐values obtained from t‐tests are also shown.
Here, we analyze the functional contribution of Cdc48 to cell cycle entry and find that this AAA‐ATPase chaperone plays a key positive role at Start. Using a cell‐free system, we show that Cln3 is released from the ER in its ubiquitinated form in a Cdc48‐dependent manner. Cdk phosphosites in Cdc48 modulate its affinity for specific processing cofactors with deubiquitination activities affecting G1‐cyclin fate, thus defining a key step at Start that compromises G1‐cyclin function at the decision to enter or not the cell cycle.
Results
Cdc48 plays a positive role at Start and hinders Cln3 degradation
To identify putative regulators of chaperone activity with a specific role in G1, we performed a bioinformatics analysis on a compiled set of chaperone interactors previously identified in multiple proteomics studies, and selected those displaying genetic or physical interactions to cyclin Cln3 (Figs 1B and EV1A). Notably, among shared interactors of the major chaperone Hsp90 and Hsp70 systems we found Bre5 and Ubp3, which form a deubiquitinase complex and act in concert with the Cdc48 segregase. As the most upstream activator of Start, Cln3 is important for timely execution of Start and proper cell size control. Thus, we first tested whether Cdc48 is required for timely entry into the cell cycle. Newborn daughter cells carrying cdc48‐3 or cdc48‐6 thermosensitive alleles displayed a noticeable delay in budding and a concomitant increase in cell volume at budding when grown in G1 at the restrictive temperature (Fig 1C and D). Very similar results were obtained by rapid and efficient downregulation of Cdc48 with an auxin‐inducible degron (Figs 1C and D, and EV1B and C). Conversely, duplicating the copy number of CDC48 and substrate‐recognizing cofactors UFD1 and NPL4 produced a strong decrease in budding volume (Fig 1E), which was not observed in cells lacking Cln3. These data suggested that the Cdc48 segregase plays a positive role in the Start network, possibly by modulating Cln3 activity.
Figure EV1. Chaperone target proteins in the Cln3 interactome and genetic interactions of CDC48 in cell cycle entry and size determination.
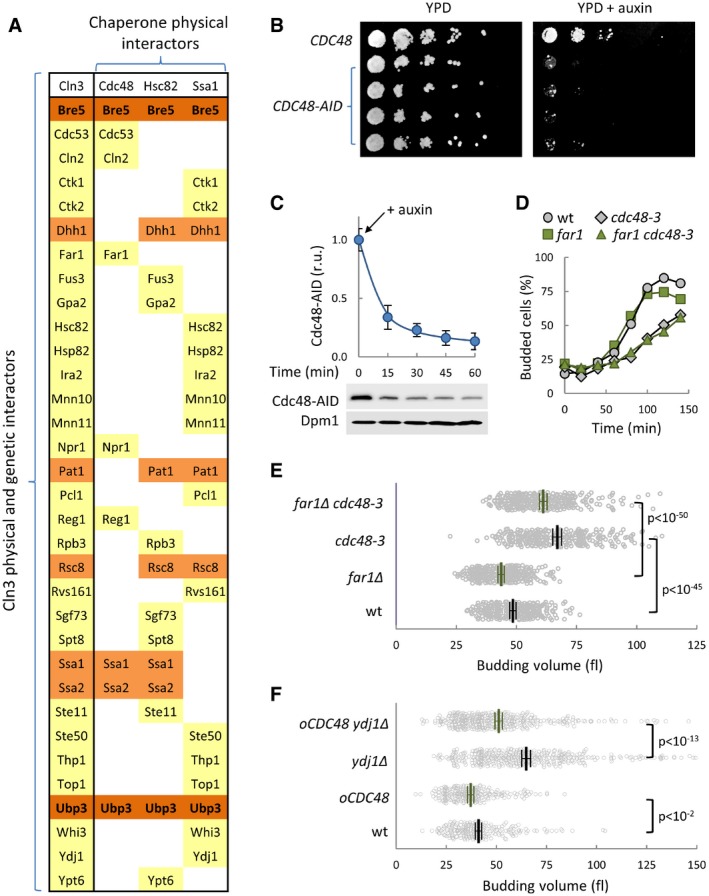
- Physical interactors of Ssa1, Hsc82, and Cdc48 that display genetic or physical interactions to Cln3 (SGD Project. http://www.yeastgenome.org 07/07/2017).
- Serial dilutions of four independent isolates expressing CDC48‐AID or CDC48 were plated and incubated for growth at 30°C for 2 days in the presence or absence of auxin.
- Cdc48‐AID levels in cells at different times from auxin addition. Dpm1 served as loading control and quantified levels with the confidence limits (α = 0.05) for the mean are plotted at the top.
- Budding frequencies of newborn daughter cells with the indicated genotypes during growth at the restrictive temperature (37°C).
- Individual volumes at budding of cells with the indicated genotypes. Mean values (N > 200, thick lines), confidence limits (α = 0.05, thin lines) for the mean and P values obtained from t‐tests are also shown.
- Individual volumes at budding of wild‐type and ydj1∆ cells overexpressing Cdc48 (oCDC48) or not. Mean values (N > 200, thick lines), confidence limits (α = 0.05, thin lines) for the mean, and P values obtained from t‐tests are also shown.
Source data are available online for this figure.
Cdc48 has been shown to regulate Far1 degradation during the G1/S transition (Fu et al, 2003). However, Far1 does not have a prominent role in cell cycle progression under unperturbed conditions and mainly acts to arrest cells in G1 in the presence of pheromone. As expected, we observed that cdc48‐3 cells displayed similar delays in G1 and increases in budding volume in both wild‐type and Far1‐deficient cells (Fig EV1D and E).
Cdc48 acts in concert with chaperones of the Hsp70‐Hsp40 family in ERAD (Vembar & Brodsky, 2008), and Ydj1 (an Hsp40 chaperone) is important for efficient ER release and proper activity of Cdc28‐Cln3 complexes at Start (Vergés et al, 2007; Ferrezuelo et al, 2012). Thus, we asked whether Cdc48 overexpression could suppress Cln3‐associated defects caused by loss of Ydj1. While ydj1Δ cells showed a large cell size phenotype (Vergés et al, 2007), expression of CDC48 from the GAL1p promoter considerably reduced the budding size of ydj1Δ cells (Fig EV1F). Notably, the relative reduction in cell size was clearly larger in ydj1Δ cells than that observed in wild‐type cells, which would point to convergent roles for Cdc48 and Ydj1 chaperones at Start.
Cln3 is an extremely short‐lived protein that is degraded by the proteasome in a ubiquitin‐dependent manner (Yaglom et al, 1995), and Cdc48 binds ubiquitinated proteins to facilitate proteasomal degradation, recycling, or functional release (Jentsch & Rumpf, 2007; Meyer et al, 2012). Thus, we decided to discern which of these functions of Cdc48 would be more relevant for Cln3. As shown in Fig 2A, cdc48‐3 cells displayed much lower levels of endogenously expressed Cln3‐3HA than wild‐type cells at the restrictive temperature, and Cdc48 overexpression lead to a concomitant increase in steady‐state levels of Cln3‐3HA (Fig 2B). CLN3‐3HA mRNA levels did not decrease in cdc48‐3 cells compared to wild type (Fig EV2A), and the hyperstable Cln3‐1 mutant did not change its levels in cdc48‐3 cells at the restrictive temperature (Fig EV2B), indicating that Cdc48 only acts at a post‐translational level on Cln3. Accordingly, Cln3 half‐life as measured by protein levels in the presence of cycloheximide was sharply reduced in cdc48‐3 cells at the restrictive temperature compared to wild‐type cells (Fig 2C and D). Thus, these data show that Cdc48 prevents Cln3 degradation, and reinforce the notion of a positive role of Cdc48 at Start.
Figure 2. Cdc48 prevents degradation and stimulates nuclear accumulation of Cln3.
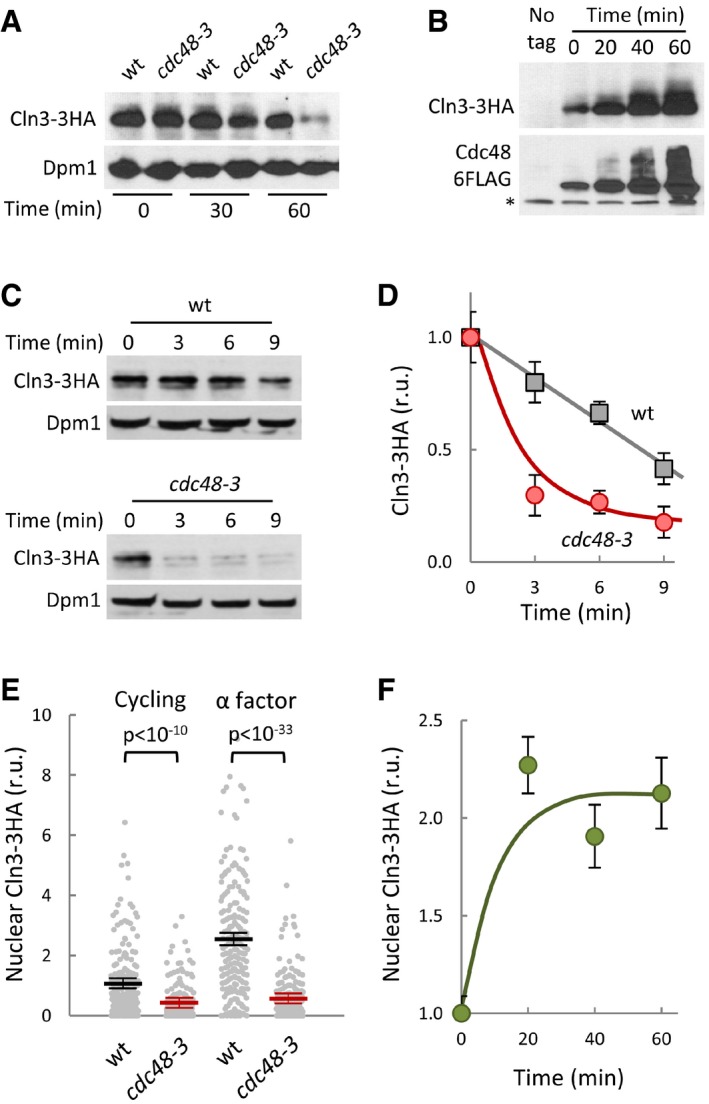
- Cln3‐3HA levels in wild‐type and cdc48‐3 cells at the indicated times after transferring cells to the restrictive temperature (37°). Dpm1 is shown as loading control.
- Cln3‐3HA levels at the indicated times after addition of β‐estradiol to induce the GAL1 promoter in GAL1p‐CDC48‐6FLAG cells expressing the Gal4‐hER‐VP16 (GEV) transactivator. The bottom panel shows Cdc48‐FLAG levels and a cross‐reacting band (*) that serves as loading control.
- Cln3‐3HA stability in Cdc48‐deficient cells. After being transferred to the restrictive temperature (37°C) for 30 min, cycloheximide was added to wild‐type and cdc48‐3 cells, which were collected at the indicated times to determine Cln3‐3HA levels. Dpm1 is shown as a loading control.
- Quantification of Cln3‐3HA levels in wild‐type (squares) and cdc48‐3 (circles) cells from immunoblot analysis as in (C). Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
- Nuclear accumulation of Cln3‐3HA in individual wild‐type or cdc48‐3 cells either cycling or arrested in late G1 with α factor as measured by semiautomated quantification of immunofluorescence levels in both nuclear and cytoplasmic compartments after 60 min at the restrictive temperature. Values were made relative to the average value obtained from wild‐type cells. Mean values (N = 200, thick horizontal lines), confidence limits (α = 0.05, thin lines) for the mean and P‐values obtained from t‐tests are also shown.
- Nuclear accumulation of Cln3‐3HA at the indicated times after overexpressing Cdc48 as in (B). Immunofluorescence analysis and calculations of relative mean values (N = 200) and confidence limits (α = 0.05) for the mean are as in (E).
Source data are available online for this figure.
Figure EV2. Cdc48 regulates Cln3 levels at a post‐translational level.
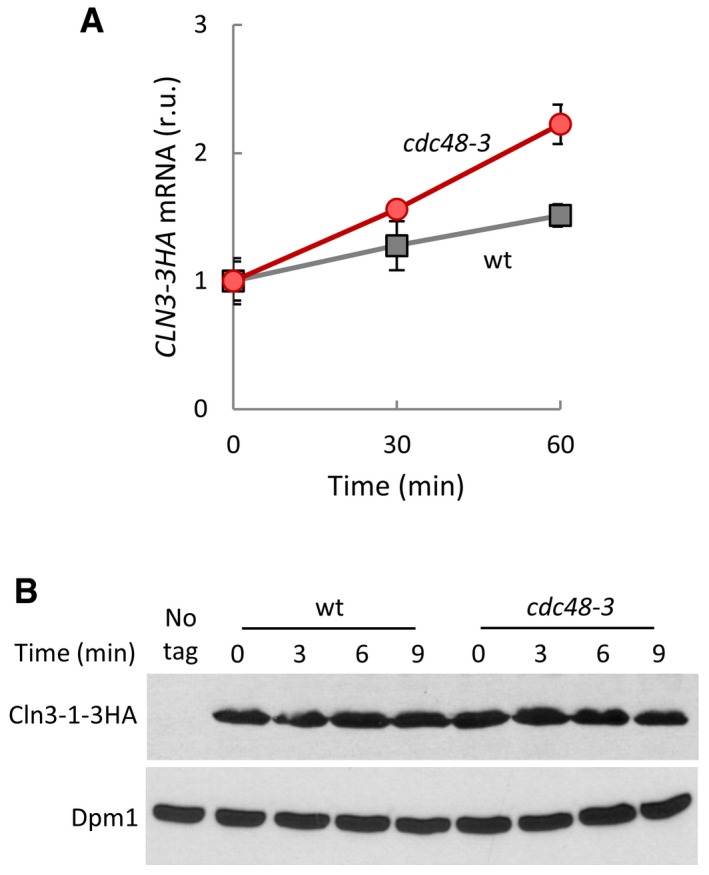
- CLN3‐3HA mRNA levels in wild‐type (wt) and cdc48‐3 cells at the indicated times after transferring cells to the restrictive temperature (37°C). Values were made relative to HXK1 mRNA and time 0. Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
- Cln3‐1‐3HA stability in Cdc48‐deficient cells. After being transferred to the restrictive temperature (37°C) for 30 min, wild‐type (wt) and cdc48‐3 cells expressing the hyperstable CLN3‐1‐3HA allele at endogenous levels were added cycloheximide and collected at the indicated times to determine protein levels. Dpm1 is shown as a loading control.
Source data are available online for this figure.
Consistent with its role in G1/S transcription activation, the ability of Cln3 to accumulate in the nucleus is a key functional trait (Edgington & Futcher, 2001; Miller & Cross, 2001; Wang et al, 2004). When we analyzed the nuclear levels of endogenously expressed Cln3‐3HA by immunofluorescence, we observed a significant decrease in cdc48‐3 cells compared to wild‐type cells (Fig 2E) 60 min after transfer to the restrictive temperature, when the total Cln3‐3HA protein is not yet greatly affected (Fig 2A). This effect was even more pronounced under conditions where nuclear accumulation of Cln3 is maximal, that is, in late‐G1 cells arrested by α‐factor (Vergés et al, 2007). As observed for total levels, nuclear accumulation of Cln3‐3HA also increased after overexpression of Cdc48 (Fig 2F), suggesting a limiting role for this segregase‐chaperone in both Cln3 stability and nuclear accumulation.
Cln3 is released from the ER in its ubiquitinated form
In a previous study, we adapted a protein mobilization method (Rape et al, 2001) to obtain a preliminary characterization of the Cln3 ER‐release mechanism in cell‐free extracts (Vergés et al, 2007), and here, we used cell extracts from a ydj1Δ strain that accumulate high levels of Cln3 in the ER‐bound fraction (Fig 3A, left panel), which were complemented with purified 6His‐Ydj1 before the assays. Notably, and consistent with the fact that Cln3 is degraded by the ubiquitin pathway (Yaglom et al, 1995) in a Ydj1‐dependent manner (Yaglom et al, 1996), we observed high levels of αHA cross‐reacting species in the high‐molecular‐weight range, which correspond to ubiquitinated forms of Cln3‐3HA as assessed by binding to beads with a ubiquitin‐binding domain (Fig 3A, right panel). After incubating cell extracts in the presence of purified Ydj1 (Fig EV3A) and an ATP‐regenerating system we observed that ubiquitinated Cln3‐3HA was mobilized into the soluble fraction with a much higher efficiency compared to unmodified Cln3‐3HA (Fig 3A, middle panel).
Figure 3. Cln3 is released from the ER by ubiquitination.
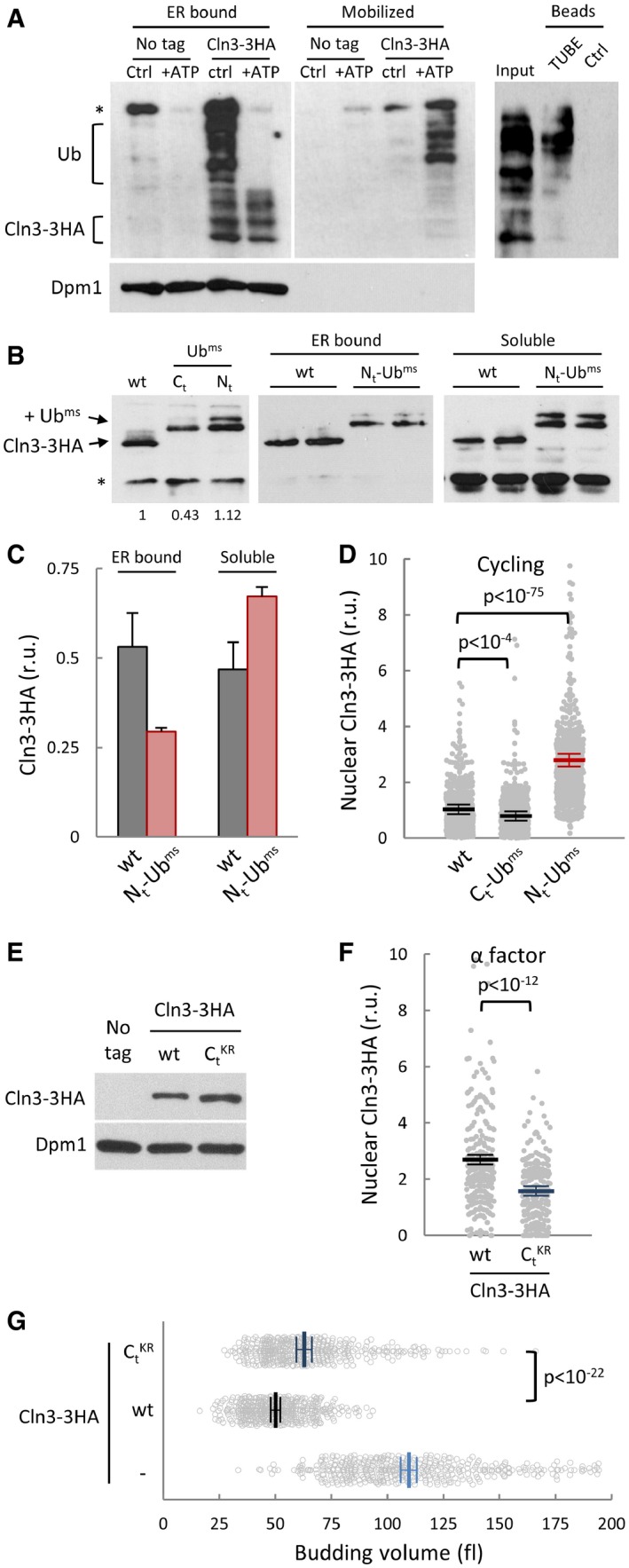
- Cell extracts from ydj1∆ CLN3‐3HA cells were complemented with 10 ng/μl purified Ydj1 protein and incubated for 30 min at 25°C (Ctrl) or in the presence of an ATP‐regeneration system (+ATP). Membrane (left panel) and mobilized soluble (middle panel) fractions were obtained to quantify released levels of Cln3‐3HA by immunoblotting. A high‐molecular‐weight αHA cross‐reacting band is marked (*), and Dpm1, an ER‐membrane protein, is shown as control for release specificity. Mobilized soluble fraction (Input) was added to either ubiquitin‐binding (TUBE) or control (Ctrl) beads and bound proteins analyzed by immunoblotting (right panel). High‐molecular‐weight species corresponding to ubiquitinated Cln3‐3HA are indicated (Ub).
- Protein levels of wild‐type Cln3‐3HA and monomeric stable ubiquitin (Ubms, see text for details) fusions at the N‐terminus (Nt‐Ubms) or C‐terminus (Ct‐Ubms) of Cln3‐3HA were analyzed by immunoblotting (left panel). A low‐molecular‐weight αHA cross‐reacting band is marked (*). Membrane (middle panel) and soluble (right panel) fractions were obtained to quantify levels of wild type and Nt‐Ubms Cln3‐3HA by immunoblotting.
- Quantification of wild‐type and Nt‐Ubms Cln3‐3HA levels from immunoblot analysis as in (B). Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
- Nuclear accumulation of wild‐type, Ct‐Ubms, and Nt‐Ubms Cln3‐3HA in individual cycling cells was analyzed by immunofluorescence as in Fig 2E. Relative mean values (N = 500, thick horizontal lines), confidence limits (α = 0.05, thin lines) for the mean, and P‐values obtained from t‐tests are also shown.
- Protein levels of 3HA‐tagged wild‐type and a KR substitution mutant in the last 180 () amino acids of Cln3 were analyzed by immunoblotting. Dpm1 is shown as a loading control.
- Nuclear accumulation of wild type and Cln3‐3HA in individual cells arrested in late G1 with α factor was analyzed by immunofluorescence as in Fig 2E. Relative mean values (N > 200, thick horizontal lines), confidence limits (α = 0.05, thin lines) for the mean and P‐values obtained from t‐tests are also shown.
- Individual volumes at budding of cln3 cells expressing wild type or Cln3‐3HA or none. Mean values (N > 400, thick vertical lines), confidence limits (α = 0.05, thin lines) for the mean, and P‐values obtained from t‐tests are also shown.
Source data are available online for this figure.
Figure EV3. Mobilization, nuclear accumulation, and interaction of Cln3 with Cdc48.
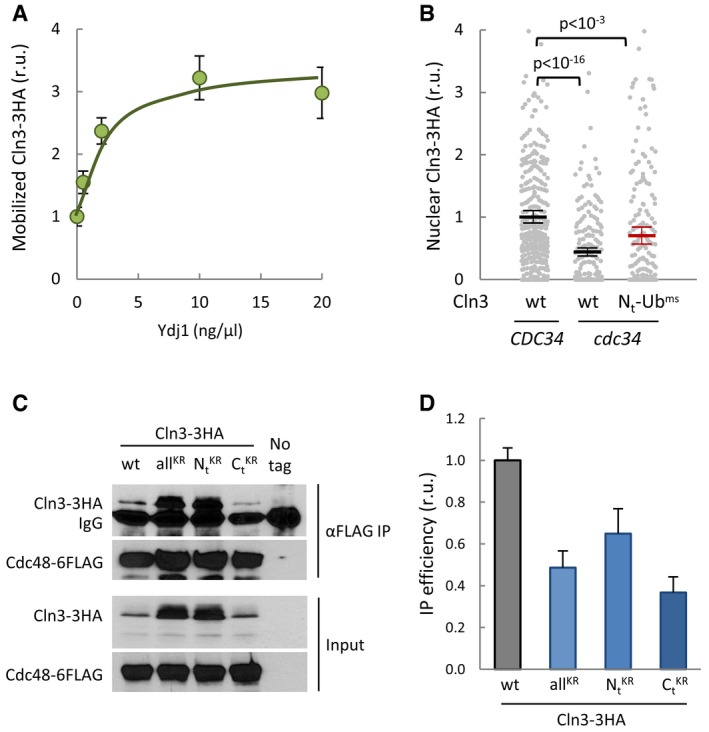
- Cell extracts from ydj1∆ CLN3‐3HA cells were incubated for 30 min at 27°C in the presence of an ATP‐regeneration system and the indicated concentrations of purified Ydj1. Mobilized soluble fractions were obtained to analyze released Cln3‐3HA by immunoblotting. Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
- Nuclear accumulation of wild‐type and Nt‐Ubms Cln3‐3HA in individual wild‐type CDC34 or thermosensitive cdc34 cells growing at the restrictive temperature was analyzed by immunofluorescence as in Fig 2E. Relative mean values (N = 500, thick horizontal lines), confidence limits (α = 0.05, thin lines) for the mean and P values obtained from t‐tests are also shown.
- Cell extracts (Input) and αFLAG immunoprecipitates (αFLAG IP) of cells expressing Cdc48‐6FLAG and wild‐type, allKR, , or Cln3‐3HA proteins were analyzed by immunoblotting with either αHA or αFLAG antibodies. The αFLAG heavy chain (IgG) band is also marked.
- Quantification of Cln3‐3HA levels from immunoblots in (C). Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
Source data are available online for this figure.
To analyze the role of ubiquitin in the ER‐release mechanism, we fused Cln3‐3HA to a monomeric stable ubiquitin (Ubms; Ndoja et al, 2014) lacking the C‐terminal glycines to avoid enzymatic removal but containing two amino acid substitutions (K11R and K48R) that prevent major polyubiquitination events (Komander & Rape, 2012) and avoid major effects on protein stability. In any event, since the presence of a ubiquitin peptide could have different effects as N or C‐terminal fusions, we first analyzed overall protein levels in both possible configurations. A fusion to the C‐terminus of Cln3‐3HA caused a minor but noticeable decrease in protein levels (Fig 3B). Since C‐terminal Ubims is immediately adjacent to the PEST regions of Cln3 in this construct, it could facilitate Ub‐binding‐dependent polyubiquitination at nearby residues in Cln3 and, hence, increase degradation. On the contrary, the N‐terminal fusion was expressed at levels very similar to wild‐type Cln3‐3HA (Fig 3B) and we therefore chose this construct for further analysis. Reinforcing the notion that the presence of ubiquitin facilitates release of Cln3 from the ER, we observed that Ubms‐Cln3‐3HA was present at higher levels in the soluble fraction of crude cell extracts compared to Cln3‐3HA (Fig 3B and C). UbiK48R does not affect stability of a fused heterologous protein but is still monoubiquitinated in K29 (Johnson et al, 1995), which could explain the additional slowly migrating band shown in Fig 3B by our Nt‐Ubims fusion containing K11R and K48R mutations. Remarkably, this form was even more soluble in relative terms than the faster migrating form. Moreover, Ubms‐Cln3‐3HA was also detected at higher levels in the nucleus of cycling yeast cells (Fig 3D). We had found that Cdc34, an E2 ubiquitinating enzyme important for Cln3 degradation (Yaglom et al, 1995), also was required for nuclear accumulation of Cln3 in late G1 (Vergés et al, 2007), perhaps by facilitating its release from the ER. Hence, we tested whether Ubms‐Cln3‐3HA did require Cdc34 for nuclear accumulation and found that the presence of a ubiquitin monomer on Cln3 suppressed most of its dependence on Cdc34 to accumulate in the nucleus (Fig EV3B). While the C‐terminal regions of Cln3 contain the most important determinants for ubiquitin‐mediated degradation (Yaglom et al, 1995), the lysines targeted by ubiquitination are unknown. Thus, we introduced lysine to arginine substitutions across the whole C‐terminal degron of Cln3. Although the Cln3‐‐3HA mutant protein attained higher levels than wt (Fig 3E), it displayed a lower nuclear signal (Fig 3F) and only partially complemented the cln3Δ strain (Fig 3G). Overall, these data reinforce the notion that ubiquitin plays a positive role in the release from the ER and nuclear accumulation of Cln3.
Cdc48 interacts with Cln3 and is required for ER release
Cdc48 plays a key role in the mechanisms that release transcription factor Spt23 from the ER to allow its accumulation in the nucleus (Rape et al, 2001). Thus, we wanted to test whether Cdc48, in addition to its role hindering Cln3 degradation, also participates in releasing the G1 cyclin from the ER. Likely due to its extremely short half‐life, we were unable to detect ubiquitinated forms of Cln3‐3HA in whole cell extracts from wild‐type cells. Nonetheless, since overexpression of Cdc48 significantly increased Cln3‐3HA protein levels (Fig 2B), we asked whether these conditions would allow the detection of ubiquitinated forms of Cln3. Co‐immunoprecipitation experiments indicated that Cdc48‐6FLAG and Cln3‐3HA physically interact in vivo (Fig 4A). Remarkably, the immunoprecipitated fraction was enriched in high‐molecular‐weight forms of Cln3‐3HA compared to the whole cell extract used as input. Moreover, as shown in Fig 4B, high‐molecular‐weight species specifically present in the αHA immunoprecipitate were clearly recognized by an αUb antibody. Finally, we found that Cdc48‐6FLAG co‐immunoprecipitation efficiencies sharply decreased when analyzing Cln3 mutants with different sets of KR substitutions (Fig EV3C and D). Cdc48 pulldowns also contained non‐ubiquitinated forms of Cln3‐3HA, an observation that has also been made for Far1 (Fu et al, 2003), Cdc5 (Cao et al, 2003), and p120 Mga2 (Shcherbik & Haines, 2007). Moreover, both sumoylated and unmodified Rad52 have been found in Cdc48 pulldowns (Bergink et al, 2013). Once ubiquitinated target proteins are bound to Cdc48 through ATPase‐dependent conformational changes, ubiquitin removal by associated deubiquitinating enzymes could explain the presence of non‐ubiquitinated target proteins in Cdc48 pulldowns.
Figure 4. Cdc48 interacts with Cln3 and plays a key role in release from the ER .
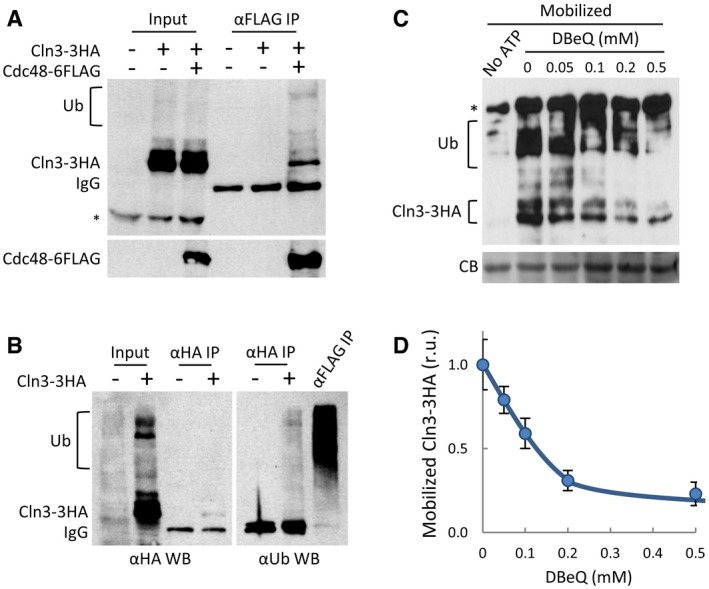
- Cell extracts (Input) and αFLAG immunoprecipitates (αFLAG IP) of cells expressing Cln3‐3HA and Cdc48‐6FLAG proteins were analyzed by immunoblotting with either αHA (top panel) or αFLAG (bottom panel) antibodies. High‐molecular‐weight species corresponding to ubiquitinated Cln3‐3HA are indicated (Ub). αFLAG heavy chain (IgG) and high‐molecular‐weight αHA cross‐reacting (*) bands are also marked.
- Cell extracts (Input), and αHA (αHA IP) immunoprecipitates of cells expressing Cln3‐3HA or not were analyzed by immunoblotting with either αHA (left panel) or αUbiquitin (right panel) antibodies. An αFLAG immunoprecipitate (αFLAG IP) is shown as a positive control to identify all ubiquitinated proteins bound to Cdc48. High‐molecular‐weight species corresponding to ubiquitinated proteins are indicated (Ub). Antibody heavy chain (IgG) bands are also marked.
- Cell extracts from ydj1∆ CLN3‐3HA cells as in Fig 3A were incubated for 30 min at 25°C in the presence of purified Ydj1, an ATP‐regeneration system, and the indicated DBeQ concentrations. Mobilized soluble fractions were obtained to analyze released Cln3‐3HA by immunoblotting. High‐molecular‐weight species corresponding to ubiquitinated Cln3‐3HA are indicated (Ub). A high‐molecular‐weight αHA cross‐reacting band is marked (*), and a prominent Coomassie Blue (CB) band is shown as loading control.
- Quantification of mobilized Cln3‐3HA as a function of DBeQ concentration from experiments as in (C). Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
Source data are available online for this figure.
To test the role of Cdc48 in Cln3 release from the ER, we used again the protein mobilization assay and quantified the mobilized soluble Cln3‐3HA levels in the presence of DBeQ, a selective and ATP‐competitive inhibitor of Cdc48. Figure 4C shows that DBeQ readily hampered mobilization of Cln3‐3HA into the soluble fraction in a dose‐dependent manner, reaching a fivefold decrease in the levels of released Cln3‐3HA when using DBeQ at 0.5 mM (Fig 4D). Very similar results were obtained when using NMS‐873 (Fig EV4A and B), an allosteric inhibitor of Cdc48 (Magnaghi et al, 2013) unrelated to DBeQ. Moreover, extracts from cells that had been depleted from Cdc48 by the auxin‐inducible degron also displayed lower efficiencies of Cln3‐3HA release (Fig EV4C and D). ER‐release assays in cell‐free extracts were done in the presence of proteasome inhibitors, thus ruling out the participation of the proteasome cleaving an anchoring protein as is the case for Spt23 (Rape et al, 2001).
Figure EV4. Cdc48 interacts with Cln3 and plays a key role in release from the ER .
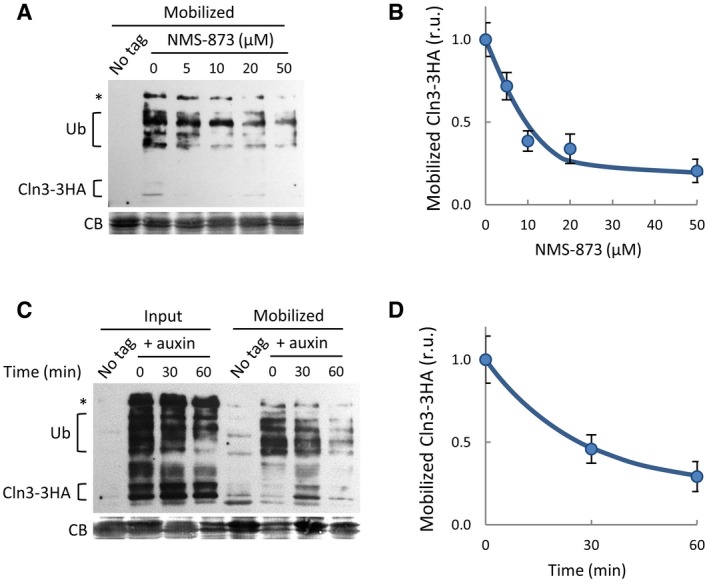
- Mobilized soluble fractions were obtained as in Fig 3A in the presence of NMS‐873 at the indicated concentrations to analyze released Cln3‐3HA by immunoblotting. High‐molecular‐weight species corresponding to ubiquitinated Cln3‐3HA are indicated (Ub). A high‐molecular‐weight αHA cross‐reacting band is marked (*), and a prominent Coomassie Blue (CB) band is shown as loading control.
- Quantification of mobilized Cln3‐3HA as a function of NMS‐873 concentration from experiments as in (A). Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
- Mobilized soluble fractions were obtained as in Fig 3A with extracts from Ydj1‐deficient cells expressing Cdc48‐AID collected at different times after auxin addition. High‐molecular‐weight species corresponding to ubiquitinated Cln3‐3HA are indicated (Ub). A high‐molecular‐weight αHA cross‐reacting band is marked (*), and a prominent Coomassie Blue (CB) band is shown as loading control.
- Quantification of mobilized Cln3‐3HA as a function of Cdc48‐AID levels from experiments as in (C). Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
Source data are available online for this figure.
Cln3 contains a hydrophobic domain (HD) that is not present in the other two G1 cyclins, Cln1 and Cln2 (Fig 5A). Compared to wild type, a Cln3 protein that lacks the HD did not accumulate as efficiently in ER‐containing fractions obtained from sucrose gradients (Fig 5B). Moreover, fusing HD to GFP caused this soluble reporter protein to display a punctate cytoplasmic pattern (Fig 5C) strikingly similar to that shown by endogenous Cln3‐3HA (Vergés et al, 2007), and to switch its distribution toward dense ER fractions (Fig 5D). This hydrophobic domain is predicted as being composed of two consecutive transmembrane helices (Persson & Argos, 1994), suggesting that it acts as a direct anchor to the ER membrane. Intriguingly, Cln3ΔHD only partially complements a Cln3‐deficient mutant, which indicates that the hydrophobic domain has additional functional roles. In any event, since Cdc48 has been shown to participate in the extraction of membrane‐embedded proteins for their degradation during ERAD (Jentsch & Rumpf, 2007; van den Boom & Meyer, 2018), our data would provide a mechanistic explanation to the role of Cdc48 in releasing Cln3 from the ER, in this case to prevent degradation and allow nuclear accumulation of this G1 cyclin for activating the G1/S regulon at Start.
Figure 5. A hydrophobic domain in Cln3 mediates retention at the ER .
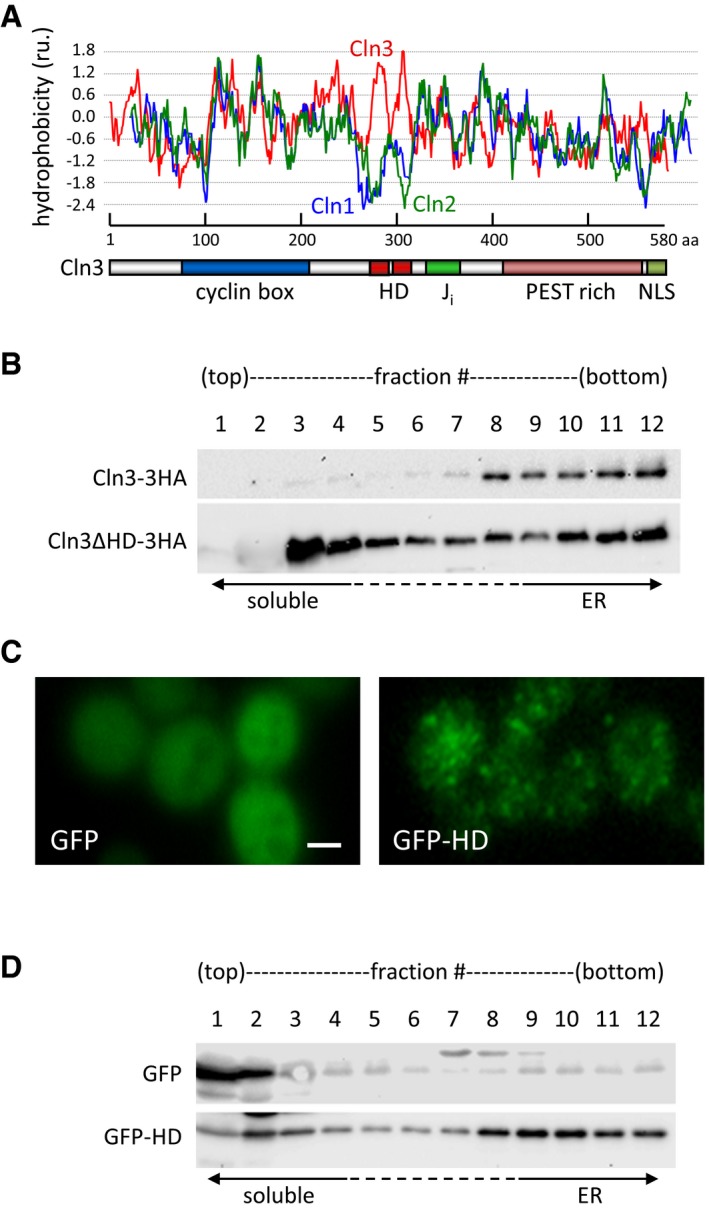
- Hydrophobicity profile of aligned G1 cyclins, Cln1, Cln2, and Cln3. The graph shows Kyte & Doolittle scale mean hydrophobicity values (scan window = 13). A scheme at the bottom shows the different domains in Cln3 (cyclin box, J i, PEST, and NLS), as well as the identified hydrophobic domain (HD).
- Total extracts from cells expressing Cln3‐3HA or Cln3ΔHD‐3HA at endogenous levels were fractionated in 20–60% sucrose gradients, and fractions obtained were analyzed by immunoblotting with αHA antibodies.
- Representative images of cells expressing GFP or GFP‐HD. Scale bar is 2 μm.
- Total extracts from cells expressing GFP or GFP‐HD were fractionated in 20–60% sucrose gradients, and fractions obtained were analyzed by immunoblotting with αGFP antibodies.
Source data are available online for this figure.
Cdk phosphosites in Cdc48 modulate its role in Cln3 fate
Two putative Cdk phosphosites in the second AAA‐ATPase domain of Cdc48 (Fig 6A and B) are largely conserved from yeast to humans (Fig 6C). Confirming the specificity of these putative phosphosites, we found that Cdk4‐Ccnd1 efficiently phosphorylated in vitro a yeast Cdc48 fragment that includes both sites (Fig 6D), but not a double S519A T674A mutant. Both sites are efficient targets of the Cdk, as introduction of a single mutation reduced, but not abolished, the phosphorylation signal. To analyze phosphorylation efficiencies of S519 in G1 vs. cycling cells, we first analyzed yeast cells either progressing through the cell cycle or arrested in G1 with α factor. Applying mass spectrometry analysis to Cdc48 pulldowns, we observed a significant increase in S519 phosphorylation in cycling cells compared to G1‐arrested cells (Figs 6E and EV5). To confirm these data, we analyzed cells either arrested in G1 by G1‐cyclin depletion or 20 min after CLN3 induction to trigger Start. As shown in Fig 6E, Cdc48 obtained from cells executing Start displayed a ca. fourfold increase in S519 phosphorylation. Cdc48 phosphorylation at S519 by Cdc28 has been reported in a high‐throughput experiment where yeast cells had been arrested in mitosis with high Clb/Cdk1 activity (Holt et al, 2009). Taken together, these data would indicate that Cdc48 is phosphorylated by both Cln/Cdc28 and Clb/Cdc28, from late G1 until anaphase.
Figure 6. Cdc48 is regulated by Cdk phosphorylation.
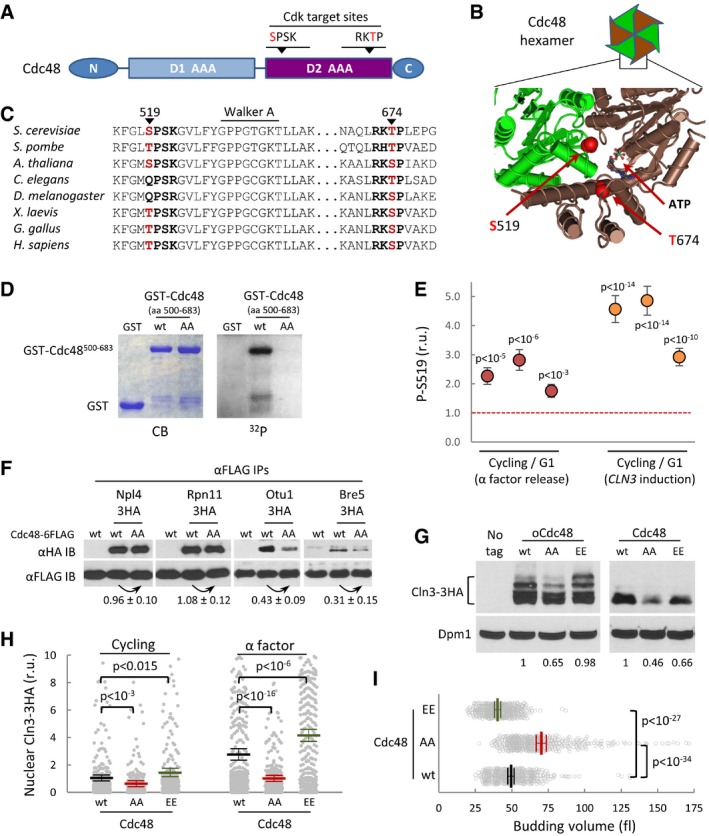
- Schematic representation of the Cdc48 protein, with special reference to the position of two Cdk phosphosites within the second AAA‐ATPase domain (D2).
- Spatial location of S519 and T674 Cdk phosphosites at the D2 interface of two monomers in the Cdc48 hexameric ring (PDB 3CF1).
- Amino acid sequence alignment of the Cdk phosphosite regions of Cdc48 from various organisms ranging from yeast to mammals.
- In vitro kinase assay of wild‐type and double S519A T674A mutant (AA) yeast Cdc48. Specified GST fusions were incubated with Cdk4‐cyclin D1 in the presence of [γ32P]‐ATP, subsequently analyzed by SDS–PAGE, and stained (CB) or detected by autoradiography (32P).
- Phosphorylation levels of S519 in vivo by mass spectrometry analysis. Cycling and G1 cells obtained with α factor (red circles) or by G1‐cyclin depletion and CLN3 induction (orange circles) were used to determine relative S519 phosphorylation levels as described under Materials and Methods, and obtained ratios for three independent experiments are plotted. Error bars correspond to the standard deviation estimated from measurements of control peptide ratios in individual samples (N = 3). The null hypothesis of a true ratio equal to 1 was tested and the resulting P‐values are indicated assuming that control and problem peptide ratios are homoscedastic and normally distributed.
- αFLAG immunoprecipitates (αFLAG IP) of cells expressing wild‐type (wt) or non‐phosphorylatable (AA) Cdc48‐6FLAG and the indicated HA‐tagged Cdc48 cofactors were analyzed by immunoblotting with either αHA or αFLAG antibodies. Co‐immunoprecipitated levels of the analyzed cofactors were made relative to immunoprecipitated wild‐type or non‐phosphorylatable Cdc48‐FLAG, and numbers indicate the mean (N = 3) fold change found in non‐phosphorylatable relative to wild‐type Cdc48 samples. Confidence limits for the mean (α = 0.05) are also indicated.
- Cln3‐3HA levels in cells expressing wild‐type (wt), non‐phosphorylatable (AA), or phosphomimetic (EE) Cdc48 proteins at endogenous levels or from the GAL1p promoter as in Fig 2B. Dpm1 is shown as a loading control. Relative levels of Cln3‐3HA quantified from the image are indicated at the bottom.
- Nuclear accumulation of Cln3‐3HA in individual cells expressing wild‐type (wt), non‐phosphorylatable (AA), or phosphomimetic (EE) Cdc48 proteins at endogenous levels. Immunofluorescence analysis was performed in cycling or late‐G1 arrested cells as in Fig 2E. Relative mean values (N > 275, thick horizontal lines), confidence limits (α = 0.05, thin lines) for the mean and P‐values obtained from t‐tests are also shown.
- Individual volumes at budding of cells expressing wild‐type (wt), non‐phosphorylatable (AA), or phosphomimetic (EE) Cdc48 proteins at endogenous levels. Mean values (N = 450, thick vertical lines), confidence limits (α = 0.05, thin lines) for the mean, and P‐values obtained from t‐tests are also shown.
Source data are available online for this figure.
Figure EV5. Identification of Cdc48 peptides containing pS519 and pS770 by mass spectrometry analysis.
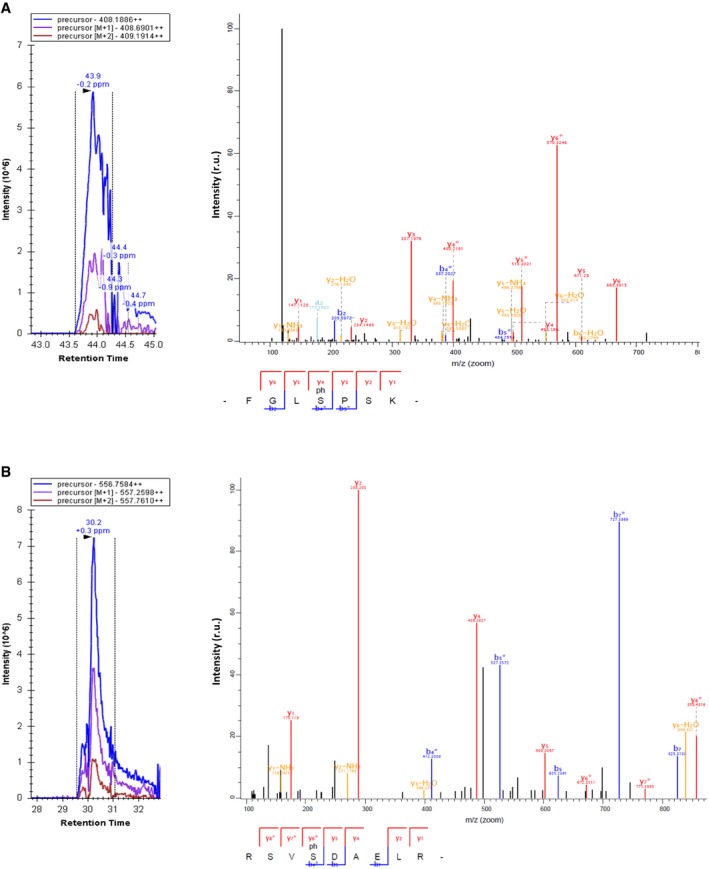
-
A, BCycling cells expressing GST‐Cdc48 at endogenous levels were used to prepare cell extracts and purify GST‐Cdc48 for MS analysis. Representative ion chromatograms (left panels) and fragmentation spectra (right panels) of ions corresponding to pS519 (A) and pS770 (B) peptides are shown.
The two identified phosphosites lie at the second AAA‐ATPase domain, close to the C‐terminal region of Cdc48, where it interacts with substrate‐processing cofactors (Rumpf & Jentsch, 2006; Ossareh‐Nazari et al, 2010). Thus, we asked whether the Cdk phosphosites affect the interaction between Cdc48 and cofactors that could explain the observed effects on Cln3 fate. Notably, interaction efficiencies of non‐phosphorylatable Cdc48 to deubiquitinases Bre5 and Otu1 were clearly reduced compared to wild‐type Cdc48 (Fig 6F). By contrary, interaction to a substrate‐recruiting factor (Npl4) and a proteasomal deubiquitinating protein (Rpn11) was unaffected. Reinforcing a role in modulating Cln3 deubiquitination, we found that cells lacking Bre5 or Otu1 displayed higher steady‐state levels of ubiquitinated Cln3 (Fig EV6A). These data point to the notion that Cdc48 activity is shifted toward releasing functions by Cdk‐dependent phosphorylation.
Figure EV6. Cln3 fate and Cdc48 phosphorylation mutant proteins.
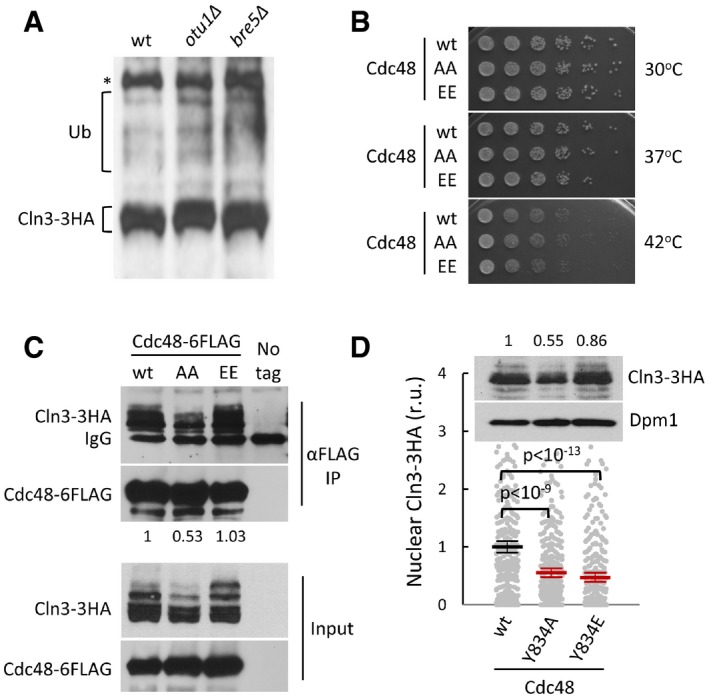
- Cell extracts from wild‐type, otu1∆ and bre5∆ cells expressing CLN3‐3HA were analyzed by immunoblotting. High‐molecular‐weight species corresponding to ubiquitinated Cln3‐3HA are indicated (Ub). A high‐molecular‐weight αHA cross‐reacting band is marked (*).
- Cells expressing wild‐type (wt), non‐phosphorylatable (AA), or phosphomimetic (EE) Cdc48 proteins at endogenous levels were diluted, spotted onto YPD plates and grown at 30, 37 or 42°C.
- Cell extracts (Input) and αFLAG immunoprecipitates (αFLAG IP) of cells expressing Cln3‐3HA and wild‐type (wt), non‐phosphorylatable (AA), or phosphomimetic (EE) Cdc48‐6FLAG proteins from the GAL1p promoter as in Fig 6G (same experiment) were analyzed by immunoblotting with either αHA or αFLAG antibodies. The αFLAG heavy chain (IgG) band is also marked. Indicated numbers refer to the relative levels of Cln3‐3HA in immunoprecipitated samples as quantified from image.
- Nuclear accumulation of Cln3‐3HA in individual cells expressing wild‐type (wt), tyrosine non‐phosphorylatable (Y834A), or tyrosine phosphomimetic (Y834E) Cdc48 proteins at endogenous levels. Immunofluorescence analysis was performed in cycling cells as in Fig 2E. Relative mean values (N > 275, thick horizontal lines), confidence limits (α = 0.05, thin lines) for the mean, and P values obtained from t‐tests are also shown. An immunoblot showing overall levels of Cln3‐3HA in cells from this experiment is shown at the top and relative amounts are indicated. Dpm1 was detected as loading control.
Source data are available online for this figure.
Next, we asked whether these phosphosites modulate the role of Cdc48 in G1‐cyclin fate. Cells expressing at endogenous levels the non‐phosphorylatable AA or the phosphomimetic EE mutant as the sole source of Cdc48 grew with similar rates to the wild type and did not show noticeable growth defects at high temperature (Fig EV6B), indicating that these mutations do not grossly affect the function of the essential Cdc48 protein. However, the presence of non‐phosphorylatable Cdc48, either at endogenous or overexpressed levels, reduced the steady‐state levels of Cln3‐3HA protein (Fig 6G) and caused a reduction of its nuclear signal (Fig 6H) in both cycling and late‐G1 cells arrested by α‐factor, where nuclear accumulation of Cln3 is maximal (Vergés et al, 2007). Accordingly, the non‐phosphorylatable Cdc48 mutant displayed the lowest efficiencies of interaction with Cln3‐3HA (Fig EV6C). In contrast, a S519E T674E phosphomimetic mutant did not show noticeable differences in overall levels of Cln3‐3HA, but did cause a considerable increase in its nuclear levels (Fig 6G and H). Finally, while the non‐phosphorylatable Cdc48 mutant protein increased the budding size similarly to the cdc48‐3 mutation, the phosphomimetic form of Cdc48 clearly reduced cell size at budding (Fig 6I), suggesting that phosphorylation of Cdc48 at these Cdk sites stimulates its positive role in G1‐cyclin activity for cell cycle entry.
Phosphorylation of Cdc48 at a tyrosine residue (Y834) has been shown to be important for nuclear accumulation of this segregase during entry into the cell cycle (Madeo et al, 1998). We found that a non‐phosphorylatable Y834A Cdc48 mutant displayed lower overall and nuclear levels of Cln3‐3HA (Fig EV6D), suggesting an additional role of Cdc48 in the stabilization of Cln3 in the nucleus. On the contrary, a phosphomimetic Y834E mutant that accumulates in the nucleus throughout the cell cycle decreased the relative nuclear levels of Cln3‐3HA with no major effects on its overall levels, which would reinforce a cytoplasmic role for Cdc48 in ER release and nuclear accumulation of Cln3 before cell cycle entry.
Cdc48 regulates mammalian cyclin D1 in a Cdk phosphosite‐dependent manner
Cdc48 is overexpressed in many human cancers (Deshaies, 2014), and its inhibition arrests lung carcinoma cells in G1 (Valle et al, 2011). We compromised Cdc48 function in mouse 3T3 cells by shRNA‐mediated downregulation (Fig EV7A and B) and DBeQ inhibition, and in both cases observed a strong decrease in the nuclear levels of Ccnd1 (Fig 7A and B). Similar to our findings in yeast cells, Cdc48 inhibition by DBeQ severely shortened the half‐life of Ccnd1 (Fig 7C and D) while, confirming a major role at the post‐translational level, CCND1 mRNA levels were not affected by the short DBeQ treatment used (Fig EV7C). Very similar results were obtained when using NMS‐873 (Fig EV7D and E), an allosteric inhibitor of Cdc48 (Magnaghi et al, 2013) functionally unrelated to DBeQ. Overexpression of human Cdc48 produced a notable increase in the nuclear levels of Ccnd1 (Fig 7E), but this effect was not observed when a non‐phosphorylatable mutant of Cdc48 was used. Supporting the limiting role of Cdc48 in Ccnd1‐dependent events, overexpression of wild‐type Cdc48 increased the level of pRB phosphorylation at S780, a Cdk4‐specific target, in both nuclear foci number (Ruiz‐Miro et al, 2011; Fig 7F) and nuclear fluorescence levels (Fig EV7F). By contrast, non‐phosphorylatable Cdc48 produced no effects in nuclear levels of phosphorylated pRB. Finally, since NMS‐873 is not as toxic as DBeQ to 3T3 cells when treated for periods of time up to 24 h, we were able to analyze its effects during cell cycle entry of 3T3 cells arrested in G1 by serum deprivation and found that inhibition of Cdc48 caused a clear delay in cell cycle entry as assessed by DNA content analysis (Fig EV7G). These data extend the role of Cdc48 in G1 progression to mammalian cells, where its segregase activity would be also modulated by Cdk activity to prevent G1‐cyclin degradation and facilitate its nuclear accumulation to trigger Start.
Figure EV7. Cdc48 also controls stability and nuclear functions of cyclin D1 in mammalian cells.
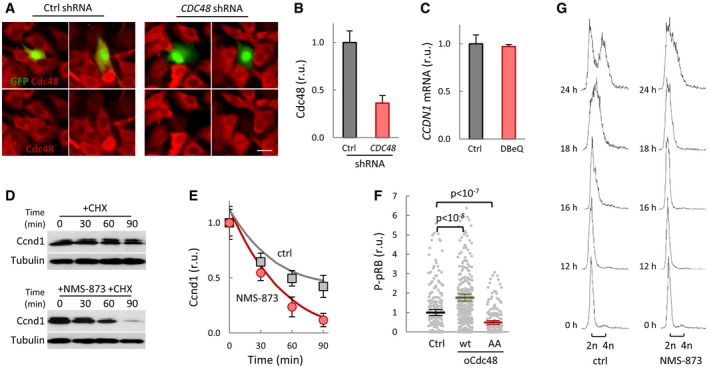
- Immunofluorescence analysis of Cdc48 (red signal) in mouse 3T3 cells transfected with control or Cdc48 shRNAs. GFP (green) was used as a transfection reporter. Scale bar, 20 μm.
- Quantification of Cdc48 levels in 3T3 cells transfected with control or Cdc48 shRNAs as in (A). Values were made relative to the average obtained from control cells. Mean values (N > 50) and confidence limits (α = 0.05) for the mean are shown.
- CCND1 mRNA levels in 3T3 cells in the absence (Ctrl) or presence of 10 μM DBeQ for 90 min. Values were made relative to GAPDH mRNA and control sample. Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
- Ccnd1 stability in 3T3 cells. After being treated with or without 2 μM NMS‐873 for 2 h, cells were added cycloheximide and collected at the indicated times to determine Ccnd1 levels. Tubulin is shown as a loading control.
- Quantification of Ccnd1 stability in control (squares) and NMS‐873 treated (circles) cells from immunoblot analysis as in (D) at the indicated times after cycloheximide addition. Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
- P‐S780‐pRB levels in 3T3 cells overexpressing wild‐type (wt) or non‐phosphorylatable (AA) Cdc48, or none as control. Values were made relative to the average obtained from control cells. Mean values (N > 200, thick horizontal lines), confidence limits (α = 0.05, thin lines) for the mean, and P values obtained from t‐tests are also shown.
- Serum‐starved 3T3 cells were stimulated to enter the cell cycle by the addition of 10% FCS in the presence or absence of 2 μM NMS‐873, and samples were taken at the indicated times to obtain DNA content profiles by FACS.
Source data are available online for this figure.
Figure 7. Cdc48 also controls stability and nuclear functions of cyclin D1 in mammalian cells.
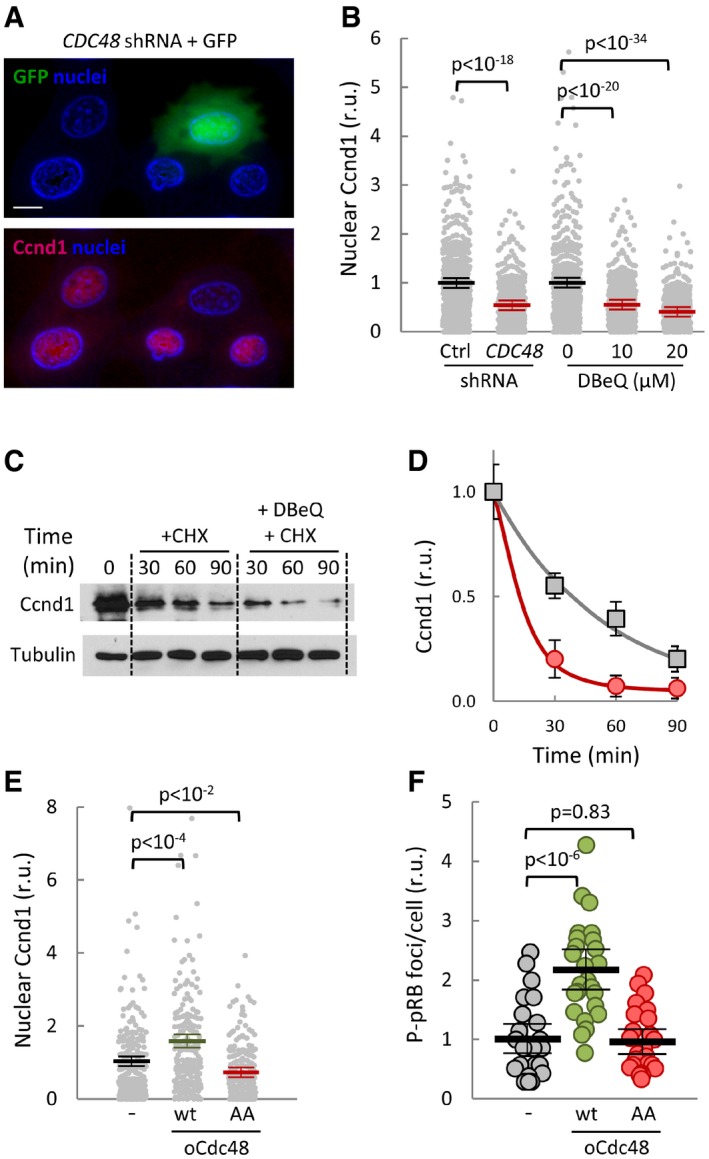
- Immunofluorescence analysis of cyclin D1 (Ccnd1, red signal) in mouse 3T3 cells transfected with Cdc48 shRNA. GFP (green) was used as a transfection reporter. A first‐derivative image of Hoechst‐stained nuclei (blue) is shown to enhance nuclear limits. Scale bar, 5 μm.
- Quantification of Ccnd1 nuclear levels in 3T3 cells transfected with control (Ctrl) or Cdc48 shRNAs, or treated with the indicated concentrations of DBeQ for 90 min. Values were made relative to the average obtained from untreated cells. Mean values (N > 350, thick horizontal lines), confidence limits (α = 0.05, thin lines) for the mean, and P‐values obtained from t‐tests are also shown.
- After being treated with or without 20 μM DBeQ, cells were added cycloheximide and collected at the indicated times to determine Ccnd1 levels. Tubulin is shown as a loading control.
- Quantification of Ccnd1 stability in control (squares) and DBeQ‐treated (circles) cells from immunoblot analysis as in (C) at the indicated times after cycloheximide addition. Mean values (N = 3) and confidence limits (α = 0.05) for the mean are shown.
- Nuclear accumulation of Ccnd1 in 3T3 cells overexpressing wild‐type (wt) or non‐phosphorylatable (AA) Cdc48, or none as control. Values were made relative to the average obtained from control cells. Mean values (N > 200), confidence limits, and P‐values are as in (B).
- Nuclear P‐S780‐pRB foci number in 3T3 cells overexpressing wild‐type (wt) or non‐phosphorylatable (AA) Cdc48, or none as control. Values were made relative to the average obtained from control cells. Mean values (N = 25, thick horizontal lines), confidence limits (α = 0.05, thin lines) for the mean, and P‐values obtained from t‐tests are also shown.
Source data are available online for this figure.
Discussion
The extremely short half‐life of G1 cyclins is thought to be a key property in allowing rapid but reversible exit from the cell cycle under unfavorable conditions. We have found that central to this decision is Cdc48, a ring‐shaped enzyme that undergoes ATP‐dependent conformational changes to extract ubiquitinated proteins from membranes and complexes for proteasomal degradation, recycling or functional release. Here, we show that Cdc48 releases ubiquitinated Cln3 from the ER and prevents its degradation to stimulate cell cycle entry. In budding yeast, Cdc48 had been found to be important for G1 progression under heat stress (Hsieh & Chen, 2011). By contrast, due to its ability to interact with ubiquitinated Cln2, Cdc48 had been proposed to have a role in the degradation of this G1 cyclin (Archambault et al, 2004). Our results reconcile these observations and point to the idea that ubiquitination and functional release of Cln2 could also play a role in the positive feedback loop that activates the G1/S regulon (Skotheim et al, 2008).
Through an increasing number of different recruiting and processing factors, Cdc48 is able to perform entirely antagonistic functions on different proteins by connecting their specific recruitment with multiubiquitination and proteasomal targeting, or deubiquitination and functional release (Jentsch & Rumpf, 2007; Meyer et al, 2012). However, the molecular determinants that allow Cdc48 to achieve regulatable combinations of these specific cofactors are not well understood. Although phosphorylation of Cdc48 has been observed in some instances, the functional relevance of specific effects on interactions in vivo has not been demonstrated (Klein et al, 2005; Zhao et al, 2007). Whereas in vitro phosphorylation by Cdk1 does not seem to affect hexamer formation (Mayr et al, 1999), our data indicate that Cdk phosphosites in Cdc48 would stimulate binding of specific processing cofactors that prevent degradation of Cln3, thus explaining the positive effect of Cdk modulation on Cdc48 as a G1‐cyclin segregase. Strikingly, while distantly located from T674 in the same monomer, S519 is very close to the T674 residue in the adjacent monomer, suggesting that phosphorylation of these two residues may also regulate Cdc48 activity by modulating conformational dynamics of the hexameric complex. Our MS data indicate that < 1% of all Cdc48 molecules are phosphorylated at S519 during G1 progression, which would suggest local and temporal modification of Cdc48 by the low number of G1 Cdk‐cyclin complexes present in the cell before Start (Tyers et al, 1993; Cross et al, 2002).
The Ssa1‐Ydj1 complex, a major Hsp70‐Hsp40 chaperone system in yeast, is also regulated by Cdk‐dependent phosphorylation (Truman et al, 2012), although the functional consequences on G1‐cyclin activity are totally opposed and act at complementary cell cycle periods. Our data point to the notion that low Cdk activity during G1 progression phosphorylates Cdc48 to enforce G1‐cyclin stability and ER release to trigger Start, where a burst of Cdk activity is produced and maintained until exit from mitosis. In turn, once in S phase, this high Cdk activity phosphorylates Ssa1 to displace Ydj1 and favor Cln3 degradation (Truman et al, 2012). Cln3 and cyclin D1 synthesis is rather constant during the cell cycle, but these excluding mechanisms would allow G1 cyclins to be mostly active in a window from mitotic exit to Start, thus establishing an additional layer of regulation to control cell cycle entry.
Our results extend to mammalian cells the Cdk‐dependent role of Cdc48 in G1‐cyclin fate. Cdc48 has a role in degradation of the replication licensing factor Cdt1 during S phase (Franz et al, 2011; Raman et al, 2011), but a role in G1 had not been established in mammals. Here, we show that Cdc48 acts in mouse cells to prevent cyclin D1 degradation and stimulate its nuclear accumulation to phosphorylate pRb. Supporting our data, cyclin D1 interacts with Cdc48 (Jirawatnotai et al, 2011) and is degraded via ubiquitination‐mediated pathways in the cytoplasm (Lin et al, 2006). In addition, Cdc48 is phosphorylated at the same two Cdk‐target residues of yeast Cdc48 (Mori‐Konya et al, 2009), and we have found that a non‐phosphorylatable Cdc48 mutant cannot stimulate nuclear accumulation of cyclin D1 and pRB phosphorylation. Cdc48 overexpression is a common trait of many human cancers (Deshaies, 2014), and its inhibition causes G1 arrest in lung carcinoma cells (Valle et al, 2011). However, these observations remain mostly mysterious at the molecular level. IκB kinases increase NFκB‐dependent cyclin D1 expression by signaling the IκB inhibitor for degradation (Cao et al, 2001), through a ubiquitin‐dependent process that requires Cdc48 (Li et al, 2014). However, a direct effect of Cdc48 activity on cyclin D1 expression has not been demonstrated. Instead, our data point to the idea that Cdc48 overexpression in tumors would lead to increased cyclin D1 protein levels mainly by preventing degradation and facilitating nuclear accumulation of this G1 cyclin.
Cdk‐dependent modulation of Cdc48 suggests the existence of a coherent feedforward module acting during G1 progression to control entry into the cell cycle (Fig 8). Contrary to feedback‐based regulation, feedforward systems allow fast state switching (Alon, 2007), and provide with stable and yet rapidly reversible states in the G1 arrest caused by sexual pheromones (Doncic & Skotheim, 2013). Thus, G1‐Cdk self‐modulation through Cdc48 would rapidly arrest cells in G1 when environmental conditions compromise chaperone function, but would also promptly reactivate G1 progression if propitious circumstances were to be restored.
Figure 8. Cdc48 and ubiquitin set G1‐cyclin fate to destruction or nuclear accumulation to trigger Start.
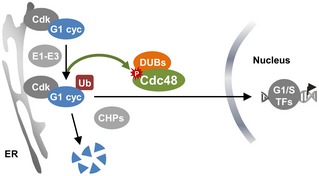
Ubiquitination of G1 cyclin Cln3 is a requirement for its release from the ER by Ssa1‐Ydj1 chaperones (CHPs) and also for its proteasomal destruction. The Cdc48 segregase tilts the process toward stabilization and release from the ER, thus allowing nuclear accumulation of the G1 cyclin to trigger Start.
Materials and Methods
Growth conditions and strains
Yeast cells were grown under exponential conditions for seven to eight generations in SC medium with 2% glucose at 30°C unless stated otherwise. Small newly born cells were isolated from Ficoll gradients (Mitchison, 1988). Late G1‐arrested cells were obtained by treating exponential cultures at OD600 = 0.5 with 5 μg/ml α factor for 105 min at 30°C. GAL1p‐driven gene expression was induced by addition of 2% galactose to cultures grown in 2% raffinose at OD600 = 0.5. When indicated, GAL1p‐driven gene expression was induced with β‐estradiol in cells expressing the Gal4‐hER‐VP16 (GEV) transactivator as described (Yahya et al, 2014). Auxin was added to 1 mM to induce degradation of the cdc48‐AID gene product (Morawska & Ulrich, 2013). In experiments to determine the half‐life of Cln3 and cyclin D1, protein synthesis was inhibited by addition of 25 μg/ml cycloheximide. Yeast strains and plasmids used are detailed in Tables 1 and 2. Methods used for chromosomal gene transplacement and PCR‐based directed mutagenesis have been described (Wang et al, 2004; Ferrezuelo et al, 2012).
Table 1.
Strains used and genotypes
| Strain | Origin |
|---|---|
| CML128 (MATa leu2‐3,112 ura3‐52 trp1‐1 his4‐1 can r) | Gallego et al (1997) |
| CML133 (tTA::LEU2) | Gallego et al (1997) |
| CML203 (CLN3‐3HA::GEN) | Gallego et al (1997) |
| CML211 (cln3::LEU2) | Gallego et al (1997) |
| CML344 (tTA::LEU2 cdc34‐2) | Gallego et al (1997) |
| MAG220 (GAL4‐ER‐VP16::URA3) | This study |
| MAG642 (GAL4‐ER‐VP16::URA3 ydj1::NAT) | This study |
| MAG643 (GAL4‐ER‐VP16::URA3 ydj1::NAT CLN3‐3HA::GEN) | This study |
| MAG925 (CLN3‐3HA::GEN GAL1 p ‐CDC48::NAT) | This study |
| MAG927 (GAL1 p ‐CDC48::NAT) | This study |
| MAG1279 (NPL4‐3HA::GEN) | This study |
| MAG1282 (OTU1‐3HA::GEN) | This study |
| MAG1283 (BRE5‐3HA::GEN) | This study |
| MAG1285 (RPN11‐3HA::GEN) | This study |
| MAG1470 (CLN3‐3HA::GEN CDC48‐AID::NAT OsTIR1::URA3) | This study |
| MAG2021 (ydj1::HPH CLN3‐3HA::GEN CDC48‐AID::NAT OsTIR1::URA3) | This study |
| BY4742 (MATα leu2Δ0 ura3Δ0 his3Δ0 lys2Δ0) | Brachmann et al (1998) |
| cdc48‐3 (cdc48‐3 in BY4742) | Tran et al (2011) |
| MAG201 (cdc48‐3 CLN3‐3HA in BY4742) | This study |
| MAG202 (CLN3‐3HA in BY4742) | This study |
| MAG1980 (far1::HPH cdc48‐3) | This study |
| MAG1981 (far1::HPH) | This study |
| DBY2064 (MATα lys2‐801 his4‐619) | D. Botstein |
| KFY205 (cdc48‐6ts in DBY2064) | K‐U. Fröhlich |
| BF305 (GAL1 p ‐CLN3::URA3 cln1::HIS3 cln2::TRP1) | B. Futcher |
Table 2.
Plasmids used and relevant genetic details
| Plasmid | Origin |
|---|---|
| pCM194 (ARS‐CEN URA3 CLN3‐3HA) | Gallego et al (1997) |
| pCM357 (ARS‐CEN URA3 tet p ‐GFP) | Gallego et al (1997) |
| pCYC41 (ARS‐CEN URA3 CLN3‐1‐3HA) | Wang et al (2004) |
| pCYC354 (ARS‐CEN URA3 CLN3ΔHD‐3HA) | Vergés et al (2007) |
| pCYC418 (ARS‐CEN URA3 tet p ‐GFP‐CLN3HD) | Vergés et al (2007) |
| pMAG149 (ARS‐CEN TRP1 CDC48 UFD1 NPL4) | This study |
| pMAG526 (ARS‐CEN TRP1 GAL1 p ‐CDC48) | This study |
| pMAG862 (ARS‐CEN URA3 CLN3‐3HA‐UBI K11R,K48R) | This study |
| pMAG864 (ARS‐CEN URA3 UBI K11R,K48R ‐CLN3‐3HA) | This study |
| pMAG1217 (ARS‐CEN URA3 CLN3 allKR ‐3HA) K(1–580)R substitutions | This study |
| pMAG1219 (ARS‐CEN URA3 CLN3 NtKR ‐3HA) K(1–400)R substitutions | This study |
| pMAG1221 (ARS‐CEN URA3 CLN3 CtKR ‐3HA) K(401–580)R substitutions | This study |
| pMAG934 (ARS‐CEN TRP1 GAL1 p ‐CDC48‐6FLAG) | This study |
| pMAG962 (ARS‐CEN TRP1 GAL1 p ‐CDC48 AA ‐6FLAG) | This study |
| pMAG966 (ARS‐CEN TRP1 GAL1 p ‐CDC48 EE ‐6FLAG) | This study |
| pMAG1027 (ARS‐CEN LEU2 CDC48) | This study |
| pMAG1028 (ARS‐CEN LEU2 CDC48 AA) | This study |
| pMAG1030 (ARS‐CEN LEU2 CDC48 EE) | This study |
| pMAG1388 (ARS‐CEN LEU2 CDC48 Y834A) | This study |
| pMAG1389 (ARS‐CEN LEU2 CDC48 Y834E) | This study |
| pMAG1505 (ARS‐CEN LEU2 CDC48‐6FLAG) | This study |
| pMAG1507 (ARS‐CEN LEU2 CDC48 AA ‐6FLAG) | This study |
| pMAG1663 (2 μm URA3 GAL1 p ‐GST‐CDC48) | This study |
| pMAG1665 (2 μm URA3 GAL1 p ‐GST‐CDC48 AA) | This study |
| pMAG1023 (ColE1 bla lacI q tac p ‐GST‐CDC48 500–683) | This study |
| pMAG1025 (ColE1 bla lacI q tac p ‐GST‐CDC48 500–683AA) | This study |
| pMAG665 (ColE1 bla NEO CMV p ‐HsCDC48) | This study |
| pMAG938 (ColE1 bla NEO CMV p ‐HsCDC48 AA) | This study |
| pMAG940 (ColE1 bla NEO CMV p ‐HsCDC48 EE) | This study |
Human 293T and mouse 3T3 cells were grown in Dulbecco's modified Eagle's medium (DMEM) containing glutamine supplemented with antibiotics and 10% FCS. Cycling cells were collected 18 h after replating. Transfection with CDC48‐directed shRNA (TRCN0000008413, Sigma) and CMVp‐CDC48 constructs was performed as described (Ruiz‐Miro et al, 2011).
Protein mobilization, immunoprecipitation, affinity purification, and kinase assays
Protein mobilization assays were performed in cell‐free extracts as described (Rape et al, 2001). Briefly, Ydj1‐deficient cells (50 OD600) were collected, resuspended in cold 250 μl STE10 (10% sucrose, 20 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM DTT with protease and phosphatase inhibitors), and vortexed in the presence of glass beads until 50% cells were broken. After adding 500 μl STE10, cells were vortexed for 5 s and spun at 1.8 krpm (300 g) for 2 min. The supernatant (500 μl) was transferred to a clean tube, added 10 μl 0.1 M ATP, 25 μl 1 M P‐creatine and 25 units creatine kinase, and complemented with 5 μg of 6His‐Ydj1 purified as described (Vergés et al, 2007). After incubating cell extracts at 25°C for 30 min, membrane and soluble fractions were obtained by centrifugation at 13.2 krpm (16,000 g) for 30 min and analyzed by SDS–PAGE and immunoblotting as described below. Other subcellular fractionations were as described (Vergés et al, 2007). FLAG‐tagged Cdc48 was immunoprecipitated with αFLAG (clone M2, Sigma) beads (Wang et al, 2004). Immunoblot analysis (Wang et al, 2004) was performed with antibodies against HA (12CA5, Roche), FLAG (M2, Sigma), ubiquitin (P4G7, Santa Cruz), Ccnd1 (DCS‐6, BD Pharmingen), Cdc48/p97 (ab11433, Abcam), GFP (A. Aragay, IBMB), Dpm1 (5C5, Molecular Probes) or tubulin (DM‐1A, Sigma). Ubiquitinated proteins and GST‐Cdc48 fusions were affinity purified with agarose‐TUBE1 (Lifesensors) and glutathione beads (GE Healthcare), respectively, following the instructions of the manufacturers. Kinase assays with Ccnd1‐Cdk4 were performed as described (Fernández et al, 2011).
Immunofluorescence
Immunofluorescence of endogenous levels of Cln3‐3HA was done by a signal‐amplification method (Wang et al, 2004) with rat‐αHA (clone 3F10, Roche) and goat‐αrat and rabbit‐αgoat Alexa555‐labeled antibodies (Molecular Probes) on methanol‐permeabilized cells. Immunofluorescence analysis in mouse 3T3 cells with antibodies against Ccnd1 (clone DCS‐6, BD Pharmingen), and P‐S780 pRB (Cell Signaling) has already been described (Ruiz‐Miro et al, 2011). To analyze localization of Cln3‐3HA and Ccnd1, we used N2CJ, a plugin for ImageJ (Wayne Rasband, NIH), to perform accurate user‐assisted immunofluorescence quantification in both cytoplasmic and nuclear compartments of cells (Yahya et al, 2014). A large number of cells were analyzed to obtain robust statistics, and experiments were repeated at least twice to confirm trends and significant differences observed.
Mass spectrometry analysis of Cdc48 phosphorylation
In experiments using α factor, cells expressing Cdc48‐GST were arrested in G1 by treating exponential cultures at OD600 = 0.5 with 5 μg/ml α factor for 105 min at 30°C, and released for 30 min after filtration and resuspension in fresh medium. In G1‐cyclin depletion experiments, cln1,2 GAL1p‐CLN3 cells expressing Cdc48‐6FLAG were arrested in G1 by growth in 2% glucose‐rich medium (YPD) for 2 h at 30°C. On the other hand, cells temporarily arrested in 2% raffinose‐rich medium for 2 h at 30°C were transferred to 2% galactose‐rich medium for 20 min at 30°C to obtain cells executing Start. GST and FLAG‐tagged Cdc48 proteins were purified as described above, digested with trypsin and analyzed by nano‐LC‐MS/MS using a Orbitrap Fusion LumosTM Tribrid instrument. Nano‐LC was carried out in a C18 analytical column (Acclaim PepMap® RSLC 75 μm × 50 cm, 2 μm, 100 Å, Thermo Scientific) with a 60 min gradient from 1 to 35% B (A = 0.1% FA in water, B = 0.1% FA in CH3CN). The column outlet was directly connected to an Advion TriVersa NanoMate (Advion BioSciences, Ithaca, NY, USA) fitted on an Orbitrap Fusion Lumos™ Tribrid (Thermo Scientific). The mass spectrometer was operated in a data‐dependent acquisition (DDA) mode. In each data collection cycle, one full MS scan was acquired in the Orbitrap (120k resolution) and the most abundant ions were selected for fragmentation by higher‐energy collisional dissociation (HCD) and analyzed in the Orbitrap (30k resolution). MS/MS spectra were searched against the SwissProt (Saccharomyces cerevisiae, release 2017_01) and contaminants database using MaxQuant v1.5.7.4 with Andromeda search engine. Peptide and protein identifications were filtered at a false discovery rate (FDR) of 1% based on the number of hits against the reversed sequence database. Ratio between peptides containing pS519 and pS770, a non Cdk target (Holt et al, 2009), were calculated from the extracted ion chromatogram areas (Fig EV5).
Miscellaneous
Cell volume at bud emergence was determined from bright‐field images with the aid of BudJ (Ferrezuelo et al, 2012), an ImageJ (Wayne Rasband, NIH) plugin that can be obtained from http://www.ibmb.csic.es/groups/spatial-control-of-cell-cycle-entry. Real‐time RT–PCR analysis of CLN3 and Ccnd1 mRNA levels was carried out with TaqMan probes following the instructions of the manufacturer (Applied Biosystems). PCRs were run and analyzed with an iCycler iQ real‐time detection system (Bio‐Rad).
Statistical analysis
Median or mean data give equally significant differences by parametric and nonparametric tests in cell volume analysis (Ferrezuelo et al, 2012), but for nuclear localization measurements, we prefer using mean values to include variations in the subpopulation with higher levels of nuclear Cln3, which are the most relevant for entry into the cell cycle (Wang et al, 2004). Visual inspection of confidence interval overlapping in all samples allows the reader to estimate directly the significance of the differences at P < 0.05. However, to facilitate a more detailed assessment of differences between relevant samples we have added the specific P‐values obtained from two‐tailed t‐tests in all panels.
Author contributions
All authors designed the experiments and analyzed the data. EP, GY, and AF built genetic constructs and strains, and performed the experiments. MA wrote the manuscript. All the authors discussed the data and approved the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank E. Rebollo, M. Vilaseca, M. Gay, and L. Villarreal for technical assistance. We also thank S. Gutiérrez for kinase assays, K.U. Fröhlich and B. Futcher for strains, A. Aragay for a rabbit αGFP antibody, C. Rose for editing the manuscript, and C. Gallego, D. F. Moreno, and B. Crosas for helpful comments. This work was funded by the Ministry of Economy and Competitiveness of Spain (BFU2013‐47710‐R), Consolider‐Ingenio 2010 (CSD2007‐15), and the European Union (FEDER).
The EMBO Journal (2018) 37: e98724
References
- Aldea M, Gari E, Colomina N (2007) Control of cell cycle and cell growth by molecular chaperones. Cell Cycle 6: 2599–2603 [DOI] [PubMed] [Google Scholar]
- Alon U (2007) Network motifs: theory and experimental approaches. Nat Rev Genet 8: 450–461 [DOI] [PubMed] [Google Scholar]
- Archambault V, Chang EJ, Drapkin BJ, Cross FR, Chait BT, Rout MP (2004) Targeted proteomic study of the cyclin‐Cdk module. Mol Cell 14: 699–711 [DOI] [PubMed] [Google Scholar]
- Bergink S, Ammon T, Kern M, Schermelleh L, Leonhardt H, Jentsch S (2013) Role of Cdc48/p97 as a SUMO‐targeted segregase curbing Rad51–Rad52 interaction. Nat Cell Biol 15: 526–532 [DOI] [PubMed] [Google Scholar]
- Bertoli C, Skotheim JM, de Bruin RAM (2013) Control of cell cycle transcription during G1 and S phases. Nat Rev Mol Cell Biol 14: 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar NO, Rapoport TA (2017) Molecular mechanism of substrate processing by the Cdc48 ATPase complex. Cell 169: 722–735.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Boom J, Meyer H (2018) VCP/p97‐mediated unfolding as a principle in protein homeostasis and signaling. Mol Cell 69: 182–194 [DOI] [PubMed] [Google Scholar]
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD (1998) Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR‐mediated gene disruption and other applications. Yeast 14: 115–132 [DOI] [PubMed] [Google Scholar]
- Cao Y, Bonizzi G, Seagroves TN, Greten FR, Johnson R, Schmidt EV, Karin M (2001) IKKα provides an essential link between RANK signaling and cyclin D1 expression during mammary gland development. Cell 107: 763–775 [DOI] [PubMed] [Google Scholar]
- Cao K, Nakajima R, Meyer HH, Zheng Y (2003) The AAA‐ATPase Cdc48/p97 regulates spindle disassembly at the end of mitosis. Cell 115: 355–367 [DOI] [PubMed] [Google Scholar]
- Charvin G, Oikonomou C, Siggia ED, Cross FR (2010) Origin of irreversibility of cell cycle start in budding yeast. PLoS Biol 8: e1000284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross FR, Archambault V, Miller M, Klovstad M (2002) Testing a mathematical model of the yeast cell cycle. Mol Biol Cell 13: 52–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dargemont C, Ossareh‐Nazari B (2012) Cdc48/p97, a key actor in the interplay between autophagy and ubiquitin/proteasome catabolic pathways. Biochim Biophys Acta 1823: 138–144 [DOI] [PubMed] [Google Scholar]
- DeLaBarre B, Brunger AT (2005) Nucleotide dependent motion and mechanism of action of p97/VCP. J Mol Biol 347: 437–452 [DOI] [PubMed] [Google Scholar]
- Deshaies RJ (2014) Proteotoxic crisis, the ubiquitin‐proteasome system, and cancer therapy. BMC Biol 12: 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl JA, Yang W, Rimerman RA, Xiao H, Emili A (2003) Hsc70 regulates accumulation of cyclin D1 and cyclin D1‐dependent protein kinase. Mol Cell Biol 23: 1764–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncic A, Skotheim JM (2013) Feedforward regulation ensures stability and rapid reversibility of a cellular state. Mol Cell 50: 856–868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgington NP, Futcher B (2001) Relationship between the function and the location of G1 cyclins in S. cerevisiae . J Cell Sci 114: 4599–4611 [DOI] [PubMed] [Google Scholar]
- Elsasser S, Finley D (2005) Delivery of ubiquitinated substrates to protein‐unfolding machines. Nat Cell Biol 7: 742–749 [DOI] [PubMed] [Google Scholar]
- Eser U, Falleur‐Fettig M, Johnson A, Skotheim JM (2011) Commitment to a cellular transition precedes genome‐wide transcriptional change. Mol Cell 43: 515–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández RMH, Ruiz‐Miró M, Dolcet X, Aldea M, Garí E, Fernandez RMH, Ruiz‐Miro M, Dolcet X, Aldea M, Gari E (2011) Cyclin D1 interacts and collaborates with Ral GTPases enhancing cell detachment and motility. Oncogene 30: 1936–1946 [DOI] [PubMed] [Google Scholar]
- Ferrezuelo F, Colomina N, Futcher B, Aldea MM (2010) The transcriptional network activated by Cln3 cyclin at the G1‐to‐S transition of the yeast cell cycle. Genome Biol 11: R67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrezuelo F, Colomina N, Palmisano A, Garí E, Gallego C, Csikász‐Nagy A, Aldea M (2012) The critical size is set at a single‐cell level by growth rate to attain homeostasis and adaptation. Nat Commun 3: 1012 [DOI] [PubMed] [Google Scholar]
- Franz A, Orth M, Pirson PA, Sonneville R, Blow JJ, Gartner A, Stemmann O, Hoppe T (2011) Cdc48/p97 coordinates CDT‐1 degradation with GINS chromatin dissociation to ensure faithful DNA replication. Mol Cell 44: 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Ng C, Feng D, Liang C (2003) Cdc48p is required for the cell cycle commitment point at Start via degradation of the G1‐CDK inhibitor Far1p. J Cell Biol 163: 21–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego C, Garí E, Colomina N, Herrero E, Aldea M (1997) The Cln3 cyclin is down‐regulated by translational repression and degradation during the G1 arrest caused by nitrogen deprivation in budding yeast. EMBO J 16: 7196–7206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwell LH, Culotti J, Pringle JR, Reid BJ (1974) Genetic control of the cell division cycle in yeast. Science 183: 46–51 [DOI] [PubMed] [Google Scholar]
- Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO (2009) Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 325: 1682–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh M‐T, Chen R‐H (2011) Cdc48 and cofactors Npl4‐Ufd1 are important for G1 progression during heat stress by maintaining cell wall integrity in Saccharomyces cerevisiae . PLoS One 6: e18988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch S, Rumpf S (2007) Cdc48 (p97): a ‘molecular gearbox’ in the ubiquitin pathway? Trends Biochem Sci 32: 6–11 [DOI] [PubMed] [Google Scholar]
- Jirawatnotai S, Hu Y, Michowski W, Elias JE, Becks L, Bienvenu F, Zagozdzon A, Goswami T, Wang YE, Clark AB, Kunkel TA, van Harn T, Xia B, Correll M, Quackenbush J, Livingston DM, Gygi SP, Sicinski P (2011) A function for cyclin D1 in DNA repair uncovered by interactome analyses in human cancers. Nature 474: 230–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ES, Ma PC, Ota IM, Varshavsky A (1995) A proteolytic pathway that recognizes ubiquitin as a degradation signal. J Biol Chem 270: 17442–17456 [DOI] [PubMed] [Google Scholar]
- Johnson A, Skotheim JM (2013) Start and the restriction point. Curr Opin Cell Biol 25: 717–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen P, Tyers M (2004) How cells coordinate growth and division. Curr Biol 14: R1014–R1027 [DOI] [PubMed] [Google Scholar]
- Klein JB, Barati MT, Wu R, Gozal D, Sachleben LR, Kausar H, Trent JO, Gozal E, Rane MJ (2005) Akt‐mediated valosin‐containing protein 97 phosphorylation regulates its association with ubiquitinated proteins. J Biol Chem 280: 31870–31881 [DOI] [PubMed] [Google Scholar]
- Komander D, Rape M (2012) The ubiquitin code. Annu Rev Biochem 81: 203–229 [DOI] [PubMed] [Google Scholar]
- Li J‐M, Wu H, Zhang W, Blackburn MR, Jin J (2014) The p97‐UFD1L‐NPL4 protein complex mediates cytokine‐induced IkBa proteolysis. Mol Cell Biol 34: 335–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DI, Barbash O, Kumar KGS, Weber JD, Harper JW, Klein‐Szanto AJP, Rustgi A, Fuchs SY, Diehl JA (2006) Phosphorylation‐dependent ubiquitination of cyclin D1 by the SCF(FBX4‐alphaB crystallin) complex. Mol Cell 24: 355–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeo F, Schlauer J, Zischka H, Mecke D, Fröhlich KU (1998) Tyrosine phosphorylation regulates cell cycle‐dependent nuclear localization of Cdc48p. Mol Biol Cell 9: 131–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnaghi P, D'Alessio R, Valsasina B, Avanzi N, Rizzi S, Asa D, Gasparri F, Cozzi L, Cucchi U, Orrenius C, Polucci P, Ballinari D, Perrera C, Leone A, Cervi G, Casale E, Xiao Y, Wong C, Anderson DJ, Galvani A et al (2013) Covalent and allosteric inhibitors of the ATPase VCP/p97 induce cancer cell death. Nat Chem Biol 9: 586–592 [DOI] [PubMed] [Google Scholar]
- Maric M, Maculins T, De Piccoli G, Labib K (2014) Cdc48 and a ubiquitin ligase drive disassembly of the CMG helicase at the end of DNA replication. Science 346: 1253596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr PS, Allan VJ, Woodman PG (1999) Phosphorylation of p97(VCP) and p47 in vitro by p34cdc2 kinase. Eur J Cell Biol 78: 224–232 [DOI] [PubMed] [Google Scholar]
- Mérai Z, Chumak N, García‐Aguilar M, Hsieh T‐F, Nishimura T, Schoft VK, Bindics J, Slusarz L, Arnoux S, Opravil S, Mechtler K, Zilberman D, Fischer RL, Tamaru H (2014) The AAA‐ATPase molecular chaperone Cdc48/p97 disassembles sumoylated centromeres, decondenses heterochromatin, and activates ribosomal RNA genes. Proc Natl Acad Sci USA 111: 16166–16171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer H, Bug M, Bremer S (2012) Emerging functions of the VCP/p97 AAA‐ATPase in the ubiquitin system. Nat Cell Biol 14: 117–123 [DOI] [PubMed] [Google Scholar]
- Miller ME, Cross FR (2001) Mechanisms controlling subcellular localization of the G1 cyclins Cln2p and Cln3p in budding yeast. Mol Cell Biol 21: 6292–6311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison JM (1988) Synchronous cultures and age fractionation In Yeast, a practical approach, Campbell I, Duffus J. (eds), pp 51–64.Oxford, UK: IRL Press; [Google Scholar]
- Morawska M, Ulrich HD (2013) An expanded tool kit for the auxin‐inducible degron system in budding yeast. Yeast 30: 341–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori‐Konya C, Kato N, Maeda R, Yasuda K, Higashimae N, Noguchi M, Koike M, Kimura Y, Ohizumi H, Hori S, Kakizuka A (2009) p97/valosin‐containing protein (VCP) is highly modulated by phosphorylation and acetylation. Genes Cells 14: 483–497 [DOI] [PubMed] [Google Scholar]
- Ndoja A, Cohen RE, Yao T (2014) Ubiquitin signals proteolysis‐independent stripping of transcription factors. Mol Cell 53: 893–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossareh‐Nazari B, Bonizec M, Cohen M, Dokudovskaya S, Delalande F, Schaeffer C, Van Dorsselaer A, Dargemont C (2010) Cdc48 and Ufd3, new partners of the ubiquitin protease Ubp3, are required for ribophagy. EMBO Rep 11: 548–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson B, Argos P (1994) Prediction of transmembrane segments in proteins utilising multiple sequence alignments. J Mol Biol 237: 182–192 [DOI] [PubMed] [Google Scholar]
- Raman M, Havens CG, Walter JC, Harper JW (2011) A genome‐wide screen identifies p97 as an essential regulator of DNA damage‐dependent CDT1 destruction. Mol Cell 44: 72–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S (2001) Mobilization of processed, membrane‐tethered SPT23 transcription factor by CDC48(UFD1/NPL4), a ubiquitin‐selective chaperone. Cell 107: 667–677 [DOI] [PubMed] [Google Scholar]
- Ruiz‐Miro M, Colomina N, Fernandez RMH, Gari E, Gallego C, Aldea M (2011) Translokin (Cep57) interacts with cyclin D1 and prevents its nuclear accumulation in quiescent fibroblasts. Traffic 12: 549–562 [DOI] [PubMed] [Google Scholar]
- Rumpf S, Jentsch S (2006) Functional division of substrate processing cofactors of the ubiquitin‐selective Cdc48 chaperone. Mol Cell 21: 261–269 [DOI] [PubMed] [Google Scholar]
- Schmoller KM, Turner JJ, Kõivomägi M, Skotheim JM (2015) Dilution of the cell cycle inhibitor Whi5 controls budding yeast cell size. Nature 526: 268–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbik N, Haines DS (2007) Cdc48p(Npl4p/Ufd1p) binds and segregates membrane‐anchored/tethered complexes via a polyubiquitin signal present on the anchors. Mol Cell 25: 385–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skotheim JM, Di Talia S, Siggia ED, Cross FR (2008) Positive feedback of G1 cyclins ensures coherent cell cycle entry. Nature 454: 291–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran JR, Tomsic LR, Brodsky JL (2011) A Cdc48p‐associated factor modulates endoplasmic reticulum‐associated degradation, cell stress, and ubiquitinated protein homeostasis. J Biol Chem 286: 5744–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truman AW, Kristjansdottir K, Wolfgeher D, Hasin N, Polier S, Zhang H, Perrett S, Prodromou C, Jones GW, Kron SJ (2012) CDK‐dependent Hsp70 phosphorylation controls G1 cyclin abundance and cell‐cycle progression. Cell 151: 1308–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyers M, Tokiwa G, Futcher B (1993) Comparison of the Saccharomyces cerevisiae G1 cyclins: Cln3 may be an upstream activator of Cln1, Cln2 and other cyclins. EMBO J 12: 1955–1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valle CW, Min T, Bodas M, Mazur S, Begum S, Tang D, Vij N (2011) Critical role of VCP/p97 in the pathogenesis and progression of non‐small cell lung carcinoma. PLoS One 6: e29073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vembar SS, Brodsky JL (2008) One step at a time: endoplasmic reticulum‐associated degradation. Nat Rev Mol Cell Biol 9: 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergés E, Colomina N, Garí E, Gallego C, Aldea M (2007) Cyclin Cln3 is retained at the ER and released by the J chaperone Ydj1 in late G1 to trigger cell cycle entry. Mol Cell 26: 649–662 [DOI] [PubMed] [Google Scholar]
- Wang HY, Garí E, Vergés E, Gallego C, Aldea M (2004) Recruitment of Cdc28 by Whi3 restricts nuclear accumulation of the G1 cyclin‐Cdk complex to late G1. EMBO J 23: 180–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaglom J, Linskens MHK, Sadis S, Rubin DM, Futcher B, Finley D (1995) p34 Cdc28 ‐mediated control of Cln3 cyclin degradation. Mol Cell Biol 15: 731–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaglom JA, Goldberg AL, Finley D, Sherman MY (1996) The molecular chaperone Ydj1 is required for the p34CDC28‐dependent phosphorylation of the cyclin Cln3 that signals its degradation. Mol Cell Biol 16: 3679–3684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahya G, Parisi E, Flores A, Gallego C, Aldea M (2014) A Whi7‐anchored loop controls the G1 Cdk‐cyclin complex at Start. Mol Cell 53: 115–126 [DOI] [PubMed] [Google Scholar]
- Zhao G, Zhou X, Wang L, Li G, Schindelin H, Lennarz WJ (2007) Studies on peptide:N‐glycanase‐p97 interaction suggest that p97 phosphorylation modulates endoplasmic reticulum‐associated degradation. Proc Natl Acad Sci USA 104: 8785–8790 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Source Data for Figure 7