

Abstract
Membrane proteins are aggregation‐prone in aqueous environments, and their biogenesis poses acute challenges to cellular protein homeostasis. How the chaperone network effectively protects integral membrane proteins during their post‐translational targeting is not well understood. Here, biochemical reconstitutions showed that the yeast cytosolic Hsp70 is responsible for capturing newly synthesized tail‐anchored membrane proteins (TAs) in the soluble form. Moreover, direct interaction of Hsp70 with the cochaperone Sgt2 initiates a sequential series of TA relays to the dedicated TA targeting factor Get3. In contrast to direct loading of TAs to downstream chaperones, stepwise substrate loading via Hsp70 maintains the solubility and targeting competence of TAs, ensuring their efficient delivery to the endoplasmic reticulum (ER). Inactivation of cytosolic Hsp70 severely impairs TA translocation in vivo. Our results demonstrate a new role of cytosolic Hsp70 in directly assisting the targeting of an essential class of integral membrane proteins and provide a paradigm for how “substrate funneling” through a chaperone cascade preserves the conformational quality of nascent membrane proteins during their biogenesis.
Keywords: Hsp70, membrane protein biogenesis, molecular chaperone, protein aggregation, tail‐anchored proteins
Subject Categories: Membrane & Intracellular Transport, Protein Biosynthesis & Quality Control
Introduction
Proper protein homeostasis is essential to all cells and requires the correct folding, localization, and quality control of all cellular proteins. Among molecular chaperones, the ubiquitous and highly conserved Hsp70s play a dominant role in regulating the proteome. Cytosolic Hsp70s are abundant and associate with over 20% of the proteome (Thulasiraman et al, 1999). They interact with client proteins at multiple stages of their life cycles, from the unfolded state, to partially folded intermediates and aggregated proteins (Clerico et al, 2015). As such, Hsp70s impact diverse aspects of protein homeostasis, from de novo folding, to trafficking and degradation (Mayer & Bukau, 2005; Clerico et al, 2015). Impaired activity of cytosolic Hsp70s is associated with a wide range of diseases, including neurodegeneration, type II diabetes, and cancer (Brehme et al, 2014; Knowles et al, 2014).
Previous works have focused on the ability of Hsp70 to chaperone non‐membrane proteins; whether cytosolic Hsp70s also protect integral membrane proteins and how its chaperone activity is linked to membrane protein biogenesis remain unclear. This is important, as the post‐translational targeting of membrane proteins poses one of the most acute challenges to the proteostasis network. Before arrival at the target membrane, improper exposure of hydrophobic transmembrane domains (TMDs) in the cytosol could lead to rapid and irreversible aggregation of membrane proteins. This disrupts the targeting process and causes mislocalization of the membrane protein that could lead to extensive proteostatic stress (Hartl et al, 2011; Shao & Hegde, 2011). Cellular machineries that mediate post‐translational membrane protein targeting must effectively compete with these off‐pathway processes and maintain newly synthesized membrane proteins in a soluble, translocation‐competent conformation; how this is accomplished is not well understood for most protein targeting pathways.
Here, we address this question for tail‐anchored proteins (TAs), an essential class of membrane proteins defined by a single TMD at the C‐terminus. TAs comprise 3–5% of the eukaryotic membrane proteome and play essential roles in numerous processes, including membrane fission/fusion, vesicular trafficking, protein translocation, quality control, and apoptosis (Kalbfleisch et al, 2007; Hegde & Keenan, 2011). In yeast and mammalian cells, TAs harboring highly hydrophobic TMDs are targeted to the ER by a conserved guided entry of tail‐anchored protein (GET) pathway (Schuldiner et al, 2008; Jonikas et al, 2009; Wang et al, 2010), in which the ATPase Get3 (or mammalian TRC40) receives TAs from the cochaperone Sgt2 (or mammalian SGTA) with help of the scaffolding Get4/5 (or mammalian BAG6) complex (Fig 1; Hegde & Keenan, 2011). Get3 subsequently delivers TAs to the Get1/2 (or mammalian WRB/CAML) receptors at the ER membrane, via which the TA is released from Get3 and inserted into the membrane (Fig 1; Hegde & Keenan, 2011; Wang et al, 2014; Colombo et al, 2016).
Figure 1. Working model of the GET pathway in yeast.
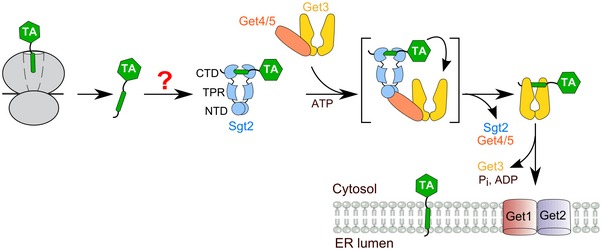
Newly synthesized TAs are captured by Sgt2 via an unknown mechanism (“?” mark) after their release from the ribosome. TAs are transferred from Sgt2 to the ATPase Get3 with the help of the scaffolding Get4/5 complex. Get3 delivers TAs to the ER membrane by interacting with the Get1/2 receptor complex. Sgt2 contains three functional domains: the N‐terminal domain (NTD) that mediates its homodimerization, the tetratricopeptide repeat (TPR) domain that interacts with heat‐shock proteins, and the C‐terminal domain (CTD) that contains the TA‐binding site.
Due to their topology, the TMDs of TAs cannot be recognized by biogenesis factors until they are released from the ribosome, demanding an obligatorily post‐translational mechanism for their capture and delivery. Sgt2 is the most upstream factor identified thus far in the GET pathway that can post‐translationally bind TAs (Wang et al, 2010). Nevertheless, how TAs are loaded onto Sgt2 as a soluble and functional complex remains unclear (Mateja et al, 2015; Shao et al, 2017). Generation of a functional Sgt2·TA complex has mostly relied on in vitro translation (IVT) in cell lysates that contain endogenous chaperones (Wang et al, 2010; Rao et al, 2016) or the use of super‐physiological concentrations of Sgt2 (Mateja et al, 2015), raising questions as to whether the currently known factors in the GET pathway are sufficient for efficient TA capture and delivery. Intriguingly, Sgt2 contains a conserved tetratricopeptide repeat (TPR) domain that associates with multiple heat‐shock proteins including Hsp70, Hsp90, and Hsp104 (Wang et al, 2010; Chartron et al, 2011; Krysztofinska et al, 2017). Heat‐shock cognate protein 70 (Hsc70) has been found to associate with TAs translated in mammalian lysate (Abell et al, 2007), and supplementing Hsc70 enhanced TA insertion into the ER in semi‐permeabilized cells (Rabu et al, 2008). However, the involvement of Hsp70 in TA biogenesis and its relationship to the GET pathway have remained elusive.
In this work, we address these questions using a combination of biochemical reconstitution and in vivo targeting assays. Our results show that the currently known cytosolic factors in the GET pathway are insufficient to effectively chaperone newly synthesized TAs. Instead, the yeast cytosolic Hsp70, Ssa1, acts upstream of Sgt2 to capture newly synthesized TAs in a soluble form and to transfer TAs to Sgt2 via an interaction between Ssa1 and the Sgt2‐TPR domain. Compared to direct TA loading onto Sgt2, this stepwise TA loading mechanism maintains the solubility and functional competence of nascent TAs during their targeting to the ER. These results demonstrate how substrate funneling through a chaperone cascade ensures the conformational quality of membrane proteins during their post‐translational targeting, and reveal a new role of cytosolic Hsp70 in directly assisting the targeting of an essential class of membrane proteins.
Results
Cytosolic Hsp70 is required to capture TAs in a soluble form
We first asked whether Sgt2 efficiently chaperones TAs. As model TA substrates, we used a non‐cleavable SUMO domain fused to the C‐terminal targeting sequences encompassing the TMDs of Sbh1p or Bos1p (here referred to as Sbh1 and Bos1, respectively; Fig EV1A; Wang et al, 2010; Rao et al, 2016). An N‐terminal Strep‐tag enabled the purification of the model substrates, and a C‐terminal opsin tag enabled measurements of their insertion into the ER. We recombinantly purified Sbh1 and Bos1 solubilized in 0.05% LDAO (N,N‐dimethyldodecylamine N‐oxide). Dilution of either substrate into aqueous buffer, which removes detergent micelles, led to their rapid aggregation as monitored by turbidity assays (Fig 2A and B, black lines). The presence of Sgt2 did not suppress the aggregation of Sbh1 (Fig 2A) and only reduced Bos1 aggregation 20–40% (Fig 2B) even at micromolar concentrations. The Sgt2‐dependent increases in the turbidity signal of Sbh1 (Fig 2A) were likely due to association of Sgt2 with aggregated TAs (see Fig 2C). These results suggest that Sgt2 is insufficient to prevent the rapid aggregation of TAs in aqueous solution.
Figure EV1. Sgt2 prevents TA aggregation only at super‐physiological concentrations.
-
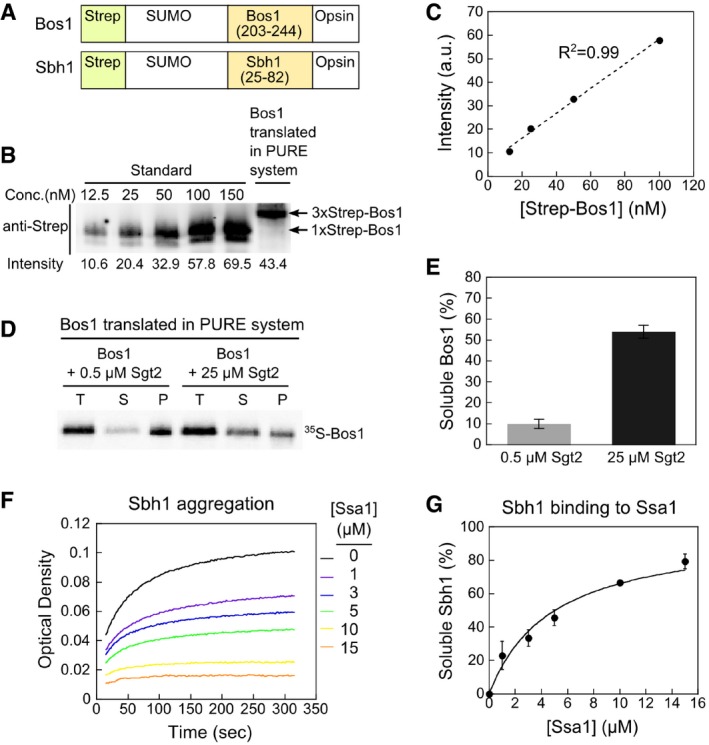
ASchematic of the model TA substrates used in in vitro assays. A Strep‐tagged non‐cleavable SUMO protein was fused to Bos1 residues 203–244 or Sbh1 residues 25–82 that encompasses their targeting sequence, which includes an N‐terminal linker to the cytosolic domain, the TMD, and the C‐terminal sequence element that regulates insertion into the ER (Rao et al, 2016). An opsin tag was attached to C‐terminus of model TAs. For IVT, a 3xStrep tag was fused to SUMO in Bos1 instead of 1xStrep tag.
-
B, CQuantitative Western blot analysis of Bos1 synthesized in the PURE‐IVT system. Known amounts of purified Strep‐Bos1 were used to construct a standard curve (panel B) for quantification of the concentration of 3xStrep‐Bos1 translated in the PURE system. Proteins were detected with a secondary antibody labeled with a near‐infrared fluorescent dye, IRDye. The 3xStrep‐Bos1 synthesized in PURE‐IVT was 71 nM, which is sub‐stoichiometric to the chaperones used in this work.
-
D, ESedimentation analysis of 35S‐labeled Bos1 synthesized by PURE‐IVT supplemented with 0.5 μM or 25 μM Sgt2. After ultracentrifugation, total input (T), soluble (S), and pellet (P) fractions were resolved by SDS–PAGE and visualized by autoradiography. The quantification of soluble Bos1 at indicated Sgt2 concentrations is shown in (E). All values are mean ± SD, with n ≥ 3.
-
F, GSsa1 suppresses Sbh1 aggregation in a dose‐dependent manner in the turbidity assay. Panel (G) shows the quantification of the data in (F) and replicates. The line was a fit of the data to equation (1) and gave a K soluble value of 4.8 ± 0.9 μM. All values are reported as mean ± SD, with n = 2.
Figure 2. Ssa1 efficiently prevents TA aggregation, whereas the currently known GET components do not.
-
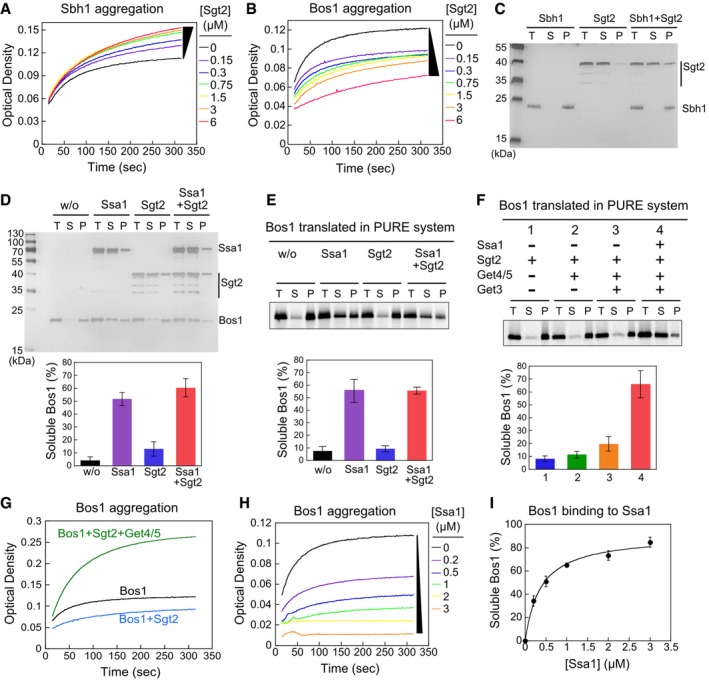
A, BTime courses of Sbh1 (A) and Bos1 (B) aggregation measured by the turbidity assay.
-
C, DSedimentation analysis of recombinant Sbh1 (C) and Bos1 (D). 1.5 μM Sbh1 or Bos1 was incubated in assay buffer with or without the indicated factors and ultracentrifuged at 390,880 g for 30 min. Total input (T), soluble (S), and pellet (P) fractions were resolved by SDS–PAGE and visualized by silver stain. 1.5 μM Sgt2 was used in (C), and 3 μM Ssa1 and/or Sgt2 was used in (D) where indicated. The lower panel in (D) shows quantification of soluble Bos1.
-
E, FSedimentation analysis of 35S‐labeled Bos1 synthesized by PURE‐IVT. Where indicated, translations reactions were supplemented with 0.5 μM Sgt2, 0.25 μM Get4/5, 0.75 μM Get3, and/or 3 μM Ssa1. The lower panels show quantification of soluble Bos1 under the indicated reaction conditions.
-
GGet4/5 exacerbates Bos1 aggregation in the turbidity assay. Reactions used 1.5 μM recombinant Bos1, 1.5 μM Sgt2 without (blue) and with (green) 1.5 μM Get4/5 present.
-
HTime courses of Bos1 aggregation in the presence of Ssa1.
-
ISsa1 solubilizes Bos1 in a dose‐dependent manner. The line was a fit of the data to equation (1).
Two independent experiments corroborated that Sgt2 does not efficiently chaperone TAs. First, we used sedimentation assays to test the solubility of purified TAs in aqueous buffer and found that TAs were predominantly in the pellet fraction irrespective of Sgt2 addition (Fig 2C and D). Second, we assessed the solubility of newly synthesized TAs by performing IVT using the PURE system, which contains no endogenous chaperones (Shimizu et al, 2001) and thus allowed us to characterize the contribution of individual cytosolic factors to the solubility of newly synthesized substrate proteins. Sedimentation after IVT showed that 35S‐labeled Bos1 remained in the insoluble fraction when translated in the presence of physiological concentrations of Sgt2 (~0.5 μM; Ghaemmaghami et al, 2003; Fig 2E). The concentration of Bos1 synthesized in the PURE‐IVT system was estimated to be 71 nM (Fig EV1B and C); therefore, this deficiency was not due to super‐stoichiometric amounts of Bos1 relative to Sgt2. As observed previously (Mateja et al, 2015), Sgt2 solubilized up to 54% Bos1 only at a super‐physiological concentration (25 μM) (Fig EV1D and E). Finally, inclusion of the other known cytosolic factors in the GET pathway, Get4/5 and Get3 (Ghaemmaghami et al, 2003), improved the solubility of Bos1 less than twofold in the sedimentation assay (Fig 2F, lanes 1–3). The presence of Get4/5 together with Sgt2 exacerbated TA aggregation in the turbidity assay (Fig 2G), excluding the possibility that interaction with Get4/5 improves the chaperone activity of Sgt2. Coupled with the low cellular abundance of Sgt2 (Ghaemmaghami et al, 2003), these results strongly suggest that Sgt2 and the other known cytosolic factors of the GET pathway are not sufficient to maintain the solubility of newly synthesized TAs.
We hypothesized that another chaperone upstream of Sgt2 is required to fulfill this role. Sgt2 contains a conserved TPR domain that binds multiple heat‐shock proteins (Wang et al, 2010; Chartron et al, 2011; Krysztofinska et al, 2017). Among them, Hsp70 has been proposed to deliver TAs to Sgt2 (Chartron et al, 2011). Hsp70 is highly abundant in the cytosol and binds short stretches of hydrophobic amino acids on client proteins with rapid association and dissociation kinetics (Clerico et al, 2015). Ssa1, an abundant (~12 μM; Ghaemmaghami et al, 2003) and constitutively expressed cytosolic Hsp70 in yeast, associates with numerous newly synthesized proteins (Albanese et al, 2006). The other Sgt2‐associated chaperones, Hsp104 and Hsp90, act downstream of Hsp70 and other chaperones and are not known to be the first factors that capture newly synthesized proteins (Albanese et al, 2006; Abell et al, 2007). Thus, Ssa1 could be a potential candidate to rapidly capture TAs and prevent their aggregation.
To test this hypothesis, we assessed the ability of purified Ssa1 to prevent TA aggregation. In sedimentation assays, the presence of Ssa1 shifted > 55% of both recombinant and in vitro‐translated Bos1 to the soluble fraction (Fig 2D and E). When Ssa1 was supplemented in the PURE‐IVT reaction together with Sgt2, Get4/5, and Get3, > 70% of Bos1 was recovered in the soluble fraction (Fig 2F, lane 4). Finally, Ssa1 prevented both Bos1 and Sbh1 from aggregation in a dose‐dependent manner in turbidity assays (Figs 2H and EV1F). The Ssa1 concentration required for half‐maximal solubilization of Bos1 and Sbh1 (K soluble) was 0.37 ± 0.07 μM and 4.8 ± 0.9 μM, respectively (Figs 2I and EV1G), comparable to the reported dissociation constants of Hsp70s for short hydrophobic peptides (0.26–6.5 μM; Xu et al, 2012). Thus, Ssa1 is more effective in chaperoning nascent TAs than the known cytosolic factors in the GET pathway.
Sequential substrate relays in an Hsp70‐Sgt2‐Get3 cascade
We next asked whether TAs can be transferred from Ssa1 to Sgt2. To this end, we developed an assay based on Förster resonance energy transfer (FRET). As the donor dye, we labeled an engineered single cysteine near the TA‐TMD with N‐(7‐Dimethylamino‐4‐Methylcoumarin‐3‐yl) Maleimide (CM; Fig 3A and Appendix Fig S1A). As the acceptor dye, we labeled BODIPY‐FL (BFL) at the Sgt2 C‐terminus near its substrate‐binding domain via sortase‐mediated ligation (Fig 3A and Appendix Fig S1B and C). When a preformed Bos1CM·Ssa1 complex was challenged with Sgt2BFL (Fig 3B), we observed a 30% reduction in CM fluorescence when the transfer reaction was carried out with Sgt2BFL but not with unlabeled Sgt2 (Fig 3C, red versus cyan), indicating that the fluorescence change arises from FRET rather than environmental sensitivity of CM. As expected from competition between Sgt2BFL and unlabeled Sgt2, inclusion of a 10‐fold excess of unlabeled Sgt2 abolished the fluorescence change (Fig 3C, purple). To further exclude the possibility that the observed FRET in Fig 3C arises from proximity of the dye pair in a Bos1CM·Ssa1·Sgt2BFL complex while Bos1CM was bound to Ssa1, we labeled CM at an engineered single cysteine, D430C, in the substrate‐binding domain (SBD) of Ssa1 (Fig 3D and E). Ssa1‐D430C protected Bos1 from aggregation (K soluble = 0.41 ± 0.05 μM) as effectively as wild‐type Ssa1 (K soluble = 0.37 ± 0.07 μM; Fig 3F versus Fig 2I), as reported by Banerjee et al (2016). No FRET was observed when TA transfer was carried out with Ssa1‐SBDCM and Sgt2BFL (Fig 3G). Finally, we stringently purified the Sgt2BFL·Bos1CM complex after the transfer reaction and quantified the FRET efficiency between the dye pair in this complex to be 30% (Fig 3H), identical to the FRET efficiency observed at the end of the transfer reaction (Fig 3C). Together, these results show that the observed FRET in the TA transfer assay reports on successful loading of Bos1 onto Sgt2.
Figure 3. A FRET assay to measure TA transfer from Ssa1 to Sgt2.
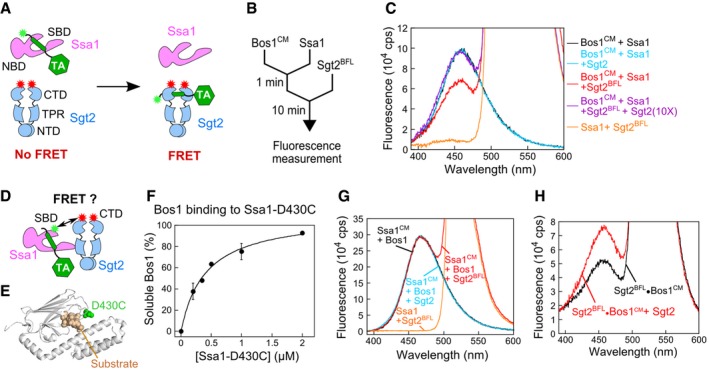
- Scheme of the FRET assay to monitor TA loading onto Sgt2. Green and red asterisks denote donor and acceptor dyes, respectively.
- Reaction setup for transfer of TA from Ssa1 to Sgt2.
- Fluorescence emission spectra of Bos1CM (donor), Sgt2BFL (acceptor), or both, in combination with the indicated factors. Reconstitutions used 50 nM Bos1CM, 5 μM Ssa1, 2 mM ATP, and 500 nM Sgt2 or Sgt2BFL where indicated.
- Scheme of the FRET assay to assess proximity between the Ssa1‐SBD and the Sgt2 C‐terminus. CM (donor) was labeled at an engineered single cysteine, C430, in the SBD of Ssa1 (denoted as Ssa1CM). Sgt2BFL was the same as that used in panel (A).
- Location of C430 in Ssa1‐SBD for fluorescence labeling. The SBD of E. coli Hsp70 homologue, DnaK, is in gray (PDB 4EZO). The residue corresponding to D430 in Ssa1 is in green, and the bound peptide (RPPYLPRP) is in wheat.
- Ssa1(D430C) prevents Bos1 aggregation as effectively as wild‐type Ssa1 in the turbidity assay. Values were reported as mean ± SD, with n = 2.
- No FRET was observed between Ssa1CM and Sgt2BFL. Fluorescence emission spectra were collected at the end of the TA transfer reaction as in (C), except that Ssa1 instead of Bos1 was CM‐labeled. The black line shows the spectrum of a sample without Sgt2 added (donor only), and the orange line shows the spectrum of a sample containing unlabeled Ssa1 and 500 nM Sgt2BFL (acceptor only).
- Donor fluorescence emission spectra for the purified Sgt2BFL·Bos1CM complex before (black) and after (red) chasing with a 7.5‐fold molar excess of unlabeled Sgt2. The Sgt2BFL·Bos1CM complex was generated as in (B) and stringently purified via the affinity tag on Sgt2. Further increasing the concentration of unlabeled Sgt2 did not lead to additional increases in donor fluorescence, indicating that the competition is largely complete.
Equilibrium titrations using this assay showed that the Sgt2 concentrations required for 50% transfer (K transfer) of Bos1 and Sbh1 in the presence of 2–5 μM Ssa1 were 30.1 ± 4.4 nM and 40.2 ± 3.4 nM, respectively (Fig 4A, black and green), suggesting that the equilibrium of the transfer reaction is ~102‐fold in favor of Sgt2. Further, the transfer of both TA substrates was complete within ~100 s (Fig 4B). Importantly, the R171A, R175A mutations in the Sgt2 TPR domain, which disrupt the Sgt2–Ssa1 interaction (Wang et al, 2010; Chartron et al, 2011), substantially reduced the efficiency of TA loading on Sgt2 (Fig 4A, purple and blue; Sgt2‐TPRmt), indicating that efficient TA transfer requires the direct interaction between Ssa1 and Sgt2. Thus, TAs bound to Ssa1 are efficiently transferred onto Sgt2 in an Ssa1·TA·Sgt2 complex.
Figure 4. Ssa1 efficiently transfers TAs to Sgt2 in an Ssal·TA·Sgt2 complex.
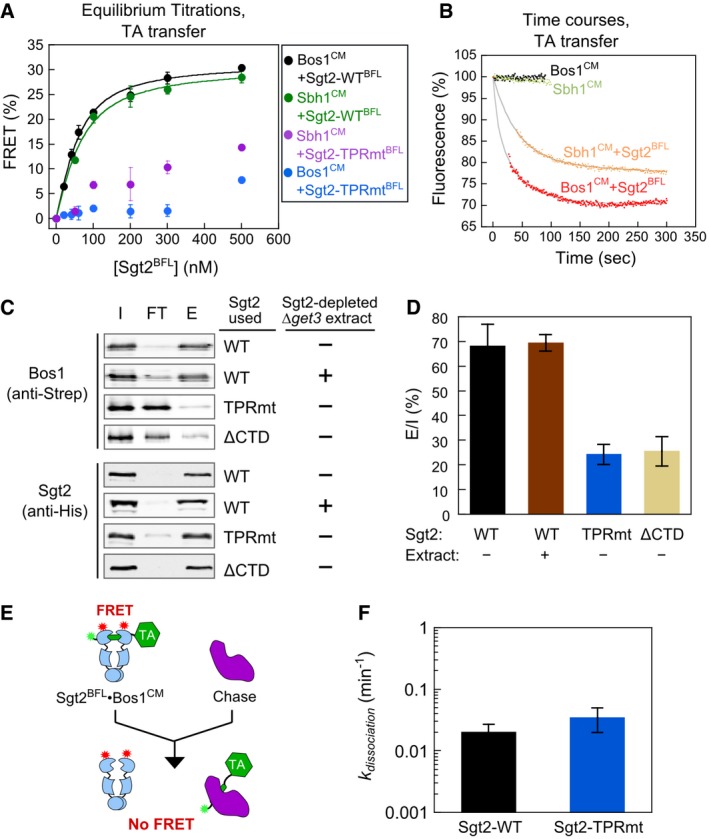
- Equilibrium titrations of TA transfer from Ssa1 to Sgt2‐WTBFL or Sgt2‐TPRmtBFL, carried out as outlined in Fig 3B at indicated Sgt2 concentrations. The reactions with Bos1CM used 5 μM Ssa1 and 2 mM ATP, and the reactions with Sbh1CM used 2 μM Ssa1 and 2 mM ATP during the pre‐incubation. The lines are fits of the data with Sgt2‐WTBFL to equation (3). Values are mean ± SD, with n = 3 (Bos1) and n = 2 (Sbh1).
- Time courses of TA transfer from Ssa1 to Sgt2. After 1 min of pre‐incubation of 50 nM Bos1CM or Sbh1CM with 5 μM Ssa1, 500 nM (red) or 100 nM Sgt2BFL (orange) was added, respectively. The data at early times (27˜39 s) were missing due to manual mixing time. Gray lines are fits of the data to equation (4). The black and green traces were from control samples that did not receive Sgt2BFL and controlled for photobleaching of the donor dye over time.
- Representative Western blot analyses of the TA transfer reaction monitored using His6‐Sgt2 pull‐downs. After pre‐incubation of 300 nM Strep‐Bos1 with 5 μM Ssa1, 500 nM wild‐type or mutant Sgt2‐WT was added. In the “+ extract” reaction, Sgt2‐depleted ∆get3 extract was supplemented during addition of Sgt2‐WT. I, FT, and E denote total input, flowthrough, and elution, respectively.
- Quantification of Bos1 that eluted with wild‐type or mutant Sgt2 from the data in (C) and replicates. Values are reported as mean ± SD, with n ≥ 3 except for the extract sample (n = 2).
- Scheme of pulse‐chase experiments to measure the kinetic stability of the Sgt2·Bos1 complex. Wild‐type and mutant His6‐Sgt2BFL·Bos1CM complexes were reconstituted as outlined in Fig 3B and purified using metal affinity resin. A higher concentration of Sgt2BFL‐TPRmt than wild‐type Sgt2BFL was used during reconstitution to generate sufficient amounts of mutant Sgt2·TA complexes for analysis.
- Dissociation rate constants of Sgt2‐WT·Bos1 and Sgt2‐TPRmt·Bos1 complexes reconstituted via transfer from Ssa1. Values are mean ± SD, with n = 4.
To independently test this model, we carried out the transfer reaction as outlined in Fig 3B but analyzed the reaction by stringent affinity capture of His6‐Sgt2 (Fig EV2A) and immunoblotting for TAs associated with it. Robust transfer of Bos1 onto Sgt2 was observed using this transfer assay (~70% of input Bos1; Fig 4C and D). Quantitative Western blot analysis of the affinity‐captured samples showed that 180 nM Bos1 co‐purified with Sgt2, whereas the residual Ssa1 that co‐purified with Sgt2 were 8.1 and 31 nM without and with TA present, respectively (Fig EV2A–C). Thus, over 83% of the Bos1 detected in this transfer assay was directly bound to Sgt2.
Figure EV2. Ssa1 that co‐purified with Sgt2 in the His6‐pull‐down assay accounted for < 17% of Sgt2‐associated TA.
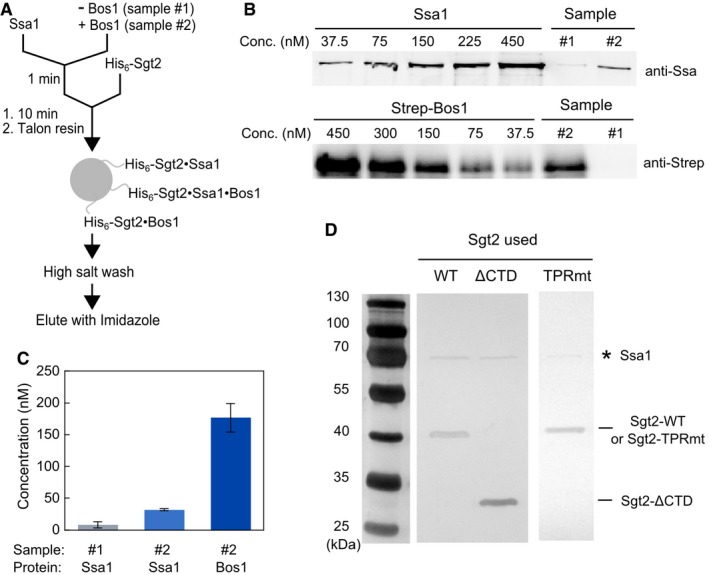
- Scheme of the His6‐Sgt2 pull‐down assay to monitor TA loading onto Sgt2 used for the experiments in Fig 4C. 5 μM Ssa1 and 2 mM ATP were pre‐incubated in the absence (sample #1) or presence (sample #2) of 300 nM Strep‐Bos1 for 1 min, and further incubated for 10 min after addition of 500 nM His6‐Sgt2. The mixture was incubated with Talon resin at 4°C for 10 min. All three possible complexes that can be immobilized on the resin via His6‐Sgt2 are depicted. After washing the resin with high‐salt (500 mM NaCl) buffer, bound proteins were eluted with 300 mM imidazole.
- Quantitative Western blot analysis of the amount of Ssa1 and Bos1 in the elution fractions from samples #1 and #2 in (A), using known concentrations of purified Ssa1 and strep‐Bos1. Proteins were detected with their corresponding secondary antibodies labeled with IRDye.
- Quantified concentrations of Ssa1 and Bos1 that co‐purified with Sgt2 in the elution fractions: 8.2 nM Ssa1 in the absence of Bos1 (sample #1), and 32 nM Ssa1 and 180 nM Bos1 in the presence of Bos1 (sample #2). Values are reported as mean ± SD, with n ≥ 2.
- Silver‐stained images of Sgt2·TA complexes reconstituted and purified as in panel (A) for wild‐type and mutant Sgt2. * denotes the Ssa1 band. Note that this gel was overdeveloped to visualize the presence of Ssa1.
Source data are available online for this figure.
Consistent with the observations in the FRET‐based transfer assay, the point mutations in the TPR domain of Sgt2 reduced the amount of Sgt2‐associated Bos1 to ~20% (Fig 4C and D). As an additional negative control, deletion of the C‐terminal substrate‐binding domain of Sgt2 (Sgt2∆CTD) also substantially reduced Bos1 association with Sgt2 (Fig 4C and D). As described in the previous paragraph, the small amounts of Bos1 that co‐eluted with mutants Sgt2‐TPRmt and Sgt2∆CTD may be attributed to residual Ssa1 that remained during the purification (Fig EV2D). The defect of Sgt2‐TPRmt in TA capture was not detected previously (Wang et al, 2010), likely due to the different substrate proteins tested in this and the previous work (Bos1 and Sbh1 here versus Sec22 in Wang et al (2010)). Finally, inclusion of Sgt2‐depleted ∆get3 yeast lysate did not affect the efficiency of this transfer (Fig 4C and D), suggesting that other TPR domain‐containing proteins in the cytosol do not interfere with TA transfer from Ssa1 to Sgt2.
To exclude the possibility that Sgt2‐TPRmt is impaired in interaction with TA, we measured the kinetic stability of Sgt2‐WT·Bos1 and Sgt2‐TPRmt·Bos1 complexes. Preformed, purified His6‐Sgt2BFL·Bos1CM complexes were chased with an external membrane protein chaperone, chloroplast SRP43 (cpSRP43; Liang et al, 2016), and the kinetics of Bos1 dissociation from Sgt2 was monitored by the loss of FRET (Fig 4E). We confirmed that a superactive variant of cpSRP43 (Liang et al, 2016) effectively suppressed TA aggregation in aqueous solution (Fig EV3A), indicating that it can bind and trap TA substrates. The dissociation rate constants of Bos1 from Sgt2‐WT and Sgt2‐TPRmt were similar and > 20‐fold slower than the rate of the Ssa1‐to‐Sgt2 TA transfer, for both Sgt2·Bos1 complexes reconstituted via transfer from Ssa1 (Fig 4F) and complexes prepared from IVT in E. coli extract (Rao et al, 2016; Fig EV3B). Therefore, the reduced TA capture by Sgt2‐TPRmt in Figs 3 and 4 can be attributed to less efficient TA loading from Ssa1 onto Sgt2. Together, the results of the FRET and pull‐down assays show that TA substrates captured by Ssa1 can be efficiently and directly transferred to Sgt2 and that the transfer is most efficient in an Ssa1·TA·Sgt2 complex.
Figure EV3. cpSRP43 can serve as a trap for TA proteins.
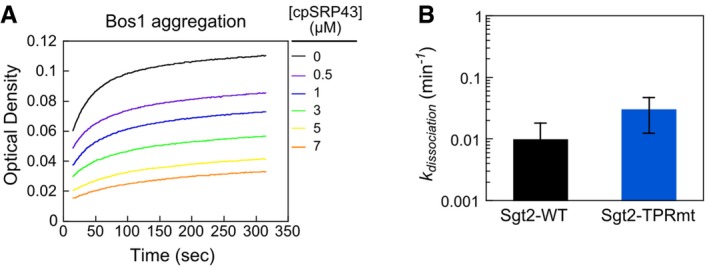
- cpSRP43 prevents Bos1 aggregation in a dose‐dependent manner, as analyzed by the turbidity assay.
- The intrinsic dissociation rate constants of Bos1 from the Sgt2‐WT·Bos1 and Sgt2‐TPRmt·Bos1 complexes reconstituted via IVT in E. coli extract. Bos1CM was translated in the E. coli S30 translation extract coupled to amber suppression, which co‐translationally incorporates the non‐natural fluorescent amino acid L‐(7‐hydroxycoumarin‐4‐yl)ethylglycine (CM) into Bos1 near the TMD, in the presence of 2 μM His6‐Sgt2‐WTBFL or His6‐Sgt2‐TPRmtBFL, as described in (Rao et al, 2016). The resulting His6‐Sgt2‐WTBFL·Bos1CM or His6‐Sgt2‐TPRmtBFL·Bos1CM complexes were purified using metal affinity resin (Rao et al, 2016). Bos1CM release from Sgt2‐WTBFL or Sgt2‐TPRmtBFL was measured as outlined in Fig 4E. Values are mean ± SEM, with n = 2.
To test the directional flow of TA once it is bound to Sgt2, we prepared His6‐Sgt2·Bos1 complexes via transfer from Ssa1 and immobilized His6‐Sgt2 on a metal affinity resin (Appendix Fig S2). The immobilized Sgt2·Bos1 complex was challenged with either Ssa1 or Get3, the targeting factor downstream of Sgt2 in the GET pathway (Fig 5A, Elution I). If Bos1 was transferred from Sgt2 to these factors, Bos1 should be detected in this elution. Over 99% of Bos1 remained bound to Sgt2 on the resin after incubation with Ssa1 (Fig 5B, lane 1). In contrast, over 80% of Bos1 was eluted by Get3 in the presence of Get4/5 (Fig 5B, lanes 2–3), consistent with Get4/5‐dependent TA transfer from Sgt2 to Get3 established previously (Wang et al, 2010; Mateja et al, 2015; Shao et al, 2017). Thus, transfer of TA in the reverse direction, from Sgt2 to Ssa1, is unfavorable.
Figure 5. Ssa1 enables efficient and unidirectional TA transfer to Sgt2 and then to Get3, and this stepwise transfer ensures the solubility and targeting competence of TAs.
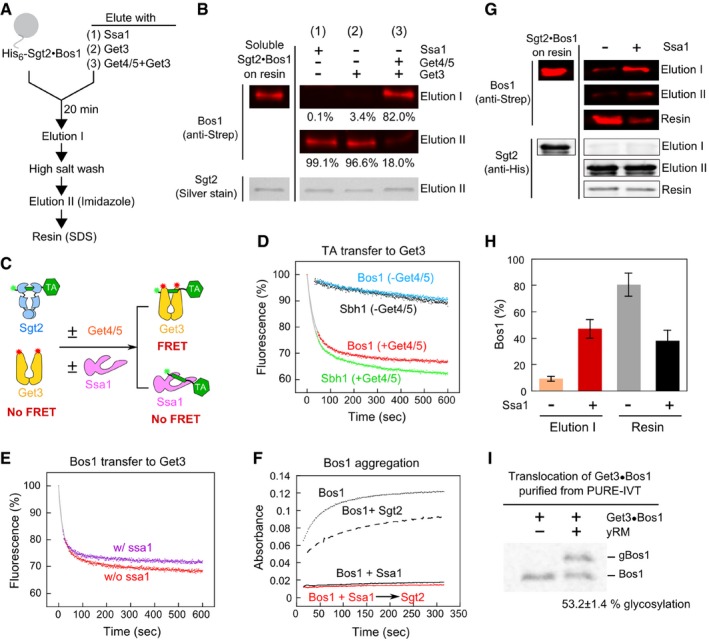
- Schematic of on‐bead TA transfer from Sgt2 to Ssa1 or Get3. Reconstituted Sgt2·Bos1 complex was immobilized on metal affinity resin as shown in Appendix Fig S2. After incubation with 3 μM Ssa1 (reaction 1), 2 μM Get3 (reaction 2), or 2 μM Get3 and 1 μM Get4/5 (reaction 3) for 20 min, Elution I was collected. After extensive washes with high salt (500 mM NaCl)‐containing assay buffer, the proteins that remain bound to the resin were eluted with imidazole (Elution II). For the experiment in (G), aggregated proteins irreversibly bound to the resin were further released with SDS (resin). All reactions contained 2 mM ATP.
- Elution fractions from the reactions in (A) were resolved by SDS–PAGE and visualized by Western blot (against strep‐Bos1) and silver stain (for Sgt2). The numbers denote %Bos1 in each fraction. To estimate the total amount of soluble Sgt2·Bos1 complex (first column), the resin was directly eluted with imidazole‐containing buffer without Elution I.
- Scheme of the FRET‐based transfer assay to monitor TA transfer from Sgt2 to Get3. Green and red asterisks denote donor and acceptor dyes, respectively.
- Time courses of TA transfer from Sgt2 to Get3 in the presence and absence of Get4/5. The transfer reactions were initiated by addition of 200 nM Get3ATTO, 2 mM ATP in the absence and presence of 200 nM Get4/5. Grey lines are fits of the data to equation (4). The fluorescence in the absence of transfer (100%) was determined prior to addition of Get3 and Get4/5.
- Time courses of TA transfer from Sgt2 to Get3, measured using the FRET assay in (C), in the presence and absence of 3 μM Ssa1. The transfer reaction was initiated by addition of a mixture of 500 nM Get3BFL, 500 nM Get4/5, and 2 mM ATP to 200 nM preformed Sgt2·Bos1CM, and donor fluorescence was monitored over time. The grey lines are fits of the data to equation (4).
- Solubility of Bos1 mixed with Ssa1 (3 μM) before (black) and after (red) addition of 1.5 μM Sgt2. The dotted and dashed lines are Bos1 alone and Bos1 directly mixed with Sgt2, respectively.
- Sgt2·Bos1 complexes reconstituted in the absence and presence of Ssa1 were analyzed for the efficiency of TA transfer to Get3 as outlined in (A). In Elution I, Sgt2·Bos1 immobilized on metal affinity resin was eluted with 2 μM Get3 and 1 μM Get4/5 in the presence of 2 mM ATP. Elution I, Elution II, and Resin fractions were resolved by SDS–PAGE and visualized by Western blot against Strep‐Bos1 and His6‐Sgt2. To estimate the total amount of immobilized Sgt2·Bos1 (first column), the resin was directly eluted with SDS.
- Quantification of the data in (G) and replicates. %Bos1 in both Elution I and Resin fractions was quantified, and values are reported as mean ± SD, with n = 3.
- Representative autoradiograph of TA insertion reactions with Get3·Bos1 generated and purified from chaperone‐supplemented PURE‐IVT. 35S‐labeled Bos1 was synthesized by PURE‐IVT in the presence of 6 μM Ssa1, 0.5 μM Sgt2, 0.5 μM Get4/5, and 0.75 μM Get3‐FLAG. The Get3·Bos1 complex was purified using anti‐FLAG magnetic beads and presented to 3 μl yeast rough microsomes (yRM) in a total reaction volume of 15 μl. Translocation efficiency was reported below the image (mean ± SD, n = 3).
To independently support these findings, we used an established assay in which TA loading onto Get3 was monitored by FRET between Bos1CM and BODIPY‐ or ATTO488‐labeled Get3 (Get3BFL or Get3ATTO; Fig 5C; Rao et al, 2016). Consistent with the result in Fig 5B, both Bos1 and Sbh1 were rapidly transferred from Sgt2 to Get3 in a Get4/5‐dependent manner (Fig 5D). If back transfer of TA to Ssa1 were significant, Ssa1 would reduce the transfer of TA from Sgt2 to Get3 (Fig 5C). However, a sixfold molar excess of Ssa1 over Get3 did not interfere with this transfer (Fig 5E), indicating that Ssa1 cannot efficiently compete with Get3 for TA binding once TA was loaded onto Sgt2. This observation, together with the favorable equilibrium of TA transfer from Ssa1 to Sgt2 (Fig 4A) and the lack of back transfer to Ssa1 (Fig 5), establishes that TA transfer from Ssa1 to Sgt2 is energetically downhill and thus places Ssa1 upstream of Sgt2.
Stepwise substrate loading via Hsp70 ensures the solubility and targeting competence of TAs
The results above, together with previous work (Rao et al, 2016), establish at least two sequential TA transfers in the GET pathway, from Ssa1 to Sgt2, and then to Get3. Moreover, both TA transfers are energetically downhill (this work and Rao et al (2016)), indicating that Sgt2 and Get3 form more stable complexes with the TA than their upstream chaperones. This raises an intriguing question: What is the advantage of using this complex mechanism of substrate loading? An attractive hypothesis is that stepwise substrate loading via Hsp70 ensures the solubility and targeting competence of nascent TAs. In support of this model, turbidity assays showed that, if Bos1 was first mixed with Ssa1 followed by transfer onto Sgt2, the TA substrate no longer forms large aggregates, in contrast to the extensive aggregation observed when Bos1 was directly mixed with Sgt2 (Fig 5F).
To test whether TA loading via Ssa1 improves the targeting competence of TAs, we generated His6‐Sgt2·Bos1 complexes by direct loading of Bos1 onto Sgt2 or via transfer from Ssa1. We tested the efficiency of these complexes in undergoing the subsequent step in the pathway, Get4/5‐dependent TA transfer from Sgt2 to Get3, using the on‐bead transfer assay (Fig. 5A). In addition to the elution fractions, which represent soluble Sgt2·TA complexes, we also measured the amount of Bos1 irreversibly bound to the resin, presumably due to aggregation (Fig 5A, Resin). The Sgt2·Bos1 complex prepared via transfer from Ssa1 transferred > 47% of Bos1 to Get3 (Fig 5G and H). In contrast, the Sgt2·Bos1 complex generated by direct loading displayed substantially reduced TA transfer to Get3 (9.3% ± 1.8), whereas over 80% of Bos1 formed irreversible aggregates on the resin (Fig 5G and H). Together with the turbidity data (Fig 5F), these results suggest that TA loading via Hsp70 is important for generating a soluble and functionally competent Sgt2·TA complex.
In addition, the FRET‐based assay to monitor the transfer of TACM from Sgt2 to Get3ATTO (Fig 5C; Rao et al (2016)) showed that TA was rapidly transferred from Sgt2·TACM, reconstituted via transfer from Ssa1, to Get3ATTO in the presence of Get4/5, with half‐times (t 1/2) of 24.8 and 24.1 s for Bos1 and Sbh1, respectively (Fig 5D). These transfer kinetics are comparable to that observed with the Sgt2·Bos1 complex prepared from IVT in cell lysate (t 1/2 = 17.7 s; Rao et al (2016)) and demonstrate the kinetic competence of the Sgt2·TA complexes. The ability of Get4/5 to accelerate this transfer event was also observed with the reconstituted Sgt2·TACM (Fig 5D). Finally, the Get3·Bos1 complex generated from PURE‐IVT in the presence of near physiological concentrations of the required cytosolic factors (Ssa1, Sgt2, Get4/5, and Get3) was highly competent for insertion into yeast rough microsomes (yRM; 53.2 ± 1.4%; Fig 5I). Thus, Ssa1 provides a minimal chaperone necessary and sufficient for maintaining TAs in a transfer and translocation‐competent conformation.
Cytosolic Hsp70 is important for the biogenesis of GET substrates in vivo
We next tested whether Ssa1 is important for the targeting and insertion of TAs into the ER membrane in vivo. To this end, we used an established temperature‐sensitive strain, ssa1 ts (ssa1 ts ssa2Δssa3Δssa4Δ), in which Ssa1 inactivation could be observed as early as 2 min and was complete within 10 min after shift of ssa1 ts to nonpermissive temperature (37°C; Becker et al, 1996). In contrast, significant changes in the activity and degradation of Hsp70 client proteins at steady state were observed in this strain after 60 min of temperature shift (Kim et al, 1998; McClellan et al, 2005; Roy et al, 2015). Thus, ssa1 ts allows direct effects of Hsp70–client interactions to be examined with minimal loss of existing protein targeting and translocation machineries, activation of stress pathways, or other perturbations on the global proteome status in the cell.
As model TA substrates for in vivo measurements, we used BirA fused to the C‐terminal targeting sequence of Bos1 (Fig 6A, BirA‐Bos1) or other tested substrates. Substrate proteins were expressed from a low copy plasmid in yeast cells to minimize overload on the targeting pathway. An engineered opsin tag at the C‐terminus of the model substrates allows their efficient glycosylation upon insertion into the ER lumen (Yabal et al, 2003; Wang et al, 2010; Rao et al, 2016), providing a quantitative readout for the targeting and insertion of substrate proteins. The insertion kinetics of newly synthesized TAs in cells was monitored using a pulse‐chase assay coupled to immunoprecipitation of HA‐tagged substrate proteins. We first showed that a major translocation defect was observed with BirA‐Bos1 in ∆get3 cells in both steady‐state and pulse‐chase analyses (Figs 6B and EV4A and B), confirming BirA‐Bos1 as a GET‐dependent substrate. ∆sgt2 cells exhibited a more modest defect in mediating rapid Bos1 targeting and insertion into the ER (Fig EV4C and D), consistent with previous observations (Kohl et al, 2011; Kiktev et al, 2012; Yeh et al, 2014). As reported, alternative pathways could enable the targeting of TAs when Sgt2 or Get3 was deleted (Aviram et al, 2016; Haßdenteufel et al, 2017), which may account for the observed TA targeting at steady state.
Figure 6. Ssa1 facilitates the post‐translational targeting of TAs to the ER.
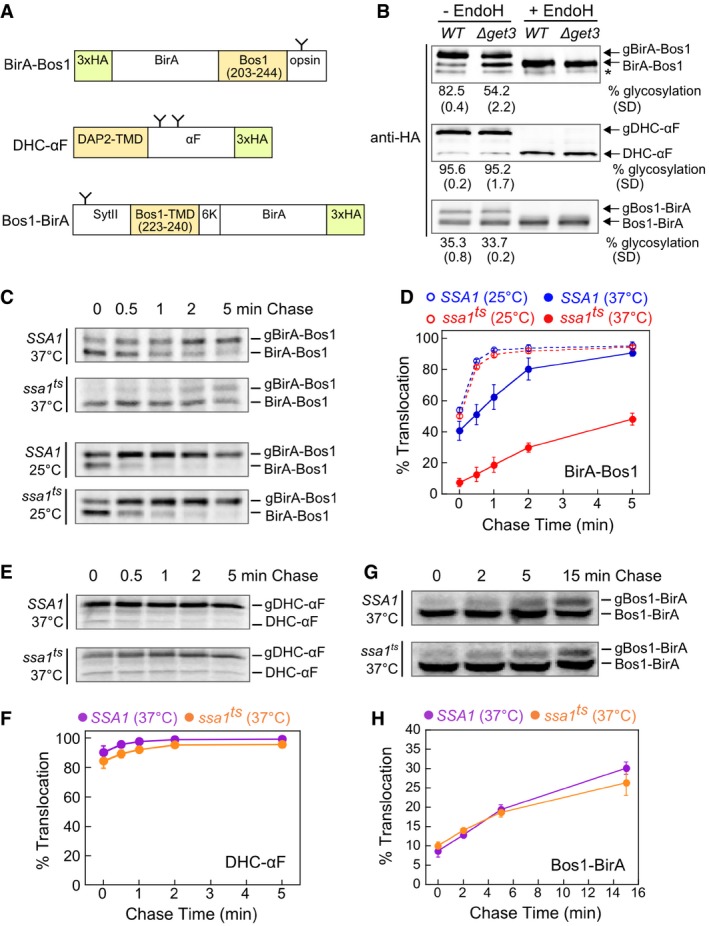
-
ASchematic representation of model substrates for the in vivo targeting assay (see details in Materials and Methods). The C‐terminal opsin tag or the native glycosylation sites in ppαF (Ng et al, 1996) and SytII (N32; Kida et al, 2000) allow substrate insertion into the ER in the correct topology to be detected by glycosylation. The glycosylation sites on the model substrates are depicted as “Y”. N‐ or C‐terminal 3xHA tags allow immunoprecipitation or Western blot detection of the substrates. The residues of Bos1 used in the substrates are shown in the diagram. To enforce type I topology of the Bos1‐BirA substrate, additional sequences were introduced at the N‐terminus (mouse synaptotagmin II (SytII) residues 1–18, 20–45, 61–65) and C‐terminus (insertion of six lysines) of the Bos1‐TMD.
-
BSteady‐state translocation levels of BirA‐Bos1, DHC‐αF, and Bos1‐BirA in WT and ∆get3 cells. Glycosylated (g) and non‐glycosylated proteins were resolved by SDS–PAGE, detected by anti‐HA antibody, and corroborated by endoglycosidase H (endoH) treatment. Quantifications of glycosylation efficiency are shown below the blot images. * denotes a non‐specific band detected by anti‐HA antibody.
-
CPulse‐chase analysis of the translocation of metabolically labeled BirA‐Bos1 in SSA1 (blue) and ssa1 ts (red) cells at 25°C (dashed lines and open circles) and 37°C (solid lines and closed circles).
-
DQuantification of the data in (C) and replicates.
-
E–HPulse‐chase analysis of the translocation of metabolically labeled DHC‐αF (E) and Bos1‐BirA (G) in SSA1 (purple) and ssa1 ts (orange) cells at 37°C. Panels (F) and (H) show the quantification of the data in (E) and (G), respectively, and their replicates.
Figure EV4. BirA‐Bos1 was inserted into the ER membrane in a GET‐dependent manner.
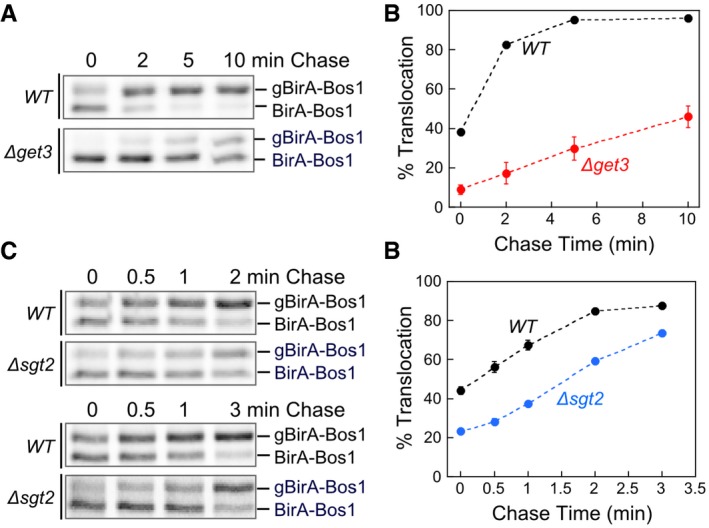
- Representative autoradiograms for pulse‐chase analysis of the translocation of metabolically labeled BirA‐Bos1 in WT (black) and ∆get3 (red) cells.
- Quantification of the data in (A) and replicates.
- Representative autoradiograms for pulse‐chase analysis of the translocation of metabolically labeled BirA‐Bos1 in WT (black) and ∆sgt2 (blue) cells.
- Quantification of the data in (C) and replicates.
Using the pulse‐chase assay, we tested the effect of cytosolic Hsp70 inactivation on the insertion kinetics of newly synthesized BirA‐Bos1. At permissive temperature (25°C), BirA‐Bos1 was efficiently inserted into the ER in both ssa1 ts cells and the corresponding wild‐type strain, SSA1 (SSA1ssa2Δssa3Δssa4Δ; Becker et al, 1996), with the insertion reactions complete within 1 min of chase (Fig 6C and D). In contrast, after shifting to the nonpermissive temperature for 5 min, insertion of BirA‐Bos1 was substantially delayed in ssa1 ts cells compared to SSA1 cells (Fig 6C and D). At steady state, the defect of Bos1 insertion in ssa1 ts cells was comparable to that observed in Δget3 cells. When BirA was fused to the TMDs of other GET‐dependent TAs, such as Sed5 and Pep12 (Schuldiner et al, 2008; Mateja et al, 2015), significant defects in translocation kinetics were also observed in ssa1 ts cells at nonpermissive temperature (Fig EV5). These results strongly suggest that cytosolic Hsp70 is important for the in vivo targeting of a variety of TAs harboring hydrophobic TMDs. We also note that the short timescale of the pulse‐chase assay (5–7 min) coupled with the rapid inactivation of Ssa1 in the ssa1 ts strain ensures that specific targeting defects of TAs were observed prior to disruptions of the cellular proteome, which were detected an hour after the temperature shift (Kim et al, 1998; McClellan et al, 2005; Roy et al, 2015). Therefore, cytosolic Hsp70 plays an important role in the ER‐targeting of TAs in vivo.
Figure EV5. Ssa1 facilitates the targeting of multiple GET‐dependent TAs to the ER.
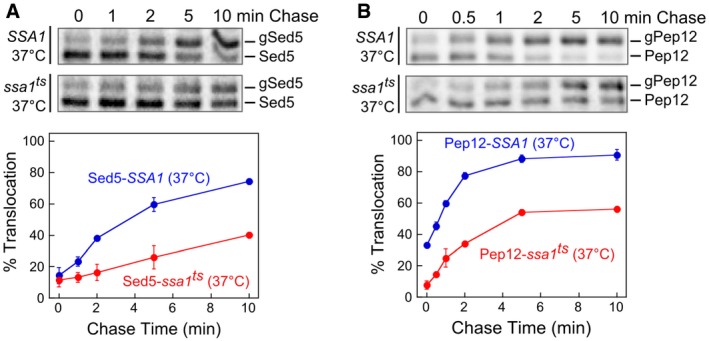
-
A, BThe top panels show representative autoradiograms for pulse‐chase analysis of the translocation of metabolically labeled Sed5 (A) and Pep12 (B) in SSA1 (blue) and ssa1 ts (red) cells at 37°C. The bottom panels show the quantifications of translocation efficiencies at indicated time points. All values are mean ± SD, with n = 2 (biological replicates). Error bars are shown but may not be visible in some cases.
To exclude the possibility that the observed TA insertion defects arise from a general folding deficiency in ssa1 ts cells, we tested a control substrate in which the Bos1‐TMD is placed at the N‐terminus of BirA (Bos1‐BirA; Fig 6A) to convert it into a GET‐independent substrate (Fig 6B). In addition, we tested an engineered substrate, DHCαF (Fig 6A), in which the signal sequence of prepro‐α‐factor is replaced by the hydrophobic core of the dipeptidyl aminopeptidase B (DAP2) signal sequence to convert it into a signal recognition particle (SRP)‐dependent (Ng et al, 1996), Get3‐independent substrate (Fig 6B). No significant differences between SSA1 and ssa1 ts cells were detected in the ER‐targeting and translocation of either substrate at nonpermissive temperature (Fig 6E–H). In addition, a previous study examined the import of six precursor proteins to the ER and three to mitochondria under similar experimental conditions, and found that ssa1 ts only affected the import of two ER‐destined and one mitochondria‐destined substrates at nonpermissive temperature (Becker et al, 1996). These substrate specificities are unlikely to arise from pleiotropic effects of Ssa1 inactivation. Finally, the total amount of protein substrates remained largely the same throughout the chase (Fig 6), consistent with the lack of stress‐activated degradation under our experimental conditions. Together, these observations strongly suggest that the ER‐insertion defects of TA substrates in ssa1 ts cells result directly from disruption of Ssa1 participation in the targeting of newly synthesized TAs.
We further asked whether Ssa1 is important for loading newly synthesized TAs onto Sgt2 in vivo. To this end, we tested whether disrupting the Hsp70–Sgt2 interaction in Sgt2‐TPRmt impacts TA capture by Sgt2. As TAs are rapidly transferred to Get3 and inserted into the ER once they are loaded onto Sgt2 (Figs 5 and 6; Rao et al, 2016), we used ∆get3 strains harboring wild‐type Sgt2 and mutant Sgt2‐TPRmt to facilitate detection of TA accumulation on Sgt2. Bos1 was transiently expressed in SGT2FLAG∆get3 (Wang et al, 2010) and sgt2TPRmtFLAG∆get3 strains, and the amount of Bos1 associated with Sgt2 was quantified and compared after immunoprecipitation of C‐terminally 3xFLAG‐tagged Sgt2 (Fig 7A). TA association with wild‐type Sgt2, albeit transient due to re‐routing of substrates to alternative pathways, can be robustly detected in the lysate of SGT2FLAG∆get3 cells (Fig 7B). In contrast, this association was undetectable in sgt2TPRmtFLAG∆get3 cells (Fig 7B and C). In agreement with these in vivo results, when Bos1 translated in Ssa1‐supplemented PURE‐IVT was incubated with yeast lysate from sgt2TPRmtFLAG ∆get3 cells, the amount of TAs bound to Sgt2 was substantially reduced compared to the results obtained with the SGT2FLAG∆get3 lysate (Fig 7D–F). As described earlier, the reduced TA association with Sgt2‐TPRmt is not due to a weaker interaction with TA (Fig 4F). These results suggest that the interaction of the Sgt2 TPR domain with heat‐shock proteins plays an important role in inducing the influx of TA substrates into the GET pathway in the cellular environment.
Figure 7. Sgt2‐TPR is important for TA loading onto Sgt2 in vivo .
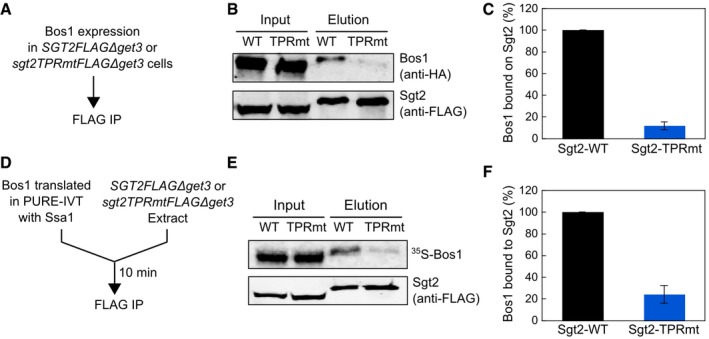
- Scheme of the FLAG‐IP assays to measure the efficiency of TA capture by Sgt2‐WT or Sgt2‐TRPmt in vivo. TAs bound to Sgt2 were isolated by FLAG‐IP as described in Materials and Methods.
- Representative images of the Sgt2‐FLAG‐IP analysis of Sgt2‐bound TAs in vivo. 3xHA‐Bos1 and Sgt2‐3xFLAG were detected by Western blot analysis.
- Quantification of the relative amount of Sgt2‐bound Bos1 from the data in (B) and replicates. Values are reported as mean ± SD, with n = 3–4.
- Scheme of the FLAG‐IP assays to measure the efficiency of TA capture by Sgt2‐WT or Sgt2‐TRPmt in yeast extract. TAs bound to Sgt2 were isolated by FLAG‐IP as described in Materials and Methods.
- Representative images of the Sgt2‐FLAG‐IP analysis of Sgt2‐bound TAs in yeast extract. 35S‐Bos1 was detected by autoradiography. Sgt2‐3xFLAG was detected by Western blot analysis.
- Quantification of the relative amount of Sgt2‐bound Bos1 from the data in (E) and replicates. Values are reported as mean ± SD, with n = 3–4.
Source data are available online for this figure.
Discussion
In this work, biochemical reconstitutions demonstrated a stepwise substrate relay mechanism for newly synthesized TAs in an Hsp70‐Sgt2‐Get3 cascade during their post‐translational targeting to the ER membrane. Although both Sgt2 and Get3 can form stable complexes with TAs (Mateja et al, 2015; Rao et al, 2016; Shao et al, 2017; Wang et al, 2010; and this work), their substrate‐binding kinetics at physiological concentrations appear to be insufficient to compete with the rapid and irreversible aggregation of TAs in aqueous environments (this work and Mariappan et al (2010)). Hsp70, by virtue of its abundance and rapid client association kinetics, effectively competes with off‐pathway processes and is essential for capturing nascent TAs in a soluble and translocation‐competent conformation (Fig 8, step 1). Via its interaction with the Sgt2 TPR domain, Hsp70 relays the TA substrates to Sgt2 (this work) and then to the Get3 ATPase (Wang et al, 2010) for delivery to the ER (Fig 8, steps 2–6). The TA transfer measurements from this and an earlier work (Rao et al, 2016) further showed that both the Ssa1‐to‐Sgt2 and Sgt2‐to‐Get3 TA relays are energetically downhill, with the transfer equilibrium ~30–100 fold in favor of the next TA‐binding chaperone. Thus, substrate capture and loading in the GET pathway are governed by a combination of kinetic constraints and thermodynamic driving forces.
Figure 8. Revised model of TA targeting in yeast.
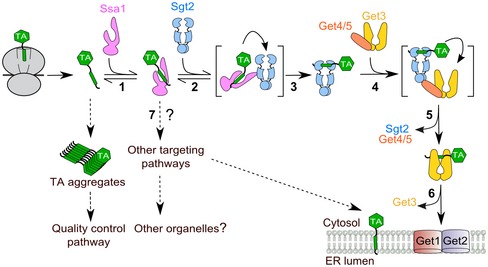
Ssa1 captures TAs upon their release from the ribosome (step 1) and effectively competes with aggregation and potential quality control pathways (dashed arrows). Soluble TAs are transferred to Sgt2 (steps 2–3) and then to Get3 (steps 4–5), which delivers TAs to the Get1/2 receptors for insertion into the ER membrane (step 6). Both TA transfer events in the GET pathway are facilitated by interacting motifs or proteins that bridge the upstream and downstream chaperones (transfer complexes in brackets). TAs bound to Ssa1 might also enter alternative targeting pathways (step 7, dashed arrows) that deliver the substrates to the ER or to other organelles (Aviram et al, 2016; Guna et al, 2018).
Intriguingly, stepwise substrate loading via Hsp70 appears to confer a “memory” on the conformational quality of TA substrates. In contrast to direct loading of TA substrates onto Sgt2, which tends to result in aggregated and inactive complexes, loading of substrates via Hsp70 generated soluble and functionally competent Sgt2·TA and Get3·TA complexes. In the simplest model, Ssa1 could generate this memory by lowering the concentration of free TAs in solution, thus preventing the aggregation of TAs prior to their capture by Sgt2. However, the results of this and previous works suggest a more active mechanism, as both of the substrate relay events in the GET pathway are facilitated by concerted mechanisms in which the upstream and downstream TA‐binding factors are tethered in a complex (Fig 8, transfer complexes in brackets; this work and Rao et al, 2016). We propose that these concerted transfer mechanisms could minimize the cytosolic re‐exposure of TAs and thus preserve their solubility and functional competence during the relays, while also providing a kinetically more favorable route for substrate loading onto the targeting factor. Analogous “substrate funneling” through chaperone cascades may provide an effective strategy for the post‐translational targeting of membrane proteins in general (Shao et al, 2017).
The roles of yeast Hsp70 in TA biogenesis described here may be conserved in mammalian cells. Although SGTA is the most upstream factor identified thus far in the mammalian GET pathway (Shao et al, 2017), the 30‐fold higher abundance of Hsc70 in human cells compared to SGTA (Kulak et al, 2014) and its rapid substrate‐binding kinetics render it likely that nascent TAs engage Hsc70 prior to SGTA. Newly synthesized TAs have been observed to associate with Hsc70 in rabbit reticulocyte lysates (Abell et al, 2007; Colombo et al, 2009). Further, the physical association between Hsp70 and the TPR domain of Sgt2 was also conserved in their mammalian homologues (Angeletti et al, 2002). Nevertheless, the precise roles and mechanisms of action of Hsc70 in TA targeting remain to be determined.
Although previous work has implicated cytosolic Hsp70 in preprotein translocation to the ER and mitochondria (Deshaies et al, 1988), it was unclear whether this represents a general function of Hsp70. Among the nine preprotein substrates examined, the import of only two ER‐destined and one mitochondria‐destined substrates were affected upon Ssa1 inactivation (Becker et al, 1996). This work provides evidence for a general role of Hsp70 in directing the localization of an essential class of integral membrane proteins. Notably, TPR‐containing cochaperones have been identified on multiple organelles, including Pex5 (peroxisomal biogenesis factor 5) involved in the biogenesis of peroxisomal matrix proteins (Harano et al, 2001; Harper et al, 2003), Tom70 that mediates the import of mitochondria inner membrane proteins (Young et al, 2003b), and Sec71/72 that form a tetrameric complex with Sec62/Sec63 and mediate post‐translational translocation at the ER membrane (Tripathi et al, 2017). The interaction of Hsp70 with the TPR domain of Tom70 has been shown to mediate the mitochondrial import of the peptide transporter Mdl1 and ADP/ATP carrier proteins (Young et al, 2003b). These observations raise the possibility of analogous substrate relay mechanisms that could assist in the localization of nascent membrane proteins to diverse cellular organelles.
Our data showed that mutations in the Sgt2 TPR domain impair TA loading onto this cochaperone in vitro and in vivo (Figs 3, 4 and 7), suggesting that the physical interaction of Sg2 with Hsp70 modulates substrate influx into the GET pathway. Compared to deletion of other GET pathway genes, however, Δsgt2 yeast cells exhibit a weak genetic link to Get3, weak defects in cell growth and in TA insertion in vivo (Kohl et al, 2011; Yeh et al, 2014 and Fig EV4). Accumulating data indicate the presence of multiple redundant pathways for TA targeting to the ER in yeast (Kiktev et al, 2012; Aviram et al, 2016; Casson et al, 2017; Guna et al, 2018), which may contribute to the weak phenotype of Δsgt2 cells as TAs can be re‐routed to alternative targeting pathways (Fig 8, step 7). Re‐routing of TA substrates upon deletion of Sgt2 is supported by the observation that the model TA, Sbh1p, forms cytosolic aggregates with Get3 and Get4/5 in ∆get2 cells, whereas ∆sgt2∆get2 cells restored the insertion of Sbh1p (Kiktev et al, 2012). An analogous phenomenon has been observed in the Hsp70‐dependent targeting of mitochondria membrane proteins, during which mutation in the Tom70 TPR domain (R171A) only exhibited a strong growth phenotype in ∆Tom70∆Tom20 cells but not in ∆Tom70 cells (Young et al, 2003b). In contrast to Δsgt2, inactivation of Ssa1 caused a much stronger defect in TA insertion, suggesting that the role of Hsp70 in preserving the conformational quality of TAs is not restricted to the GET pathway in vivo and may extend to alternative TA targeting pathways (Fig 8, step 7). This hypothesis, as well as the precise role of Sgt2 in vivo, remains to be tested.
While cytosolic Hsp70s are known to interact with diverse client proteins, the functional diversity of Hsp70s can be further expanded by its interaction with downstream cochaperones (Young et al, 2003a; Kampinga & Craig, 2010). For example, the TPR‐containing cochaperone, Sti1/Hop, mediates the relay of kinase substrates from Hsp70 to Hsp90, enabling the latter to complete the folding of numerous kinases in the cell (Wegele et al, 2006; Alvira et al, 2014). When associated with aggregated client proteins, Hsp70 could further recruit and collaborate with the Hsp100 family of chaperones to remodel protein aggregates (Winkler et al, 2012; Lee et al, 2013; Rosenzweig et al, 2013). This work adds an important new branch—the localization of an essential class of integral membrane proteins—to the protein biogenesis pathways that initiate from Hsp70. The interactions of Hsp70 with diverse downstream chaperones or cochaperones likely provide a versatile triaging mechanism via which distinct classes of client proteins can be directed to specific biogenesis pathways within the cell. This hypothesis and its mechanistic details await to be tested.
Materials and Methods
Strains and plasmids
BY4741 (WT) and ∆get3 (BY4741 YDL100C::KanMX) strains were purchased from American Type Culture Collection (ATCC). SSA1 (MATα leu2‐3,112 his3‐11 ura3‐52 trp1∆1 lys2 ssa2::LEU2 ssa3::TRP1 ssa4::LYS2) and ssa1 ts (MATα leu2‐3,112 his3‐11 ura3‐52 trp1∆1 lys2 ssa1‐45:URA3 ssa2::LEU2 ssa3::TRP1 ssa4::LYS2) strains were kindly provided by Dr. Elizabeth A. Craig (Becker et al, 1996). SGT2FLAG∆get3 (BY4741 Δget3::ura Sgt2FLAG::kan) and sgt2TPRmtFLAG (BY4741 sgt2R171A, R175AFLAG::kan) strains were kindly provided by Dr. Vladimir Denic (Wang et al, 2010). Using homologous recombination, we deleted the Get3 gene from sgt2TPRmtFLAG yeast to generate the strain sgt2TPRmtFLAG∆get3 (BY4741 Δget3::his sgt2R171A, R175AFLAG::kan).
Vector pRS316/GPD‐PGK (a gift from Dr. Raymond Deshaies) was used for expression of model substrates in yeast for pulse‐chase measurements. To generate the plasmid pRS316‐3xHA‐BirA‐Bos1TMD‐opsin (for expression of BirA‐Bos1), a DNA fragment encoding the following sequences, from the N‐ to the C‐terminus, were cloned between the GPD promoter and PGK terminator of pRS316: an N‐terminal 3xHA‐BirA, residues 203‐244 of Bos1 encompassing its targeting sequence (TMD, 20 amino acids N‐terminal to the TMD and KWLR C‐terminal to the TMD), a GS linker (GSGGSGS), and an opsin tag at the extreme C‐terminus (MNGTEGPNFYVPFSNKTVD). The DNA fragments encoding BirA and Bos1TMD were swapped to generate pRS316‐Bos1TMD‐BirA‐3xHA (for expression of Bos1‐BirA). In addition, to enforce type I topology in the model substrate Bos1‐BirA (Kida et al, 2000), DNA sequences encoding mouse synaptotagmin II (SytII) residues 1–18, 20–45, and 61–65 were inserted at the N‐terminus of Bos1‐TMD (residues 223–240 of Bos 1), and five additional lysines (underlined) were inserted into the sequences C‐terminal to the Bos1‐TMD (KKWLRKKKK). The 3xHA tag was moved to the C‐terminus of Bos1‐BirA. To generate pRS316‐DHC‐αF‐3xHA, DNA encoding DHC‐αF [a gift from Dr. Maya Schuldiner (Aviram et al, 2016)] was inserted into the same site in pRS316, and a 3xHA tag was fused to the C‐terminus. For induction using galactose, DNA encoding BirA‐Bos1 was inserted into either the pESC‐ura or pESC‐his vector under control of the GAL10 promoter.
Plasmids for recombinant protein expression and purification were constructed as follows. For expression of Bos1 and Sbh1, a DNA fragment encoding a Strep‐tag (WSHPQFEK), SUMO with a mutated UplA cleavage site, Bos1 residues 203–244 or Sbh1 residues 25–82, and an opsin tag was cloned into pET28b to generate pStrep‐SUMO‐Bos1 (or Sbh1)‐opsin. pHis6‐SUMO‐Ssa1 for expression of Ssa1 was a gift from Dr. Elke Deuerling. For expression of tagless Sortase, a PreScission recognition sequence (LEVLFQG) was inserted into pQE30‐His6‐Sortase (a gift from Dr. Raymond Deshaies) to generate pHis6‐PreScission‐Sortase A. All mutant plasmids used in this study were generated using QuikChange mutagenesis (Agilent Technologies).
Protein expression and purification
The expression and purification of recombinant His6‐Sgt2, His6‐Get4/5, and Get3 were performed as described in Rao et al (2016) and Rome et al (2013).
Strep‐tagged Bos1 and Sbh1 were expressed in E. coli BL21 Star (DE3) (Thermo Fisher Scientific Inc.). Protein expression was induced at OD600 of 0.6 with 0.1 mM isopropyl β‐D‐1‐thiogalactopyranoside (IPTG) at 18°C overnight. Cells were resuspended in buffer A [50 mM Tris (pH 8.0), 300 mM NaCl, 10% glycerol, and 2 mM β‐mercaptoethanol (β‐ME)] supplemented with protease inhibitor cocktail (Roche). After sonication, the suspension was supplemented with 0.5% LDAO (N,N‐dimethyldodecylamine N‐oxide; Avanti Polar Lipids, Inc.), incubated at room temperature for 30 min, followed by incubation at 4°C for 30 min. Clarified lysate was loaded onto Strep‐tactin resin (IBA Lifesciences). The resin was washed with buffer A supplemented with 0.05% LDAO, eluted with the same buffer supplemented with 15 mM d‐Desthiobiotin (Sigma‐Aldrich), and dialyzed in buffer B (50 mM HEPES (pH 7.5), 150 mM NaCl, 10% glycerol, 0.05% LDAO).
His6‐SUMO‐Ssa1 or His6‐SUMO‐Ssa1‐D430C were expressed in BL21‐CodonPlus (DE3)‐RIL (Agilent Technologies) at 18°C overnight after induction with 0.5 mM IPTG. Cells were resuspended in buffer C (20 mM Tris (pH 8.0), 500 mM NaCl, 10% glycerol, 2 mM β‐ME, 20 mM imidazole) supplemented with 2 mM ATP, 5 mM MgCl2, and protease inhibitor cocktail. Clarified lysate was loaded onto Ni Sepharose resin (GE Healthcare). The column was washed with buffer C and eluted with buffer C containing 300 mM imidazole. The fusion protein was digested with His6‐SUMO protease during dialysis (20 mM HEPES (pH 7.5), 50 mM NaCl, 5% glycerol). His6‐SUMO and His6‐SUMO protease were removed by Ni Sepharose resin. Hisless Ssa1 was further purified using MonoQ 10/100 GL (GE Healthcare).
His6‐PreScission‐Sortase A was expressed in E. coli BL21 Star (DE3; Thermo Fisher Scientific Inc.) with 0.5 mM IPTG at 18°C overnight and purified as with His6‐SUMO‐Ssa1 with following modifications: Cells were lysed in buffer D (20 mM Tris (pH 8.0), 300 mM NaCl, 10% glycerol, 25 mM imidazole) supplemented with protease inhibitor cocktail. After affinity purification with Ni‐NTA (Qiagen), dialysis and digestion with His6‐PreScission protease (a gift from Dr. André Hoelz) were performed in storage buffer (50 mM Tris (pH 7.5), 150 mM NaCl, 20% glycerol) at 4°C overnight. Flowthrough fractions containing Hisless Sortase A were collected after passing the digestion mixture over Ni‐NTA resin.
Fluorescence labeling
Bos1 (Strep‐SUMO‐Bos1‐opsin) and Sbh1 (Strep‐SUMO‐Sbh1‐opsin) do not contain any cysteines. An engineered cysteine was introduced seven residues downstream of the TMD in these proteins. Single cysteine mutants of TAs were expressed and purified as described above. Before elution, TA‐bound Strep‐tactin was incubated with 2 mM DTT in buffer B at 4°C for 20 min and then washed with buffer B containing 0.2 mM TCEP. The resin was incubated with buffer B containing 2 mM TCEP and a 10‐fold molar excess of CM (N‐(7‐Dimethylamino‐4‐Methylcoumarin‐3‐yl))Maleimide; Thermo Fisher Scientific Inc.) on a rotator at 4°C overnight. Unconjugated dye was removed by washing the resin with buffer B. Labeled TAs were eluted with buffer B containing 15 mM d‐Desthiobiotin and dialyzed in buffer B.
Sgt2 was C‐terminally labeled using Sortase‐mediated ligation (Guimaraes et al, 2013). The GGGC peptide was synthesized from GenScript. The peptide and BODIPY‐FL maleimide (Thermo Fisher Scientific Inc.) were incubated in PBS buffer at a 3:1 molar ratio with rotation at 25°C overnight. GGGC‐BODIPY‐FL was purified by reversed‐phase HPLC. The lyophilized BODIPY‐FL‐conjugated GGGC peptide was dissolved in DMSO and stored at −80°C. The sortase recognition sequence LPATGG was fused to the C‐terminus of His6‐Sgt2, and His6‐Sgt2‐LPATGG was purified as described (Rao et al, 2016). Sortase‐mediated ligation was performed as described in Guimaraes et al (2013). Briefly, 40 μM His6‐Sgt2‐LPATGG, 500 μM GGGC‐BODIPY‐FL, and 50 μM Sortase A were incubated in Labeling buffer (50 mM Tris (pH 7.5), 150 mM NaCl, 10 mM CaCl2) at 25°C for 3 h. Labeled His6‐Sgt2‐BODIPY‐FL (Sgt2BFL) was purified using Talon resin (Clontech). ATTO488‐labeled Get3 (Get3ATTO) was prepared as described in Rao et al (2016), except that ATTO488 maleimide (ATTO‐TEC) was used instead of BODIPY‐FL maleimide.
Ssa1 was labeled at an engineered single cysteine via maleimide chemistry. Wild‐type Ssa1 has three native cysteines (C15, C264, and C303), among which only C15 is conserved. Based on sequence alignment, we mutated C264 and C303 in Ssa1 to the residues at corresponding positions in DnaK (Ala and Val, respectively). In addition, Ssa1‐C15 was mutated to Ala to generate a cysless Ssa1, in which a single cysteine was engineered at residue 430. Purified Ssa1‐D430C were incubated with a 10‐fold molar excess of CM in the presence of 4 mM TCEP for 7 h at 4°C. Free dyes were removed from labeled protein by chromatography on Sephadex G‐25 (Sigma‐Aldrich).
Turbidity assay
TAs solubilized in 0.05% LDAO were rapidly (within 15 s) diluted into assay buffer (20 mM K‐HEPES (pH 7.5), 150 mM KOAc, 5 mM Mg(OAc)2) to a final concentration of 1.5 μM and a final LDAO concentration of < 0.001% in the absence and presence of indicated concentrations of cytosolic factors. The optical density at 360 nm was recorded using a spectrophotometer (Beckman Coulter). Observed % soluble TA (S obsd) was calculated from the difference in optimal readings at 5 min between reactions with and without Ssa1 normalized to that of Bos1 alone. The data were plotted as a function of Ssa1 concentration and fit to equation (1)
| (1) |
in which S Max is the % soluble TA at saturating Ssa1 concentrations, and K soluble is the concentration of Ssa1 required to reach half of S Max.
In vitro translation in the PURE system
In vitro translation was performed following manufacturer's instructions (New England Biolabs). 6.5 μl PURE‐IVT reactions were incubated in the absence or presence of specified cytosolic factors for 90 min at 30°C. The reaction was diluted with assay buffer (50 μl) and ultracentrifuged for 1 h at 390,880 g, 4°C. The supernatant and pellet fractions were resolved on SDS–PAGE and analyzed by autoradiography.
Equilibrium measurements of TA transfer from Ssa1 to Sgt2
50 nM Bos1CM was mixed with 5 μM Ssa1 in GET buffer (50 mM K‐HEPES (pH 7.5), 150 mM KOAc, 5 mM Mg(OAc)2, 10% glycerol, 1 mM DTT) supplemented with 2 mM ATP. After 1‐min incubation at room temperature, wild‐type or mutant Sgt2BFL was added at indicated concentrations, and the reaction mixture was incubated for an additional 5 min. Fluorescence measurements were performed on a Fluorolog‐3‐22 spectrofluorometer (HORIBA Scientific) using an excitation wavelength of 360 nm and emission wavelength of 460 nm. FRET efficiency (E) was calculated according to equation (2),
| (2) |
where F D and F DA are the donor fluorescence in the absence and presence of the acceptor dye, respectively. Observed FRET efficiency (E obsd) was measured as a function of Sgt2 concentration, and the data were fit to equation (3),
| (3) |
in which E Max is the FRET efficient at saturating Sgt2 concentrations, and K Transfer is the concentration of Sgt2 required to reach half of E Max at the Ssa1 concentration used (5 μM).
Reconstitution of Sgt2·TACM and Sgt2BFL·TACM via transfer from Ssa1
0.3 μM recombinant TACM was mixed with 5 μM Ssa1 in assay buffer containing 2 mM ATP and 2 mM β‐ME and incubated for 1 min at room temperature. 0.5 μM His6‐Sgt2 was added in the reaction and incubated for 10 min. To form the Sgt2‐TPRmtBFL·Bos1CM complex, 1 μM His6‐Sgt2‐TPRmtBFL and 0.5 μM Bos1CM were used. The reaction was incubated with Talon resin at 4°C for 15 min. After washing the column with the assay buffer containing 500 mM NaCl, Sgt2·TACM complexes were eluted with 300 mM imidazole, dissolved in the assay buffer and then concentrated using 10K kDa Amicon® Ultra‐4 Centrifugal Filter (EMD Millipore). The complexes were flash‐frozen in liquid nitrogen and stored at −80°C.
Fluorescence assay for TA transfer from Sgt2 to Get3
Sgt2·TACM, Get4/5, and Get3BFL or Get3ATTO were ultracentrifuged at 390,880 g for 30 min at 4°C prior to all measurements. Time courses of TA transfer from Sgt2 to Get3 were measured using a Fluorolog‐3‐22 spectrofluorometer as described (Rao et al, 2016). Briefly, 50 nM Sgt2·TACM was supplemented with 150 nM unlabeled Sgt2, and transfer reactions were initiated by addition of 200 nM Get3ATTO, 200 nM Get4/5 where applicable, and 2 mM ATP. Observed fluorescence time traces (F obsd) were normalized to that at t = 0 and fit to equation (4),
| (4) |
in which F e is the normalized donor fluorescence when the reaction reaches equilibrium, k fast and k slow are the rate constants of the fast and slow phases in the transfer reaction, respectively, and ∆F fast and ∆F slow are the amplitudes of the fast and slow phases, respectively. The overall rate constant of the TA transfer reaction, k obsd (= ln2/t 1/2), was calculated from equation (5),
| (5) |
Kinetic stability of the Sgt2·Bos1 complex
Dissociation rate constants of Sgt2·Bos1 complexes were measured by chasing ~200 nM preformed Sgt2BFL·Bos1CM or Sgt2TPRmtBFL·Bos1CM complexes with 5–21 μM cpSRP43. The time courses of donor fluorescence change were monitored. Observed fluorescence time traces (F obsd) were normalized to that at t = 0 and fit to equation (6),
| (6) |
in which F e, ∆F fast, ∆F slow, k fast, and k slow are the same as defined in equation (4). Observed dissociation rate constant (k dis,obsd) was determined using equation (5). With Sgt2·TA complexes reconstituted via transfer from Ssa1, k dis,obsd values were independent of chase concentration and reports on the intrinsic TA dissociation rate constant. With Sgt2·TA complexes generated by IVT in E. coli extract, k dis,obsd values rose linearly with chase concentration and were extrapolated to zero chase concentration to obtain the intrinsic TA dissociation rate constants.
Pulse‐chase experiments
Yeast cells were grown in selective media (SD‐Ura) to mid‐log phase, washed with SD‐Ura‐Met‐Cys media, and resuspended in SD‐Ura‐Met‐Cys media at 6 OD600 units per ml. Cells were incubated at 25°C for 20 min and then incubated at either 25 or 37°C for an additional 5 min. Cells were pulse‐labeled with 100 μCi/ml EasyTag™ EXPRESS35S Protein Labeling Mix (Perkin Elmer) for 2 min and chased with 8 mM cold methionine and 1 mM cysteine. Aliquots of cells were flash‐frozen in liquid nitrogen at indicated times during chase.
Cells were washed with water and treated with 0.3 M NaOH for 3 min, and twice with 20 mM Tris (pH 8.0) and 150 mM NaCl. Cell pellets were resuspended in lysis buffer (20 mM Tris (pH 8.0), 150 mM NaCl, 2% SDS) supplemented with protease inhibitor cocktail at 65°C for 15 min. After centrifugation at 15,000 g for 10 min, the clarified lysate was diluted over 20‐fold with HA IP buffer (20 mM Tris (pH 8.0), 150 mM NaCl, 1% Triton X‐100) and incubated with anti‐HA magnetic beads (Thermo Fisher Scientific Inc.) at 25°C for 10 min. The beads were sequentially washed with: W1 buffer (20 mM Tris (pH 8.0), 150 mM NaCl, 1% Triton X‐100, 2M urea), W2 buffer (20 mM Tris (pH 8.0), 500 mM NaCl, 1% Triton X‐100), W3 buffer (20 mM Tris (pH 8.0), 150 mM NaCl, 0.1% SDS) and W4 buffer (20 mM Tris (pH 8.0), 150 mM NaCl). Immunoprecipitated proteins were eluted with 1 × SDS‐sample buffer, resolved by SDS–PAGE and quantified by autoradiography. Translocation efficiency was calculated as [I glycosylated/(I non‐glycosylated+I glycosylated)]*100, where I denotes the intensity of the band of interest.
Native FLAG immunoprecipitation (IP)
For in vivo IP, BirA‐Bos1 was transiently expressed in SGT2FLAG∆get3 or sgt2TPRmtFLAG∆get3 cells under the GAL10 promoter for 0.5–1.5 h. Cells were lysed by glass beads in the FLAG‐IP buffer (20 mM K‐HEPES (pH 7.5), 100 mM KOAc, 2 mM Mg(OAc), 14% glycerol) containing protease inhibitor cocktail and 1 mM DTT. Lysate was clarified by centrifugation at 9,000 g, 4°C for 10 min and incubated with anti‐FLAG magnetic beads (Sigma‐Aldrich) at 4°C for 1.5 h. Beads were washed three times with 10 column volumes of the FLAG‐IP buffer, and bound proteins were eluted with 1 mg/ml 3xFLAG peptide (Sigma‐Aldrich). For FLAG‐IP in yeast extracts, clarified yeast lysate was further centrifuged at 104,300 g, 4°C for 30 min, and the supernatant was separated using G25 size‐exclusion chromatography as described in Schuldiner et al (2008). 35S‐Bos1 generated by PURE‐IVT supplemented with 6 μM Ssa1 was incubated with the extract at 25°C for 10 min before immunoprecipitation.
Western blot analysis
Protein samples from in vitro and in vivo assays were separated on 10 or 12.5% Tris‐glycine gels and then transferred to nitrocellulose membrane (Bio‐Rad). Primary antibodies used were anti‐His tag (1:5,000, cat# A00186, GenScript), anti‐Strep II tag (1:3,000, Cat# ab76949, Abcam), and anti‐HA tag (1:3,000, cat#A01244, GenScript). Ssa1 antibody was kindly provided by Dr. Elizabeth A. Craig (1:3,000). For secondary antibodies, IRDye® 800CW Goat anti‐Mouse IgG (1:10,000, cat# P/N 925‐32211, LI‐COR Inc.) and RDye® 800CW Goat anti‐Mouse IgG (1:10,000, cat# P/N 925‐32210, LI‐COR Inc.) were used. The membranes were scanned by Odyssey Imager (LI‐COR Inc.)
Author contributions
HC and SS designed experiment; HC performed experiments and analyzed the data; HC and SS wrote the paper.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 6
Source Data for Figure 7
Acknowledgements
We thank E. A. Craig for the Ssa antibody and the SSA1 and ssa1 ts strains, V. Denic for the SGT2FLAG∆get3 and sgt2TPRmtFLAG strains, U. S. Chio, M. Rao, A. Montequin, F.C. Liang, A. Siegel for reagents, and the Dougherty lab for use of HPLC. We thank Bil Clemons, Ray Deshaies, and members of the Shan, Clemons, and Deshaies laboratories for discussions and critical comments on the manuscript. This work was supported by NIH grant GM107368 and grant from the Weston Havens Foundation to S.S.
The EMBO Journal (2018) 37: e99264
References
- Abell BM, Rabu C, Leznicki P, Young JC, High S (2007) Post‐translational integration of tail‐anchored proteins is facilitated by defined molecular chaperones. J Cell Sci 120: 1743–1751 [DOI] [PubMed] [Google Scholar]
- Albanese V, Yam AY, Baughman J, Parnot C, Frydman J (2006) Systems analyses reveal two chaperone networks with distinct functions in eukaryotic cells. Cell 124: 75–88 [DOI] [PubMed] [Google Scholar]
- Alvira S, Cuéllar J, Röhl A, Yamamoto S, Itoh H, Alfonso C, Rivas G, Buchner J, Valpuesta JM (2014) Structural characterization of the substrate transfer mechanism in Hsp70/Hsp90 folding machinery mediated by Hop. Nat Commun 5: 5484 [DOI] [PubMed] [Google Scholar]
- Angeletti PC, Walker D, Panganiban AT (2002) Small glutamine‐rich protein/viral protein U‐binding protein is a novel cochaperone that affects heat shock protein 70 activity. Cell Stress Chaperones 7: 258–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aviram N, Ast T, Costa EA, Arakel EC, Chuartzman SG, Jan CH, Hassdenteufel S, Dudek J, Jung M, Schorr S, Zimmermann R, Schwappach B, Weissman JS, Schuldiner M (2016) The SND proteins constitute an alternative targeting route to the endoplasmic reticulum. Nature 540: 134–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee R, Jayaraj GG, Peter JJ, Kumar V, Mapa K (2016) Monitoring conformational heterogeneity of the lid of DnaK substrate‐binding domain during its chaperone cycle. FEBS J 283: 2853–2868 [DOI] [PubMed] [Google Scholar]
- Becker J, Walter W, Yan W, Craig EA (1996) Functional interaction of cytosolic hsp70 and a DnaJ‐related protein, Ydj1p, in protein translocation in vivo . Mol Cell Biol 16: 4378–4386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehme M, Voisine C, Rolland T, Wachi S, Soper JH, Zhu Y, Orton K, Villella A, Garza D, Vidal M, Ge H, Morimoto RI (2014) A chaperome subnetwork safeguards proteostasis in aging and neurodegenerative disease. Cell Rep 9: 1135–1150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casson J, McKenna M, Haßdenteufel S, Aviram N, Zimmerman R, High S (2017) Multiple pathways facilitate the biogenesis of mammalian tail‐anchored proteins. J Cell Sci 130: 3851–3861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chartron JW, Gonzalez GM, Clemons WM Jr (2011) A structural model of the Sgt2 protein and its interactions with chaperones and the Get4/Get5 complex. J Biol Chem 286: 34325–34334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerico EM, Tilitsky JM, Meng W, Gierasch LM (2015) How hsp70 molecular machines interact with their substrates to mediate diverse physiological functions. J Mol Biol 427: 1575–1588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo SF, Longhi R, Borgese N (2009) The role of cytosolic proteins in the insertion of tail‐anchored proteins into phospholipid bilayers. J Cell Sci 122: 2383–2392 [DOI] [PubMed] [Google Scholar]
- Colombo SF, Cardani S, Maroli A, Vitiello A, Soffientini P, Crespi A, Bram RJ, Benfante R, Borgese N (2016) Tail‐anchored protein insertion in mammals. Function and reciprocal interactions of the two subunits of thE TRC4O receptor. J Biol Chem 291: 18855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshaies RJ, Koch BD, Werner‐Washburne M, Craig EA, Schekman R (1988) A subfamily of stress proteins facilitates translocation of secretory and mitochondrial precursor polypeptides. Nature 332: 800–805 [DOI] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS (2003) Global analysis of protein expression in yeast. Nature 425: 737–741 [DOI] [PubMed] [Google Scholar]
- Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AE, Ploegh HL (2013) Site‐specific C‐terminal and internal loop labeling of proteins using sortase‐mediated reactions. Nat Protoc 8: 1787–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guna A, Volkmar N, Christianson JC, Hegde RS (2018) The ER membrane protein complex is a transmembrane domain insertase. Science 359: 470–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harano T, Nose S, Uezu R, Shimizu N, Fujiki Y (2001) Hsp70 regulates the interaction between the peroxisome targeting signal type 1 (PTS1)‐receptor Pex5p and PTS1. Biochem J 357: 157–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper CC, Berg JM, Gould SJ (2003) PEX5 binds the PTS1 independently of Hsp70 and the peroxin PEX12. J Biol Chem 278: 7897–7901 [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, Hayer‐Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475: 324–332 [DOI] [PubMed] [Google Scholar]
- Haßdenteufel S, Sicking M, Schorr S, Aviram N, Fecher‐Trost C, Schuldiner M, Jung M, Zimmermann R, Lang S (2017) hSnd2 protein represents an alternative targeting factor to the endoplasmic reticulum in human cells. FEBS Lett 591: 3211–3224 [DOI] [PubMed] [Google Scholar]
- Hegde RS, Keenan RJ (2011) Tail‐anchored membrane protein insertion into the endoplasmic reticulum. Nat Rev Mol Cell Biol 12: 787–798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonikas MC, Collins SR, Denic V, Oh E, Quan EM, Schmid V, Weibezahn J, Schwappach B, Walter P, Weissman JS, Schuldiner M (2009) Comprehensive characterization of genes required for protein folding in the endoplasmic reticulum. Science 323: 1693–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalbfleisch T, Cambon A, Wattenberg BW (2007) A bioinformatics approach to identifying tail‐anchored proteins in the human genome. Traffic 8: 1687–1694 [DOI] [PubMed] [Google Scholar]
- Kampinga HH, Craig EA (2010) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11: 579–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida Y, Sakaguchi M, Fukuda M, Mikoshiba K, Mihara K (2000) Membrane topogenesis of a type I signal‐anchor protein, mouse synaptotagmin II, on the endoplasmic reticulum. J Cell Biol 150: 719–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiktev DA, Patterson JC, Muller S, Bariar B, Pan T, Chernoff YO (2012) Regulation of chaperone effects on a yeast prion by cochaperone Sgt2. Mol Cell Biol 32: 4960–4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Schilke B, Craig EA, Horwich AL (1998) Folding in vivo of a newly translated yeast cytosolic enzyme is mediated by the SSA class of cytosolic yeast Hsp70 proteins. Proc Natl Acad Sci USA 95: 12860–12865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles TP, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15: 384–396 [DOI] [PubMed] [Google Scholar]
- Kohl C, Tessarz P, von der Malsburg K, Zahn R, Bukau B, Mogk A (2011) Cooperative and independent activities of Sgt2 and Get5 in the targeting of tail‐anchored proteins. Biol Chem 392: 601–608 [DOI] [PubMed] [Google Scholar]
- Krysztofinska EM, Evans NJ, Thapaliya A, Murray JW, Morgan RML, Martinez‐Lumbreras S, Isaacson RL (2017) Structure and interactions of the TPR domain of Sgt2 with yeast chaperones and Ybr137wp. Front Mol Biosci 4: 68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M (2014) Minimal, encapsulated proteomic‐sample processing applied to copy‐number estimation in eukaryotic cells. Nat Methods 11: 319–324 [DOI] [PubMed] [Google Scholar]
- Lee J, Kim JH, Biter AB, Sielaff B, Lee S, Tsai FTF (2013) Heat shock protein (Hsp) 70 is an activator of the Hsp104 motor. Proc Natl Acad Sci USA 110: 8513–8518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang FC, Kroon G, McAvoy CZ, Chi C, Wright PE, Shan SO (2016) Conformational dynamics of a membrane protein chaperone enables spatially regulated substrate capture and release. Proc Natl Acad Sci USA 113: E1615–E1624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariappan M, Li X, Stefanovic S, Sharma A, Mateja A, Keenan RJ, Hegde RS (2010) A ribosome‐associating factor chaperones tail‐anchored membrane proteins. Nature 466: 1120–1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateja A, Paduch M, Chang HY, Szydlowska A, Kossiakoff AA, Hegde RS, Keenan RJ (2015) Structure of the Get3 targeting factor in complex with its membrane protein cargo. Science 347: 1152–1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer MP, Bukau B (2005) Hsp70 chaperones: cellular functions and molecular mechanism. Cell Mol Life Sci 62: 670–684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClellan AJ, Scott MD, Frydman J (2005) Folding and quality control of the VHL tumor suppressor proceed through distinct chaperone pathways. Cell 121: 739–748 [DOI] [PubMed] [Google Scholar]
- Ng DT, Brown JD, Walter P (1996) Signal sequences specify the targeting route to the endoplasmic reticulum membrane. J Cell Biol 134: 269–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabu C, Wipf P, Brodsky JL, High S (2008) A precursor‐specific role for Hsp40/Hsc70 during tail‐anchored protein integration at the endoplasmic reticulum. J Biol Chem 283: 27504–27513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao M, Okreglak V, Chio US, Cho H, Walter P, Shan SO (2016) Multiple selection filters ensure accurate tail‐anchored membrane protein targeting. Elife 5: e21301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rome ME, Rao M, Clemons WM, Shan SO (2013) Precise timing of ATPase activation drives targeting of tail‐anchored proteins. Proc Natl Acad Sci USA 110: 7666–7671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenzweig R, Moradi S, Zarrine‐Afsar A, Glover JR, Kay LE (2013) Unraveling the mechanism of protein disaggregation through a ClpB‐DnaK interaction. Science 339: 1080–1083 [DOI] [PubMed] [Google Scholar]
- Roy J, Mitra S, Sengupta K, Mandal AK (2015) Hsp70 clears misfolded kinases that partitioned into distinct quality‐control compartments. Mol Biol Cell 26: 1583–1600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, Metz J, Schmid V, Denic V, Rakwalska M, Schmitt HD, Schwappach B, Weissman JS (2008) The GET complex mediates insertion of tail‐anchored proteins into the ER membrane. Cell 134: 634–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Hegde RS (2011) Membrane protein insertion at the endoplasmic reticulum. Annu Rev Cell Dev Biol 27: 25–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao S, Rodrigo‐Brenni MC, Kivlen MH, Hegde RS (2017) Mechanistic basis for a molecular triage reaction. Science 355: 298–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Inoue A, Tomari Y, Suzuki T, Yokogawa T, Nishikawa K, Ueda T (2001) Cell‐free translation reconstituted with purified components. Nat Biotechnol 19: 751–755 [DOI] [PubMed] [Google Scholar]
- Thulasiraman V, Yang CF, Frydman J (1999) In vivo newly translated polypeptides are sequestered in a protected folding environment. EMBO J 18: 85–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi A, Mandon EC, Gilmore R, Rapoport TA (2017) Two alternative binding mechanisms connect the protein translocation Sec71/Sec72 complex with heat shock proteins. J Biol Chem 292: 8007–8018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Brown EC, Mak G, Zhuang J, Denic V (2010) A chaperone cascade sorts proteins for posttranslational membrane insertion into the endoplasmic reticulum. Mol Cell 40: 159–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Chan C, Weir NR, Denic V (2014) The Get1/2 transmembrane complex is an endoplasmic‐reticulum membrane protein insertase. Nature 512: 441–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegele H, Wandinger SK, Schmid AB, Reinstein J, Buchner J (2006) Substrate transfer from the chaperone Hsp70 to Hsp90. J Mol Biol 356: 802–811 [DOI] [PubMed] [Google Scholar]
- Winkler J, Tyedmers J, Bukau B, Mogk A (2012) Hsp70 targets Hsp100 chaperones to substrates for protein disaggregation and prion fragmentation. J Cell Biol 198: 387–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Sarbeng EB, Vorvis C, Kumar DP, Zhou L, Liu Q (2012) Unique peptide substrate binding properties of 110‐kDa heat‐shock protein (Hsp110) determine its distinct chaperone activity. J Biol Chem 287: 5661–5672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabal M, Brambillasca S, Soffientini P, Pedrazzini E, Borgese N, Makarow M (2003) Translocation of the C‐terminus of a tail‐anchored protein across the ER membrane in yeast mutants defective in signal peptide‐driven translocation. J Biol Chem 278: 3489–3496 [DOI] [PubMed] [Google Scholar]
- Yeh YH, Lin TW, Li YC, Tung JY, Lin CY, Hsiao CD (2014) Structural and functional characterization of ybr137wp implicates its involvement in the targeting of tail‐anchored proteins to membranes. Mol Cell Biol 34: 4500–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young JC, Barral JM, Hartl FU (2003a) More than folding: localized functions of cytosolic chaperones. Trends Biochem Sci 28: 541–547 [DOI] [PubMed] [Google Scholar]
- Young JC, Hoogenraad NJ, Hartl FU (2003b) Molecular chaperones Hsp90 and Hsp70 deliver preproteins to the mitochondrial import receptor Tom70. Cell 112: 41–50 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Review Process File
Source Data for Figure 2
Source Data for Figure 6
Source Data for Figure 7