

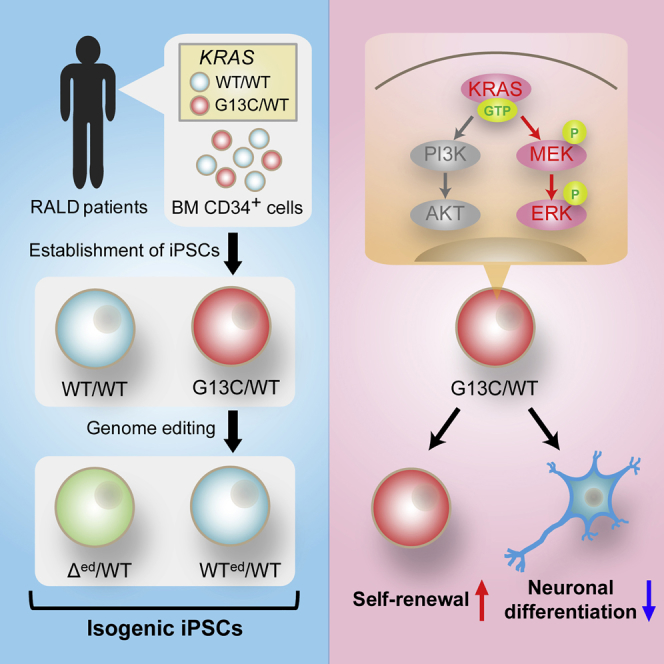
Summary
Oncogenic KRAS mutations in hematopoietic stem cells cause RAS-associated autoimmune lymphoproliferative syndrome-like disease (RALD). KRAS plays essential roles in stemness maintenance in some types of stem cells. However, its roles in pluripotent stem cells (PSCs) are poorly understood. Here, we investigated the roles of KRAS on stemness in the context of induced PSCs (iPSCs). We used KRAS mutant (G13C/WT) and wild-type isogenic (WT/WT) iPSCs from the same RALD patients, as well as wild-type (WTed/WT) and heterozygous knockout (Δed/WT) iPSCs, both obtained by genome editing from the same G13C/WT clone. Compared with WT iPSCs, G13C/WT iPSCs displayed enforced retention of self-renewal and suppressed capacity for neuronal differentiation, while Δed/WT iPSCs showed normalized cellular characteristics similar to those of isogenic WTed/WT cells. The KRAS-ERK pathway, but not the KRAS-PI3K pathway, was shown to govern these G13C/WT-specific phenotypes, indicating the strong impact of the KRAS-ERK signaling upon self-renewal and differentiation propensity in human iPSCs.
Keywords: iPSCs, KRAS, RAS-associated autoimmune lymphoproliferative syndrome-like disease, self-renewal, stemness, differentiation, MAPK pathway
Graphical Abstract
Highlights
-
•
Isogenic wild-type and KRAS mutant iPSCs were established from RALD patients
-
•
Oncogenic KRAS enforces retention of self-renewal in human iPSCs
-
•
Oncogenic KRAS suppresses neuronal differentiation from human iPSCs
-
•
KRAS-ERK signaling pathway governs the KRAS mutant-specific phenotypes
In this article, Kubara, Yamazaki, and colleagues show that, using RALD patient-derived iPSCs, oncogenic KRAS activates mainly the MEK-ERK pathway, but not the PI3K-AKT pathway, leading to enforced retention of self-renewal and suppressed capacity for undergoing neuronal differentiation in human iPSCs.
Introduction
The small GTPase RAS family proteins (KRAS, NRAS, and HRAS) are controlled through the exchange of the GDP-bound form for the GTP-bound one, which then allows RAS to bind various effectors, such as RAF, phosphoinositide 3-kinase (PI3K), and RALGDS (Castellano and Downward, 2010, D'Adamo et al., 1997, Moodie et al., 1993, Vivanco and Sawyers, 2002, Vojtek et al., 1993). RAS-associated signaling pathways play important roles in multiple cellular functions, such as cell growth, migration, adhesion, survival, and differentiation. The mutations at hotspots (G12, G13, and G61) in the RAS genes cause accumulation of the GTP-bound form due to defective intrinsic GTP hydrolysis activity and resistance to GTPase-activating proteins (Prior et al., 2012). These oncogenic mutations in the RAS genes are observed in approximately 30% of all human cancers. KRAS is one of the most widely known oncogenes and is frequently found to be mutated in colorectal, pancreatic, and lung cancers (Adjei, 2001).
Oncogenic KRAS has been reported to play a significant role in stem cell activities in some types of cancers. For example, it has been shown that oncogenic KRAS in colon cancers enhances the embryonic stem (ES) cell-like program during human colon cancer initiation from adenoma to carcinoma, and activates cancer stem cell (CSC) properties in APC-mutated cells through the MAPK pathway (Le Rolle et al., 2016, Moon et al., 2014). In addition, oncogenic KRAS has been reported to enhance stemness in CSCs in pancreatic cancers through the PI3K/AKT/mammalian target of rapamycin pathways (Matsubara et al., 2013).
The mutations in the RAS pathway are known to be involved not only in cancers, but also in other disorders including a series of congenital diseases and an acquired hemato-immunological disease, namely, RAS-associated autoimmune lymphoproliferative syndrome (ALPS)-like disease (RALD). RALD has been reported as a disease affecting the hemato-immune system, caused by a somatic KRAS or NRAS mutation in hematopoietic lineage cells. RALD patients exhibit ALPS- and/or juvenile myelomonocytic leukemia-like symptoms, including autoimmune cytopenia, lymphadenopathy, and hepatosplenomegaly (Niemela et al., 2011, Shiota et al., 2015, Takagi et al., 2011). Moreover, a RALD patient exhibiting intestinal Behcet's disease-like phenotypes was reported (Moritake et al., 2016). In RALD, individual patients have clones with KRAS or NRAS mutation and wild-type clones together in hematopoietic lineage cells in a mosaic state, allowing the generation of a set of isogenic induced pluripotent stem cell (iPSC) clones from the same patients. RALD patient-derived iPSCs therefore represent a unique experimental tool that is useful for studying basic RAS biology, particularly the roles of KRAS on stemness maintenance in the context of PSCs.
In the culture of human embryonic stem cells (ESCs) and iPSCs, basic fibroblast growth factor (bFGF) is essential to maintain their stemness through activating the MAPK and PI3K pathways. If human ESCs and iPSCs are cultured without bFGF, they lose their stemness and start to differentiate (Chen et al., 2011, Ding et al., 2010, Lanner and Rossant, 2010, Levenstein et al., 2006, Li et al., 2007). These observations clearly demonstrate the importance of bFGF-mediated signaling for the maintenance of human iPSCs and ESCs. However, it remains largely unknown how the status of effector molecules including KRAS located downstream in bFGF signals affects stemness maintenance in human iPSCs.
Here, we investigated the roles of KRAS on stemness maintenance in the context of human iPSCs by using isogenic KRAS mutant (G13C/WT) and wild-type (WT/WT) iPSCs, generated from two RALD patients with the same somatic KRAS mutation. By genome-editing techniques, we succeeded in generation of “gene-corrected” wild-type iPSCs (WTed/WT) and heterozygous knockout iPSCs (Δed/WT), both of which could serve as relevant controls for the experiments. Using this series of isogenic iPSCs, we determined how the status of KRAS could impact upon stemness maintenance in human iPSCs and differentiation propensity under permissive conditions.
Results
Establishment of iPSC Clones from RALD Patients
We generated iPSCs from CD34+ hematopoietic stem/progenitor cells of two RALD patients with the same somatic G13C heterozygous mutation in the KRAS gene (Tables S1 and S2). We obtained mutant (G13C/WT) and isogenic wild-type (WT/WT) iPSC clones from each patient as confirmed by direct sequencing (Figure 1A). The presence of oncogenic mutations other than KRAS was excluded by whole exome sequencing (Table S3). Karyotyping showed that all RALD patient-derived iPSC clones exhibited a normal 46XY karyotype (Figure 1B). All iPSC clones expressed the markers, OCT4, NANOG, TRA-1-60, and SSEA4 (Figure 1C).
Figure 1.

Establishment and Characterization of iPSC Clones Generated from Two RALD Patients
(A) KRAS sequences of wild-type (WT/WT) and mutant (G13C/WT) iPSC lines derived from two RALD patients (cases no. 1 and no. 2). A position of mutation (G to T) is indicated by red letters.
(B) Karyotypes of WT/WT and G13C/WT iPSC lines derived from case no. 2 (RALD patient).
(C) Immunocytochemistry of iPSC markers (OCT4, NANOG, TRA-1-60, and SSEA4) in WT/WT and G13C/WT iPSC clones. Ho, Hoechst 33342. Scale bar, 100 μm.
To assess how the status of KRAS affects stemness and differentiation in these iPSC clones, we investigated changes in gene expression levels of stemness and lineage markers after in vitro embryoid body (EB)-mediated differentiation for 16 days. In the suspension culture to induce differentiation, all iPSC clones formed EBs (Figures 2A and S1A). However, RNA sequencing (RNA-seq) analysis showed that there were clear differences in stemness and lineage marker expression between WT/WT and G13C/WT genotypes in case no. 2 (Figure 2B). Following 16-day differentiation, mRNA levels of stemness markers decreased over time in the WT/WT genotype, whereas they seemed to remain high in the G13C/WT cells, especially R2-1 clone. In general, mRNA levels of lineage markers were elevated upon differentiation in both WT/WT and G13C/WT cells. The expression levels of endodermal and early mesodermal markers were higher in the G13C/WT cells than in the WT/WT counterparts, whereas those of ectodermal markers in the G13C/WT genotype were lower. The same trend in expression differences of stemness (POU5F1 and NANOG), mesodermal (EOMES and T), and ectodermal markers (PAX6 and ASCL1) was confirmed in qRT-PCR analysis of case no. 2 (Figure 2C). However, mRNA levels were comparable in two genotypes regarding endodermal markers (FOXA2 and SOX17) after differentiation. Although PAX6 mRNA levels after differentiation were comparable between groups, the clear difference in two genotypes was also observed on POU5F1, NANOG, and ASCL1 in qRT-PCR analysis of case no. 1 (Figure S1B). Thus, we focused on the marked differences of stemness and ectodermal markers in the two genotypes.
Figure 2.

Different Differentiation Propensity between WT/WT and G13C/WT iPSCs Generated from Two RALD Patients
(A) Embryoid body formation of WT/WT and G13C/WT iPSC clones from case no. 2. Scale bar, 200 μm.
(B) RNA-seq data showing gene expression levels of stemness and lineage markers from case no. 2 samples before and after 16-day in vitro differentiation. Undiff. and Diff., undifferentiated iPSCs and differentiated cells, respectively.
(C) qRT-PCR analysis of 16-day in vitro differentiated cells from WT/WT and G13C/WT iPSC clones derived from case no. 2 (n = 3 independent experiments; mean ± SEM).
(D) Immnunocytochemistry of βIII-Tubulin and MAP2 in 16-day in vitro differentiated cells from WT/WT and G13C/WT iPSC clones derived from case no. 2. Scale bar, 50 μm.
See also Figures S1–S3.
Considering that both PAX6 and ASCL1 are transcription factors essential for neurogenesis (Ali et al., 2014, Castro and Guillemot, 2011, Osumi et al., 2008), we compared expression of neuronal markers βIII-Tubulin and MAP2 between WT/WT and G13C/WT iPSCs after 16-day differentiation (Figure 2D). In differentiated cells harboring the WT/WT genotype, many βIII-Tubulin- and MAP2-double-positive neurons with long extending neurites could be seen. In contrast, there were fewer βIII-Tubulin- and MAP2-double-positive neurons differentiated from G13C/WT iPSCs. There were some neurites positive for βIII-Tubulin but negative for MAP2 in clone R2-1. In the case of clone R2-3, neurites of βIII-Tubulin- and MAP2-double-positive cells were few in number and very short in length. Similar observations were also made from immunostaining of case no. 1-derived iPSC line differentiated cells. βIII-Tubulin-positive neurons were seen in differentiated C1-1 cells, whereas no positive cells were detected in differentiated R1-1, R1-2, or R1-3 cells (Figure S1C). These results indicate that oncogenic KRAS impacts upon self-renewal capacity and differentiation propensity in human iPSCs.
Rescue of the KRAS Mutation by Genome Editing
To validate whether the phenotypes described above were attributable to the specific KRAS genotype, we attempted to generate “gene-corrected” wild-type iPSCs from a G13C/WT clone. For genome editing, we used the CRISPR/Cas9 system for one G13C/WT iPSC clone from RALD patient no. 1 (clone R1-2) (Figure S2A). Through homologous recombination, we were able to obtain genome-edited wild-type homozygous clones (clone; C8 and H2, WTed/WT; “ed” meaning genome-edited). In addition, we could obtain heterozygous knockout clones (Δed/WT) from the same genome-editing experiment (clone; A1 and D2, Figure S2B). We checked the top five off-target candidate sites of the single guide RNA used in this procedure and observed no off-target cleavage (Figure S2C). Western blotting analysis confirmed that KRAS expression levels were comparable in WTed/WT and G13C/WT clones (clone; F4 and G4, which are non-edited clones). KRAS expression was lower in Δed/WT clones compared with the other two cell types, consistent with the haploinsufficient state (Figure S2D).
These clones were subjected to 16-day in vitro differentiation and examined for the expression of stemness genes and three-germ layer markers. In the suspension culture to induce differentiation, all clones formed EBs (Figure S3A). Analysis by qRT-PCR showed that expression levels of POU5F1 and NANOG remained high in G13C/WT cells (F4 and G4) after 16-day differentiation, although they decreased in WTed/WT (clone; C8 and H2) and Δed/WT cells (clone; A1 and D2). Transcription of ASCL1 mRNA in G13C/WT cells was not induced upon differentiation, whereas it showed clear induction in WTed/WT and Δed/WT cells (Figure S3B). Expression of the neuronal lineage markers, βIII-Tubulin and MAP2, also became more intense in WTed/WT clones in comparison with G13C/WT mutant cells (Figure S3C). These results demonstrated that gene-correction in the KRAS allele was enough to normalize the phenotypes inherent to iPSCs carrying the G13C/WT genotype.
Enforced Retention of Self-Renewal by G13C/WT iPSCs in the Absence of bFGF
Based on the results of in vitro differentiation assays, we speculated that there could be different expression patterns of genes relating to stemness, self-renewal, and pluripotency, between WT/WT and G13C/WT iPSCs. RALD patient-derived iPSCs were cultured with or without bFGF for 5 days, after which global gene expression was monitored by microarray. Scatterplots between the two combinations of iPSC lines and culture conditions are depicted in Figure 3A. In the presence of bFGF, there was a high degree of correlation in gene expression pattern between WT/WT and G13C/WT iPSCs (Figure 3A, upper left). However, the extent of correlation between WT/WT and G13C/WT cells decreased when they were cultured without bFGF for 5 days (Figure 3A, upper right). Comparison in gene expression for WT/WT iPSCs between two conditions, i.e., with and without bFGF, showed marked changes, characterized by downregulation of stemness genes (POU5F1 and NANOG) and upregulation of ectodermal genes (PAX6) in the absence of bFGF (Figure 3A, lower left). In sharp contrast, there were no changes in gene expression observed in G13C/WT cells even when they were cultured without bFGF (Figure 3A, lower right).
Figure 3.

Microarray Analysis of Isogenic WT/WT and G13C/WT KRAS Mutant iPSCs from a RALD Patient and Those Cultured without bFGF for 5 Days
(A) Scatterplots with coefficients of correlation (R) for whole genes in WT/WT and G13C/WT iPSCs cultured with (w/) or without (w/o) bFGF for 5 days. Two clones per genotype were used: C2-1 and C2-2 for WT/WT; R2-1 and R2-2 for G13C/WT, derived from case no. 2. In this analysis, data of the same genotypes were averaged. Positions of POU5F1 (two probes), NANOG, and PAX6 (two probes) are indicated. The green lines indicate the diagonal and 2-fold changes between the two samples.
(B) A heatmap with hierarchical clustering for stemness and lineage marker expression in the same samples as described above.
Figure 3B is a heatmap of stemness and linage markers. Stemness markers, including POU5F1, NANOG, and MYC, were expressed at a high level in both WT/WT and G13C/WT iPSC lines cultured in the presence of bFGF (four lanes in the middle).
When cells were cultured without bFGF, the expression of these genes diminished in the case of the WT/WT genotype (two lanes on left, C2-1 and C2-2; w/o). On the contrary, they remain highly expressed, almost unchanged in the G13C/WT genotype (two lanes on right, R2-1 and R2-2; w/o). The dendrogram indicated that the expression pattern was more closely related between G13C/WT samples cultured with bFGF and those without bFGF than between G13C/WT iPSCs and WT/WT iPSCs both in culture with bFGF. This was also reflected in the results of the scatterplots in Figure 3A. Among linage markers, ectodermal marker expression tended to be elevated even after 5-day culture without bFGF in WT/WT cells (two lanes on left, C2-1 and C2-2; w/o), but it did not in bFGF-depleted G13C/WT cells (third and fourth lanes from left, R2-1 and R2-2, w/o). Taken together, these microarray data demonstrate enforced retention of self-renewal of G13C/WT iPSCs in the absence of bFGF.
Correlation between KRAS Genotypes and Stemness Maintenance Potentials Quantified by an Imaging Approach
To analyze the enforced retention of self-renewal at the protein level, the expression of stemness markers was confirmed by immunocytochemistry staining. Upon bFGF removal, cell morphology changed to a more flattened appearance and the immunocytochemical expression of stemness markers (OCT4 and NANOG) dramatically decreased in WT/WT iPSCs (Figure 4A, left panel, C1-1, C2-1, and C2-2). In contrast, cell morphology and stemness marker expression in G13C/WT iPSCs remained almost unaffected (R1-2, R2-1, and R2-3). Regarding other stemness markers, TRA-1-60 also decreased in expression, whereas SSEA4 did not do so much in WT/WT iPSCs by the depletion of bFGF for 5 days (Figure 4A, right panel). Quantitative imaging analysis of bFGF-deprived cells showed that OCT4+ rates of G13C/WT clones (R1-1, -2, and -3) remained higher than 75% of vehicle treatment, whereas those of the WT/WT clone C1-1 reduced to levels less than 25% (Figure 4B). Similar results were obtained in iPSC clones derived from case no. 2 (Figure 4C). Combining the data of the three RALD iPSC clones for each genotype from case no. 2, we examined the effects of genotype and bFGF depletion on OCT4 expression by using two-way ANOVA, and obtained statistically significant results (Table S4).
Figure 4.

Enforced Retention of Self-Renewal of KRAS G13C/WT Mutant iPSCs in the Absence of bFGF, Revealed by Immunocytochemical Reactivity for Stemness Markers, Colony Formation, and Alkaline Phosphatase Staining
(A) Immunocytochemistry of stemness markers (OCT4, NANOG, TRA-1-60, and SSEA-4) in WT/WT and G13C/WT iPSC clones from RALD patients cultured without bFGF for 5 days. Scale bar, 100 μm.
(B and C) Quantitative imaging analysis for OCT4 in iPSC clones from RALD patients, cases no. 1 (B) and no. 2 (C), respectively (n = 8 independent experiments; mean ± SEM; ∗∗∗p < 0.001; two-way ANOVA followed by Bonferroni's multiple comparison test).
(D) Whole six-well (left) and magnified images (right) of alkaline phosphatase (ALP)-stained WT/WT (C2-1) and G13C/WT (R2-1) cells. Scale bar, 100 μm.
(E) Quantification of ALP-positive (ALP+) colony number in (D) (n = 3 independent experiments; mean ± SEM; ∗∗p < 0.01; ∗∗∗p < 0.001; two-way ANOVA followed by Bonferroni's multiple comparison test).
We next compared oncogenic KRAS genotype-associated changes in OCT4+ rates in genome-edited iPSCs after removal of bFGF. The OCT4+ area of WTed/WT cells was drastically reduced compared with G13C/WT cells. Interestingly, the OCT4+ area of Δed/WT cells was significantly reduced compared with that of WTed/WT cells (Figure S3D). The detailed summary of statistical analysis is shown in Table S4.
We then investigated if mutant iPSCs retaining stemness marker expression after bFGF depletion also maintained pluripotency. To this end, the iPSCs pre-depleted of bFGF for 5 days were reseeded as single cells and cultured for another 7 days back in bFGF-replete culture. As shown in Figure 4D, G13C/WT cells formed many iPSC colonies which were alkaline phosphatase (ALP)-positive, whereas only a few colonies appeared in WT/WT cells (Figure 4D). ALP+ colony numbers in G13C/WT genotype were significantly higher than those in WT/WT genotype (Figure 4E). These results confirmed oncogenic KRAS genotype-associated enhancement in retention of self-renewal capacity in iPSCs.
Biochemical Analysis on KRAS Activity and Downstream ERK and AKT Pathways
To determine the mechanisms underlying the KRAS mutant-specific phenotypes as described above, we first examined the existing amount of KRAS-GTP by GST-RAF1 pull-down assays. We observed significantly larger amounts of KRAS-GTP in G13C/WT iPSCs compared with WT/WT iPSCs under both conditions with and without bFGF (Figures 5A and 5B). Importantly, KRAS-GTP levels still remained high in G13C/WT iPSCs, but not in WT/WT iPSCs, even in the absence of bFGF, indicating constitutive activation of KRAS in G13C/WT iPSCs (Figures 5A and 5B). Consistent with these results, the same pattern was observed when genome-edited WTed/WT clones were compared with G13C/WT clones (Figures S4A and S4B). Furthermore, KRAS-GTP levels in Δed/WT clones were significantly reduced compared with those in WTed/WT clones in the absence of bFGF (Figures S4A and S4B).
Figure 5.

Biochemical Analysis on the ERK and AKT Pathways Activity in Enforced Retention of Self-Renewal of KRAS G13C/WT iPSCs
(A) GST-RAF1 pull-down assays of RALD patient-derived iPSC (WT/WT and G13C/WT) clones from case no. 1 and no. 2 cultured with (w/) or without (w/o) bFGF for 3 days.
(B) Densitometric analysis of western blotting results shown in (A). Each point indicates an individual clone's value (n = 4–6 independent clones; mean ± SEM; ∗∗∗p < 0.001; Student's t test or Mann-Whitney test).
(C) Western blot analysis of WT/WT and G13C/WT iPSC clones stimulated for the indicated time course after the removal of bFGF for 3 days. ERK and AKT were analyzed for their phosphorylation. β-Tubulin was used as an internal control.
(D) Western blot analysis of WT/WT and G13C/WT clones cultured w/ or w/o bFGF for 2 days. Phosphorylation of ERK and AKT was analyzed as in (C).
(E and F) Densitometric analysis of ERK (E) and AKT (F), respectively, in (D). Each point indicates an individual clone's value (n = 4–6 independent clones; mean ± SEM; ∗∗∗p < 0.001; Student's t test or Mann-Whitney test). GST, glutathione S-transferase; RBD, Ras-binding domain.
See also Figure S4.
Next, we analyzed the phosphorylation levels of ERK and AKT in G13C/WT and WT/WT iPSCs to determine downstream signaling activity in KRAS-signaling pathways. Upon bFGF stimulation for the indicated times after its removal, the ERK pathway was prominently upregulated in both cell types, whereas it showed only slight upregulation of pAKT (Figure 5C). To see if sustained activation of the downstream pathways from KRAS occurred in G13C/WT iPSCs even after bFGF deprivation, we compared phosphorylation levels of ERK and AKT after culturing cells with or without bFGF. Although there was only a marginal increase in pERK in G13C/WT iPSCs compared with WT/WT counterparts in the presence of bFGF (p = 0.0936), significantly higher pERK levels were maintained in the mutant clones than the control in the absence of bFGF (p < 0.001) (Figures 5D and 5E). On the other hand, there was no significant difference in pAKT levels between WT/WT and G13C/WT iPSCs in either case with or without bFGF (Figures 5D and 5F). Using genome-edited iPSC clones, retention of pERK expression in the absence of bFGF was also confirmed for G13C/WT clones when compared with WTed/WT clones. Notably, Δed/WT clones showed pERK levels that were significantly lower than those in WTed/WT clones in the absence of bFGF (p < 0.05), which is similar to aforementioned results and consistent with the idea that these artificial control clones exhibit haploinsufficiency and thus “loss-of-function” phenotypes (Figures S4C and S4D). These results indicate that bFGF stimulation appears to act through the ERK signaling pathway and that there is sustained activity of KRAS-ERK signaling in G13C/WT iPSCs despite bFGF withdrawal.
Pharmacological Analysis on the Involvement of the MEK-ERK and PI3K Pathways in the Enforced Retention of Self-Renewal and Suppressed Neuronal Differentiation Propensity of G13C/WT iPSCs
Finally, to investigate which downstream pathway from KRAS was responsible for the retained OCT4 expression in the mutant clones, we performed pharmacological intervention using kinase-specific inhibitors targeting the RAF-MEK and PI3K pathways (Favata et al., 1998, Hall-Jackson et al., 1999, Montagut et al., 2008, Powis et al., 1994, Sebolt-Leopold et al., 1999, Vlahos et al., 1994) (Figure S5A). As expected, the retention of OCT4 expression after bFGF removal in G13C/WT iPSCs was reversed by treatment with MEK inhibitors (PD184352 and U0126) (Figures 6A and S5B). Western blot analysis showed that pERK levels were diminished by MEK inhibitors at the same concentrations that induced the inhibition of OCT4 expression, while pAKT levels were not affected (Figure 6B). As for RAF inhibitors, AZD628 decreased the OCT4+ rate, whereas ZM336372 led to an increased OCT4+ rate (Figures 6C and S5C). Consistent with these results, AZ628 decreased pERK levels, but ZM336372 treatment led to an increase in pERK (Figure 6D). Clone R2-1 also displayed the same results as clone R1-2 (Figures S5E and S5F).
Figure 6.

Pharmacological Analysis on the Involvement of the RAF-MEK-ERK and PI3K-AKT Pathways in Enforced Retention of Self-Renewal of KRAS G13C/WT iPSCs
(A and B) Effects of MEK inhibitors (PD184352 and U0126) on OCT4+ area (A) (n = 3 independent experiments; mean ± SEM) and phosphorylation of ERK and AKT (B), respectively, in G13C/WT iPSCs (clone R1-2).
(C and D) Effects of RAF inhibitors (ZM336372 and AZ628) on OCT4+ area (C) (n = 3 independent experiments; mean ± SEM) and phosphorylation of ERK and AKT (D), respectively, in G13C/WT iPSCs (clone R1-2).
(E) Representative fluorescent images of G13C/WT iPSCs (clone R1-2) treated with the compounds at indicated concentrations. Vehicle, 0.1% DMSO. Scale bar, 100 μm.
When these compounds diminished OCT4+ areas, a part of OCT4– cells exhibited flattened morphology especially in the treatment of MEK inhibitors, which resembled that of WT/WT iPSCs cultured without bFGF (Figure 6E). Regarding PD184352, the concentrations that induced 20%, 50%, and 80% (IC20, IC50, and IC80) inhibition relative to vehicle were determined for all clones (Figure S5G; Table S5). Among PI3K inhibitors, LY294002 showed a partial decrease in the OCT4+ rate, whereas wortmannin did not affect that, although it prominently decreased pAKT levels (Figures S5D, S5H, and S5I).
To ask whether the MEK-ERK pathway was also responsible for the suppressed neuronal differentiation observed in G13C/WT iPSCs, mutant iPSCs (clones R1-2 and R2-1) were treated with PD184352 during 16-day in vitro differentiation. qRT-PCR analysis showed that mRNA expression of ASCL1 increased following treatment with PD184352 in a concentration-dependent manner (Figure 7A). Immunoreactive βIII-Tubulin appeared in greater density in differentiated cells from G13C/WT iPSCs following treatment with PD184352 in a concentration-dependent manner (Figure 7B). These data support the notion that KRAS mutant-specific phenotypes depend mostly on the RAF-MEK-ERK pathway, but not the PI3K-AKT pathway.
Figure 7.

Effects of a MEK Inhibitor on Suppressed Neuronal Differentiation in G13C/WT iPSCs
(A) qRT-PCR analysis of a neuronal lineage marker (ASCL1) in 16-day differentiated cells from WT/WT (clones C1-1 and C2-1) and G13C/WT iPSCs (clones R1-2 and R2-1) treated with a MEK inhibitor (PD184352) at the concentrations of IC20, IC50, and IC80 values as described in Table S5, or 0.1% DMSO (vehicle) (n = 3 independent experiments; mean ± SEM; ∗p < 0.05; ∗∗p < 0.01, ∗∗∗p < 0.001; one-way ANOVA followed by Dunnett's test).
(B) Immnunocytochemistry of βIII-Tubulin in 16-day differentiated cells from WT/WT and G13C/WT iPSCs treated with PD184352 as described above. Scale bar, 100 μm. PD, PD184352.
Discussion
In the present study, we established KRAS mutant and isogenic wild-type iPSC clones from two RALD patients sharing the same somatic heterozygous KRAS G13C mutation. In comparison with the isogenic iPSC clones, we showed that activated KRAS with the G13C mutation conferred retained self-renewal of undifferentiated cells and difficulty in differentiation into neurons under permissive conditions. In addition, we generated genome-edited wild-type (WTed/WT) and heterozygous knockout (Δed/WT) iPSCs from a G13C/WT clone. The phenotypes of parental G13C/WT iPSCs were rescued in WTed/WT iPSCs, and lower self-renewal and higher neuronal differentiation potentials compared with WTed/WT iPSCs was observed in Δed/WT iPSCs. Accordingly, we concluded that the phenotypes are KRAS G13C mutation specific.
First, we found retained high expression of stemness markers in G13C/WT iPSCs, which was observed not only when cells were cultured without bFGF for 5 days, but also in 16-day in vitro differentiation. Global gene expression analysis identified differential expression profiles between G13C/WT and WT/WT iPSCs in genes relating to self-renewal of undifferentiated cells, including POU5F1 and NANOG. Their expression was retained at a high level despite the removal of bFGF, suggesting enforced retention of self-renewal of G13C/WT iPSCs. Furthermore, clear differences in the size of OCT4+ cell populations between WT/WT and G13C/WT iPSCs could be detected in the 5-day bFGF-depletion assays. The capacity for retaining OCT4 expression was removed by rescuing the G13C allele (WTed/WT), and lowered with the G13C-specific knockout (Δed/WT). Thus, we concluded that the enforced retention of self-renewal was a KRAS mutation-specific phenotype. In general, bFGF is an essential factor for maintaining human ESC and iPSC pluripotency through downstream MEK-ERK and PI3K-AKT pathways (Ding et al., 2010, Lanner and Rossant, 2010, Li et al., 2007). We postulated that the retained self-renewal of undifferentiated cells observed in G13C/WT iPSCs was caused by the constitutive activation of KRAS and its downstream signals even though iPSCs were cultured without bFGF. We attempted to detect KRAS mutation-specific hyper-activation of the KRAS pathways. As expected, RAF1 pull-down assays showed hyper-activated KRAS in G13C/WT iPSCs in the absence of bFGF. Western blot analysis showed that the ERK pathway was largely upregulated, but only slight upregulation was observed in the AKT pathway upon temporal stimulation with bFGF. Moreover, pERK was more upregulated in G13C/WT iPSCs compared with WT/WT iPSCs, but such a trend was not observed in the case of pAKT when cultured without bFGF for a few days. Taken together, the MEK-ERK pathway, but not the PI3K pathway, was considered to be the main pathway involved in the bFGF signal resulting in KRAS mutant-specific phenotypes.
To further investigate whether the ERK pathway was responsible for the enforced retention of self-renewal, we performed pharmacological analysis using specific kinase inhibitors. MEK inhibitors (PD184352 and U0126) decreased OCT4+ area rates of G13C/WT iPSCs cultured in the absence of bFGF in a concentration-dependent manner. In terms of RAF inhibitors, paradoxical results were obtained between AZ628 and ZM336372; AZ628 lowered OCT4+ area rates, whereas ZM336372 elevated them. It has been reported that some RAF inhibitors block MEK-ERK signaling in cells with mutant BRAF, but enhance signaling in cells with wild-type BRAF. These RAF inhibitors include PLX4720 (vemurafenib), GDC-0879 (BRAF inhibitors), and ZM336372 (a CRAF inhibitor) (Hall-Jackson et al., 1999, Hatzivassiliou et al., 2010, Poulikakos et al., 2010). Interestingly, among the RAF inhibitor examined, AZ628, a pan-RAF inhibitor, does not induce phosphorylation of MEK or hyper-proliferation of KRASWT/BRAFWT cells (Hatzivassiliou et al., 2010). Our data from G13C/WT iPSCs were in agreement with these previous reports. The PI3K-AKT pathway is also known to be important in maintaining stemness of human ESCs (Ding et al., 2010, Li et al., 2007). The addition of LY294002 in particular led to a decrease in OCT4+ area rates, while wortmannin did not affect them, although pAKT was effectively lowered by wortmannin treatment. Recently, LY294002 has been reported to act as not only a PI3K inhibitor, but also as a bromodomain and extraterminal domain (BET) inhibitor (Dittmann et al., 2014). BRD4, a member of the BET family of proteins, is required for maintenance in human ESCs through the regulation of pluripotency genes, including POU5F1 and NANOG (Di Micco et al., 2014). Assuming that the effects of LY294002 on OCT4+ area rates were caused by BET inhibition, the involvement of PI3K in the retained self-renewal of undifferentiated cells would be moderate under our assay conditions. This is because wortmannin, another chemotype PI3K inhibitor, did not decrease OCT4+ area rates at all. Therefore, it could be demonstrated that the enforced retention of self-renewal in G13C/WT iPSCs derived from RALD patients was caused mainly by retained KRAS activation and subsequent prolonged hyper-activation of the RAF-MEK-ERK signals.
In this study, we found suppression of neuronal differentiation from G13C/WT iPSCs as shown in 16-day in vitro differentiation experiments. So far, mutations in the RAS pathway have been reported to be implicated in neural diseases. The RASopathies, such as Noonan syndrome, Costello syndrome (CS), cardio-facio-cutaneous syndrome, and neurofibromatosis type 1 (NF-1), are developmental syndromes caused by germline mutations in genes involved in the RAS-MEK pathways. These disorders are all characterized by neurological symptoms, such as mental retardation, cognitive impairment, and increased anxiety. These abnormalities are thought to originate from dysregulated differentiation of neuronal progenitor cells, based on structural CNS characteristic to the RASopathy patients, and the results obtained from animal model studies (Axelrad et al., 2011, Costa et al., 2002). In neural differentiation, bFGF-ERK signaling is known to inhibit PAX6 induction and act as an inhibitory signal to neural induction in human ESCs (Greber et al., 2010). Moreover, activated RAS signaling caused by mutations of KRAS or NF-1 has been reported to lead to enhanced proliferation of neuronal progenitor cells and aberrant differentiation into neurons in mouse models (e.g., shorter neurites) (Bender et al., 2015, Brown et al., 2012, Rooney et al., 2016). Recently, Rooney et al. reported the generation of CS patient-derived iPSCs harboring a heterozygous constitutively active G12S mutation in HRAS. By demonstrating an increased population of PAX6+/MAP2− cells and shorter neurite length in neurons differentiated from HRAS-mutated iPSCs, they suggested that activated HRAS led to an extended neuronal progenitor phase and difficulty in maturation of neurons (Rooney et al., 2016). In this study, we manifested the decreased expression of the neuronal markers, particularly ASCL1, and reduced βIII-Tubulin+/MAP2+ cells in G13C/WT iPSC lines cultured for 16 days in a differentiation-permissive condition. Despite taking into consideration the differences in experimental settings between studies, our results would further strengthen the idea that constitutive RAS activation in general likely has the potential to affect fates of stem cells under differentiation-permissive conditions. We speculated that the perturbation of neuronal differentiation in G13C/WT iPSCs occurred mainly due to sustained KRAS activity despite withdrawing bFGF. The inhibited neuronal differentiation was restored completely by the rescue of the G13C allele using genome-editing technology or partially by the treatment of an MEK inhibitor, PD184352. Our findings are in agreement with the previous paper reporting that FGF signaling inhibits neural induction, and inhibition of FGF/ERK upregulates a neuroectodermal fate determinant in human ESCs (Greber et al., 2011).
Overall, our data indicated the significance of KRAS status for the maintenance of iPSC stemness, which would eventually affect aspects such as self-renewal and differentiation propensity. The roles of oncogenic KRAS in stemness maintenance have been implicated in some types of stem cells. Oncogenic KRAS plays important roles in the imposition of ESC-like transcriptional profiles and upregulation of CSC markers, leading to the tumor initiation and development shown in colorectal and pancreatic cancers (Le Rolle et al., 2016, Matsubara et al., 2013, Moon et al., 2014). In a model of retinoic acid (RA)-induced stem cell differentiation to endoderm, oncogenic KRAS confers on cells resistance to differentiation associated with the maintenance of stem cell characteristics despite RA treatment (Quinlan et al., 2008). In addition, oncogenic KRAS promotes self-renewal properties and tumor initiation by suppressing non-canonical WNT signaling (Wang et al., 2015). These studies suggest that the involvement of oncogenic KRAS in stemness maintenance might be a common feature among various types of stem cells, especially in CSCs. Taken together, the RALD patient-derived isogenic iPSCs would be applicable to research not only on pathology of RALD but also certain aspects of CSCs as a general tool for studying oncogenic KRAS-driven stem cell dysfunctions. Using this unique and invaluable tool, the development of pharmacological treatment will likely be achieved for RALD, CSCs, and other oncogenic KRAS-related diseases in the future.
Experimental Procedures
Ethics
Bone marrow cell samples from patients were used in accordance with the Declaration of Helsinki. Written informed consent for samples to be used for research purposes was obtained from the patient or patient’s parents. The study was approved by the Ethics Committee of The Institute of Medical Science, The University of Tokyo (protocol number, 25-3-0701), the Ethics Committee of Tokyo Medical and Dental University (approval numbers, 676), and the Eisai Research Ethics Committee (approval numbers, 2014-0238, 2014-0259, 2015-0238, 2015-0259, 2016-0238, and 2016-0259). The use of viral vectors was approved by the ethics committees of The Institute of Medical Science, The University of Tokyo.
Establishment of iPSCs from RALD Patients
Establishment of iPSCs from CD34+ hematopoietic stem/progenitor cells was performed as described previously (Lin et al., 2015, Takayama et al., 2010) at The Institute of Medical Science, The University of Tokyo. Mononuclear cells were extracted by Ficoll density gradient separation from whole bone marrow aspirate obtained from the two individual RALD patients. CD34+ cells were isolated from this mononuclear fraction by immunomagnetic separation using a CD34 MicroBead Kit (Miltenyi Biotec) according to the manufacturer's protocol. Non-integrating Sendai virus (SeV) vectors harboring human POU5F1, SOX2, KLF4, and c-MYC (Nishimura et al., 2011) were used to transduce isolated CD34+ cells for reprogramming into RALD patient-derived iPSCs. Lipofectamine RNAi Max (Thermo Fisher Scientific) was used to transfect established RALD patient-derived iPSCs with small interfering RNA L527 for the removal of SeV vectors. Established clones were screened for mutations in the KRAS gene by sequencing genomic DNA. A proportion of CD34+ cells obtained from both patients did not carry any KRAS mutation. The resulting iPSC clones free of the mutation were retained and used as isogenic controls. Karyotyping was performed at Nihon Gene Research Laboratories (Sendai, Japan).
iPSC Maintenance
RALD patient-derived iPSC clones were introduced into Eisai Tsukuba Laboratories from The Institute of Medical Science. Detailed information of the clones is given in Table S2. In the Eisai Tsukuba Laboratories, iPSCs were maintained on vitronectin (Thermo Fisher Scientific)-coated plates in a culture medium for iPSCs, StemFit (Ajinomoto) added with StemFit's supplement package including bFGF and 1× penicillin-streptomycin (P/S; Wako). At passage, iPSCs were dissociated with TrypLE Select (Thermo Fisher Scientific) and cultured with 10 μM Y-27632 (Wako), and the medium was changed to Y-27632-free StemFit the next day. The medium was replaced every 2 days, and iPSCs were passaged every 3 or 4 days. Cell culture was carried out at 37°C in a humidified atmosphere of 20% O2. In this study, we used iPSCs at the passage numbers less than 30 after the introduction into Eisai.
In Vitro EB-Mediated Differentiation
In vitro differentiation was performed basically according to procedures as described previously (Takahashi et al., 2007). iPSCs were harvested after cell dissociation with TrypLE Select, and seeded to six-well ultra-low attachment plates (Corning) in bFGF-free StemFit containing 10 μM Y-27632 (Wako) to produce EBs. On the following day, the medium was changed to a fresh one without Y-27632. After the 8-day suspension culture, EBs were collected and transferred to 0.1% gelatin (Sigma)-coated plates to initiate adherent culture. Cells were allowed to differentiate in bFGF-free StemFit containing 10% fetal bovine serum (Thermo Fisher Scientific) for additional 8 days. The media were exchanged basically every 2 days.
Immunocytochemistry
Cells were fixed by 4% paraformaldehyde (Wako) for 30 min at room temperature. They were blocked and permeabilized in 4% (w/v) Block Ace (AbD Serotec)/cell staining buffer (20 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 0.2% Triton X-100) for 30 min at room temperature. Then, cells were incubated with primary antibodies in 0.4% (w/v) Block Ace/cell staining buffer overnight at 4°C, and subsequently incubated with appropriate secondary antibodies with Hoechst 33342 (10 μg/mL; Sigma-Aldrich) in 0.4% (w/v) Block Ace/cell staining buffer for 1.5 hr at room temperature. Detailed conditions of antibodies are given in Table S6. Fluorescence images were acquired on an IX73 (Olympus) or IN-Cell Analyzer (GE Healthcare).
bFGF-Depletion Assays
Cells were dissociated by TrypLE Select, and 2 × 103 cells were seeded on vitronectin-coated 96-well plates in StemFit containing Y-27632 (10 μM) with or without bFGF and cultured for 5 days. Medium was changed on day 1, to remove Y-27632, and on day 3. Immunocytochemistry of stemness markers was performed as described above. In quantitation, OCT4+ and nucleus (Hoechst 33342-stained) areas were measured by an IN-Cell Developer (GE Healthcare). OCT4+ rates and expected total cell numbers were calculated as follows:
-
-
OCT4+ rate (%) = total OCT4+ area/Hoechst-positive area × 100
-
-
Expected total cell numbers = total Hoechst-positive area/median of individual Hoechst-positive area
Colony-Formation Assays and ALP Staining of Reseeded iPSCs that Had Been Cultured without bFGF
RALD iPSCs (C2-1 and C2-2 of WT/WT clones; R2-1 and R2-2 of G13C/WT clones) were plated and cultured in StemFit without bFGF for 5 days. After harvesting cells, they were reseeded into six-well plates at 1.0 × 104 cells/well (n = 3), and cultured in StemFit with bFGF. When colonies were formed 7 days after reseeding, cells were stained for ALP using nitrotetrazolium blue chloride (400 μM; Sigma-Aldrich) and 5-bromo-4-chloro-3-indolyl phosphate p-toluidine salt (400 μM; Sigma-Aldrich) in ALP buffer (100 mM NaCl, 100 mM Tris-HCl, and 50 mM MgCl2 [pH 9.5]). ALP+ colony number was measured using BZ-X710 (Keyence).
Pharmacological Examinations
We examined effects of specific inhibitors related to RAS pathways (Favata et al., 1998, Hall-Jackson et al., 1999, Montagut et al., 2008, Powis et al., 1994, Sebolt-Leopold et al., 1999, Vlahos et al., 1994) on bFGF-depletion assays and in vitro differentiation. PD184532 and wortmannin were purchased from Sigma. U0126 was obtained from Wako. ZM336372, AZ628, and LY294002 were purchased from Selleck. To exclude the effect of cell toxicity, the concentrations of each compound were selected at the range of lower than 80% cell growth inhibition judged by the percentage of nucleus number relative to the vehicle treatment in bFGF-depletion assays (Figures S5B–S5D).
Author Contributions
K.K., K.Y., M.I., K.T., M.T., and M.O. conceived and designed the study. K.K., K.Y., Y.I., and T.N. performed the experiments and analyzed the data. K.N., M.O., M.N., H.-T.L., M.T., and M.O. contributed to generate the patient-derived iPSCs. K.K., K.Y., H.-T.L., M.T., and M.O. wrote the manuscript. M.I., K.T., T.M., M.T., and M.O. supervised the project.
Acknowledgments
We thank Drs. Sadakazu Miyashita, Koji Sagane, and Toru Arai for useful advice and comments, and Drs. Haruna Takagi and Kentaro Takahashi for technical assistance. Patient care is provided by Dr. Hiroshi Moritake. This study was partially supported by grants from the Program for Intractable Disease Research Utilizing Disease-specific iPS Cells funded by the Japan Science and Technology Agency (JST)/Japan Agency for Medical Research and Development (A-MED) under grant numbers JP16bm0609006/JP17bm0804004/JP17ek0109233.
Published: July 5, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, five figures, and seven tables and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.06.008.
Contributor Information
Kenji Kubara, Email: k-kubara@hhc.eisai.co.jp.
Kazuto Yamazaki, Email: k5-yamazaki@hhc.eisai.co.jp.
Accession Numbers
Whole exome sequencing data are available in the Japanese Genotype-phenotype Archive (JGA, http://trace.ddbj.nig.ac.jp/jga), which is hosted by the DNA Data Bank of Japan (DDBJ), under accession number JGAS00000000136. RNA-seq and microarray data are available in the GEO under accession numbers GEO: GSE111345 and GSE94141, respectively.
Supplemental Information
References
- Adjei A.A. Blocking oncogenic Ras signaling for cancer therapy. J. Natl. Cancer Inst. 2001;93:1062–1074. doi: 10.1093/jnci/93.14.1062. [DOI] [PubMed] [Google Scholar]
- Ali F.R., Cheng K., Kirwan P., Metcalfe S., Livesey F.J., Barker R.A., Philpott A. The phosphorylation status of Ascl1 is a key determinant of neuronal differentiation and maturation in vivo and in vitro. Development. 2014;141:2216–2224. doi: 10.1242/dev.106377. [DOI] [PubMed] [Google Scholar]
- Axelrad M.E., Schwartz D.D., Katzenstein J.M., Hopkins E., Gripp K.W. Neurocognitive, adaptive, and behavioral functioning of individuals with Costello syndrome: a review. Am. J. Med. Genet. C Semin. Med. Genet. 2011;157C:115–122. doi: 10.1002/ajmg.c.30299. [DOI] [PubMed] [Google Scholar]
- Bender R.H., Haigis K.M., Gutmann D.H. Activated k-ras, but not h-ras or N-ras, regulates brain neural stem cell proliferation in a raf/rb-dependent manner. Stem Cells. 2015;33:1998–2010. doi: 10.1002/stem.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J.A., Diggs-Andrews K.A., Gianino S.M., Gutmann D.H. Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol. Cell. Neurosci. 2012;49:13–22. doi: 10.1016/j.mcn.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano E., Downward J. Role of RAS in the regulation of PI 3-kinase. Curr. Top Microbiol. Immunol. 2010;346:143–169. doi: 10.1007/82_2010_56. [DOI] [PubMed] [Google Scholar]
- Castro D.S., Guillemot F. Old and new functions of proneural factors revealed by the genome-wide characterization of their transcriptional targets. Cell Cycle. 2011;10:4026–4031. doi: 10.4161/cc.10.23.18578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G., Gulbranson D.R., Hou Z., Bolin J.M., Ruotti V., Probasco M.D., Smuga-Otto K., Howden S.E., Diol N.R., Propson N.E. Chemically defined conditions for human iPSC derivation and culture. Nat. Methods. 2011;8:424–429. doi: 10.1038/nmeth.1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa R.M., Federov N.B., Kogan J.H., Murphy G.G., Stern J., Ohno M., Kucherlapati R., Jacks T., Silva A.J. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- D'Adamo D.R., Novick S., Kahn J.M., Leonardi P., Pellicer A. rsc: a novel oncogene with structural and functional homology with the gene family of exchange factors for Ral. Oncogene. 1997;14:1295–1305. doi: 10.1038/sj.onc.1200950. [DOI] [PubMed] [Google Scholar]
- Di Micco R., Fontanals-Cirera B., Low V., Ntziachristos P., Yuen S.K., Lovell C.D., Dolgalev I., Yonekubo Y., Zhang G., Rusinova E. Control of embryonic stem cell identity by BRD4-dependent transcriptional elongation of super-enhancer-associated pluripotency genes. Cell Rep. 2014;9:234–247. doi: 10.1016/j.celrep.2014.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding V.M., Ling L., Natarajan S., Yap M.G., Cool S.M., Choo A.B. FGF-2 modulates Wnt signaling in undifferentiated hESC and iPS cells through activated PI3-K/GSK3beta signaling. J. Cell. Physiol. 2010;225:417–428. doi: 10.1002/jcp.22214. [DOI] [PubMed] [Google Scholar]
- Dittmann A., Werner T., Chung C.W., Savitski M.M., Falth Savitski M., Grandi P., Hopf C., Lindon M., Neubauer G., Prinjha R.K. The commonly used PI3-kinase probe LY294002 is an inhibitor of BET bromodomains. ACS Chem. Biol. 2014;9:495–502. doi: 10.1021/cb400789e. [DOI] [PubMed] [Google Scholar]
- Favata M.F., Horiuchi K.Y., Manos E.J., Daulerio A.J., Stradley D.A., Feeser W.S., Van Dyk D.E., Pitts W.J., Earl R.A., Hobbs F. Identification of a novel inhibitor of mitogen-activated protein kinase kinase. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- Greber B., Wu G., Bernemann C., Joo J.Y., Han D.W., Ko K., Tapia N., Sabour D., Sterneckert J., Tesar P. Conserved and divergent roles of FGF signaling in mouse epiblast stem cells and human embryonic stem cells. Cell Stem Cell. 2010;6:215–226. doi: 10.1016/j.stem.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Greber B., Coulon P., Zhang M., Moritz S., Frank S., Muller-Molina A.J., Arauzo-Bravo M.J., Han D.W., Pape H.C., Scholer H.R. FGF signalling inhibits neural induction in human embryonic stem cells. EMBO J. 2011;30:4874–4884. doi: 10.1038/emboj.2011.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall-Jackson C.A., Eyers P.A., Cohen P., Goedert M., Boyle F.T., Hewitt N., Plant H., Hedge P. Paradoxical activation of Raf by a novel Raf inhibitor. Chem. Biol. 1999;6:559–568. doi: 10.1016/s1074-5521(99)80088-x. [DOI] [PubMed] [Google Scholar]
- Hatzivassiliou G., Song K., Yen I., Brandhuber B.J., Anderson D.J., Alvarado R., Ludlam M.J., Stokoe D., Gloor S.L., Vigers G. RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature. 2010;464:431–435. doi: 10.1038/nature08833. [DOI] [PubMed] [Google Scholar]
- Lanner F., Rossant J. The role of FGF/Erk signaling in pluripotent cells. Development. 2010;137:3351–3360. doi: 10.1242/dev.050146. [DOI] [PubMed] [Google Scholar]
- Le Rolle A.F., Chiu T.K., Zeng Z., Shia J., Weiser M.R., Paty P.B., Chiu V.K. Oncogenic KRAS activates an embryonic stem cell-like program in human colon cancer initiation. Oncotarget. 2016;7:2159–2174. doi: 10.18632/oncotarget.6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levenstein M.E., Ludwig T.E., Xu R.H., Llanas R.A., VanDenHeuvel-Kramer K., Manning D., Thomson J.A. Basic fibroblast growth factor support of human embryonic stem cell self-renewal. Stem Cells. 2006;24:568–574. doi: 10.1634/stemcells.2005-0247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Wang G., Wang C., Zhao Y., Zhang H., Tan Z., Song Z., Ding M., Deng H. MEK/ERK signaling contributes to the maintenance of human embryonic stem cell self-renewal. Differentiation. 2007;75:299–307. doi: 10.1111/j.1432-0436.2006.00143.x. [DOI] [PubMed] [Google Scholar]
- Lin H.T., Masaki H., Yamaguchi T., Wada T., Yachie A., Nishimura K., Ohtaka M., Nakanishi M., Nakauchi H., Otsu M. An assessment of the effects of ectopic gp91phox expression in XCGD iPSC-derived neutrophils. Mol. Ther. Methods Clin. Dev. 2015;2:15046. doi: 10.1038/mtm.2015.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsubara S., Ding Q., Miyazaki Y., Kuwahata T., Tsukasa K., Takao S. mTOR plays critical roles in pancreatic cancer stem cells through specific and stemness-related functions. Sci. Rep. 2013;3:3230. doi: 10.1038/srep03230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagut C., Sharma S.V., Shioda T., McDermott U., Ulman M., Ulkus L.E., Dias-Santagata D., Stubbs H., Lee D.Y., Singh A. Elevated CRAF as a potential mechanism of acquired resistance to BRAF inhibition in melanoma. Cancer Res. 2008;68:4853–4861. doi: 10.1158/0008-5472.CAN-07-6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moodie S.A., Willumsen B.M., Weber M.J., Wolfman A. Complexes of Ras.GTP with Raf-1 and mitogen-activated protein kinase kinase. Science. 1993;260:1658–1661. doi: 10.1126/science.8503013. [DOI] [PubMed] [Google Scholar]
- Moon B.S., Jeong W.J., Park J., Kim T.I., Min do S., Choi K.Y. Role of oncogenic K-Ras in cancer stem cell activation by aberrant Wnt/beta-catenin signaling. J. Natl. Cancer Inst. 2014;106:djt373. doi: 10.1093/jnci/djt373. [DOI] [PubMed] [Google Scholar]
- Moritake H., Takagi M., Kinoshita M., Ohara O., Yamamoto S., Moriguchi S., Nunoi H. Autoimmunity including intestinal Behcet disease bearing the KRAS mutation in lymphocytes: a case report. Pediatrics. 2016;137:e20152891. doi: 10.1542/peds.2015-2891. [DOI] [PubMed] [Google Scholar]
- Niemela J.E., Lu L., Fleisher T.A., Davis J., Caminha I., Natter M., Beer L.A., Dowdell K.C., Pittaluga S., Raffeld M. Somatic KRAS mutations associated with a human nonmalignant syndrome of autoimmunity and abnormal leukocyte homeostasis. Blood. 2011;117:2883–2886. doi: 10.1182/blood-2010-07-295501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura K., Sano M., Ohtaka M., Furuta B., Umemura Y., Nakajima Y., Ikehara Y., Kobayashi T., Segawa H., Takayasu S. Development of defective and persistent Sendai virus vector: a unique gene delivery/expression system ideal for cell reprogramming. J. Biol. Chem. 2011;286:4760–4771. doi: 10.1074/jbc.M110.183780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osumi N., Shinohara H., Numayama-Tsuruta K., Maekawa M. Concise review: Pax6 transcription factor contributes to both embryonic and adult neurogenesis as a multifunctional regulator. Stem Cells. 2008;26:1663–1672. doi: 10.1634/stemcells.2007-0884. [DOI] [PubMed] [Google Scholar]
- Poulikakos P.I., Zhang C., Bollag G., Shokat K.M., Rosen N. RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature. 2010;464:427–430. doi: 10.1038/nature08902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis G., Bonjouklian R., Berggren M.M., Gallegos A., Abraham R., Ashendel C., Zalkow L., Matter W.F., Dodge J., Grindey G. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994;54:2419–2423. [PubMed] [Google Scholar]
- Prior I.A., Lewis P.D., Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res. 2012;72:2457–2467. doi: 10.1158/0008-5472.CAN-11-2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan M.P., Quatela S.E., Philips M.R., Settleman J. Activated Kras, but not Hras or Nras, may initiate tumors of endodermal origin via stem cell expansion. Mol. Cell. Biol. 2008;28:2659–2674. doi: 10.1128/MCB.01661-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney G.E., Goodwin A.F., Depeille P., Sharir A., Schofield C.M., Yeh E., Roose J.P., Klein O.D., Rauen K.A., Weiss L.A. Human iPS cell-derived neurons uncover the impact of increased Ras signaling in Costello syndrome. J. Neurosci. 2016;36:142–152. doi: 10.1523/JNEUROSCI.1547-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebolt-Leopold J.S., Dudley D.T., Herrera R., Van Becelaere K., Wiland A., Gowan R.C., Tecle H., Barrett S.D., Bridges A., Przybranowski S. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nat. Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- Shiota M., Yang X., Kubokawa M., Morishima T., Tanaka K., Mikami M., Yoshida K., Kikuchi M., Izawa K., Nishikomori R. Somatic mosaicism for a NRAS mutation associates with disparate clinical features in RAS-associated leukoproliferative disease: a report of two cases. J. Clin. Immunol. 2015;35:454–458. doi: 10.1007/s10875-015-0163-3. [DOI] [PubMed] [Google Scholar]
- Takagi M., Shinoda K., Piao J., Mitsuiki N., Matsuda K., Muramatsu H., Doisaki S., Nagasawa M., Morio T., Kasahara Y. Autoimmune lymphoproliferative syndrome-like disease with somatic KRAS mutation. Blood. 2011;117:2887–2890. doi: 10.1182/blood-2010-08-301515. [DOI] [PubMed] [Google Scholar]
- Takahashi K., Tanabe K., Ohnuki M., Narita M., Ichisaka T., Tomoda K., Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Takayama N., Nishimura S., Nakamura S., Shimizu T., Ohnishi R., Endo H., Yamaguchi T., Otsu M., Nishimura K., Nakanishi M. Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J. Exp. Med. 2010;207:2817–2830. doi: 10.1084/jem.20100844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vivanco I., Sawyers C.L. The phosphatidylinositol 3-kinase AKT pathway in human cancer. Nat. Rev. Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- Vlahos C.J., Matter W.F., Hui K.Y., Brown R.F. A specific inhibitor of phosphatidylinositol 3-kinase, 2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002) J. Biol. Chem. 1994;269:5241–5248. [PubMed] [Google Scholar]
- Vojtek A.B., Hollenberg S.M., Cooper J.A. Mammalian Ras interacts directly with the serine/threonine kinase Raf. Cell. 1993;74:205–214. doi: 10.1016/0092-8674(93)90307-c. [DOI] [PubMed] [Google Scholar]
- Wang M.T., Holderfield M., Galeas J., Delrosario R., To M.D., Balmain A., McCormick F. K-Ras promotes tumorigenicity through suppression of non-canonical Wnt signaling. Cell. 2015;163:1237–1251. doi: 10.1016/j.cell.2015.10.041. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.