

Abstract
T-cell-based immunotherapies represent a growing medical paradigm that has the potential to revolutionize contemporary cancer treatments. However, manufacturing bottlenecks related to the enrichment of therapeutically optimal T-cell subpopulations from leukopak samples impede scale-up and scale-out efforts. This is mainly attributed to the challenges that current cell purification platforms face in balancing the quantitative sorting capacity needed to isolate specific T-cell subsets with the scalability to meet manufacturing throughputs. In this work, we report a continuous-flow, quantitative cell enrichment platform based on a technique known as ratcheting cytometry that can perform complex, multicomponent purification targeting various subpopulations of magnetically labeled T cells directly from apheresis or peripheral blood mononuclear cell (PBMC) samples. The integrated ratcheting cytometry instrument and cartridge demonstrated enrichment of T cells directly from concentrated apheresis samples with a 97% purity and an 85% recovery of magnetically tagged cells. Magnetic sorting of different T-cell subpopulations was also accomplished on chip by multiplexing cell surface targets onto particles with differing magnetic strengths. We believe that ratcheting cytometry’s quantitative capacity and throughput scalability represents an excellent technology candidate to alleviate cell therapy manufacturing bottlenecks.
Keywords: ratcheting cytometry, cell therapy manufacturing, immunotherapy
Introduction
Adoptive cell therapies taking advantage of engineered chimeric antigen receptors (CARs) or T-cell receptors (TCRs) have shown incredible potential as “living drugs” that achieve personalized immunotherapies for cancer patients.1–5 The fundamental concept behind these immunotherapies involves the genetic modification of human T cells to express targeting moieties against tumor antigens. Adoptive T-cell therapy is regarded as a new cancer treatment paradigm with four major initial public offerings and two recent new drug application approvals,6,7 as well as the first two FDA approvals for the marketing of CAR-T therapies, Yescarta and Kmyriah.8 However, scale-up and scale-out for T-cell therapies remain a challenge due to the complex nature of the manufacturing workflow,9 which is a significant contributor to its high price point.10 One major bottleneck and cost driver is the purification of T cells or T-cell subpopulations from highly concentrated samples of white blood cells, known as apheresis samples or leukopaks, needed to engineer the CAR-T or TCR cells.11,12 In order to derive potent therapeutic cells, high-purity T-cell enrichment is essential, as it ensures that each batch consists of therapeutically active cell types. Furthermore, studies have shown that therapies derived from well-defined sets of T-cell subpopulations, such as CD4 and CD8 subtypes, have superior potency and sustained reaction compared with therapies derived from heterogenous populations.13,14 Unfortunately, current cell purification platforms struggle to balance the throughput and quantitative power needed to precisely enrich target T-cell populations at a scale tenable for cell therapy scale-up/scale-out. For both patient-derived (autologous) and donor-derived (allogenic) therapies, the most commonly utilized cell purification platforms are magnetic-assisted cell separation (MACS) and fluorescence-assisted cell separation (FACS). While robust, MACS separates cells above a single threshold for a single surface antigen, lacking the capability to perform orthogonal enrichment based on multiple cell surface markers or multiple levels of these markers. This is problematic when the target cell population shares expression with other cell types or when multiple subpopulations need to be sequestered to derive a potent cell therapy. This often necessitates multiple separations to be performed serially, which substantially decreases throughput and increases consumable cost per run. Furthermore, the high-density packed bed of superparamagnetic beads utilized in MACS columns can easily clog and often requires a continuous centrifugation or Ficoll gradient step before passing through the column.15 This not only adds time and cost to the platform but also can limit T-cell yield, which is detrimental when T-cell counts are low, as is the case with patients who have undergone several rounds of chemotherapy. In contrast, FACS-based techniques are highly quantitative and can precisely enrich T-cell subpopulations based on multiple markers. However, standard FACS systems are too low throughput (~103 cells/s)16 to achieve manufacturing-scale quantities on the order of 105–106 cell/s. Additionally, FACS approaches are difficult to integrate into a closed system for aseptic operation, which is critical for current good manufacturing practice (cGMP) compliance. Microfluidic technologies have shown promise in precision cell sorting and have even demonstrated integrated magnetic separation of multiple cell subpopulations using microscale fluid flow with ferromagnetic structures.17 However, these platforms face inherent scaling challenges due to their complex fabrication and physical throughput limitations, which make manufacturing-scale implementation challenging and costly. Furthermore, microfluidic channels are highly susceptible to clogging, which can be problematic in working with complex biomatrices, which often contain large cell aggregates. For this reason, microfluidic technologies are generally more effective in rapidly processing smaller sample volumes for analysis and quality control assays. Indeed, a purification platform that could enrich large quantities of T cells or T-cell subpopulations rapidly and efficiently, directly from complex biomatrices, would alleviate a critical manufacturing bottleneck for T-cell immunotherapies.
Here, we report a continuous-flow, quantitative, surface expression–based cell enrichment platform using a technique known as ratcheting cytometry,18–20 which can perform complex, multicomponent, purification workflows targeting various subpopulations of magnetically labeled T cells directly from apheresis or peripheral blood mononuclear cell (PBMC) samples. Furthermore, the prototype system demonstrated higher throughput than standard FACS, with the ability to reach manufacturing-scale throughput consistent with MACS technologies. The platform consists of a semiautomated instrument that pumps complex biomatrix solutions containing immunomagnetically tagged T cells through a closed cartridge for parallelized and quantitative magnetic purification at discrete locations along the cartridge. Leveraging well-established immunomagnetic cell labeling techniques traditionally used in MACS, the ratcheting cytometry platform innovatively allows for sorting based on differential levels of antigen and multisurface target magnetic sorting with a variety of magnetic bead types. In this way, the ratcheting cytometry platform can separate cells based on the level of bound magnetic content, what we refer to as quantitative magnetic sorting. These capabilities are similar to those provided by traditional flow cytometry but can be easily scaled to high-throughput capacity due to the platform’s highly parallelized nature. The system demonstrated continuous positive selection of immunomagnetically tagged T cells based on CD3 directly from apheresis samples, without the need for a centrifugation step, with a 97% purity, 85% efficiency, and throughput of 6.5 × 104 cells/s. Additionally, the system demonstrated simultaneous isolation of CD4 and CD8 T-cell sub-populations from the same sample by multiplexing with multiple magnetic particle sizes and iron oxide contents targeted at separate surface antigens. Two magnetic particle types with varying magnetite contents (1 μm diameter, 26% Fe3O4, and 80% Fe3O4) were functionalized with antibodies specific to CD4 and CD8 T-cell subpopulations (αCD4 to 0.5 μm and αCD8 to 1 μm), which were successfully separated to distinct locations within the cartridge due to the differing magnetic contents for the labeled cell populations. Additionally, we performed multisurface target magnetic sorting based on both CD3 (0.5 μm particle) and CD8 (1 μm particle) with the system to isolate CD3(+) and CD8(+) T-cell subpopulations.
Continuous, Quantitative, and Closed-Cartridge Purification Using Ratcheting Cytometry
Ratcheting cytometry is based on a micromanipulation technique known as magnetic ratcheting18–20 in which magnetic particles or magnetically labeled cells can be precisely manipulated and transported across a chip composed of ferromagnetic micropillar arrays that are subjected to a directionally cycled (ratcheted) magnetic field (Suppl. Fig. S1). Our ratcheting chip consists of a glass or plastic substrate upon which 2D arrays of 4 μm diameter ferromagnetic micropillars were constructed via an electroplating process. These pillars were then coated with polystyrene to create a planarizing layer (Suppl. Fig. S2). When subjected to a directionally cycling magnetic field provided by a wheel of radially arrayed neodymium ferrite magnets, arranged in a directional orientation known as a Halbach array (Suppl. Fig. 1), each micropillar dynamically magnetizes in alignment to the instantaneous applied field. In this way, the micropillar array generates an oscillating microscale field that causes magnetic particles or immunomagnetically tagged cells to magnetophoretically migrate to the point of highest magnetization and continuously traverse across the array as the magnetization direction shifts (Suppl. Video S1). In our previous work,19 we demonstrated that introducing a gradient in the micropillar pitch (i.e., the distance between ferromagnetic pillars) enables quantitative sorting of cells based on the number of magnetic particles bound to their surface. In this way, cells with increasing amounts of bound magnetic particles will separate and trap at increasingly distant spatial locations across the chip, at a location corresponding to a specific critical pitch (Fig. 1A,B).
Figure 1.
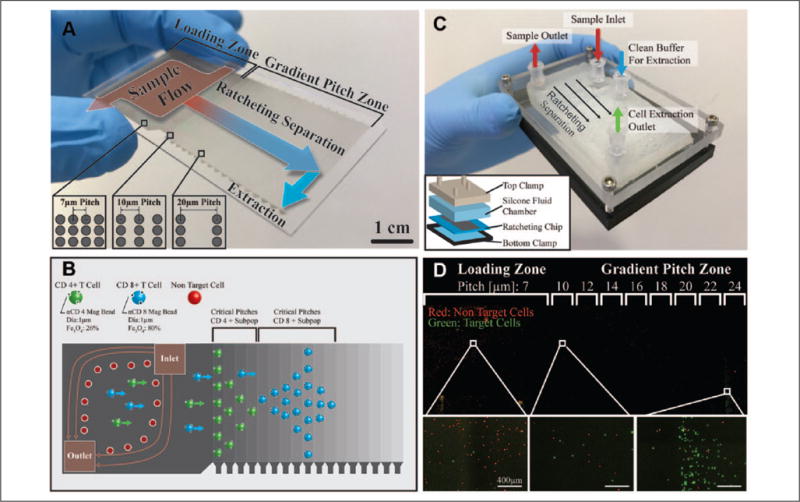
(A) Quantitative magnetic separations of cells can be achieved with chips composed of arrays of ferromagnetic micropillars with gradient pitch and subjected to a directionally cycled magnetic field. When magnetized, the micropillars within the chip instantaneously align to the bulk field, introducing shifting field maxima in which magnetically tagged cells migrate at a rate proportional to the rotational frequency of the applied field. Above a certain pitch between adjacent micropillars, cells will reach a critical pitch where they are unable to traverse to the next pillar, a process critically depending on the magnetic content bound to the cell. In this way, cells can be sorted quantitatively based on cell-bound magnetic content in the gradient pitch zones of the chip. Samples containing immunomagnetically tagged cells can be continuously flowed over the loading zone (7 μm pitch) and pulled onto the micropillar arrays where they can be quantitatively separated and concentrated at different locations based on bound magnetic content in the gradient pitch zone (10–42 μm pitch, 2 μm increments) for off-chip extraction. (B) By multiplexing antibody targets with particles of varying magnetic strengths, as determined by Fe3O4 content, different cell subpopulations will separate to different critical pitches, thereby concentrating to different localities within the chip. Cells labeled with low-Fe3O4-content particles will trap at lower pillar pitches than cells tagged with high-Fe3O4-content particles. In this way, cell purification can be achieved on multiple markers to purify several subpopulations simultaneously. (C) To achieve continuous and aseptic operation, a fluidic chamber is clamped over the ratcheting chip to form a closed cartridge. Fluidic interfaces using luer lock connections to form ports for sample introduction and cell extraction. (D) Ratcheting cartridge operation demonstrated with a mixture of CD3(−) HL60 cells (red) and CD3(+) Jurkat E6-1 cells (green), which have been tagged with an αCD3 4.5 μm magnetic particle. Green target cells are extracted out of the flow, transported across the loading zone, and then separated and concentrated for extraction in the gradient pitch zone.
In this work, we demonstrate that ratcheting cytometry can be readily scaled for high-throughput operation, scaling the cell processing throughput by 2000-fold from our previous work and adding instrument components toward a fully automated and scaled prototype. Additionally, we show the sorting of multiple T-cell subpopulations based on multiple cell surface markers simultaneously, which to our knowledge has not been achieved in a magnetic sorting context at throughputs higher than flow cytometry. As part of this effort, we developed a scaled gradient pitch ratcheting chip, 3 × 6.5 cm (Fig. 1A), to accommodate the continuous purification of millions of cells simultaneously, toward a cell therapy manufacturing scale. The micropillar array geometry consists of a 3 × 3 cm loading zone (7 μm pitch), leading to a 3 × 3.5 cm gradient pitch zone consisting of sections of incrementing pitch (pitch ranges from 10 to 42 μm, with 2 μm increments) (Suppl. Fig. S3). To enable continuous processing of these larger cell quantities, the new setup also introduces a flow of immunomagnetically tagged cell samples across the loading zone, where they are pulled onto the micropillars by application of the ratcheting magnetic field. Cells then transport across the loading zone, separate, and trap at a critical pitch within the gradient pitch zone, where they can be extracted off the chip (Fig. 1B). The ratcheting chip forms the bottom substrate of a closed-cartridge (Fig. 1C) assembly composed of two clamping sections that compress a soft silicone plastic fluidic chamber over the ratcheting chip. Standard luer lock fluidic connections link to the top piece and silicone chamber, allowing separations within a closed system. Figure 1D demonstrates the ratcheting cartridge operation with a mixture of red CD3(−) HL60 cells and green CD3(+) Jurkat E6-1 cells tagged with a 4.5 μm αCD3 magnetic particle. CD3(+) target cells show separation from nontarget cells and concentrate at the 24 μm pitch under a 10 Hz ratcheting field, where they can be extracted with a sterile luer lock syringe.
Materials and Methods
Ratcheting Cartridge and Instrument Design and Characterization
Chip Fabrication
Glass wafers (University Wafer, 100 mm) were cleaned with piranha solution and coated with a Ti-Cu-Ti metal seed layer via e-beam evaporation. SPR220 resist was used to make electroplating molds for ferromagnetic permalloy micropillar arrays, as described in previous work.19 Following electroplating, the resist was stripped with standard acetone wash and the Ti-Cu-Ti layers released using 1% hydrofluoric acid and copper etchant. The wafer was then diced into chips, coated with hexamethyldisilazane (HMDS), and spin-coated with polystyrene dissolved in toluene at 5% w/v.
Fluidic Cartridge Fabrication
The fluid cartridge that overlays the ratcheting chip consists of a larger sample injection chamber (5 mm depth) leading into a thinner chamber (1 mm depth) for sample extraction (Suppl. Fig. S4A). During injection and extraction of target cells, sample flows into the larger chamber through inlet A and out of the chamber through outlet B (Suppl. Fig. S4B). Since the sample injection chamber is deeper, it biases the flow direction to prevent flow of nontarget cells into the extraction region, where magnetically tagged target cells will be pulled onto the ratcheting chip and separated to the extraction regions (Suppl. Fig. S4C). Of note is that in this prototype design, only one extraction port was present to perform extraction of a single cell population. This was to demonstrate proof of concept that target cells could be easily extracted from the cartridge. Future development is needed, and is currently underway, to enable extraction from multiple ports to extract multiple cell populations simultaneously. To fabricate the fluidic cartridges, Smooth Sil 910 silicon rubber compound (Smooth-On) was mixed at a 1:1 ratio of part A and part B and poured into a 3D printed mold for a fluidic chamber to fit over the ratcheting chips. This mold was allowed to cure overnight and then was cut to shape. To assemble the cartridge with the ratcheting chip, a clamping assembly was fabricated by 3D printing a bottom frame that held a 3 × 5 in. glass slide with threaded holes that interfaced with a machined polycarbonate top clamp with 10–32 threaded leur lock connectors. The ratcheting chips were aligned with the silicone fluid chamber and sandwiched between the two clamping frames to form a complete cartridge (Fig. 1C). In this way, ratcheting separations on the chip could be imaged since the cartridge had a transparent bottom. Cartridges were filled with a solution of 2% Pluronic F127 in deionized water for at least 45 min before use. Solutions and sample were added into the chip using the peristaltic pump or manually using a syringe.
Continuous-Injection Ratcheting Instrument
The ratcheting instrument (Fig. 2A) consisted of an integrated peristaltic pump to continuously draw sample through the cartridge and two stepper motor subsystems. These stepper motor subsystems were designed to (1) drive the magnetic wheel, which generates the directionally cycled magnetic field for ratcheting separation, and (2) control the ratcheting vector offset for simultaneous separation and concentration of target cells. The system framework was either custom fabricated or 3D printed in such a way that the instrument and chip were tilted 45° from horizontal (Fig. 2B) in order to prevent the flow of nontarget cells into the cartridge extraction port. The ratcheting offset, defined as the relative angle between the chip longitudinal axis and the direction of ratcheting (Suppl. Fig. S5), was set to 10°, which enabled optimal ratcheting separation and concentration to the extraction port.
Figure 2.
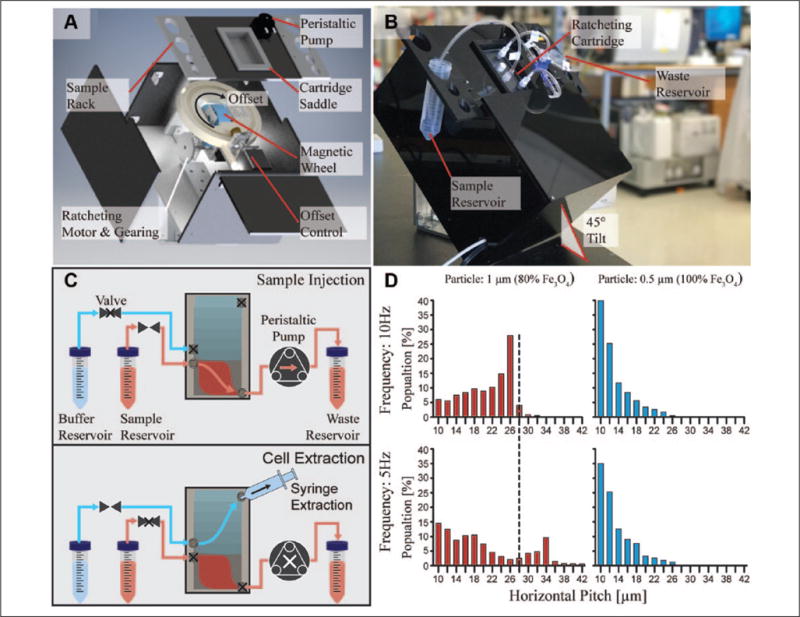
(A) An exploded view of the ratcheting cytometry instrument illustrates its subsystems, which were designed to generate the ratcheting magnetic field and pump magnetically tagged specimens through the cartridge via a peristaltic pump. The ratcheting subsystem consists of a stepper motor with gearing to drive the magnetic wheel, as well as a motorized framework to control the ratcheting angle offset relative to the cartridge. The framework that rests directly above the ratcheting subassembly includes the cartridge saddle, a sample rack to hold centrifuge tubes containing sample/buffer, and a peristaltic pump to draw flow. (B) A ratcheting cartridge placed in the cartridge saddle was then connected to sample/buffer reservoirs and the peristaltic pump to perform separation operations. (C) The ratcheting separation workflow can be broken down into sample injection and cell extraction steps. First, the valve to the sample reservoir is opened to enable sample flow through the cartridge provided by the peristaltic pump. Simultaneously to the sample flow, the ratcheting wheel is rotated to drive separation of target cells into the gradient pitch region. After sample injection, the pump is stopped and the valve to the fluid reservoir is closed. Cells are hydrodynamically extracted by attaching a syringe to the cartridge extraction port and opening the valve to the clean buffer reservoir. (D) Ratcheting distributions of T cells magnetically tagged with two different particle types functionalized with either αCD8 or αCD3 include a 1 μm 80% Fe3O4 particle (αCD8) and a 0.5 μm 100% Fe3O4 particle (αCD3) to equilibrate to different pitch ranges, depending on their particle magnetic content and driving frequency. T cells labeled with the 1 μm 80% Fe3O4 particle separate between the 10 and 32 μm pitches under a 10 Hz ratchet, with a peak at the 26 μm pitch. In contrast, cells tagged with the 0.5 μm 100% Fe3O4 particle trap between the 10 and 24 μm pitches, with a peak at the 10 μm pitch. When the frequency is lowered, the 1 μm particle shows a significant shift down the chip while the 0.5 μm particle remains relatively unaffected.
The magnetic wheel consisted of an array of four N52-grade rare-earth wedge-shaped neodymium ferrite magnets (KJ Magnetics, Pipersville, PA) with alternating magnetization directions relative to the wheel’s radial axis. This field arrangement, known as a Halbach array, generates the directionally cycling magnetic field needed to drive ratcheting separation (Suppl. Fig. S1). The wheel was driven at frequencies ranging between 5 and 10 Hz and controlled by a custom-designed Arduino software and Java interface, which also controlled the ratcheting offset subsystem, as well as the peristaltic pump, which was set to inject at a flow rate of 520 μL/min. In order to calibrate the working distance between the surface of the magnetic wheel and the ratcheting chip, iron filings were placed on a glass slide within the cartridge saddle while the wheel was cycling in order to visualize the field direction (Suppl. Video S2). Using plastic spacers, the working distance was increased until the field demonstrated continuous operation, which yielded an optimal working distance of 23 mm corresponding to a peak field strength of 200 mT. These empirical findings were validated by 3D numerical simulations of the magnetic wheel performed with the Comsol 4.2 magnetostatics module (Suppl. Fig. S6). The magnetic wheel geometry was imported into Comsol, and the field strengths and directions of each wedge magnet were set according to the manufacturer’s specification. From the simulations, the magnetic flux density vector was measured around the perimeter of the wheel at various working distances. This showed that working distances between 5 and 15 mm demonstrated abrupt changes in the magnetic field direction due to the near-field effects when too close to the Halbach array, which is not ideal for continuous ratcheting operation. Working distances between 20 and 25 mm demonstrated continuous field cycling, which is consistent with the field visualization using iron filings. To perform separations, the ratcheting cartridge was first placed in the cartridge saddle (Fig. 2A), which is positioned directly centerline to the magnetic wheel and connected via ¼ in. standard polyvinyl chloride (PVC) tubing in order to draw solutions from 50 mL centrifuge tubes secured in the sample rack (Fig. 2B).
Direct CD3(+) Cell Isolation from Nontarget Cell Background or Apheresis Sample
Ratcheting separations were first evaluated with controlled mixtures of CD3(+) Jurkat E6-1 cells (ATCC) spiked into a background of CD3(−) HL60 cells (ATCC) at a ratio of 1:10 CD3(+) to CD3(−) at concentrations ranging between 2 and 5 × 107 cells/mL, approximate to the working concentration of a leukopak. Before mixing, Jurkat cells were stained with Calcein AM (green) and the HL60 cells were stained with Calcein AM (red). Both were centrifuged and washed three times to prevent cross labeling. The mixtures were magnetically labeled with Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher, Waltham, MA) 4.5 μm magnetic particles at a 1:1 Jurkat cell-to-bead ratio at room temperature for 60 min while being rotationally stirred at 20 rpm. The samples were then flowed through the ratcheting cytometry instrument under a 10 Hz ratchet. Apheresis samples were obtained from the UCLA CFAR Virology Core by eluting cells from platelet donation Trima filters using 30 mL of phosphate-buffered saline (PBS). Using a hemocytometer and 10× lysis buffer (eBioscience), the red blood cell concentration was determined to be ~1 × 109 cells/mL and the total nucleated cell concentration was 2.8–5.1 × 107 cells/mL. The CD3(+) population (43%–67%) was analyzed on a BD FACSCanto II using a CD3–fluorescein isothiocyanate (FITC) antibody (clone BW264/56, Miltenyi, Bergisch Gladbach, Germany) and DRAQ5 (BD, Franklin Lakes, NJ). Using the T-cell count, a 1:1 ratio of Dynabeads Human T-Activator CD3/CD28 (Thermo Fisher) was added to 1 mL of the apheresis product and diluted to 1.5 mL using PBS. This mixture was then incubated at room temperature for 60 min with rotation at 20 rpm. Before injection into the cartridge, a small aliquot of the magnetically tagged sample was stained with Hoechst nuclear stain (Thermo Fisher), CD4-FITC (clone VIT4, Miltenyi), and CD8-FITC (clone BW135/80, Miltenyi) to determine the magnetic labeling efficiency of T cells. Due to the fact that the Dynabeads fluoresced at the 568 nm wavelength, the magnetic labeling efficiency was determined to range between 50% and 65% and was calculated by enumerating the number of CD4(+) CD8(+) cells that had beads bound on their surface.
Prior to sample injection, 2% Pluronic F127 solution was removed from the assembled cartridge, rinsed with deionized water, and prefilled with running buffer (40 mM Pluronic F127 in PBS) before use. Standard ¼ in. PVC tubing with leur lock connections was used to connect sample and buffer reservoirs to the cartridge and pump. Care was taken to mitigate bubble formation in the cartridge and interfaces. To initiate separation, ratcheting was initiated at either 5 or 10 Hz and the peristaltic pump was activated to draw solution at a flow rate of 520 μL/min. Once the sample finished injecting, an additional 5 mL of running buffer was flushed through the cartridge. After the flush was completed, ratcheting was continued for 3 min for a total of 15 min of run time to concentrate CD3(+) cells to the extraction port. Once concentrated, the cartridge was then manually removed from the saddle to prevent the magnetic field from holding cells on the chip. The valve to the sample reservoir was closed and the buffer reservoir valve was opened. A syringe was connected to the upper-right-side luer lock (extraction port) and withdrawn to remove the target cells from the chip (Fig. 2C, Suppl. Fig. S4). Due to the high cell concentration of the extracted cells, an aliquot of cells was diluted 1000× and labeled with Hoechst nuclear stain (Thermo Fisher), CD4-FITC (clone VIT4, Miltenyi), and CD8-FITC (clone BW135/80, Miltenyi), and then plated into a 48-well plate for fluorescent imaging. Purity was determined by measuring the ratio of CD4(+) and CD8(+) cells to the total nucleated cell count. The device capture efficiency was determined by enumerating the number of target cells extracted and dividing by the total number of magnetically tagged target cells. All imaging was performed using a Nikon Eclipse Ti fluorescent microscope and processed with ImageJ. The entire chip was imaged using image stitching functionality under 4× magnification.
Multitarget Magnetic Separations from PBMCs
Particle functionalization with biotinylated antibody was carried out on several particle sizes, including 0.5 μm diameter (Micromod, Rostock, Germany), 1.0 μm diameter (Thermo Fisher), or 1.0 μm diameter (Chemicell, Berlin, Germany) streptavidin functionalized magnetic particles. This was done by adding approximately 1 × 108 of a particular particle diameter to 1 mL of particle functionalization buffer (0.5% w/v bovine serum albumin [BSA] and 2 mM EDTA in PBS) and 10 μL of biotinylated antibody, such as CD3 (clone REA613, Miltenyi), CD4 (clone M-T466, Miltenyi), or CD8 (clone REA734, Miltenyi), followed by an incubation at room temperature for 30 min under a 20 rpm rotation. Beads were pulled down using a magnet, washed with functionalization buffer twice, and stored at 2°C until ready for use. Of note is that the bead counts were based on the manufacturer’s specification, which prescribed a bead concentration per batch.
PBMCs obtained from the UCLA CFAR Virology Core were thawed using RPMI complete media (10% fetal bovine serum [FBS] and 1% pen-strep) and counted using a hemocytometer. Functionalized beads were added to 1 × 106 cells at a 10:1 bead–total cell ratio and incubated at room temperature for 60 min with 20 rpm rotation. For the CD4 and CD8 experiments, cells were then labeled with Calcein AM (Thermo Fisher) for 5 min before running. For CD3 and CD8 experiments, cells were labeled with Calcein Red-Orange AM (Thermo Fisher) for 5 min at room temperature before running. Once magnetically labeled, samples were injected through the cartridge in the same manner as previously described, without the cell extraction steps, and yielded a capture efficiency of 60%–75% for each cell type. After 30 min of total ratcheting time, the cartridge was removed from the system and imaged on a Nikon Eclipse Ti fluorescent microscope. The resulting images of the chip were processed using ImageJ.
Results and Discussion
Ratcheting cytometry represents a novel approach to cell purification that balances sorting attributes similar to those of flow cytometry with the scalability and robustness of MACS separations. To adapt the technology toward cell therapy manufacture, four design attributes were considered during prototype development: (1) closed-system operation, (2) continuous-flow functionality, (3) quantitative cell sorting, and (4) quick/high-throughput processing. The capacity to perform separations within a closed system is essential for aseptic purification to meet cGMP compliance. To demonstrate feasibility, a fluidically enclosed ratcheting cartridge was developed with standard leur lock fluidic connections (Fig. 1C) to interface with sample tubing and pumping functionality. Second, continuous-flow operation was also considered a driving parameter to build toward a system capable of processing highly concentrated clinical apheresis samples or leukopaks. Generally, leukopak volumes range between 200 and 300 mL, with nucleated cell counts on the order of 1010 cells. As such, the ratcheting cytometry instrument was constructed with an integrated peristaltic pump to continuously process samples through the cartridge while simultaneously rotating the magnetic wheel to drive separation. In this way, large sample volumes and cell counts can be purified efficiently, without the need for batch processing or a centrifugation step. Furthermore, ratcheting cytometry’s quantitative sorting ability was a valuable feature for parallel separations of T-cell subpopulations needed to derive precision therapies. And finally, rapid sample processing was a driving parameter to meet throughputs acceptable for cell therapy manufacturing workflows. In the following sections, the ratcheting cytometry platform and its performance are discussed in light of these design parameters to demonstrate feasibility as a quantitative enrichment technology for cell therapy manufacturing.
Toward an Automated Benchtop Instrument for Continuous-Flow Ratcheting Cytometry
Considering the closed-system and continuous-flow operation parameters, the instrument was designed to drive separation by spinning a wheel of rare-earth magnets while simultaneously flowing samples via a peristaltic pump through the ratcheting cartridge. Figure 2A shows the instrument schematic consisting of a ratcheting subsystem and a peristaltic pump, both of which are centrally controlled by an Adruino Uno Rev 3 and custom-printed circuit board (not shown). This offset control was used to slightly bias the ratcheting field vector relative to the cartridge, which induces cell concentration to the edges of the cartridge for cell extraction, thereby conveying the ability to continuously separate and extract target cells off chip (Suppl. Fig. S4). It was determined that an offset angle between 10° and 15° was sufficient to bias cell transport for extraction. During sample injection, the peristaltic pump continually drew sample through the cartridge injection chamber, where magnetically tagged target cells were pulled onto the chip surface and separated to the gradient pitch zones while the nontarget cells flowed out of the injection chamber (Suppl. Fig. S4C,D). An important parameter considered was the distance between the magnetic wheel and the cartridge saddle, as this determined the shape and strength of the magnetic field that the ratcheting cartridge experiences. There is an inherent trade-off between the optimal shape of the ratcheting field and the strength to pull immunomagnetically tagged cells out of the flowing solution and onto the cartridge micropillar arrays. If the cartridge is too close, the magnetic field shape can be inconsistent due to the near-field effects of the radial Halbach array. If it is too far, the field strength will be insufficient to pull magnetically tagged cells out of the flowing sample onto the chip. This working distance was calibrated by placing iron filings on a glass slide within the cartridge saddle and offsetting the saddle with plastic spacers until a uniform and consistent directionally cycling field was visualized by the iron filings (Suppl. Video S2). The optimal distance to balance the optimal field shape with high magnetic strength was determined to be 23 mm between the cartridge saddle and the magnetic wheel surface, corresponding to a peak field strength of 200 mT. In addition to visualizing the field with iron filings, 3D numerical simulations of the magnetic wheel were performed to validate the optimal working distance. Modeling of the wheel’s magnetic field and measurement of the magnetic field vectors at varying working distances around the wheel perimeter showed that working distances between 5 and 15 mm had abrupt changes in the magnetic field direction due to the near-field effects, which were not ideal for continuous ratcheting operation. However, working distances between 20 and 25 mm demonstrated continuous field cycling, which is consistent with the field visualization using iron filings (Suppl. Fig. S6). Sample injection and ratcheting separation was the first step in the platform operational workflow (Fig. 2C), where the peristaltic pump draws specimens of magnetically tagged target cells through the ratcheting cartridge. During this step, target cells were quantitatively sorted into the gradient pitch region and concentrated to the upper right edge of the chip. Because ratcheting cytometry is an equilibrium-based separation, where cells trap and remain at their critical pitch, the entire sample was processed before extraction, without the need for batch processing. After sample injection, the pump was halted and the sample reservoir tubing line was blocked via a manual valve. Target cells were then extracted by opening the valve to the buffer reservoir, connecting a sterile syringe to the cartridge extraction port, and physically withdrawing the syringe. Due to the hydrodynamic forces, the concentrated target cells were removed from the chip and captured in the syringe for characterization and downstream use.
In addition to closed and continuous-flow capability, the platform also demonstrated quantitative cell separations in a fashion similar to flow cytometry. In flow assays, fluorophores with differing excitation and emission are used to quantify biomarkers in combination with a photomultiplier tube (PMT) voltage that can be tuned to achieve an optimal signal-to-noise ratio. In a similar fashion, the ratcheting cytometer utilized immunomagnetic particles with differing magnetic contents to multiplex on cell surface markers. Figure 2D demonstrates the difference between T cells that have been immunomagnetically labeled with CD8 on a 1 μm 80% Fe3O4 particle (2.05 pg maghemite per particle) and CD3 on a 0.5 μm 100% Fe3O4 particle (0.32 pg maghemite per particle) subjected to a 10 Hz ratcheting field. As illustrated, the cells tagged with the 1 μm 80% Fe3O4 particle separate down the ratcheting chip, with a peak at the 26 μm pitch. However, the cells tagged with the 0.5 μm 100% Fe3O4 particle equilibrate at the lower pitches, with a peak at the 10 μm pitch due to the fact that the magnetic content per particle is 6.4 times lower. Along these lines, the system demonstrates that particle magnetic content in ratcheting cytometry draws a parallel with fluorescent emission spectra in flow cytometry for quantitative separation. Additionally, the ratcheting frequency can be tuned to modify the separation distribution similarly to PMT voltage tuning in a flow cytometer. The 1 μm 80% Fe3O4 cell distribution profile demonstrated a significant expansion down the chip as the frequency was lowered from 10 to 5 Hz (Fig. 2D) and appears to show the emergence of two populations. In contrast, the 0.5 μm 100% Fe3O4 particle shows little difference, suggesting that there is an optimal particle magnetic content to achieve high-dynamic-range separations, enabling cell distribution to spread to high pillar pitch ranges. However, this frequency independence for the 0.5 μm particle could be advantageous for system calibration or when trying to increase the separation between cell populations on chip.
Continuous Positive Enrichment of CD3(+) Cells Directly from Complex Samples
After the system’s capacity for closed, continuous, and quantitative cell purification was demonstrated, the separation of target cells from complex biomatrices was evaluated to demonstrate relevance in a cell therapy manufacturing setting. As a first step, controlled mixtures of HL60 cells (CD3[–]) and Jurkat E6-1 cells (CD3[+]) were utilized as a surrogate for a clinical sample to evaluate system performance. HL60 cells, stained with Calcein Red-Orange AM (red), acted as a background to spiked CD3(+) Jurkat cells, stained with Calcein AM (green), and were mixed at cell concentrations on the order of 107 cells/mL, similar to a leukopak. The mixtures were labeled with αCD3/CD28 4.5 μm magnetic particles at a 1:1 target cell–bead ratio and flowed through the ratcheting cytometry instrument under a 10 Hz ratchet. As expected, the target cells traversed the ratcheting cartridge and were successfully separated from the HL60 background (Fig. 3A). Quantification of the target and non-target distributions shows substantial physical separation on the cartridge (Fig. 3B), where 90.5% of the target cells equilibrated between the 24 and 36 μm pitch with a 95% purity of target cells. After evaluating successful separation from a cell line–based surrogate, the ratcheting system was challenged by purifying T cells directly from leukapheresis specimens. In the same manner as used for the HL60/Jurkat surrogates, samples from leukapheresis specimens were labeled with αCD3/CD28 4.5 μm magnetic particles at a 1:1 T-cell–particle ratio, which was determined by flow cytometry. Sample volumes containing approximately 5.5 × 107 nucleated cells (~1 mL of leukopak specimen) were sampled through the system for a total processing time of 15 min and corresponding to a total cell throughput of 6.5 × 104 cells/s. While this throughput is low compared with the clinical-grade MACS platform, which is on the order of 106 cells/s, we believe it represents an unoptimized, lower bound for the system throughput for several reasons. First, cell transport across the micropillar arrays can be amplified by increasing driving frequency and/or increasing the pillar pitch up until the critical pitch value. In our previous work,18,19 we demonstrated that the cell speed is equal to the product of the pillar pitch and driving frequency; therefore, a cell ratcheting at 10 Hz across a 7 μm pitch array will transport at 70 μm/s. As shown in Figure 3A, the target cells do not begin to trap until the 24 μm pitch under a 10 Hz ratchet and are able to traverse a 22 μm pitch. If the loading zone and previous pitch values were increased to 22 μm, the cells would traverse at 220 μm/s, which yields approximately a threefold increase in throughput. Second, the chip cell capacity scales with area, whereby simply scaling the chip size can substantially increase throughput. Finally, the sample injection flow rate of 520 μL/min was a conservative value to ensure that the magnetically labeled cells had sufficient time to be magnetically pulled onto the ratcheting chip. The optimization of flow rate and cartridge geometry is a tenable method to increase the volumetric flow rate through the cartridge. Nevertheless, the demonstrated throughput shows substantial improvement compared with standard flow cytometry, as well as the feasibility to be scaled to manufacturing-scale throughputs.
Figure 3.
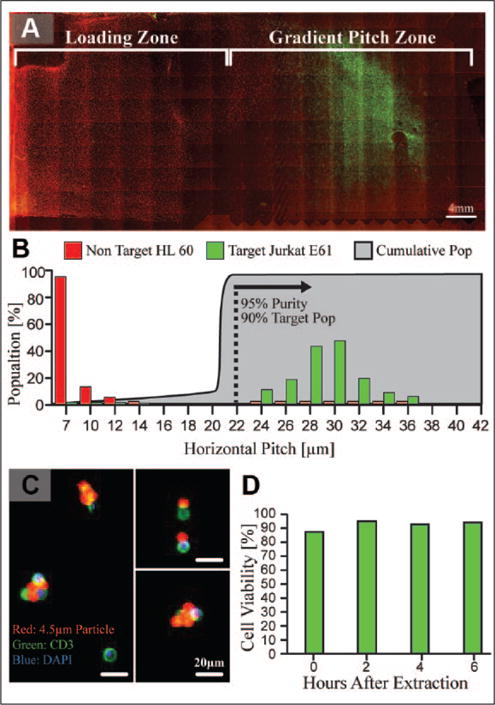
(A) Ratcheting cytometry separations performed with mixtures of CD3(−) HL60 cells (red) and CD3(+) Jurkat E6-1 cells (green) to show successful separation and concentration on chip by tagging green target cells with a 4.5 μm αCD3 magnetic particle. (B) Quantification of the separations shows a 90% recovery of target cells with a 95% purity of target cells between the 22 and 42 μm pitches under a 10 Hz ratchet. (C) Ratcheting separations performed with leukopak samples demonstrated a 97 ± 2% purity, as determined by staining extracted cells with CD3 (green) and nuclear stain (blue). Shown are snapshots of extracted cells that were diluted 1000× in order to precisely quantify the capture efficiency and purity due to the high cell and particle concentration. Magnetic particles, which fluoresced in the TRITC wavelength, were attached to CD3(+) target cells. (D) After extraction from the chip cell, viability was monitored at 2 h time points for 6 h, demonstrating a viability between 90% and 95% under standard culture conditions.
After injection and separation, T cells were extracted from the ratcheting cartridge and visualized to quantify purity and efficiency. Due to the high concentration of the extracted cells, the extracted fraction was diluted 1000×, fluorescently imaged, and counted to enumerate purity and recovery. The extracted populations exhibited a purity of 97% ± 2% and 85% recovery of magnetically tagged cells (Fig. 3C). The purity, defined as the ratio of target cells to the number of total nucleated cells, shows that the ratcheting platform has the capacity to highly enrich target cell populations, which will be critical in clean and efficient T-cell expansion, as well as in deriving potent cell therapies. It is noted that the capture efficiency is based on the extraction of magnetically tagged cells, which is normalized with the magnetic labeling efficiency that ranges between 50% and 65%. The magnetic labeling efficiency is heavily reliant on the quality of the manufacturer’s antibody, as well as bead size and concentration, which should be further optimized to operate consistently in leukopak or apheresis samples to increase the labeling efficiency of target cells. However, for the target cells that were magnetically tagged, the ratcheting system demonstrated efficient extraction. After extraction, cell viability was evaluated every 2 h for a period of 6 h after extraction and maintained a viability above 90% (Fig. 3D). Importantly, this shows that the ratcheting enrichment process does not substantially affect the cell viability, which is critical to delivering usable T cells for therapy derivation.
Quantitative Magnetic Separation of T-Cell Subpopulations by Multiplexed Magnetic Strength
In addition to processing cells based on a single marker from a complex biomatrix, the ratcheting system showed the capacity for multiplexed magnetic purification for enrichment of surface marker–specific T-cell subpopulations. As a first demonstration, CD4(+) and CD8(+) surface markers were targeted using two different 1 μm particles with varying magnetic contents. αCD4 was functionalized to a 1 μm 26% Fe3O4 particle, while αCD8 was functionalized to a 1 μm 80% Fe3O4 particle, incubated with a PBMC population, and injected through the chip. These particles were chosen due to the fact that they have a similar size but varying iron oxide contents, which will yield separation of the CD4 and CD8 subpopulations to different points on the chip. The binding efficiency of a particle to a target cell is related to the particle’s diffusion, which is a function of diameter. This creates an inherent trade-off for magnetic labeling where a larger particle has higher magnetic content but lower diffusion, and therefore a lower binding efficiency. By using two different particle types of the same diameter, we could directly compare the separation behavior of the two populations without biasing binding efficiency based on particle size. As Figure 4A shows, the PBMCs labeled with the CD8 functionalized particles equilibrated at higher pillar pitches than the CD4-labeled cells due to the sixfold difference in the magnetic content between the two particle types. In this instance, the maximum system frequency of 10 Hz was used to minimize the overlap between the two populations due to the fact that high ratcheting frequencies tend to reduce the variance of the population distributions on the chip.19 This resulted in statistically significant partitioning between the CD4 and CD8 subpopulations (p = 0.03) with some observed distribution overlap. A vast majority (98%) of the CD4(+) population equilibrated early in the pitch gradient zone (10–16 μm pitches) under a 10 Hz ratchet, while only about 11% of the CD8(+) population occupied the same pitches. Setting 16 μm as a gate to divide the two populations yields purities of 79% and 98% for the CD4 and CD8, respectively. This overlap is likely related to the binding efficiency of magnetic particles to the CD8(+) population, whereas minimally labeled CD8(+) cells will equilibrate lower on the chip. Based on the predictive model developed in our previous work, this means that the CD4 population had between 5 and 16 pg of cell-bound Fe3O4, whereas >98% of the CD8 population had between 16 and 124 pg of cell-bound Fe3O4. Further separation between these two populations could be achieved by further increasing the Fe3O4 content. For example, if the Fe3O4 content of the CD8 particles was increased from 80% to 90%, it would shift the CD8 distribution two additional pitch zones, or approximately 4 mm on the current chip design. Maximizing separation may also be addressed by optimizing the labeling conditions for the particle cocktails to ensure that each target cell is sufficiently tagged. Another approach to widen the separation between the two populations would be to use a stronger magnetic particle. As demonstrated previously, cells tagged with large 4.5 μm particles equilibrated between the 24 and 36 μm pitches, which could mitigate overlap between the two populations. Despite the minor overlap, the system demonstrated that CD4 and CD8 T-cell subpopulations can be positively selected and quantitatively subdivided in a single-step assay. This can be a transformative attribute for cell manufacturing, enabling multiple T-cell subtypes to be isolated in a single run, which is currently done with multiple MACS steps or with FACS at low throughput. Having a multiplexed sorter able to purify target T-cell subpopulations in this way could substantially increase production throughput by eliminating the need to perform multiple separations in series. Furthermore, having the ability to separate based on the level of a surface marker can further streamline the purification process. For example, both monocytes and certain T cells constitutively express CD4, but at different expression levels, where CD4(+) T cells have much higher expression than CD4(+) monocytes.21 Performing MACS without a centrifugation or density gradient step would yield a collection of monocytes, with the CD4(+) T-cell fraction resulting in a low-purity separation. However, if the low-expressing monocyte fraction could be discriminated from the highly expressing CD4 T cells, then the purification could be done directly out of the complex matrix, thereby decreasing another timely process step.
Figure 4.

(A) Ratcheting separations to enrich CD4(+) and CD8(+) T cells from a PBMC population were performed by multiplexing targets on 1 μm particles with different magnetic contents, specifically an 80% maghemite particle functionalized with αCD8 and a 26% maghemite particle functionalized with αCD4. Ratcheting separation under a 10 Hz ratchet showed statistically significant partitioning between the separately labeled populations (p = 0.03) where the CD4(+) fraction trapped between the 10 and 16 μm pitches and a majority of the CD8(+) fraction trapped between the 16 and 30 μm pitches. Setting 16 μm as a gating pitch, the 10–16 μm pitch range consisted of CD4(+) cells with a 79% purity, while the 18–30 μm pitch range housed the CD8(+) cells with a 98% purity. (B) Additionally, multiplexed ratcheting separations were also performed against CD3 and CD8 markers with a cocktail of particle sizes. A 0.5 μm 100% Fe3O4 particle type was functionalized to target CD3, and a 1 μm 26% Fe3O4 particle (red) was used to target CD8. Ratcheting distributions under a 5 Hz ratchet demonstrate the emergence of two cell populations, a CD3(+) CD8(−) population and a CD3(+) CD8(+) population, which equilibrated to different locations on the chip.
In addition to sorting two differentially expressing sub-populations, multisurface target magnetic labeling was also demonstrated using the ratcheting cytometry system. In this application, αCD3 was functionalized to a small 0.5 μm magnetic bead and αCD8 was functionalized to a 1 μm 26% Fe3O4 bead. As opposed to the previous case, we chose two different bead sizes in an effort to mitigate the occlusion of bead bound surface markers since both particle types would be binding to a target cell surface. We reasoned that CD3 would be highly expressed on a majority of the population and could occlude the CD8 bead from binding, and therefore used a smaller-diameter bead to ensure the larger CD8 bead had sufficient room to bind to the cell surface. In the same manner as previously described, the bead cocktail was incubated with a PBMC population and injected through the chip, but here it was subjected to a 5 Hz ratchet. In this case, we used a lower ratcheting frequency to maximize the spread of the cell across the chip, as we expected a more continuous cell distribution with cells that could contain combinations of 0.5 and 1 μm sizes. The cell distribution across the chip consisted of two populations where cells labeled with only 0.5 μm particles equilibrated between the 10–24 μm pitches and cells labeled with both 0.5 and 1 μm particles (shown in red) equilibrated between the 20 and 34 μm pitches (Fig. 4B). This suggests that multiplexing on bead size and/or magnetic strength can be used to target multiple surface markers simultaneously and adds another degree of freedom for quantitative sorting. However, the relationship between particle size and binding efficiency to multiple surface targets on the same cell needs to be further optimized to mitigate occlusion bias. In addition to reagent development, optimizing continuous extraction of multiple populations off the chip must be pursued. While we have demonstrated single-subpopulation extraction with CD3(+) cells, the isolation of multiple sub-populations will require further cartridge engineering. This can be accomplished by engineering multiple extraction ports and fluid cartridge geometry to continuously syphon separated cells from multiple locations on the chip. In this case, the fluid flow must be controlled carefully to minimize cross-flow between the extraction outlets and is the focus of current development. This can easily be automated using syringe pumps connected to different extraction ports and would still enable closed-system operation. We believe that the ratcheting cytometry’s ability for quantitative magnetic sorting of T-cell subpopulations in this capacity shows valuable potential for cell therapy manufacturers, giving them the capacity to obtain and control specific cell populations in a format that is scalable to meet manufacturing throughput.
Conclusions
The ratcheting cytometry platform is a continuous-flow, quantitative cell purification system that shows valuable potential as a manufacturing technology for T-cell immunotherapies. Adopting a closed-cartridge and automated instrument-based approach, the system showed competency in sorting complex leukapheresis samples, as well as multi-component and multiparameter sorting to enrich specific T-cell subpopulations. In order to bring this technology to a manufacturing scale, the system throughput must be increased, which, as discussed, can be done by modifying the micropillar array geometry and scaling the cartridge size for operational capacity on the order of 106 cells/s. Additionally, more work is needed to optimize cartridge geometry for continuous cell extraction from the chip. In this study, the extraction was accomplished by hand, but it can be easily automated by the addition of more pumps and more cell extraction ports to syphon target subpopulations that separate to different cartridge localities. For example, syringe pumps and single-use disposable syringes can be used to syphon target cells from different critical pitch zones in parallel. Overall, we believe this work established ratcheting cytometry as a viable candidate to accelerate the development of precision cell therapies and shows proof of concept for a tool that can balance quantitative sorting with manufacturing-scale throughput to address scale-up/scale-out bottlenecks.
Supplementary Material
Acknowledgments
The authors thank the Integrated Systems Nanofabrication Cleanroom at the California Nanosystems Institute for aid in chip fabrication. We would also like to thank the UCLA CFAR Virology Core for providing apheresis samples.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by NIH, PO no. 401038186 through a subcontract from General Electric Corporation.
Abbreviations
- CAR
chimeric antigen receptor
- TCR
T-cell receptor
- MACS
magnetic-assisted cell separation
- FACS
fluorescence-assisted cell separation
- PBMC
peripheral blood mononuclear cell
Footnotes
Supplementary material is available online with this article.
Declaration of Conflicting Interests
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Coleman Murray, Edward Pao, and Dino Di Carlo hold equity in Ferrologix, Inc., which has licensed technology described in this article.
References
- 1.National Institutes of Health, National Cancer Institute. CAR T-Cell Therapy: Engineering Patients’ Immune Cells to Treat Their Cancers. http://www.cancer.gov/about-cancer/treatment/research/car-t-cells (accessed Aug 1, 2017)
- 2.Jackson HJ, Rafiq S, Brentjens RJ. Driving CAR T-Cells Forward. Nat Rev Clin Oncol. 2016;13:370–383. doi: 10.1038/nrclinonc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Andrade Pereira B, Fraefel C. Novel Immunotherapeutic Approaches in Targeting Dendritic Cells with Virus Vectors. Discov Med. 2015;109:111–119. [PubMed] [Google Scholar]
- 4.Geldres C, Savoldo B, Dotti G. Chimeric Antigen Receptors for Cancer Immunotherapy. Methods Mol Biol. 2016;1393:75–86. doi: 10.1007/978-1-4939-3338-9_7. [DOI] [PubMed] [Google Scholar]
- 5.Frigault MJ, Maus MV. Chimeric Antigen Receptor-Modified T Cells Strike Back. Int Immunol. 2016;7:355–363. doi: 10.1093/intimm/dxw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brower V. The CAR T-Cell Race. The Scientist. 2015 Apr 1; http://www.the-scientist.com/?articles.view/articleNo/42462/title/The-CAR-T-Cell-Race/ (accessed March 21, 2016)
- 7.Urquhart L, Gardner J. Pharma & Biotech 2015 in Review. Evaluate Ltd.: London; 2016. (Evaluate Pharma Market Report). http://info.evaluategroup.com/rs/607-YGS-364/images/epv-pbr15.pdf (accessed Aug 29, 2017) [Google Scholar]
- 8.National Institutes of Health, National Cancer Institute. CAR T Cells: Expanding into Multiple Myeloma. National Institutes of Health: Bethesda, MD; 2017. https://www.cancer.gov/news-events/cancer-currents-blog/2017/car-t-cell-multiple-myeloma (accessed Aug 29, 2017) [Google Scholar]
- 9.Lakelin M. Maintaining an Efficient and Safe Cell-Therapy Supply Chain during Scale-Up and Scale-Out. BioPharm International. 2014 Nov 3; http://www.biopharminternational.com/maintaining-efficient-and-safe-cell-therapy-supply-chain-during-scale-and-scale-out (accessed Aug 15, 2017)
- 10.CNBC. Gilead to Buy Kite Pharma in $11.9 Billion Deal. 2017 https://www.cnbc.com/2017/08/28/gilead-to-buy-kite-pharma-for-about-11-billion-in-cash-dow-jones.html (accessed Aug 29, 2017)
- 11.Dodson B, Levine A. Challenges in the Translation and Commercialization of Cell Therapies. BMC Biotechnol. 2015;7:15–70. doi: 10.1186/s12896-015-0190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hourd P, Chandra A, Medcalf N, et al. Regulatory Challenges for the Manufacture and Scale-Out of Autologous Cell Therapies. 2014 https://www.ncbi.nlm.nih.gov/books/NBK201975/ [PubMed]
- 13.Sommermeyer D, Hudecek M, Kosasih PL, et al. Chimeric Antigen Receptor-Modified T Cells Derived from Defined CD8+ and CD4+ Subsets Confer Superior Antitumor Reactivity In Vivo. Leukemia. 2016;2:492–500. doi: 10.1038/leu.2015.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Koehl U, Esser R, Zimmermann S, et al. Ex Vivo Expansion of Highly Purified NK Cells for Immunotherapy after Haploidentical Stem Cell Transplantation in Children. Klin Padiatr. 2005;6:345–350. doi: 10.1055/s-2005-872520. [DOI] [PubMed] [Google Scholar]
- 15.Spohn G, Wiercinska E, Karpova D, et al. Automated CD34+ Cell Isolation of Peripheral Blood Stem Cell Apheresis Product. Cytotherapy. 2015;10:1465–1471. doi: 10.1016/j.jcyt.2015.04.005. [DOI] [PubMed] [Google Scholar]
- 16.Kumar A, Yu I, Mattiasson G. Cell Separation: Fundamentals, Analytical and Preparative Methods. Adv Biochem Eng Biotechnol. 2007;106:43. [Google Scholar]
- 17.Adamsa J, Kimb U, Soh HT. Multitarget Magnetic Activated Cell Sorter. Proc Natl Acad Sci USA. 2008;47:18165–18170. doi: 10.1073/pnas.0809795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Murray C. Micromagnetic Ratcheting Manipulation for Bioengineering Research Dissertation. University of California; Los Angeles: 2015. ProQuest Dissertations Publishing, 3724141. [Google Scholar]
- 19.Murray C, Pao E, Tseng P, et al. Quantitative Magnetic Separation of Particles and Cells Using Gradient Magnetic Ratcheting. Small. 2016;14:1891–1899. doi: 10.1002/smll.201502120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tay A, Murray C, Di Carlo D. Phenotypic Selection of Magnetospirillum magneticum (AMB-1) Overproducers Using Magnetic Ratcheting. Adv Funct Mater. 2017:1703106. [Google Scholar]
- 21.Lee B, Sharron M, Montaner L, et al. Quantification of CD4, CCR5, and CXCR4 Levels on Lymphocyte Subsets, Dendritic Cells, and Differentially Conditioned Monocyte-Derived Macrophages. Proc Natl Acad Sci USA. 1999;96:5215–5220. doi: 10.1073/pnas.96.9.5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.