

Abstract
The cellular sensor stimulator of interferon genes (STING) initiates type I interferon (IFN) and cytokine production following association with cyclic dinucleotides (CDNs) generated from intracellular bacteria or via a cellular synthase, cGAS, after binding microbial orself-DNA. Although essential for protecting the host against infection, unscheduled STING signaling is now known to be responsible for a variety of auto- inflammatory disorders. Here, we report a gain-of-function mutation in STING (R284S), isolated from a patient who did not require CDNs to augment activity and who manifested a constitutively active phenotype. Control of the Unc-51-like autophagy activating kinase 1 (ULK1) pathway, which has previously been shown to influence STING function, was potently able to suppress STING (R284S) activity to alleviate cytokine production. Our findings add to the growing list of inflammatory syndromes associated with spontaneous STING signaling and provide a therapeutic strategy for the treatment of STING-induced inflammatory disease.
Graphical Abstract
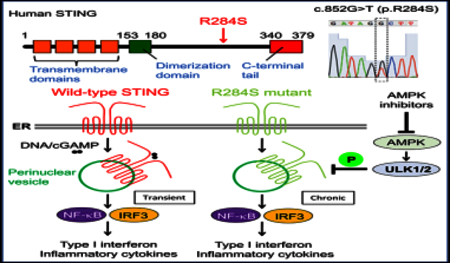
INTRODUCTION
Autoinflammatory diseases such as systemic lupus erythematosus (SLE) and arthritis affect millions worldwide, and although the exact mechanisms remain to be clarified, it is thought that deregulation of innate immune signaling likely plays a key role in manifesting these disorders. (Crow and Manel, 2015; Mohan and Putterman, 2015) Innate immune sensors such as STING, RIG-I/MDA5, or Toll-like receptors have evolved to detect microbial infection of the cell, predominantly through recognition of pathogen-derived nucleic acids, an event that triggers the transcription of numerous host defense-related proteins, including pro-inflammatory cytokines (Barber, 2015; Pandey et al., 2014). Although essential for host defense against pathogens, the activation of innate immune signaling is tightly controlled to avoid chronic pro-inflammatory disease, which can be fatal. (Barber, 2015).
The cellular sensor stimulator of interferon genes (STING) resides in the endoplasmic reticulum (ER) and is activated by cyclic dinucleotides (CDNs) such as cyclic di-guanosine monophosphate (GMP) and cyclic di-AMP secreted by intracellular bacteria following infection. (Burdette et al., 2011) Alternatively, STING can be activated by cyclic GMP-AMP (cGAMP) generated by a cellular cGAMP synthase, cGAS (MB21D1), after association with aberrant cytosolic double-stranded DNA (dsDNA) species, which can include microbial DNA, self-DNA leaked from the nucleus, or even mtDNA (Sun et al., 2013; Wu et al., 2013). Association with CDNs induces a conformational change and enables STING to traffic through the Golgi apparatus, with TANK-binding kinase 1 (TBK1) to activate the transcription factors IRF3 and nuclear factor κB (NF-κB), which stimulate the production of type interferon (IFN) and pro-inflammatory cytokines, which facilitate adaptive immunity (Ishikawa and Barber, 2008; Ishikawa et al., 2009).However, chronic activation of STING signaling has been shown to cause lethal inflammatory disease. For example, a loss-of-function mutation in the DNase TREX1 (DNaseIII) has been found to cause severe forms of SLE referred to as Aicardi-Goutieres syndrome (AGS) in human patients (Crow and Manel, 2015) The mechanistic cause was shown to be due to the accumulation of undigested self-DNA in the cytoplasm, which triggers STING signaling (Gall et al., 2012). Similarly, loss of DNaseII causes lethal pre-natal inflammatory disease in mice via STING signaling in macrophages by the undigested DNA of engulfed apoptotic cells (Ahn et al., 2012). Inflammatory disease caused by STING overactivity has also been shown to occur in patients with mutations in the ADAR and ribonuclease H2 enzyme complex. (Crow et al., 2015; Mackenzie et al., 2016; Melki et al.,2017; Pokatayev et al., 2016; Rice et al., 2013).
In addition, evidence indicates that inflammatory disease can be caused by germline missense mutations in the coding region of the STING gene itself (V147L/M, N154S, V155M, C206Y, R281Q, R284G, and S102P/F279L), which exert a gain-of-func-tion phenotype referred to as STING-associated vasculopathy with onset in infancy (SAVI) (Clarke et al., 2016; Fre´mond et al., 2016; Jeremiah et al., 2014; Liu et al., 2014; Melki et al., 2017;Picard et al., 2016; Seo et al., 2017). This disease causes skin lesions, rashes, and interstitial lung disease in children. Mutations in STING (G166E) have also been shown to be implicated in familial chilblain lupus (Ko¨ nig et al., 2017) However, we report here the identification of a germline missense mutation in exon 7 of STING (R284S) that was derived from a patient in Ecuador who died at approximately 9 months of age. Subsequent examination indicated that STING (R284S) was a potent constitutively active gain-of-function mutant that could traffic through the Golgi apparatus and trigger innate immune signaling in the absence of activating CDNs. JAK inhibitors such as tofacitinib failed to significantly repress STING (R284S) function, but use of an AMP-activated protein kinase (AMPK)/AKT inhibitor, GSK 690693, which affects Unc-51-like autophagy activating kinase 1 (ULK1) activity, robustly inhibited STING-dependent IRF3 activation. Our report underscores the growing incidence of deregulated STING signaling in manifesting acute cytokine-related disease and indicates that suppression of STING signaling activation may be useful for the treatment of a wide range of autoinflammatory disorders.
RESULTS
STING R284S Is Constitutively Active
A 9-month-old boy with fever (101.6°F/38.6°C) and a severe neck abscess was admitted to hospital and noted to have an interleukin-6 (IL-6) concentration (34 mg/dL) approximately five times above normal. However, subsequent studies showed that there was no indication of any severe bacterial infection. Neither were there any of the rashes on the cheeks, nose, or fingers that are typically observed in SAVI patients. The patient died shortly after admission with a diagnosis of pneumonia. Whole-exome sequencing was carried out, and after filtering against allele frequencies from available databases and comparison against the parents’ genome, a de novo heterozygous mutation was noted in the STING gene, c.852G/T (p.R284S), which was confirmed by Sanger sequencing. (Figure 1A) The parents did not present this variant, which occurred in a highly conserved region of STING (Figures 1B and S1). To determine whether the amino acid substitution (R284S) affected STING function, we cloned the cDNA and transiently co-transfected HEK293T cells with the variant and a luciferase gene under control of the type IIFN promoter. In this assay, expression of wild-type STING activates the type I IFN promoter, by stimulating IRF3 and NF-κB signaling, to generate luciferase. For comparison, we also transfected a known STING (N154S) variant that was reported to be hyperactive and known to cause SAVI. This analysis indicated that the STING (R284S) mutant was potently able to trigger activation of the type I IFN promoter, almost 10-fold higher than wild-type STING and almost twice as much as the SAVI variant, according to this assay (Figure 1C). This data was confirmed by demonstrating that STING (R284S) could also trigger luciferase transcription under control of the pRD III-I promoter (specifically activated by IRF3), the NF-κB promoter, or an interferon-stimulated response element (ISRE) promoter(Ishikawa and Barber, 2008; Figure 1C). We also examined whether STING (R284S) could be additionally activated in the presence of transfected human cGAS plasmid DNA (hcGAS), which generates CDNs (cGAMP) to further augment wild-type STING activity(Figure 1C). However, we did not observe that cGAS could further stimulate STING (R284S) activity significantly in this assay, in contrast to wild-type STING or the SAVI variant (N154S) (Figure 1C). Indeed, STING (R284S) appeared to be highly constitutively active. The amino acid change in the patient’s variant (R284S) did not appear to be in the known STING dimerization domain, according to structural analysis (Figure 1B). Accordingly, co-immunoprecipitation analysis suggested that STING (R284S) retained its ability to dimerize with wild-type STING (Figure 1D). Collectively, our data indicate that STING (R284S) is a hyperactive variant that does not require CDNs to be activated.
Figure 1. The STING Mutant (R284S) Is Hyperactive.
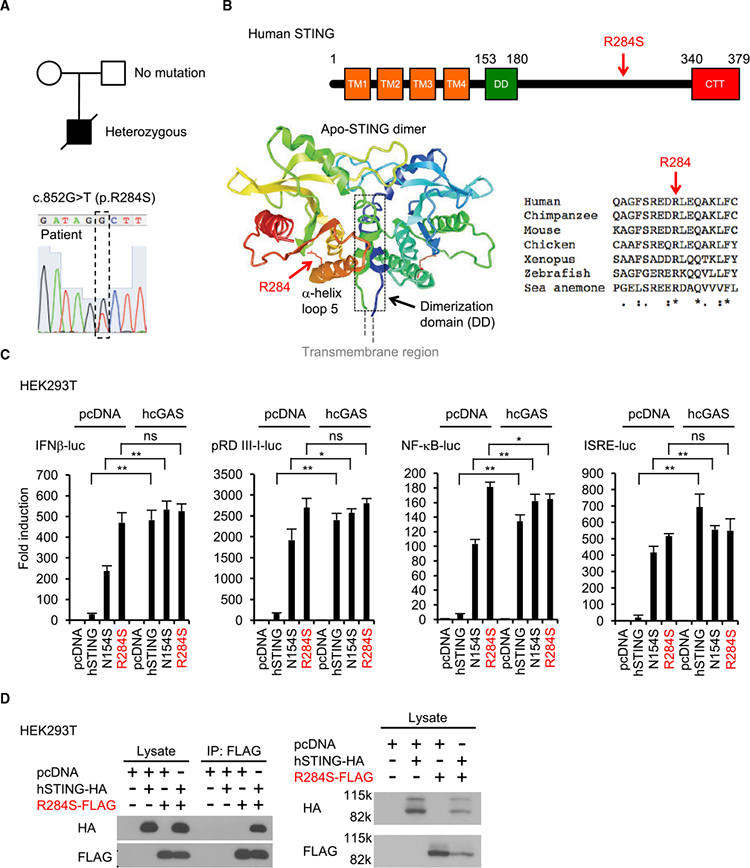
(A) The patient had a missense heterozygous mutation in the STING (TMEM173) gene (c.852G > T, p.R284S).
(B)R284 is highly conserved among different species. The R284S mutation ison the α helix loop 5 in theSTING C terminus. The crystal structure of the apo-STING dimer (PDB: 4F5W) indicates that R284 may not be involved in STING dimerization
(C) HEK293T cells were transfected with plasmid encoding human STING (hSTING) or its mutants (N154S and R284S) and the indicated reporter plasmids. Human cGAS (hcGAS) was expressed to generate cGAMP. After 24 hr, luciferase activity was measured.
(D) After transfection with the indicated plasmids, such as hSTING-HA (HA-tagged hSTING) and/or R284S-FLAG (FLAG-tagged R284S) into HEK293T cells for 24 hr, the cell lysates were incubated with anti-FLAG antibody, followed by immunoprecipitation using protein G beads. Western blots were performed for the precipitants with the indicated antibodies. The STING dimer resistant to boiling in SDS sample buffer was also detected by the indicated antibodies. Data shown here are the averages ± SD (n = 3). *p < 0.05, **p < 0.01, determined by Student’s t test. ns, not significant. See also Figure S1
STING R284S Traffics in the Absence of Agonists
To analyze STING (R284S) function further, we reconstituted primary Sting-’- mouse embryonic fibroblast (MEF) cells with wild-type STING, N154S (SAVI), or R284S using retrovirus delivery. It had been previously reported that SAVI variants leak out from the ER to likely trigger innate immune signaling (Dobbs et al., 2015;Mukai et al., 2016) We therefore similarly examined the localization of STING (R284S) in the cell by confocal microscopy. Our data indicated that wild-type STING could only traffic in the presence of transfected cytosolic dsDNA (Figure 2A). MEFs reconstituted with the SAVI variant (N154S) were also not seen to robustly traffic in the absence of cytosolic dsDNA activators, although a murine equivalent (N153S) was seen to traffic more readily (Figures 2A and S2A). However, the human STING (R284S) variant was seen to traffic to perinuclear regions without the requirement of STING agonists, again suggesting that it was permanently active (Figure 2A). We further observed that, similar to wild-type STING, STING (R284S) localized to endosomal and autophagosome-like vesicles, although did not require the presence of cytosolic dsDNA (Ishikawa et al., 2009; Saitoh et al.,2009; Figure S2B). Wild-type STING is known to escort and activate TBK1 during trafficking, an event required to activate IRF3 (Ishikawa et al., 2009). Confocal analysis further indicated that TBK1 constitutively accompanied STING (R284S) to perinuclear regions that harbor IRF3 transcription factors (Figures 2A and S2C). In addition, we confirmed that, similar to wild-type STING, STING (R284S) could interact with TBK1 (Figure S2D). Thus, STING (R284S) retains its ability to bind to TBK1. The constitutive trafficking of STING (R284S) could be blocked by brefeldin A (BFA), confirming that this variant traffics from the ER through the Golgi apparatus Figure 2B).
Figure 2. STING (R284S) Constitutively Localizes to the Peri-nuclear Area.
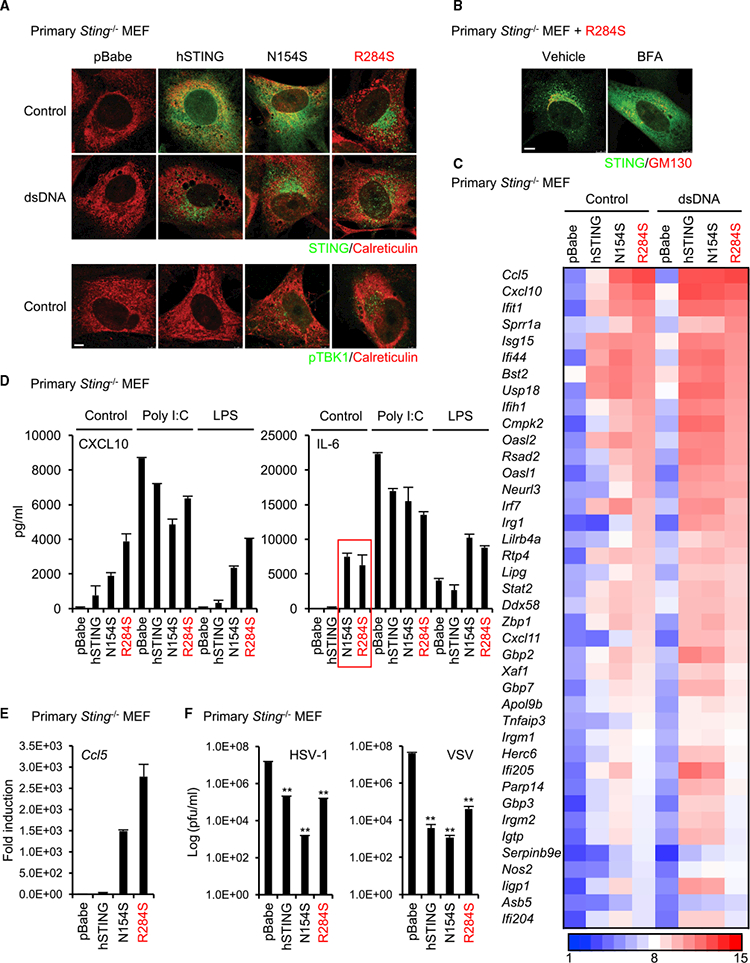
(A) Primary Sting-’- MEF cells were reconstituted with wild-type STING or its mutants (N154S and R284S) using a retrovirus. The reconstituted cells were treated with dsDNA(4 mg’mL) using Lipofectamine 2000 for 9 hr. After fixation, the cells were stained with the indicated antibodies, and then the localization of STING and phosphorylated TBK1 (pTBK1) was observed using a confocal microscope. Calreticulin is an ER marker. Scale bar, 5 μm.
(B) The reconstituted cells with R284S were treated with BFA (5 μg’mL) for 9 hr. The localization of STING was observed as described in (A). GM130 is a c/s-Golgi marker. Scale bar, 7.5 μm.
(C) Total RNA was purified from the reconstituted cells after treatment with dsDNA as described in (A) and then examined for gene expression using the Affymetrix GeneChip array. The scale represents the intensity of gene expression (log2 scale).
(D) The amounts of CXCL10 or IL-6 in the supernatants of the reconstituted cells were measured by ELISA after treatment with poly(I:C) (1 mg’mL) using Lipofectamine 2000 or LPS (10 mg/mL) for 15 hr.
(E) cDNA was synthesized from total RNA that was extracted from the reconstituted cells, and then real-time PCR was performed with the Ccl5 probe.
(F) The reconstituted cells were infected with HSV-1 or VSV at MOI = 1 for 24 hr, and then a plaque assay was performed to determine the viral titers. Data shown here are the averages ± SD (n = 3). **p < 0.01, determined by Student’s t test. See also Figure S2.
We confirmed that STING (R284S) was potently able to trigger the transcription of innate immune genes using microarray analysis. This study indicated that the STING N154 and R284S variants robustly induced innate immune gene transcription even in the absence of a cytosolic dsDNA activator in this assay (Figure 2C). The reconstitution of wild-type human STING in these cells also appeared to trigger innate immune gene activity, but not to the extent seen with the STING N154 and R284S variants. ELISA or real-time PCR confirmed the microarray results and indicated high levels of CXCL10, IL-6, and Ccl5 production in cells expressing N154S or R284S (Figures 2D and 2E). The observed high level of constitutive IL-6 production was reminiscent of the situation observed in the patient. The observed elevated levels of host defense-related proteins being produced by cells expressing STING N154S or R284S may be expected to contribute to resistance to viral infection. We thus infected the reconstituted MEFs with herpes simplex virus 1 (HSV-1, a DNA virus) or vesicular stomatitis virus (VSV, an RNA virus) and indeed noted that cells expressing the STING variants were resistant to viral replication (Figure 2F). Of interest is that it has recently been reported that the STING pathway can influence cellular senescence, a cell-autonomous growth arrest program, by inducing senescence-associated secretory phenotype (SASP) factors such as IL-6 (Dou et al., 2017; Gl€uck et al., 2017; Yanget al., 2017). Because we had observed that STING (R284S) can induce IL-6, we evaluated whether this variant can influence SASP (Figure 2D). Recon stituted Sting-’- MEFs expressing STING (R284S) were found to exhibit significantly slower growth rates compared with control cells, with plausible signs of senescence (Figures S2E-S2H). Our data thus suggest that STING (R284S) is a highly active STING variant that could conceivably influence the control of cellular senescence.
Suppression of Type IIFN Production through Negative Feedback
The activation of STING leads to the potent induction of type I IFN and pro-inflammatory genes in response to CDNs. However, the induction of Ifnbl and Ifna family genes was not robustly detected by RT-PCR or microarray analysis in Sting-/- MEF cells that were reconstituted with N154S or R284S (Figures 3A andS3A). Wild-type MEFs were seen to generate type I IFN, as determined from ELISA analysis, following transfection of cytosolic dsDNA, but little detectable type IFN was detected in Sting−/− cells reconstituted with the STING mutants, likely because of the diminutive amounts made (Figure 3B).. However, treatment of the reconstituted MEFs with dsDNA did trigger detectable type I IFN in all cells except for MEFs reconstituted with STING (R284S) (Figure 3B). In contrast, production of alternate innate immune genes such as Cxcl10 was unaffected (Figures 2C and 3C) It is possible that N154S and R284S may need to interact with wild-type STING to exert activity because all reported SAVI patients are heterozygous (as was the patient of the present study). However, Sting+/+ MEFs expressing N154S or R284S also failed to robustly secrete type I IFN, whereas CXCL10 was readily detected, possibly because of low-level transcription of the former gene It is known that the transcription of type I IFN induction is controlled via a negative feedback mechanism likely to prevent proinflammatory complications and toxicity (Ivashkiv and Donlin, 2014; Porritt andHertzog, 2015). Given the robust constitutive activity of STING (R284S), it is plausible that this mutant could be driving the negative feedback regulation of type I IFN transcription. To address this, we treated reconstituted MEF cells with BFA, which would block STING (R284S) trafficking and, hence, its activity. After removing the BFA, we monitored the ability of STING (R284S) to trigger type I IFN production. This analysis confirmed that both the N154S or R284S mutants could drive type I IFN production in the absence of cytosolic dsDNA activators and were constitutively active and able to produce type I IFN. (Figure 3D).
Figure 3. Type IIFN Production Is Suppressed, although Proinflammatory Genes Are Highly Induced by STING (R284S).
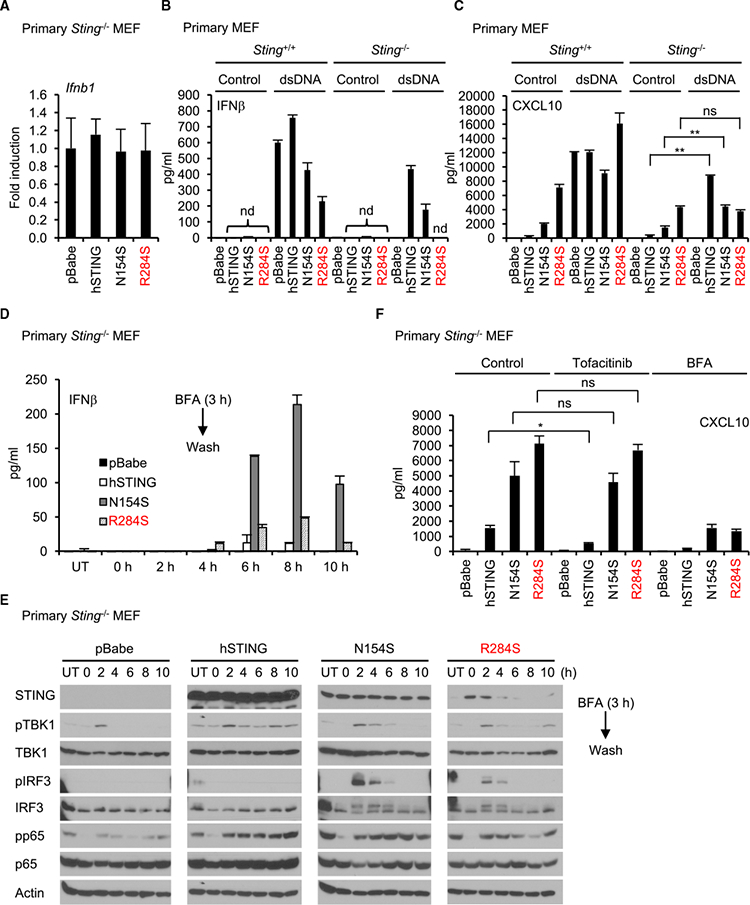
(A) Real-time PCR was performed for Ifnbl expression in reconstituted Sting-’- MEF cells.
(B and C) Wild-type human STING or its mutants (N154S and R284S) were expressed in Sting+/+ and Sting−/− MEF cells using a retrovirus. The cells were treated with dsDNA (4μg/mL) using Lipofectamine 2000 for 15 hr. The amounts of IFNβ (B) or CXCL10 (C) in the supernatants were measured by ELISA.
(D) The reconstituted cellswere incubated with BFA(5 μg/mL) for3 hrand then washed with culture medium twiceto remove BFA. The indicated times showthe time after washing. UT, untreated with BFA. The supernatants were collected at the indicated times, and IFNβwas measured by ELISA.
(E) The reconstituted cells were lysed after the supernatants were collected as described in (D), and then western blots were performed with the indicated antibodies.
(F) The reconstituted cells were treated with tofacitinib (5 μM) or BFA (5 μg/mL) for 9 hr, and then CXCL10 in the supernatants was measured by ELISA. Data shown here are the averages ± SD (n = 3). *p < 0.05, **p < 0.01, determined by Student’s t test. nd, not detected. See also Figure S3
Consistent with these results, immunoblot analysis confirmed that IRF3 phosphorylation was not readily detectable in MEFs constitutively expressing STING N154S and R284S. However, after blocking STING trafficking with BFA, IRF3 phosphorylation was readily detected 2 hr after the removal of the BFA (Figure 3E) The activation of NF-κB, as determined through analysis of p65 phosphorylation, seemed to retain modest constitutive activity, unlike IRF3. These results suggest that STING N154S and R284S are indeed able to activate IRF3 as well as NF-κB and suggest that the lack of IRF3 phosphorylation may be due to negative feedback regulation of this transcription factor (Ivashkiv and Donlin, 2014; Porritt and Hertzog, 2015). Because type I IFN requires both NF-κB as well as IRF3 for transcription, the lack of type I IFN induction may conceivably be explained by low-level IRF3 activity. In contrast, because Cxcl10 feedback regulation of this transcription harbors NF-κB activation sites in its promoter region, its expression would be less affected by high IRF3 turnover(Figure 3C). This concept is enforced by demonstrating that the JAK inhibitor tofacitinib, which inhibits IFN-stimulated gene (ISG) signaling, failed to significantly suppress CXCL10 expression(Figures 3F, S3A, and S3B). This effect may also explain why there is little association between the interferon scores in patients suffering from AGS (as determined from measuring the expression of 6 pro- inflammatory genes (IFI27, IFI44L, IFIT1, ISG15, RSAD2, and SIGLEC1) and levels of interferon activity (Rice et al., 2013).
ULK1 Regulators Are Potential Therapeutic Drugs for STING-Induced Inflammatory Diseases
We have previously shown that AMPK can influence ULK1 and the control of STING-dependent IRF3 activity (Konno et al.,2013). Constitutively phosphorylated AMPK (on AMPKa T172) ensures AMPK-dependent phosphorylation of ULK1 on S556 (S555 in murine cells), maintaining an inactive state. However, inhibition of AMPK results in the dephosphorylation of T172 and loss of ULK1 phosphorylation on S556. This releases ULK1 to phosphorylate STING on S366, an effect that inhibits STING activity. Given this, we therefore evaluated whether regulators of AMPK activity could influence STING-dependent innate immune signaling. Three putative regulators of AMPK were examined, compound C, doxorubicin (an anthracycline), and GSK 690693 (an AKT/AMPK inhibitor) (Heerding et al., 2008; Vogt et al.,2011; Wang et al., 2012). We postulated that suppression of AMPK may release ULK1 to phosphorylate STING, an event that can prevent STING-dependent IRF3 activation (Konno et al., 2013). Complete blocking of IRF3 activity may conceivably prevent the stimulation of interferon and, subsequently, ISGs (Sato et al., 2000). As a comparison, we evaluated use of the JAK inhibitor tofacitinib, which is efficacious in rodent models, as well as the ER-Golgi trafficking blocker BFA (Furumotoand Gadina, 2013; Ishikawa et al., 2009). Principally, treatment of MEFs expressing STING (R284S) with tofacitinib leads to suppression of ISGs predominantly harboring STAT1 sites, such as Usp18 and Irgm2 (indicated by blue) but less suppression of genes with NF-κB and/or IRF1 transcription sites such as Cmpk2 and Rsad2, which were more evidently suppressible by GSK 690693 (Figure 4A). In this analysis, doxorubicin, an intercalating agent, appeared to be less able to suppress innate immune gene induction compared with compound C. RT-PCR analysis confirmed that GSK 690693 was most competent at suppressing the transcription of Ifit1, Oas1, and Rsad2 induced by both STING N154S and R284S (Figure 4B). Immunoblot analysis indicated that compound C, doxorubicin, and GSK 609693 could suppress AMPK (T172) and ULK1 (S555) phosphorylation, albeit to different degrees, as well as IRF3 phosphorylation (Figure 4C). In contrast, tofacitinib more robustly suppressed STAT1 phosphorylation, as expected (Figure 4C). To extend this study, we treated human fibroblast cells (hTERT-BJ1) with GSK 690693 or tofacitinib and measured, by immunoblot, IRF3 phosphorylation in the presence and absence of a cytosolic dsDNA activator. This analysis confirmed that GSK 690693, but not tofacitinib, could inhibit dsDNA-triggered, STING-dependent IRF3 phosphorylation (Figure 4D). We further observed that GSK 690693 could suppress ULK1 phosphorylation on S556, suggesting a role for this kinase in controlling IRF3 activity (Konno et al., Figures 4D and S4A-S4C). GSK 690693 was observed to promote STING S366 phosphorylation in the presence of ULK1 (Figure S4D). The examination of hTERT-BJ1 cells confirmed that GSK 690693 could suppress the production of type I IFN as well as other pro-inflammatory genes (Figure 4E). Thus, GSK 690693 may be useful either alone or in conjunction with other accepted drugs such as tofacitinib as a treatment for “interferonopathies” mediated by chronic STING signaling (Barber, 2015).
Figure 4. AMPK Inhibitors Are Potential Therapeutic Drugs for STING-Induced Inflammatory Diseases.
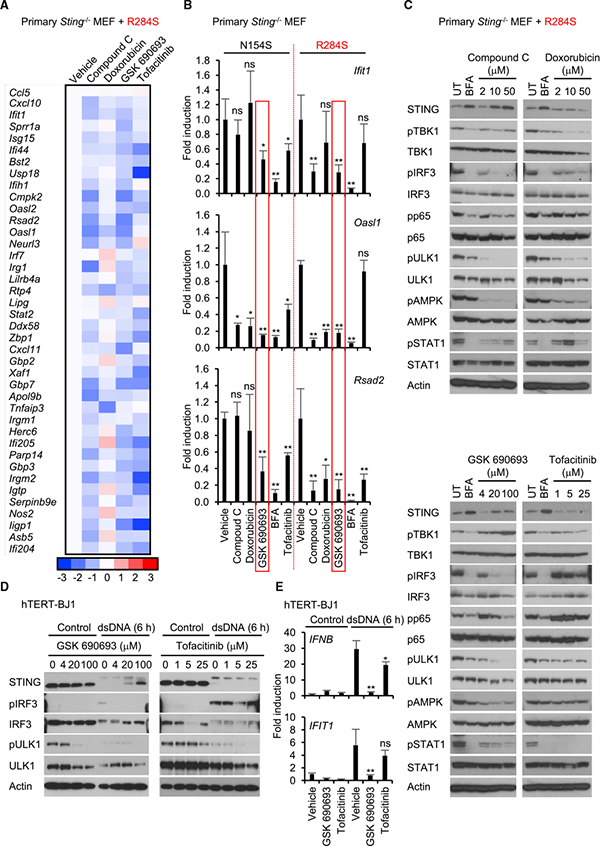
(A) The reconstituted Sting−/−MEF cells expressing R284S were treated with the indicated drugs (compound C, 10 μM; doxorubicin, 10 mM; GSK 690693,20 μM tofacitinib, 5 mM)) for 9 hr. Total RNA was purified and then examined for gene expression compared with vehicle as described in Figure 2C. The scale represents the intensity of gene expression (log2 scale).
(B) The reconstituted cells expressing N154S or R284S were treated with the indicated drugs as described in Figure 4A. Realtime PCR was performed with Ifit1, Oasl1, or Rsad2 probe.
(C) The reconstituted cells expressing R284S were treated with the drugs at the indicated concentration for 9 hr. Wester blots were performed for the cell lysates with the indicated antibodies. pULK1 and pAMPK indicate phosphorylated serine 555 on ULK1 (S555) and threonine 172 on AMPKα (T172), respectively. UT,untreated.
(D) hTERT-BJ1 cells were pre-incubated with the drugs at the indicated concentration for 1 hr. Then the cells were treated with dsDNA for 6 hr as described in Figure 2A. Western blots were performed for the cell lysates with the indicated antibodies.
(E) hTERT-BJ1 cells were pre-incubated with drugs (GSK 690693,20 μM; tofacitinib, 5 μM))for 1 hr. Then the cells were treated with dsDNA for 6 hr as described in Figure 2A. Real-time PCR was performed with the IFNB1 or IFIT1 probe. Data shown here are the averages ± SD (n = 3). *p < 0.05, **p < 0.01, determined by Student’s t test. See also Figure S4.
DISCUSSION
We report here a missense germline mutation in the coding region of the STING gene that generates a gain-of-function phenotype leading to chronic production of proinflammatory cytokines. The observed mutation appears outside of the STING dimerization domain and in the cytoplasmic region of the molecule, although it is presently unclear how the mutation initiates the active phenotype. However, it is likely that the alteration induces a conformational change, plausibly affecting post-translational events or protein-protein interactions that spontaneously enable STING trafficking and signaling without the requirement for association with agonists such as CDNs. The mutation appeared to be heterozygous in nature and manifested a gain-of-function phenotype. Although the patient died shortly after admission with a diagnosis of pneumonia, the exact cause of death is unclear. Although no autopsy was carried out on the patient, there were no signs of a responsible infectious agent detected in the patient’s abscess, suggesting sterile inflammation. The reported STING (R284S) variant appears to be more aggressive compared with a known SAVI variant (N154S), at least by in vitro analysis. However, reports of a patient exhibiting a mutation at R284 have been documented (R284G compared with R284S), without any noted lethality. It may thus be possible that the serine substitution has a more dramatic effect in vivo for reasons that remain to be fully clarified (Melki et al., 2017). This may help explain the clinical condition associated with STING (R284S) expression compared with the known SAVI-associated mutations, which do not commonly cause death in newborn infants (Liu et al., 2014). These data suggest that certain amino acid changes can more dramatically affect the activity of STING compared with others, which, in turn, may affect the severity and outcome of disease.
This case adds to the growing list of inflammation-related syndromes that involve the deregulation of STING signaling. For example, mutations in DNaseIII (TREX1) are also known to cause severe and often lethal inflammatory disease in infants (referred to as AGS), which is similar to severe forms of SLE(Crow and Manel, 2015; Mohan and Putterman, 2015). Subsequent studies have demonstrated that TREX1 functions to degrade aberrant self-DNA species that may have escaped into the cytosol following cell division(Ahn et al., 2014). Failure to degrade these nucleic acids can lead to the activation of DNA-triggered innate immune pathways, such as those controlled by STING, likely after association with cGAS and the generation of CDNs(Barber, 2015). Mice lacking TREX1 suffer from lethal inflammation-related disease that can be prevented following breeding onto a STING-deficient background(Gall et al., 2012). Other cellular genes exhibiting mutations that can cause AGS plausibly through aberrant STING signaling include the ribonuclease H2 enzyme family (RNASEH2A, RNASEH2B, and RNASEH2C), SAMHD1, and ADAR1(Crow and Manel, 2015). In animal models, loss of DNaseII also causes prenatal lethal inflammatory disease, again through self-DNA-triggered STING signaling(Ahn et al., 2012). It remains to be seen whether chronic STING signaling is involved in other types of autoimmune or inflammation-related disease.
Disorders involving the chronic production of type I IFNs and pro-inflammatory cytokines have given rise to the term interfero- nopathies) (Rodero and Crow, 2016). It has been reported that the use of JAK inhibitors such as tofacitinib may be of use as a therapy in such situations because they block harmful ISG protein production(Winthrop, 2017). However, we noticed that cells expressing STING (R284S) expressed very little type I IFN. We demonstrated, using BFA, that the cause involved negative feedback suppression of type I IFN transcription, which has evolved to prevent the deleterious consequences that sustained type I IFN production can cause in vivo(Porritt and Hertzog, 2015). Type I IFN transcription involves transient NF-κB and IRF3/7 signaling and is tightly controlled(Hertzog and Williams, 2013). Thus, the use of JAK inhibitors, although beneficial, may not provide the optimal therapeutic intervention to prevent STING- related disease. We have previously shown that stimulation of AMPK activity releases ULK1-mediated phosphorylation of STING, which inhibits STING’s ability to phosphorylate IRF3(Konno et al., 2013). Given this, we evaluated the ability of drugs that reportedly affect AMPK/ULK1 activity to influence STING signaling. Two drugs, doxorubicin and GSK 690693, have been documented as affecting AMPK activity, although alternate targets have also been reported, such as those involving the AKT/ phosphatidylinositol 3-kinase (PI3K) pathway (Heerding et al., 2008; Wang et al., 2012). Doxorubicin, an anthracycline antibiotic, is widely used for the treatment of numerous cancers, whereas GSK 690693 has been evaluated in phase I trials to treat hematological malignancies (Carol et al., 2010; Levy et al., 2009). In this report, the two drugs were noted to affect ULK1 activity, with GSK 690693 exhibiting a potent ability to repress STING (R284S)-mediated gene induction, predominantly via inhibition of STING-controlled IRF3 phosphorylation. These results may indicate that such drugs, either alone or in conjunction with JAK inhibitors, may be useful for the treatment of STING-related disease. In summary, our data describe a case of STING-mediated autoinflammatory malaise and suggest a therapeutic strategy for the treatment of such cases.
EXPERIMENTAL PROCEDURES
Cells, Reagents, and Viruses
Primary Sting+/+ and Sting−/−MEF cells, HEK293T cells, and Platinum-E retroviral packaging cells were prepared as described previously (Ishikawa and Barber, 2008; Konno et al., 2013). Primary MEF cells, HEK293T cells, and Plat- inum-E cells were cultured in DMEM (Gibco) supplemented with 10% fetal bovine serum (FBS), and antibiotics. dsDNA (IFN stimulatory DNA [ISD] 90-mer) was prepared as described previously (Konno et al., 2013). Poly(I:C) was obtained from American Biosciences. LPS, BFA, compound C, and tofa-citinib were purchased from Sigma. Doxorubicin and GSK 690693 were purchased from Santa Cruz Biotechnology. Lipofectamine 2000 and RNAiMAX were purchased from Invitrogen. Anti-STING rabbit polyclonal antibody and mouse monoclonal antibody were prepared as described previously (Ishikawa and Barber, 2008; Konno et al., 2013). The pS366 antibody was made at Thermo Fisher Scientific. Other antibodies used in this paper were as follows: β-actin (Sigma, A5441), calreticulin (Abcam, ab14234), phospho-IRF3 (Cell Signaling Technology, 4947), IRF3 (Cell Signaling Technology, 4302), phos- pho-p65 (Cell Signaling Technology, 3031), p65 (Cell Signaling Technology, 3987), phospho-TBK1 (Cell Signaling Technology, 5483), TBK1 (Abcam, ab40676), phospho-AMPK (T172) (Cell Signaling Technology, 2535), AMPK (Cell Signaling Technology, 5832), phospho-ULK1 (S556) (Cell Signaling Technology, 5869), ULK1 (Cell Signaling Technology, 4773), phospho-STAT1 (Cell Signaling Technology, 9167), STAT1 (Cell Signaling Technology, 14994), Rab5 (Cell Signaling Technology, 3547), LC3 (MBL, M152–3), hemagglutinin (HA) (Sigma, H9658), FLAG (Sigma, F1804), and GM130 (BD Biosciences, 558712, Alexa Fluor 647-conjugated). HSV-1 (Kendall Owen Smith [KOS] strain) was purchased from the ATCC. VSV was prepared as described previously (Ishikawa and Barber, 2008). TaqMan probes for Infb, Ccl5, Cxcl10, Ifitl, Oasll, Oasl2, Rsad2, Ulk1, Ulk2, and Gapdh were purchased from Applied Biosystems. ELISA kits for IFNβ and CXCL10/IL-6 were purchased from PBL Interferon Source and R&D Systems, respectively. Small interfering RNAs (siRNAs) for ULK1, ULK2, AMPKα1, and AMPKα2 were purchased from Dharmacon.
Plasmids and Mutagenesis
Reporter plasmids and thymidine kinase (TK)-luc were obtained as described previously (Ishikawa and Barber, 2008). pcDNA-hSTING, pcDNA-ULK1, pcDNA-TBK1, pBabe-hSTING, and pBabe-mSTING were made as described previously (Ishikawa and Barber, 2008; Konno et al., 2013). pCMV-hcGAS was purchased from OriGene. To make STING mutants, the QuikChange II XL Site- Directed Mutagenesis Kit (Stratagene) was used with the following primers:
hSTING_N154S-F: 5΄-GAAAAAGGGAATTTCAGCGTGGCCCATGGGC TG-3΄
hSTING_N154S-R:5΄-CAGCCCATGGGCCACGCTGAAATTCCCTTTTTC-3΄
hSTING_R284S-F:5΄-TTTAGCCGGGAGGATTCTCTTGAGCAGGCCAAA-3΄
hSTING_R284S-R:5΄-TTTGGCCTGCTCAAGAGAATCCTCCCGGCTAAA-3΄
mSTING_N153S-F: 5΄-GAAGAAAAGAAGTTATCTGTTGCCCACGGGC TG-3΄
mSTING_N153S-R: 5΄-CAGCCCGTGGGCAACAGATAACTTCTTTTCTTC-3΄
mSTING_R283S-F: 5΄-TTCAGTCGGGAGGATTCCCTTGAGCAGGCTA AA-3΄
mSTING_R283S-R: 5΄ -TTTAGCCTGCTCAAGGGAATCCTCCCGACTG AA-3΄.
Whole-Exome Sequencing
Written informed consent was obtained from the family. The research protocol was approved by the relevant institutional review boards. Whole-exome sequencing was performed as part of an ongoing collaboration with the Baylor Hopkins Center for Mendelian Genomics (BHCMG) (Stray-Pedersen et al.,2017). Exome capture was performed with the in-house-developed Baylor College of Medicine Human Genome Sequencing Center (BCM-HGSC) Core design (52 Mb, Roche NimbleGen) as described previously(Yang et al.,2013). The variant calling was performed by the ATLAS2 suite(Challis et al.,2012). To confirm the mutation, PCR was used to amplify the DNA region of interest with the primers (forward, 5-ggaccctccattctccatcc-30; reverse, 5΄-aggcggcagttgttctgag-3΄) prior to Sanger sequencing. The PCR conditions were as follows: 95°C for 5 min, 40 cycles of 94°C for 30 s/62°C for 1 min/ 72°C for 1 min, and 72°C for 10 min.
Luciferase Reporter Assay
HEK293T cells were transfected with pcDNA3-hSTING or mutants and reporter plasmids (IFNβ-luc, NF-kB-luc, pRD III-I-luc, and ISRE-luc) using Lipo-fectamine 2000. TK-luc was used for transfection control. After 24 hr, the cells were lysed in cell culture lysis reagent (Promega), and then luciferase activity was measured using luciferase assay substrate (Promega) for firefly luciferase activity (reporter plasmids) and the Renilla luciferase assay system (Promega) for Renilla luciferase activity (TK-luc).
Western Blot
Samples were separated in an acrylamide gel and then transferred to an Immobilon-P membrane (Millipore). The membrane was treated with 5% blot- ting-grade blocker (Bio-Rad) in TBS-T buffer (Tris-buffered saline [TBS] with 0.1% Tween 20) for blocking and then incubated with the indicated primary antibody. The membrane was washed three times using TBS-T buffer and then incubated with horseradish peroxidase (HRP)-conjugated anti-rabbit or mouse immunoglobulin G (IgG) (Promega). After three washes, Super Signal West Pico or Femto (Thermo Fisher Scientific) was used to develop the signal, and the membranes were exposed to Premium X-ray film (Phenix).
Immunoprecipitation Assay
HEK293T cells were transfected with the indicated plasmids for 24 hr and then lysed with TNE buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA, and 1% NP-40) with protease and phosphatase inhibitors. The cell lysates were incubated with anti-FLAG or STING antibody overnight at 40C, and then protein G beads (Thermo Fisher Scientific) was added to the lysates. After incubation for 1 hr at 40C, the precipitants were washed with TNE buffer five times and then boiled in SDS sample buffer to elute the precipitated proteins.
Immunostaining
The cells on poly-D-lysine-coated round coverslips (BD Biosciences) were fixed with 4% paraformaldehyde/PBS for 15 min and then permeabilized with 0.2% Triton X-100/PBS for 15 min. After blocking with 1% BSA/PBS for 30 min, the coverslips were incubated with the indicated primary antibodies in 1% BSA/PBS for 1 hr at room temperature (RT) in a wet chamber. After washing three times with PBS, the coverslips were incubated with Alexa Fluor 488-goat anti-rabbit IgG, Alexa Fluor 555-goat anti-mouse IgG, and Alexa Fluor 647-goat anti-chicken IgG (Invitrogen) for 1 hr at RT in a wet chamber. After washing three times with PBS, the coverslips were mounted onto glass slides with ProLong Gold antifade reagent (Invitrogen). The coverslips were observed using a Leica SP5 confocal microscope.
Gene Expression Array
Total RNA from the reconstituted MEF cells was purified using the RNeasy RNA extraction kit (QIAGEN). The GeneChip Mouse Gene 2.0 ST array (Affymetrix) was used to observe gene expression. Data collection and analysis were performed atthe Centerfor Genome Technology (CGT), John P. Hussman Institute for Human Genomics, University of Miami Miller School of Medicine.
Real-Time PCR
Total RNA was extracted using the RNeasy Mini Kit (QIAGEN), and cDNA was synthesized using the QuantiTect reverse transcription kit (QIAGEN) following the manufacturer’s instructions. Real-time PCR was performed with the indicated TaqMan probes (Applied Biosystems) using a LightCycler 2.0 (Roche) or StepOnePlus real-time PCR system (Applied Biosystems). GAPDH was used for normalization.
Plaque Assay
The cells were infected with HSV-1 or VSV at MOI = 1 for 24 hr. The infected cells were freeze-thawed three times to release HSV-1 from the cells. Vero cells were incubated with the supernatants, including viruses, for 1 hr and then cultured in 1% low-melting agarose (Invitrogen)/DMEM/1% FBS to avoid a secondary colony. After 24 hr for VSV or 48 hr for HSV-1, the cells were fixed with methanol after removing the overlaid gel and then stained with 0.1% crystal violet/30% methanol solution to visualize plaques.
Cell Growth and Senescence Assay
5 × 105 cells of the reconstituted S’ting−/− MEF cells with pBabe, hSTING, or R284S were seeded on a 10-cm dish, and the cell numbers were counted by hemocytometer after staining with trypan blue every 3 days. Senescent cells were identified with a senescence-associated β-galactosidase kit (Cell Signaling Technology, 9860).
Supplementary Material
Highlights.
A single nucleotide change in the STING gene generates an active STING variant
STING (R284S) is active in the absence of cyclic dinucleotides
ULK1 phosphorylation of STING can inhibit STING- dependent cytokine production
AMPK/ULK1 regulators may be useful to treat STING-related inflammatory disease
ACKNOWLEDGMENTS
We thank William Hulme and Anthony Griswold for gene array analysis. This work was funded in part by NIH grant AI079366.
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the microarray data reported in this paper is GEO: GSE111198.
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures and can be found with this article online at https://doi.org/10.1016Zj.celrep.2018.03.115.
AUTHOR CONTRIBUTIONS
H.K. carried out most of the experiments. A.M. and L.A.P. collected and tested patient samples. I.K.C., D.H., J.S.O., and J.R.L. conducted whole-exome sequencing. H.K. and G.N.B. wrote the manuscript.
DECLARATION OF INTERESTS
G.N.B. is a founder of STINGINN, which presently has no financial assets. A provisional patent was filed on the STING variant (R284S) through the University of Miami.
REFERENCES
- Ahn J, Gutman D, Saijo S, and Barber GN (2012). STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. USA 109, 19386–19391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn J, Ruiz P, and Barber GN (2014). Intrinsic self-DNA triggers inflammatory disease dependent on STING. J. Immunol 193, 4634–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barber GN (2015). STING: infection, inflammation and cancer. Nat. Rev. Immunol 15, 760–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdette DL, Monroe KM, Sotelo-Troha K, Iwig JS, Eckert B, Hyodo M, Hayakawa Y, and Vance RE (2011). STING is a direct innate immune sensor of cyclic di-GMP. Nature 478, 515–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carol H, Morton CL, Gorlick R, Kolb EA, Keir ST, Reynolds CP, Kang MH, Maris JM, Billups C, Smith MA, et al. (2010). Initial testing (stage 1) of the Akt inhibitor GSK690693 by the pediatric preclinical testing program. Pediatr. Blood Cancer 55, 1329–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Challis D, Yu J, Evani US, Jackson AR, Paithankar S, Coarfa C, Milosavljevic A, Gibbs RA, and Yu F (2012). An integrative variant analysis suite for whole exome next-generation sequencing data. BMC Bioinformatics 13, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke SL, Pellowe EJ, de Jesus AA, Goldbach-Mansky R, Hilliard TN, and Ramanan AV (2016). Interstitial Lung Disease Caused by STING-associated Vasculopathy with Onset in Infancy. Am. J. Respir. Crit. Care Med 194, 639–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, and Manel N (2015). Aicardi-Goutie` res syndrome and the type I interferonopathies. Nat. Rev. Immunol 15, 429–440. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, et al. (2015). Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am. J. Med. Genet. A 167A, 296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs N, Burnaevskiy N, Chen D, Gonugunta VK, Alto NM, and Yan N (2015). STING Activation by Translocation from the ER Is Associated with Infection and Autoinflammatory Disease. Cell Host Microbe 18, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fre´ mond ML, Rodero MP, Jeremiah N, Belot A, Jeziorski E, Duffy D, Bessis D, Cros G, Rice GI, Charbit B, et al. (2016). Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J. Allergy Clin. Immunol 138, 1752–1755. [DOI] [PubMed] [Google Scholar]
- Furumoto Y, and Gadina M (2013). The arrival of JAK inhibitors: advancing the treatment of immune and hematologic disorders. BioDrugs 27, 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr., Barber GN, and Stetson DB (2012). Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 36, 120–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gl€uck S, Guey B, Gulen MF, Wolter K, Kang TW, Schmacke NA, Bridgeman A, Rehwinkel J, Zender L, and Ablasse A (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. Cell Biol 19, 1061–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heerding DA, Rhodes N, Leber JD,Clark TJ,Keenan RM, Lafrance LV, Li M, Safonov IG, Takata DT, Venslavsky JW, et al. (2008). Identification of 4-(2-(4-amino-1,2,5-oxadiazol-3-yl)-1-ethyl-7-[(3S)-3-piperidinylmethyl]oxy-1Himidazo[ 4,5-c]pyridin-4-yl)-2-methyl-3-butyn-2-ol (GSK690693), a novel inhibitor of AKT kinase. J. Med. Chem 51, 5663–5679. [DOI] [PubMed] [Google Scholar]
- Hertzog PJ, and Williams BR (2013). Fine tuning type I interferon responses. Cytokine Growth Factor Rev. 24, 217–225. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, and Barber GN (2008). STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 455, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa H, Ma Z, and Barber GN (2009). STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 461, 788–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv LB, and Donlin LT (2014). Regulation of type I interferon responses. Nat. Rev. Immunol 14, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, Goudin N, Fre´ mond ML, Nitschke P, Molina TJ, et al. (2014). Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J. Clin. Invest 124, 5516–5520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko¨ nig N, Fiehn C, Wolf C, Schuster M, Cura Costa E, T€ungler V, Alvarez HA, Chara O, Engel K, Goldbach-Mansky R, et al. (2017). Familial chilblain lupus due to a gain-of-function mutation in STING. Ann. Rheum. Dis 76, 468–472. [DOI] [PubMed] [Google Scholar]
- Konno H, Konno K, and Barber GN (2013). Cyclic dinucleotides trigger ULK1 (ATG1) phosphorylation of STING to prevent sustained innate immune signaling. Cell 155, 688–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy DS, Kahana JA, and Kumar R (2009). AKT inhibitor, GSK690693, induces growth inhibition and apoptosis in acute lymphoblastic leukemia cell lines. Blood 113, 1723–1729. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, Tenbrock K, Wittkowski H, Jones OY, Kuehn HS, et al. (2014). Activated STING in a vascular and pulmonary syndrome. N. Engl. J. Med 371, 507–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, Revuelta A, Abbondati E, Rigby RE, Rabe B, et al. (2016). Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J. 35, 831–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki I, Rose Y, Uggenti C, Van Eyck L, Fre´ mond ML, Kitabayashi N, Rice GI, Jenkinson EM, Boulai A, Jeremiah N, et al. (2017). Diseaseassociated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J. Allergy Clin. Immunol 140, 543– 552. e5. [DOI] [PubMed] [Google Scholar]
- Mohan C, and Putterman C (2015). Genetics and pathogenesis of systemic lupus erythematosus and lupus nephritis. Nat. Rev. Nephrol 11, 329–341. [DOI] [PubMed] [Google Scholar]
- Mukai K, Konno H, Akiba T, Uemura T, Waguri S, Kobayashi T, Barber GN, Arai H, and Taguchi T (2016). Activation of STING requires palmitoylation at the Golgi. Nat. Commun 7, 11932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey S, Kawai T, and Akira S (2014). Microbial sensing by Toll-like receptors and intracellular nucleic acid sensors. Cold Spring Harb. Perspect. Biol 7, a016246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picard C, Thouvenin G, Kannengiesser C, Dubus JC, Jeremiah N, Rieux-Laucat F, Crestani B, Belot A, Thivolet-Be´ jui F, Secq V, et al. (2016). Severe Pulmonary Fibrosis as the First Manifestation of Interferonopathy (TMEM173 Mutation). Chest 150, e65–e71. [DOI] [PubMed] [Google Scholar]
- Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, Morris HD, Yan N, and Crouch RJ (2016). RNase H2 catalytic core Aicardi-Goutie` res syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J. Exp. Med 213, 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porritt RA, and Hertzog PJ (2015). Dynamic control of type I IFN signaling by an integrated network of negative regulators. Trends Immunol. 36, 150–160. [DOI] [PubMed] [Google Scholar]
- Rice GI, Forte GM, Szynkiewicz M, Chase DS, Aeby A, Abdel-Hamid MS, Ackroyd S, Allcock R, Bailey KM, Balottin U, et al. (2013). Assessment of interferon-related biomarkers in Aicardi-Goutie` res syndrome associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, and ADAR: a case-control study. Lancet Neurol. 12, 1159–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodero MP, and Crow YJ (2016). Type I interferon-mediated monogenic autoinflammation: The type I interferonopathies, a conceptual overview. J. Exp. Med 213, 2527–2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, Fujita N, Hayashi T, Takahara K, Satoh T, Lee H, Matsunaga K, Kageyama S, Omori H, Noda T, et al. (2009). Atg9a controls dsDNAdriven dynamic translocation of STING and the innate immune response. Proc. Natl. Acad. Sci. USA 106, 20842–20846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, and Taniguchi T (2000). Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity 13, 539–548. [DOI] [PubMed] [Google Scholar]
- Seo J, Kang JA, Suh DI, Park EB, Lee CR, Choi SA, Kim SY, Kim Y, Park SH, Ye M, et al. (2017). Tofacitinib relieves symptoms of stimulator of interferon genes (STING)-associated vasculopathy with onset in infancy caused by 2 de novo variants in TMEM173. J. Allergy Clin. Immunol 139, 1396–1399. e12. [DOI] [PubMed] [Google Scholar]
- Stray-Pedersen A, Sorte HS, Samarakoon P, Gambin T, Chinn IK, Coban Akdemir ZH, Erichsen HC, Forbes LR, Gu S, Yuan B, et al. (2017). Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J. Allergy Clin. Immunol 139, 232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun L, Wu J, Du F, Chen X, and Chen ZJ (2013). Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt J, Traynor R, and Sapkota GP (2011). The specificities of small molecule inhibitors of the TGFß and BMP pathways. Cell. Signal 23, 1831–1842. [DOI] [PubMed] [Google Scholar]
- Wang S, Song P, and Zou MH (2012). Inhibition of AMP-activated protein kinase a (AMPKa) by doxorubicin accentuates genotoxic stress and cell death in mouse embryonic fibroblasts and cardiomyocytes: role of p53 and SIRT1. J. Biol. Chem 287, 8001–8012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winthrop KL (2017). The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 13, 320. [DOI] [PubMed] [Google Scholar]
- Wu J, Sun L, Chen X, Du F, Shi H, Chen C, and Chen ZJ (2013). Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 339, 826–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Muzny DM, Reid JG, Bainbridge MN, Willis A, Ward PA, Braxton A, Beuten J, Xia F, Niu Z, et al. (2013). Clinical whole-exome sequencing for the diagnosis of mendelian disorders. N. Engl. J. Med 369, 1502–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang H, Ren J, Chen Q, and Chen ZJ (2017). cGAS is essential for cellular senescence. Proc. Natl. Acad. Sci. USA 114, E4612–E4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.