

Abstract
Herpes simplex virus (HSV) establishes a latent infection in peripheral neurons and can periodically reactivate to cause disease. Reactivation can be triggered by a variety of stimuli that can activate different cellular processes to result in increased HSV lytic gene expression and production of infectious virus. The use of model systems has contributed significantly to our understanding of how reactivation of the virus is triggered by different physiological stimuli that are correlated with recrudescence of human disease. Furthermore, these models have led to the identification of both common and distinct mechanisms of different HSV reactivation pathways. Here, we summarize how the use of these diverse model systems has led to a better understanding of the complexities of HSV reactivation, and we present potential models linking cellular signaling pathways to changes in viral gene expression.
Keywords: Herpes simplex virus, reactivation, epigenetics, NGF-deprivation, axotomy, DLK
Introduction
Herpes simplex virus (HSV) 1 and 2 are ubiquitous pathogens that persist for the life of infected individuals. The ability of these viruses to develop lifelong infections is due to the presence of a latent pool of virus in terminally differentiated neurons, most commonly in the peripheral ganglia. It is estimated that approximately 90% of individuals worldwide are infected with HSV-1, HSV-2 or both of the viruses (Arvin et al., 2007). In the United States, the estimated prevalence rate of HSV-1 and HSV-2 in people aged 14–49 is 47.8% and 11.9% respectively, with higher prevalence in women and Mexican-American and non-Hispanic black persons (McQuillan et al., 2018). HSV infection is often clinically silent. However, HSV periodically re-enters a lytic replication cycle in a process known as reactivation. In immunocompetent persons, reactivation events result in replication at the body surface that can give rise to recurrent blisters or sores, which are typically self-limiting and resolve rapidly (Roizman et al., 2013). These lesions most commonly occur at oral, nasal or ocular sites with HSV-1 infection and at the genital skin and mucosa with HSV-2 infection. HSV reactivation can also lead to significant morbidity and mortality in immunocompromised individuals, and, in rare cases, infection of the central nervous system can lead to acute viral encephalitis or a recurrent lymphocytic meningitis.
The ability of HSV to reactivate from latency and re-enter a productive replication cycle significantly contributes to its ability to cause lifelong disease that results in recurrent infection, viral shedding and transmission to new hosts. However, the molecular events that underlie the switch from latent to productive infection are not well understood. This review will examine our current understanding of how different reactivation stimuli may trigger this switch in the HSV viral life cycle and illuminate some of the remaining questions in the field.
Lytic replication and latency: contrasting viral life-cycles
The ability of HSV to establish a lifelong infection can be attributed to its capacity to undergo contrasting infectious events, termed the lytic and latent stages. Primary infection at the body surface results in productive replication in epithelial cells (Roizman et al., 2013). During this initial lytic stage of infection, over 80 viral gene products are expressed in a cascade-dependent manner. Initial viral gene expression is enhanced by delivery of viral tegument proteins, including the viral transactivator, VP16, into the nucleus. This potent transcriptional activator, induces the formation of a transcriptional regulatory complex with multiple cellular co-activators to promote transcription of the viral immediate early (IE) or α genes. Products of the IE genes include proteins required for transcription of the early (E) or β genes, which encode proteins required for viral DNA replication. The final group of genes is the late (L) or γ genes, whose expression is dependent on viral DNA replication. L genes encode structural proteins required for assembly, egress and release of the infectious HSV particle.
During primary infection, HSV is able to enter the terminal axons of neurons that innervate tissue at the initial site of infection. Although viral genomes are most frequently detected in sensory ganglia, particularly the trigeminal ganglia (HSV-1) and lumbar-sacral ganglia (HSV-2), viral DNA and reactivation-competent virus can also be isolated from sympathetic and parasympathetic neurons (Baringer and Pisani, 1994; Baringer and Swoveland, 1973; Richter et al., 2009; Warren et al., 1978). Furthermore, HSV DNA can be detected in the central nervous system, with the frequency of detection increasing with age (Beffert et al., 1998; Fraser et al., 1981; Gordon et al., 1996). Following neuronal infection, the virus can enter latent infection. However, neurons can also support lytic replication, which may be associated with neuronal death (Thompson and Sawtell, 2001). There is also evidence of prior lytic promoter activity in latently infected neurons (Proenca et al., 2008). The mechanisms that regulate entry into lytic replication versus latent infection in neurons remain largely undefined.
HSV latency is defined as the persistence of viral DNA in the absence of detectable infectious virus that retains the ability to reactivate following an appropriate stimulus. While expression of the viral lytic genes is largely repressed during latent infection, there is active transcription of the latency-associated transcript (LAT), which is composed of a primary 8.3kb unstable transcript that is spliced to give rise to a stable intron of approximately 2kb in length and multiple miRNAs (Kramer et al., 2011; Stevens et al., 1987; Umbach et al., 2008). Latency is usually defined at the level of the ganglia, but within a ganglion only a sub-population of latently infected cells will reactivate at any one time (Sawtell and Thompson, 2004). In addition, there is evidence that different stimuli can result in reactivation from different subtypes of neurons (Yanez et al., 2017). Therefore, the definition of a “latently” infected neuron may depend not only on the neuronal subtype, but also on the nature of the reactivating trigger.
Modeling HSV latency
A strength of the HSV field is the diversity of the model systems used to investigate the pathways to reactivation. Although there may be differences in the interpretation of the data based on the system or stimuli, all these systems will ultimately have relevance to basic science and human health. To elucidate the cellular signaling pathways involved in the reactivation processes, models of latency that allow faithful establishment of latency, robust reactivation and easy manipulation of signal transduction pathways are required. The most commonly used model organism in HSV research is the mouse. Infection of mice with HSV-1 results in initial lytic replication at the body surface and entry of the virus into innervating sensory and autonomic neurons. Following an initial period of acute replication in the ganglia, HSV establishes latency, and reactivation can be triggered by explant of the ganglia (Sawtell and Thompson, 2004), hyperthermic stress (Sawtell and Thompson, 1992), UV irradiation (Shimeld et al., 1996) or hormone treatment (Cook et al., 1991; Miguel et al., 2010). Overall, in vivo models of reactivation have the advantage of more accurately recapitulating the natural course of infection and incorporate host antiviral responses that may impact the state and population of latent genomes or modulate viral reactivation. However, the manipulation of cellular pathways in vivo can be challenging.
To elucidate the molecular pathways involved in HSV reactivation, in vitro models have proven to be invaluable (Thellman and Triezenberg, 2017; Wilson and Mohr, 2012). The optimal model system would utilize mature, human neurons. However, while sensory neurons can be isolated and maintained from human donors (Valtcheva et al., 2016), access to this material is limited, consistency is difficult to achieve, and the tissue may already be latently infected with HSV or varicella zoster virus (VZV). Human sensory neurons differentiated from embryonic stem cells have also been used to investigate latency and reactivation for both HSV and VZV (Markus et al., 2015; Pourchet et al., 2017). A recent study utilizing human differentiated neurons achieved latently infected cultures that could be reactivated with sodium butyrate, a histone deacetylase inhibitor (Pourchet et al., 2017). There is also an emerging interest in human cell lines that can be easily differentiated into neurons, as shown using the HD10.6 cell line, which can be differentiated into sensory neurons with nociceptive properties (Raymon et al., 1999; Thellman et al., 2017). Quiescent infection can be established in the presence of acyclovir, and reactivation can be triggered from a sub-population of neurons following depletion of nerve growth factor (Thellman et al., 2017).
Perhaps one of the best characterized in vitro systems to study HSV reactivation utilizes primary sensory or sympathetic neurons isolated from the peripheral ganglia of pre-natal rats and post-natal or adult mice (Camarena et al., 2010; Cliffe et al., 2015; Ives and Bertke, 2017; Wilcox and Johnson, 1987; Wilcox et al., 1990). Infection of these primary neuronal cultures in the presence of acyclovir or phosphonoacteic acid (PAA) results in a quiescent infection that resembles latency. Importantly, to accurately define a quiescent infection in these model systems, it is imperative to show that replicating virus remains undetectable following the removal of viral DNA replication inhibitors. When a quiescent infection is properly established, these systems exhibit all of the known molecular hallmarks of latency, including accumulation of the LAT intron, expression of latency-associated miRNAs, absence of replicating virus and undetectable levels of viral proteins (Camarena et al., 2010; Cliffe et al., 2015; Jurak et al., 2014; Wilcox and Johnson, 1987). Furthermore, these models maintain the capacity to undergo reactivation triggered by a variety of stimuli, including NGF-deprivation, dexamethasone, inhibition of protein synthesis or high intracellular levels of cAMP (Camarena et al., 2010; Cliffe et al., 2015; Linderman et al, 2017; Colgin et al., 2001; Kobayashi et al., 2012; Wilcox and Johnson, 1987). Thus, these systems provide a powerful tool to study the molecular features of latency and reactivation, such as the role of cell stress pathways or chromatin modulation, in primary neuronal populations. Some caveats to these model systems is the absence of support cells that may also impact the nature of the latent infection and/or reactivation. The use of DNA replication inhibitors to promote latency is instead utilized to compensate for missing immune components. Whether DNA replication inhibitors impact the nature of latency or reactivation mechanisms is not known. However, it is worth noting that symptomatic primary HSV-1 is often treated with anti-viral compounds (James and Whitley, 2010). Moving forward, it will be important to determine if the mode infection, age of neurons, presence of immune mediators or addition of DNA replication inhibitors alters the nature of latency or impacts events that occur during reactivation, as this will have relevance to both the model systems used and human disease.
Latent viral chromatin structure: silent but poised?
Following the establishment of latent infection, viral lytic gene expression is silenced, and the lytic gene promoters are associated with repressive heterochromatin (Knipe and Cliffe, 2008). Key experiments performed in the 1980’s indicated that latent genomes in the brain stems of infected mice have a nucleosomal structure (Deshmane and Fraser, 1989). Later studies confirmed that the latent viral genome associates with cellular histones in the trigeminal ganglia of mice (Cliffe et al., 2012; Kubat et al., 2004b; Wang et al., 2005). Coinciding with the silencing of lytic transcripts, the viral lytic gene promoters become enriched with characteristic heterochromatic histone modifications, namely histone H3 di- and tri-methylated at lysine 9 (H3K9me2/3) and H3K27me3 (Cliffe et al., 2012; 2009; Kwiatkowski et al., 2009; Nicoll et al., 2016; Wang et al., 2005). While it appears that factors intrinsic to neurons play a key role in the transcriptional silencing of the virus (Cliffe et al., 2012), viral gene products expressed during latent infection can also modulate the chromatin structure (Cliffe et al., 2009; Kwiatkowski et al., 2009; Raja et al., 2016; Wang et al., 2005). This modulation likely promotes long-term latency, while priming the genome for reactivation following the appropriate stimuli (Leib et al., 1989; Trousdale et al., 1991).
In contrast to the lytic gene promoters, the region encompassing the LAT promoter and enhancer elements are enriched with euchromatin-associated modifications, (Cliffe et al., 2009; Kubat et al., 2004a). This apparent demarcation in the nature of the chromatin likely arises due to binding sites for the cellular insulator protein CCCTC-binding factor (CTCF) on the viral genome. Interestingly, CTCF eviction coincides with reactivation (Ertel et al., 2012; Washington et al., 2018b), and depletion of CTCF in vivo promotes reactivation (Washington et al., 2018a). Furthermore, one important binding site of CTCF, known as CTRL2, lies downstream of the LAT enhancer and separates it from the nearby lytic ICP0 gene (Amelio et al., 2006b). Deletion of this site from the viral genome results in increased heterochromatin formation on the LAT enhancer region and a paradoxically small increase in LAT gene expression (Lee et al., 2018). The CTRL2 binding site deletion also decreases the mutant virus’s ability to reactivate from latency. These studies suggest that the organization of chromatin domains may play a role in both the establishment and maintenance of latency and potentially poise the viral genome for reactivation.
Although regions of the latent viral genome are associated with heterochromatin, there is evidence to suggest that it exists in a state that is primed for reactivation. In mouse models of latency, the viral genome does not contain detectible canonical CpG methylation (Dressler et al., 1987; Kubat et al., 2004b), which is associated with a particularly stable form of gene silencing. In addition, lytic promoters do not appear to be associated with H4K20me3 (Cliffe et al., 2009), which is a modification that is classically associated with transcriptionally silenced regions of stable, constitutive heterochromatin in mammalian cells (Jorgensen et al., 2013). A component of the repressive PRC1 complex, which is a histone reader often found enriched at sites of cellular H3K27me3, is present at very low levels, if at all, on the latent viral genome (Cliffe et al., 2012; Kwiatkowski et al., 2009). Furthermore, a recent study by Alfonso-Dunn et al. (2017) suggests that activation of the super-elongation complex by treatment with BET domain inhibitors enhances reactivation ex vivo, implicating RNA polymerase II promoter-proximal pausing as a rate-limiting step in HSV reactivation. Whether this is due to ‘poised’ RNA polymerase II on the latent viral genome clearly deserves more investigation. Taken together, this data indicates that portions of the viral genome are enriched with a form of heterochromatin that may be readily remodeled to permit rapid gene expression.
Triggers implicated in HSV reactivation
In order to regulate the delicate balance between latent infection and re-entry into the lytic phase, the latent HSV genome must sense neuronal cues that induce the up-regulation of viral lytic gene expression. However, the nature of these stimuli and how they can act on the viral genome to permit gene expression is not well understood. In the context of human disease, exposure to sunlight, psychological stress, fever, menstruation and surgical resection have all been associated with HSV reactivation (Chida and Mao, 2009; Hayderi et al., 2013; Padgett et al., 1998; Roizman and Whitley, 2013). It is likely that reactivation of HSV in a normal human host results from a combination of factors, including signals that act directly on latently infected neurons and suppression of the immune responses that typically prevent reactivation and clear replicating virus. The key role of the host immune response in HSV infection is clearly evident by the significant morbidity and mortality associated with HSV infection in immunocompromised individuals, and we refer readers to excellent reviews on the immune control of HSV (Divito et al., 2006; Egan et al., 2013; Kinchington et al., 2012). In this review, we will primarily focus on how stimuli can act directly on neurons or the viral genomes to result in reactivation.
When studying the mechanisms involved in latency and reactivation, it is important to make the distinction between the neuronal signaling pathways that trigger reactivation and the downstream factors that may act directly on the genome to modulate viral chromatin or regulate viral transcription. In the first case, stimuli are used to mimic a physiological response in an attempt to recapitulate natural in vivo reactivation episodes. In the second case, other triggers, such as down-regulation of a repressive protein or treatment with an HDAC inhibitor, potentially bypass up-stream cellular pathways and act directly on the viral chromatin (Neumann et al., 2007b). These types of stimuli result in efficient re-entry into lytic replication and prove useful for assessing the role of host proteins in maintaining latency. To add another level of complexity, depletion of a cellular protein or HDAC inhibition could also activate a cell stress response within the neuron and result in lytic gene expression, thus indirectly triggering reactivation (Dai et al., 2005). Here, we try to focus on the role of cellular signaling pathways that mimic natural stimulation of viral reactivation in humans.
As reviewed recently, the process of reactivation likely requires multiple steps (Cliffe and Wilson, 2017; Sawtell and Thompson, 2016a). In primary neuronal culture systems, it has been shown that there is an initial relaxing of the chromatin structure, which likely includes multiple changes that may or may not be accompanied by detectable increases in lytic gene transcription. After chromatin relaxation and transcription, viral proteins are synthesized followed by the production of infectious virus and progression to full reactivation (Kim et al., 2012; Sawtell and Thompson, 2016b). At each stage of the pathway, only a subset of neurons will progress and complete the subsequent step (Kim et al., 2012; Thompson et al., 2009), indicating that there are thresholds and multiple controls that regulate progression through reactivation. It is currently not well understood whether these thresholds relate to intrinsic neuronal responses, levels of viral protein synthesis or non-coding RNAs, or changes in the chromatin structure, but it is likely that many, if not all, of these factors contribute. As reviewed in Sawtell et al. (2016), the techniques for measuring HSV “reactivation” vary between different researchers and have contributed to some discrepancies in the literature (Sawtell and Thompson, 2016a). Importantly, while the end point of reactivation, defined as the active production of infectious viral particles, is the same, the intervening steps that regulate this process are likely different depending on the nature of the initiating signal. Below we summarize what we have learned from a variety of model systems on the process of reactivation in response to different stimuli.
Loss of neurotrophin-associated signaling
Using both animal models and in vitro systems, progress has been made in recapitulating stimuli that trigger reactivation in humans. However, there remain significant gaps in our understanding of how these stimuli correlate with reactivation of the virus that results in clinical disease. One pathway that has been found to stimulate HSV reactivation in multiple systems is nerve-growth factor (NGF) deprivation. In humans, this may occur during the developmental period, and particularly through adolescence, as the process of axon pruning helps correctly wire the developing nervous system. In addition, damage to innervated tissues that results in loss of the neurotrophin-producing cells or changes in neurotrophin synthesis may occur following periods of chronic stress (Eckart et al., 2013), changes in hormone levels (Kaur et al., 2007) or UV irradiation (Stefanato et al., 2003). NGF-deprivation was first found to trigger HSV reactivation in primary neuronal models of HSV latency using rat sympathetic neurons (Wilcox and Johnson, 1987). In vivo injection of anti-NGF serum into latently infected rabbits has also been shown to enhance reactivation of HSV (Hill et al., 1997). Furthermore, interruption of signals downstream of the NGF receptor triggered reactivation in a variety of in vitro models of HSV latency (Camarena et al., 2010; Cliffe et al., 2015; Kobayashi et al., 2012; Linderman et al., 2017), and have been shown to enhance explant mediated reactivation ex vivo (Du et al., 2011; Messer et al., 2015).
The changes in cell-signaling in response to NGF-deprivation have been well delineated in both primary sympathetic and sensory neurons. In immature neurons, NGF-deprivation leads to apoptosis due to up-regulation of the pro-apoptotic BH3-only proteins (Biswas et al., 2007). These proteins activate Bax, which results in mitochondrial outer member permeabilization (MOMP) and cytochrome c-mediated caspase activation. However, as neurons mature, they develop mechanisms to restrict both MOMP and downstream caspase activation, permitting their long-term survival (Kole et al., 2013). In murine sympathetic neurons, this occurs at around post-natal day 10. Interestingly, neuronal maturation appears to be intrinsic to neurons and occurs both in vivo and ex vivo, as seen in primary neurons isolated postnatally (Kole et al., 2013; Annis et al., 2016). Importantly, many studies have shown reactivation of the virus after restriction of the apoptotic pathway, suggesting that apoptosis itself does not play a significant role in HSV reactivation. This is consistent with observations that neither caspase activity nor MOMP are required for reactivation upon NGF-deprivation (Camarena et al., 2010; Cliffe et al., 2015). Therefore, signals up-stream of MOMP appear to be important for triggering HSV reactivation.
In an important study by Camarena et al. (2010), the authors investigated how interruption of cell signaling pathways by NGF-deprivation triggers HSV reactivation in a primary neuronal model of latency. Although NGF can signal through two major receptors present on sympathetic neurons, TrkA and p75, the authors found that NGF binding to the TrkA receptor sustained latency in rat sympathetic neurons. Signaling through TrkA results in the activation of three major signaling pathways: MEK/ERK, PLCγ and PI3K/AKT (Chao, 2003). However, only interruption of the PI3K/AKT pathway triggered reactivation of HSV (Camarena et al., 2010). Since this study, inhibition of PI3K signaling has been found to also trigger reactivation in primary murine sympathetic and sensory neurons (Cliffe et al., 2015; Hukkanen et al., 2015).
Inhibition of PI3K or AKT signaling results in inhibition of the mammalian target of rapamycin (mTOR) signaling pathway. mTOR is a serine/threonine kinase that is a component of two functionally distinct protein complexes, mTORC1 and mTORC2, which regulate a number of cellular processes in response to environmental signals, including cap-dependent translation and autophagy (Foster and Fingar, 2010). Direct inhibition of mTORC1 activity or transient inhibition of protein synthesis can result in HSV reactivation in rat primary neurons (Kobayashi et al., 2012). This implicates mTOR as another key mediator of the HSV lytic/latent switch in neurons, either directly by sustaining the synthesis of proteins required for the maintenance of latency or indirectly by activating pathways following protein synthesis inhibition. Inhibition of AKT also results in activation of the mixed lineage kinase protein, dual leucine zipper kinase (DLK) (Wu et al., 2015). DLK is a key mediator of apoptosis resulting from global NGF-deprivation, as well as axon pruning following axon-specific deprivation. Key targets of DLK are the cell stress proteins c-Jun N-terminal kinases (JNKs) (Tedeschi and Bradke, 2013). In neurons, JNKs are constitutively active and carry out many physiological roles, including dendritic arborization and synaptic plasticity (Coffey et al., 2000). In response to neurotrophic deprivation, JNKs are redirected to activate a cell stress response through mobilization of DLK and JNK interacting proteins 3 (JIP3) (Sengupta Ghosh et al., 2011). In a study using a mouse primary neuronal system of HSV reactivation, we found that activation of JNK via DLK/JIP3 was an essential step in induction of lytic gene expression (Cliffe et al., 2015). Thus, it is clear that multiple downstream cellular processes are involved in reactivation following inhibition of PI3K.
The ability to study the kinetics of lytic gene expression following interruption of NGF-signaling in tractable in vitro systems has highlighted key differences in the mechanism of viral gene expression from silenced latent genomes, as compared to de novo lytic infection. In this context, reactivation occurs as a two-stage program that overcomes a more compact viral chromatin structure and the absence of tegument factors, such as VP16 (reviewed in Cliffe and Wilson, 2017). During the first stage, termed phase I, there is a transient burst of lytic gene transcription. This first wave of synchronous lytic gene expression leads to the simultaneous synthesis of many lytic transcripts with the potential to encode IE, E, and L viral proteins (Kim et al., 2012). A similar synchronous wave of gene expression has been observed following ex vivo reactivation when explant was combined with NGF-deprivation (Du et al., 2011), indicating that the specific stimuli, and not experimental system, impacts the mechanism of reactivation. However, it is worth noting that NGF-deprivation combined with axotomy does not result in the exact same cellular response as NGF-deprivation performed on intact neurons. Axotomized neurons reach an earlier commitment to death than intact neurons and, unlike intact neurons, are unable to recover from mitochondrial membrane permeabilization (Fletcher et al., 2000). Therefore, further work is required to directly show that differences in the nature of viral gene expression is independent of the nature of the model used. Importantly, phase I is not dependent on the viral transactivator VP16 (Kim et al., 2012) in primary neurons. Furthermore, neither the synthesis of IE proteins nor viral DNA replication are necessary for the expression of viral E or L genes (Kim et al., 2012). Viral gene expression in phase I is dependent on JNK activity, but it is independent of histone demethylases that have been implicated in the removal of repressive H3K9 and H3K27 methylation marks. Instead, histones associated with viral lytic promotors are phosphorylated on S10 in a JNK-dependent manner, resulting in a H3K9me3pS10 histone methyl/phospho switch. This switch could permit increased viral gene expression without the need to recruit histone demethylases and allow gene silencing to re-occur if the genome does not progress to full reactivation (Cliffe et al., 2015).
The second phase of reactivation closely resembles de novo infection and is hypothesized to occur only if threshold amounts of key viral proteins are synthesized during phase I (Kim et al., 2012). Similar to de novo infection, viral gene expression during phase II is dependent on histone demethylase enzyme activity and may require the viral transactivator VP16. (Cliffe et al., 2015; Kim et al., 2012). Ultimately, phase II results in the amplification of viral DNA and production of infectious viral particles (Kim et al., 2012).
Axotomy/explant
One of the first clinical observations of a stimulus resulting in HSV reactivation was by Cushing in 1905, when he documented reactivation in a patient following trigeminal root surgery for trigeminal neuralgia (Cushing, 1905). Reactivation of HSV following various forms of surgical treatment for trigeminal neuralgia has now been well documented and is even considered an indication of successful surgery (Tenser, 2015). The most likely reason for surgery-triggered reactivation is damage to the trigeminal nerve and activation of cellular responses induced by axotomy. In animal models of latency, complete axotomy via explant of infected ganglia is a commonly used stimulus for reactivation (Sawtell and Thompson, 2004; Stevens and Cook, 1971) and, therefore, should closely mimic reactivation caused by mechanical injury of neurons in humans.
With respect to the pathways linked to HSV reactivation, the cellular response to axotomy has overlapping features with the NGF-deprivation pathway, but there are also some key differences. In the case of axotomy, calcium influx into the injured axon is a central trigger for changes in gene expression (Cho et al., 2013). The increase in intracellular calcium results in increased electrical activity of the neuron, as well as calcium-dependent modulation of cellular proteins (Rishal and Fainzilber, 2014). Calcium-dependent signaling triggers the rapid cellular changes required for neuronal survival, including membrane resealing, activation of cellular transcription factors and relocalizaiton of specific epigenetic modifiers (Mar et al., 2014; Rishal and Fainzilber, 2014). This likely accounts for the rapid changes in cellular gene expression seen in explant models of HSV reactivation (Sawtell and Thompson, 2004), as compared to other models.
One downstream effect of axotomy-driven calcium influx is activation of adenylate cyclase and increase in intracellular cAMP levels (Mahar and Cavalli, 2018). High intracellular levels of cAMP, which also can be achieved with cAMP mimics and activators of adenylate cyclase such as forskolin, has been shown to trigger HSV reactivation (Colgin et al., 2001; De Regge et al., 2010). This suggests that increased cAMP levels following axotomy may contribute to reactivation, although a dependence of cAMP in axotomy-induced reactivation has not yet been described. Following explant, increased intracellular calcium and cAMP can also trigger activation of DLK (Hao et al., 2016; Yan and Jin, 2012). A key sensor of local axon injury, DLK is required for many downstream signaling pathways that promote axon regeneration following axotomy. This is in contrast to its role in the NGF-deprivation pathway, where it mediates axon apoptosis and pruning (Geden and Deshmukh, 2016; Tedeschi and Bradke, 2013). Therefore, DLK seems to function as a key regulator of the axon response, promoting different outcomes depending on the nature of the stimulus. DLK may also be a key mediator of HSV reactivation, as one of its downstream targets, JNK, is required for HSV reactivation following explant (Cliffe et al., 2015). Although a direct role of DLK in explant-induced reactivation has not been shown, it will be important to investigate whether this protein acts as a common mediator of HSV reactivation in addition to its role as a central sensor of neuronal stress. If so, it is likely that there will different DLK-mediated reactivation pathways for different stimuli, just as there are different phenotypic outcomes for different mechanisms of DLK activation (Geden and Deshmukh, 2016).
In an NGF-deprivation model of reactivation, DLK/JIP-3-JNK pathway signaling mediates a histone methyl/phospho switch and enrichment of H3K9me3/pS10 on lytic gene promoters (Cliffe et al., 2015). It remains unclear if JNK plays a similar mechanistic role following explant of latently infected ganglia, and, as a whole, very little is known about the mechanisms linking cell signaling pathways and changes in viral chromatin following axotomy. However, a number of important observations about the viral chromatin have been made using the explant model, including changes in histone post-translational modification and changes in CTCF binding (Ertel et al., 2012; Washington et al., 2018b). Most notably, it has been shown that, following axotomy, there is a transient increase in the enrichment of euchromatic histone markers (acetylated H3K9/K14) at IE gene promoters (Amelio et al., 2006a). The mechanism responsible for the increased acetylation has not been determined but may be related to the global increases in histone acetylation that have been observed following axotomy and result from calcium-dependent nuclear export of HDAC5, along with HDAC3 (Cho et al., 2013; Fischle et al., 2002). Notably, reactivation can be triggered by inhibition of HDAC activity, which also results in similar changes in histone acetylation on viral promoters (Neumann et al., 2007a).
Following explant of sensory ganglia, there is also a rapid re-localization of host cell factor-1 (HCF-1) from the cytoplasm to the nucleus (Kristie et al., 1999). Once in the nucleus, HCF-1 quickly associates with gene enhancer domains of IE proteins, which was found to correlate with RNA polymerase II occupancy and gene expression (Whitlow and Kristie, 2009). Interestingly, HCF-1 occupancy at IE gene promoters was detected as early as 1hr post-explant, indicating a potential role of HCF-1 in the initiation of reaction transcription (Whitlow and Kristie, 2009). Furthermore, the activities of LSD-1 and JMJD2 proteins, which are histone K9 demethylases that interact with HCF-1, are required for the early induction of lytic gene expression (Liang et al., 2013; 2009). The HCF-interacting histone H3K4 methyltransferases may also be recruited to promote active euchromatin on IE promoters following explant (Kristie et al., 2010). VP16, which induces a transcriptional complex with HCF-1 and Oct-1 during lytic infection, is dispensable for explant-induced reactivation (Sears et al., 1991; Steiner et al., 1990). This is in contrast to reactivation in intact ganglia, in which VP16 is required (Thompson et al., 2009), again indicating that the mechanisms of reactivation likely differ depending on the nature of the trigger. Lastly, it has been shown that CTCF domains positioned around IE (ICP0 and ICP4) and LAT gene regions lose CTCF occupancy following explant-induced reactivation (Ertel et al., 2012; Washington et al., 2018b), suggesting that disruption of CTCF-mediated chromatin domains may contribute to reactivation.
Psychological stress
Psychological stress has been extensively linked to HSV-associated disease in humans (Chida and Mao, 2009; Cohen et al., 1999; Glaser and Kiecolt-Glaser, 1997). During times of high emotional stress, there is activation of the sympathetic nervous system and release of catecholamines, as well as activation of the hypothalamic pituitary adrenal axis and release of glucocorticoids (Carrasco and Van de Kar, 2003). The downstream actions of these two stress pathways have been shown to play significant roles in HSV latency and reactivation, and stress hormones have been well-utilized as tools to trigger efficient reactivation. Iontophoresis of epinephrine has been widely used to reactivate HSV-1 in different animal models (Creech and Neumann, 2010; Rootman et al., 1990; Willey et al., 1984). In addition, dexamethasone, a synthetic corticosteroid, stimulates reactivation of HSV-1 both ex vivo and in primary neuronal cultures (Cliffe et al., 2015; Du et al., 2012), and the closely related bovine herpesvirus 1 (BHV-1) can also be reactivated in latently infected calves by intravenous injection of dexamethasone (Workman et al., 2012).
Although stress hormone-mediated immunosuppression likely plays a role in recurrence of disease in humans, there is evidence that stress hormones can have a direct effect on neurons, as well as on HSV gene expression (Du et al., 2012; Ives and Bertke, 2017; Workman et al., 2012). During productive replication in non-neuronal cells, BHV-1 lytic gene expression is stimulated by dexamethasone (Zhu et al., 2017). Furthermore, differential expression of adrenergic and glucocorticoid receptors on sensory and autonomic neurons has been well-characterized, and differences in expression correlate with the effect of stress hormones on HSV-1 and HSV-2 replication (Ives and Bertke, 2017). The signaling pathways that ultimately result in viral gene expression in response to stress hormones are not fully understood. However, in a rabbit model of HSV latency, reactivation triggered by transcorneal iontophoresis of epinephrine results in an increase in enrichment of the euchromatin marker H3K4me2 at the ICP4 promoter region (Creech and Neumann, 2010). How this treatment results in the observed changes in the chromatin structure is not clear.
Binding of epinephrine to the β2-adrenergic receptor, which is expressed on sensory and sympathetic neurons, activates adenylate cyclase and results in increased levels of the second messenger cAMP (Gold et al., 1997; Vásquez and Lewis, 2003). Interestingly, forskolin, which also activates adenylate cyclase, is commonly used in cultured neurons to trigger HSV reactivation (Colgin et al., 2001; De Regge et al., 2010; Halford et al., 1996). While the cAMP-dependent signaling pathways that ultimately promote HSV reactivation have not been elucidated, a recent study using pseudorabies virus (PRV) found that the repression of an axon-specific infection could be overcome by addition of forskolin in a JNK-dependent manner (Koyuncu et al., 2017). Dexamethasone can also trigger JNK activity (Lee et al., 2014; Zhang et al., 2000), and dexamethasone-induced HSV reactivation in primary sympathetic neurons has been found to be JNK dependent (Cliffe et al., 2015). Although the contribution of adenylate cyclase and JNK activity to stress-hormone induced reactivation have not been fully elucidated, these observations suggest that cAMP and JNK may be key regulators of the HSV quiescence/lytic switch in response to stress. However, the activation mechanisms may be divergent and dependent on the stimulus.
Heat shock/Fever
Historically, fever has been one of the most strongly associated environmental triggers of HSV reactivation in humans (Roizman et al., 2013). However, like many stimuli, the underlying mechanisms and cellular pathways that induce this reactivation remain correlative, at best. Reactivation in response to fever could be due to a variety of different factors including direct heat stress to latently infected neurons and/or the resultant release of pyrogenic cytokines. In animal models, whole-body hyperthermia can mimic the effects of fever and stimulate the release of pyrogenic cytokine, IL-6. Transient hyperthermic stress can also trigger HSV-1 reactivation in vivo (Sawtell and Thompson, 1992), and there is evidence for a role of IL-6 secretion in response to heat stress in mice, which likely occurs due to glucocorticoid-mediated activation of the hypothalamic pituitary adrenal axis (Noisakran et al., 1998). Additional cytokines and prostaglandins produced during fever also have the capability of acting directly on neurons (Poon et al., 2015). IL-6 and prostaglandins can both trigger sensory neuron hyperstimulation (Black et al., 2018), which, at least for prostaglandins, appears to occur as a result of increased levels of cAMP (Emery et al., 2011). As mentioned previously, elevated cAMP levels can trigger HSV reactivation. Therefore, it will be interesting to explore the direct effects of pyrogenic cytokines on the latent viral genome and potential cell signaling pathways involved.
Heat shock of cultured neurons can trigger reactivation (Halford and Schaffer, 2001), and in non-neuronal systems viral lytic promoters can be activated in response to heat shock (Kushnir et al., 2009), suggesting indicating a direct, intrinsic role. The cellular heat shock response involves increased synthesis of the heat shock proteins (HSPs), along with activation of the mitogen-activated protein kinase family, including JNK, which as previously discussed has been linked to HSV reactivation. The signaling proteins important in triggering HSV reactivation resulting from heat-stress are not known, although in non-neuronal cells heat-stress activation of lytic-promoters has been linked to NF-Y (Kushnir et al., 2009).
An advantage of the hyperthermia model of HSV reactivation is the ability to induce in vivo reactivation. Using this model, Sawtell and Thompson (1992) provided evidence that reactivation was neuron-specific, only occurred in 1–3 neurons per trigeminal ganglia and led to degenerative changes that indicated cell death. Whether similar numbers of reactivating neurons occur in the sympathetic ganglia is not known. Subsequent in vivo studies using this model have indicated that de novo expression of VP16, but not ICP0 or ICP4, may be a requirement for the initiation of reactivation (Thompson et al., 2009; Thompson and Sawtell, 2006), which is in contrast to axotomy induced reactivation in which VP16 is dispensable, highlighting that the mechanism of reactivation may differ depending on the trigger. Interestingly, this study suggested that the VP16 promoter could be activated in neurons during acute infection in the absence of other viral proteins (Thompson et al., 2009), suggesting a neuronal specific pre-IE VP16 promoter that may be activated during reactivation. However, the mechanisms responsible for this non-canonical VP16 expression remain unknown. The identification of the precise region of the VP16 promoter important for pre-IE expression and the corresponding interacting cellular proteins will be key to narrowing down the cellular pathways that are responsible.
UV radiation
To model sunlight-induced reactivation, exposure of the cornea or skin to UV radiation can trigger reactivation in mice infected with the most pathogenic strains of HSV (Laycock et al., 1991; Shimeld et al., 1999). The molecular events that trigger reactivation in this model are not well understood, as UV radiation at the body surface results in multiple neuronal effects that could be relevant to reactivation. For example, UV radiation-induced danger signals at the body surface can lead to the changes in the levels of regulatory neuropeptides, neurotrophins, neurotransmitters and oxygen products that could signal to the innervating sensory and autonomic nerves (Roosterman et al., 2006). In addition, UV treatment in mice results in increased serum levels of cortisol (Han et al., 2017) and may act through a pathway that is similar to psychological stress-induced reactivation. Clearly the molecular pathways that trigger UV-induced reactivation deserve a more thorough investigation, especially when considering that this common insult can result in HSV-related disease in humans.
Conclusions and future directions
Significant progress has been made in understanding the mechanisms that underlie HSV reactivation, but considerable gaps remain in our knowledge of how different signaling pathways ultimately act on the latent genome. Although the activation status of cellular proteins, including mTORC1, adenylate cyclase, JNK and DLK, have been linked to reactivation under certain conditions, it is not clear whether these proteins are common regulators of the latent to lytic switch in response to different stimuli. However, these proteins represent potential targets for therapeutics. A particularly intriguing cellular protein is DLK because it appears to serve as a master regulator of axonal processes, including degeneration and regeneration (Tedeschi and Bradke, 2013). Highly expressed in neurons (Hirai et al., 2005; Kelkar et al., 2000), DLK is a key candidate for therapies that counteract neurodegeneration and may also be an ideal target to prevent HSV reactivation.
Interestingly, the outcome of DLK activation depends not only on the type of stimuli, but also on the location of the stimulus. This is especially highlighted by the differential effects of localized NGF deprivation. Global deprivation of NGF results in apoptosis of immature neurons, whereas axon-specific deprivation results in the degeneration of the deprived axon – a process known as axon pruning (Cusack et al., 2013; Geden and Deshmukh, 2016). Following NGF deprivation, mature neurons will restrict apoptosis but fail to restrict axon pruning (Cusack et al., 2013; Easton et al., 1997; Kole et al., 2013). Because axons of infected neurons are usually exposed to a different milieu from the soma, it will be important to investigate whether stimuli exhibit localization specific outcomes in terms of HSV reactivation, as well. It has been shown that axon-specific mTOR inhibition can trigger reactivation, indicating that inhibition of local protein synthesis in the axon is a sufficient stimulus for reactivation (Kobayashi et al., 2012). Determining whether axon-specific deprivation of neurotrophins can reactivate HSV, and, if so, whether HSV traffics down the same axon from which deprivation occurs would also be of significant physiological importance.
It is likely that there are many uncharacterized modulations of the viral chromatin structure that occur either prior to or concurrent with lytic gene expression, as well as other changes that are involved in the transition to full reactivation. In addition, little is known about the cellular chromatin remodelers, histone modifying factors and transcription factors that contribute to different triggers of reactivation. The removal of H3K27me3 from the viral genome is required for full reactivation (Cliffe et al., 2015; Messer et al., 2015), but which K27 histone demethylases are required and how these proteins are recruited to the viral genome are not known. Similarly, a JNK-dependent histone methyl/phospho switch has been shown to occur on viral lytic promoters following interruption of NGF-signaling (Cliffe et al., 2015), but the mechanisms regulating gene expression following histone methyl/phospho switches are still poorly defined (Fischle et al., 2003; Noh et al., 2014; Winter et al., 2007). Further investigation of how histone phosphorylation influences transcription of the viral genome will uncover mechanisms regulating HSV gene expression and provide insights on the control of cellular gene expression. Furthermore, there is clear evidence that the insulator protein CTCF and components of the polycomb repressive complex (Washington et al., 2018b) are evicted from sites on the viral genome following different reactivation stimuli, but the cause-and-effect relationships and mechanisms of these processes are less clear.
Likewise, the regulation of RNA polymerase recruitment and elongation during the different phases of reactivation has not been fully explored. Recent results suggest that inhibition of BET domain proteins enhances HSV reactivation following explant, indicating that formation of the RNA-polymerase super-elongation complex may promote reactivation (Alfonso-Dunn et al., 2017). In unpublished studies, we have also found that BET-domain inhibitors are sufficient to trigger robust reactivation in human differentiated and primary neurons. One interpretation of this data is that RNA polymerase II is present on HSV lytic promoters in a poised form and switches to an elongating form following a reactivation stimulus. However, the presence of RNA polymerase on lytic promoters during latency has not been shown. It is worth noting that BET domain proteins do exist on the Kaposi’s sarcoma associated herpesvirus genome during latency, where they have been found to regulate the three-dimensional genome structure (Mo et al., 2015; Stroud et al., 2017). Thus, BET domain proteins could potentially play a similar role in HSV latency.
There is now a growing body of evidence demonstrating heterogeneity in latently infected ganglia in terms of localization of viral genomes, copy numbers of viral DNA and expression of lytic and latent transcripts (Catez et al., 2012; Ma et al., 2014; Proenca et al., 2008; Sawtell, 1997). This likely results in different viral chromatin states that are more or less primed to reactivate depending on the stimulus. How this heterogeneity arises is unclear, but it could occur due to a variety of factors, including the exposure of the neuron to different cytokines and signaling molecules, the amount of infecting virus, or heterogeneity in neuronal populations themselves. There is great diversity in sensory and sympathetic neurons, which can differ in their physiological, anatomical, structural and molecular properties (DeLeón et al., 1994; Gold et al., 1997; Lallemend and Ernfors, 2012; Liu and Ma, 2011), and this may have significant implications for HSV infection. Certain sensory neuron subtypes, characterized by the presence of neurofilaments (NefH) and calcitonin gene related peptide α (CGRP), as well as positive staining with the A5 antibody, have the highest levels of LAT promoter activity, are less permissive for HSV-1 productive infection, and preferentially undergo early-phase reactivation (Bertke et al., 2009; 2011; Cabrera et al., 2018). Sympathetic and sensory neurons also vary in regards to their expression of different stress hormone receptors, neurotrophin receptors, and ion channels, which also influence how these neurons respond to cues during both acute infection and reactivation from latency (DeLeón et al., 1994; Gold et al., 1997; Lallemend and Ernfors, 2012; Liu and Q. Ma, 2011). Neurons acquire differences in their epigenetic signatures resulting from experience dependent activity (Mo et al., 2015; Stroud et al., 2017), and there is even evidence of DNA copy number variations, at least in the central nervous system (McConnell et al., 2013). In addition, there is evidence of heterogeneity in how individual neurons respond to axotomy (Hu et al., 2016), and this is likely the case for other triggers of reactivation. Following a given stimulus, only a subpopulation of neurons fully reactivates, perhaps suggesting that certain viral genomes or populations of neurons are not capable of progressing through to full reactivation. Understanding how heterogeneity contributes to reactivation potential of latently infected neurons will have significant therapeutic and basic science value.
In summary, a growing body of literature in the field has shed light on the mechanisms of HSV reactivation, but there is still an incredible amount that is not known about these processes. By using a variety of model systems, strains of virus and reactivation stimuli, studies have uncovered both commonalities and differences in the mechanisms of reactivation. Diversity in the neuronal populations, methods of infection and modes of detecting reactivation should be capitalized upon to understand a) how different stimuli can act on different viral genomes in distinct neuronal populations and b) why some genomes may be refractory to reactivation. As an extra-chromosomal, heterochromatin-associated piece of DNA, the HSV genome serves as a sensor of alterations in the neuronal environment and provides a trackable system to understand the resulting changes in gene expression. Ultimately, the goal is to better understand how the viral genome goes from a silent form to fully expressing lytic genes and develop therapies that target latent infection. One potential strategy for developing therapeutics could be to manipulate latency into a form that is completely refractory to reactivation stimuli.
Figure 1. Model of HSV reactivation in response to NGF-deprivation.
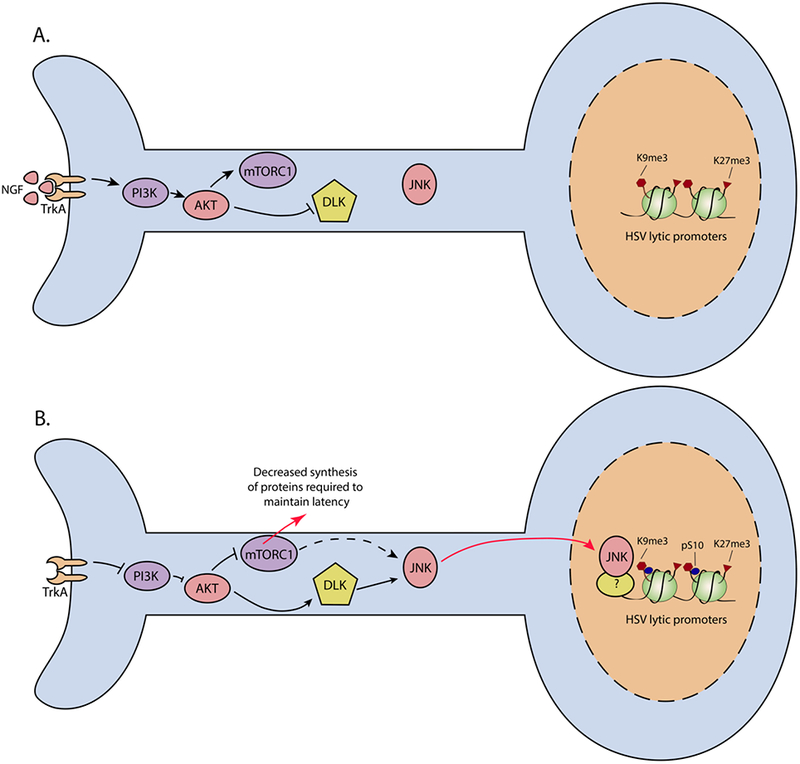
(A) In primary neurons, sustained NGF-signaling through the TrkA receptor maintains latency via activation of PI3K/AKT. AKT activation maintains mTOR activity, while inhibiting the activation of DLK. JNK is constitutively active but does not mediate a stress response. Viral lytic promoters associate with repressive histones. (B) Following loss of NGF binding to TrkA, or inhibition of PI3K activity, HSV reactivates. Inhibition of AKT results in mTOR inhibition and redirection of JNK to a cell stress role via DLK. Decreased mTOR activity could result in decreased synthesis of proteins required to maintain latency and possibly activation of JNK. JNK is found enrichened on lytic gene promoters and there is an accompanying H3K9me3pS10 histone methyl/phospho switch.
Figure 2. Model of HSV reactivation in response to axotomy/explant.
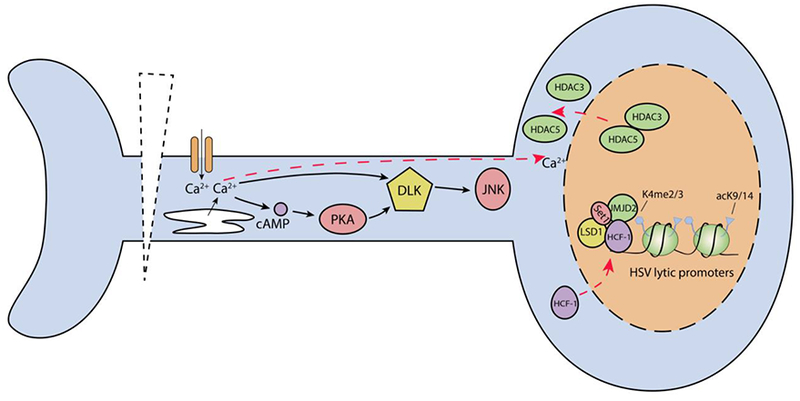
Following axotomy, there is a rapid increase in intracellular calcium levels triggering a calcium wave that is propagated to the soma. This permits rapid cellular responses, including a global increase in histone acetylation due to the export of HDAC5, along with HDAC3, from the nucleus. This may explain the increased histone acetylation observed on HSV lytic promoters following explant. Increased intracellular calcium also activates adenylate cyclase and DLK, which activates JNK. JNK activity is required for explant induced reactivation. HCF-1 has been found to rapidly translocate to the nucleus and bind to HSV IE promoters. Viral gene expression is dependent on LSD-1 and JMJD2 histone K9 demethylase activity, which are presumably recruited along with Set1, by HCF-1.
Acknowledgements
This work was supported by a grant from the National Institutes of Health to ARC (NS105630). JBS is funded by training grants T32GM007267 and T32AI007046. We thank the reviewers for their helpful comments and edits and apologize to our many colleagues whose findings could not be cited directly due to space constraints.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alfonso-Dunn R, Turner A-MW, Beltran PMJ, Arbuckle JH, Budayeva HG, Cristea IM, Kristie TM, 2017. Transcriptional Elongation of HSV Immediate Early Genes by the Super Elongation Complex Drives Lytic Infection and Reactivation from Latency. Cell Host Microbe 21, 507–517.e5. doi: 10.1016/j.chom.2017.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio AL, Giordani NV, Kubat NJ, O’Neil JE, Bloom DC, 2006a. Deacetylation of the Herpes Simplex Virus Type 1 Latency-Associated Transcript (LAT) Enhancer and a Decrease in LAT Abundance Precede an Increase in ICP0 Transcriptional Permissiveness at Early Times Postexplant. J. Virol 80, 2063–2068. doi: 10.1128/JVI.80.4.2063-2068.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amelio AL, McAnany PK, Bloom DC, 2006b. A Chromatin Insulator-Like Element in the Herpes Simplex Virus Type 1 Latency-Associated Transcript Region Binds CCCTC-Binding Factor and Displays Enhancer-Blocking and Silencing Activities. J. Virol 80, 2358–2368. doi: 10.1128/JVI.80.5.2358-2368.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Annis RP, Swahari V, Nakamura A, Xie AX, Hammond SM, Deshmukh M, 2016. Mature neurons dynamically restrict apoptosis via redundant premitochondrial brakes. FEBS J 283, 4569–4582. doi: 10.1111/febs.13944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, 2007. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. [PubMed] [Google Scholar]
- Baringer JR, Pisani P, 1994. Herpes simplex virus genomes in human nervous system tissue analyzed by polymerase chain reaction. Ann Neurol 36, 823–829. [DOI] [PubMed] [Google Scholar]
- Baringer JR, Swoveland P, 1973. Recovery of herpes-simplex virus from human trigeminal ganglions. N. Engl. J. Med 288, 648–650. doi: 10.1056/NEJM197303292881303 [DOI] [PubMed] [Google Scholar]
- Beffert U, Bertrand P, Champagne D, Gauthier S, Poirier J, 1998. HSV-1 in brain and risk of Alzheimer’s disease. Lancet 351, 1330–1331. doi: 10.1016/S0140-6736(05)79057-7 [DOI] [PubMed] [Google Scholar]
- Bertke AS, Patel A, Imai Y, Apakupakul K, Margolis TP, Krause PR, 2009. Latency-associated transcript (LAT) exon 1 controls herpes simplex virus species-specific phenotypes: reactivation in the guinea pig genital model and neuron subtype-specific latent expression of LAT. J. Virol 83, 10007–10015. doi: 10.1128/JVI.00559-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertke AS, Swanson SM, Chen J, Imai Y, Kinchington PR, Margolis TP, 2011. A5-Positive Primary Sensory Neurons Are Nonpermissive for Productive Infection with Herpes Simplex Virus 1 In Vitro. J. Virol 85, 6669–6677. doi: 10.1128/JVI.00204-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SC, Shi Y, Sproul A, Greene LA, 2007. Pro-apoptotic Bim induction in response to nerve growth factor deprivation requires simultaneous activation of three different death signaling pathways. J Biol Chem 282, 29368–29374. [DOI] [PubMed] [Google Scholar]
- Black BJ, Atmaramani R, Kumaraju R, Plagens S, Romero-Ortega M, Dussor G, Price TJ, Campbell ZT, Pancrazio JJ, 2018. Adult Mouse Sensory Neurons on Microelectrode Arrays Exhibit Increased Spontaneous and Stimulus-Evoked Activity in the Presence of Interleukin-6. J Neurophysiol jn.00158.2018. doi: 10.1152/jn.00158.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabrera JR, Charron AJ, Leib DA, 2018. Neuronal subtype determines HSV-1 Latency-Associated-Transcript (LAT) promoter activity during latency. J. Virol JVI.00430–18–34. doi: 10.1128/JVI.00430-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarena V, Kobayashi M, Kim JY, Roehm P, Perez R, Gardner J, Wilson AC, Mohr I, Chao MV, 2010. Nature and Duration of Growth Factor Signaling through Receptor Tyrosine KinasesRegulates HSV-1 Latency in Neurons. Cell Host Microbe 8, 320–330. doi: 10.1016/j.chom.2010.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrasco GA, Van de Kar LD, 2003. Neuroendocrine pharmacology of stress. Eur J Pharmacol 463, 235–272. doi: 10.1016/S0014-2999(03)01285-8 [DOI] [PubMed] [Google Scholar]
- Catez F, Picard C, Held K, Gross S, Rousseau A, Theil D, Sawtell N, Labetoulle M, Lomonte P, 2012. HSV-1 Genome Subnuclear Positioning and Associations with Host-Cell PMLNBs and Centromeres Regulate LAT Locus Transcription during Latency in Neurons. PLoS Pathog 8, e1002852. doi: 10.1371/journal.ppat.1002852.s003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao MV, 2003. Neurotrophins and their receptors: A convergence point for many signalling pathways. Nat Rev Neurosci 4, 299–309. doi: 10.1038/nrn1078 [DOI] [PubMed] [Google Scholar]
- Chida Y, Mao X, 2009. Does psychosocial stress predict symptomatic herpes simplex virus recurrence? A meta-analytic investigation on prospective studies. Brain Behav. Immun 23, 917–925. doi: 10.1016/j.bbi.2009.04.009 [DOI] [PubMed] [Google Scholar]
- Cho Y, Sloutsky R, Naegle KM, Cavalli V, 2013. Injury-Induced HDAC5 Nuclear Export Is Essential for Axon Regeneration. Cell 155, 894–908. doi: 10.1016/j.cell.2013.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Arbuckle JH, Vogel JL, Geden MJ, Rothbart SB, Cusack CL, Strahl BD, Kristie TM, Deshmukh M, 2015. Neuronal Stress Pathway Mediating a Histone Methyl/Phospho Switch Is Required for Herpes Simplex Virus Reactivation. Cell Host Microbe 18, 649–658. doi: 10.1016/j.chom.2015.11.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Coen DM, Knipe DM, 2012. Kinetics of Facultative Heterochromatin and Polycomb Group Protein Association with the Herpes Simplex Viral Genome during Establishment of Latent Infection. mBio 4, e00590–12–e00590–12. doi: 10.1128/mBio.00590-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Garber DA, Knipe DM, 2009. Transcription of the Herpes Simplex Virus Latency-Associated Transcript Promotes the Formation of Facultative Heterochromatin on Lytic Promoters. J. Virol 83, 8182–8190. doi: 10.1128/JVI.00712-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffe AR, Wilson AC, 2017. Restarting Lytic Gene Transcription at the Onset of Herpes Simplex Virus Reactivation. J. Virol 91, e01419–16–6. doi: 10.1128/JVI.01419-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey ET, Hongisto V, Dickens M, Davis RJ, Courtney MJ, 2000. Dual roles for c-Jun N-terminal kinase in developmental and stress responses in cerebellar granule neurons. J. Neurosci 20, 7602–7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen F, Kemeny ME, Kearney KA, Zegans LS, Neuhaus JM, Conant MA, 1999. Persistent stress as a predictor of genital herpes recurrence. Arch. Intern. Med 159, 2430–2436. [DOI] [PubMed] [Google Scholar]
- Colgin MA, Smith RL, Wilcox CL, 2001. Inducible Cyclic AMP Early Repressor Produces Reactivation of Latent Herpes Simplex Virus Type 1 in Neurons In Vitro. J. Virol 75, 2912–2920. doi: 10.1128/JVI.75.6.2912-2920.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SD, Paveloff MJ, Doucet JJ, Cottingham AJ, Sedarati F, Hill JM, 1991. Ocular herpes simplex virus reactivation in mice latently infected with latency-associated transcript mutants. Invest. Ophthalmol. Vis. Sci 32, 1558–1561. [PubMed] [Google Scholar]
- Creech CC, Neumann DM, 2010. Changes to Euchromatin on LAT and ICP4 Following Reactivation Are More Prevalent in an Efficiently Reactivating Strain of HSV-1. PLoS ONE 5, e15416. doi: 10.1371/journal.pone.0015416.t001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cusack CL, Swahari V, Hampton Henley W, Michael Ramsey J, Deshmukh M, 2013. Distinct pathways mediate axon degeneration during apoptosis and axon-specific pruning. Nat Commun 4, 1876. doi: 10.1038/ncomms2910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushing H, 1905. The Surgical Aspects of Major Neuralgia of the Trigeminal Nerve. A Report of Twenty Cases of Operation on the Gasserian Ganglion, with Anatomic and Physiologic Notes on the Consequence of its Removal. JAMA 44, 773–779. [Google Scholar]
- Dai Y, Rahmani M, Dent P, Grant S, 2005. Blockade of histone deacetylase inhibitor-induced RelA/p65 acetylation and NF-kappaB activation potentiates apoptosis in leukemia cells through a process mediated by oxidative damage, XIAP downregulation, and c-Jun N-terminal kinase 1 activation. Mol Cell Biol 25, 5429–5444. doi: 10.1128/MCB.25.13.5429-5444.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Regge N, Van Opdenbosch N, Nauwynck HJ, Efstathiou S, Favoreel HW, 2010. Interferon Alpha Induces Establishment of Alphaherpesvirus Latency in Sensory Neurons In Vitro. PLoS ONE 5, e13076. doi: 10.1371/journal.pone.0013076.t001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLeón M, Coveñas R, Chadi G, Narváez JA, Fuxe K, Cintra A, 1994. Subpopulations of primary sensory neurons show coexistence of neuropeptides and glucocorticoid receptors in the rat spinal and trigeminal ganglia. Brain Res 636, 338–342. [DOI] [PubMed] [Google Scholar]
- Deshmane SL, Fraser NW, 1989. During latency, herpes simplex virus type 1 DNA is associated with nucleosomes in a chromatin structure. J. Virol 63, 943–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divito S, Cherpes TL, Hendricks RL, 2006. A triple entente: virus, neurons, and CD8+ T cells maintain HSV-1 latency. Immunol. Res 36, 119–126. doi: 10.1385/IR:36:1:119 [DOI] [PubMed] [Google Scholar]
- Dressler GR, Rock DL, Fraser NW, 1987. Latent herpes simplex virus type 1 DNA is not extensively methylated in vivo. J Gen Virol 68 (Pt 6), 1761–1765. doi: 10.1099/0022-1317-68-6-1761 [DOI] [PubMed] [Google Scholar]
- Du T, Zhou G, Roizman B, 2012. Induction of apoptosis accelerates reactivation of latent HSV-1 in ganglionic organ cultures and replication in cell cultures. Proc. Natl. Acad. Sci. U.S.A 109, 14616–14621. doi: 10.1073/pnas.1212661109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du T, Zhou G, Roizman B, 2011. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc. Natl. Acad. Sci. U.S.A 108, 18820–18824. doi: 10.1073/pnas.1117203108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton RM, Deckwerth TL, Parsadanian AS, Johnson EM, 1997. Analysis of the mechanism of loss of trophic factor dependence associated with neuronal maturation: a phenotype indistinguishable from Bax deletion. J. Neurosci 17, 9656–9666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckart S, Hörtnagl H, Kronenberg G, Gertz K, Hörster H, Endres M, Hellweg R, 2013. Reduced nerve growth factor levels in stress-related brain regions of folate-deficient mice. Neurosci 245, 129–135. doi: 10.1016/j.neuroscience.2013.04.014 [DOI] [PubMed] [Google Scholar]
- Egan KP, Wu S, Wigdahl B, Jennings SR, 2013. Immunological control of herpes simplex virus infections. J Neurovirol 19, 328–345. doi: 10.1007/s13365-013-0189-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery EC, Young GT, Berrocoso EM, Chen L, McNaughton PA, 2011. HCN2 ion channels play a central role in inflammatory and neuropathic pain. Science 333, 1462–1466. doi: 10.1126/science.1206243 [DOI] [PubMed] [Google Scholar]
- Ertel MK, Cammarata AL, Hron RJ, Neumann DM, 2012. CTCF occupation of the herpes simplex virus 1 genome is disrupted at early times postreactivation in a transcription-dependent manner. J. Virol 86, 12741–12759. doi: 10.1128/JVI.01655-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, Verdin E, 2002. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell 9, 45–57. [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Allis CD, 2003. Binary switches and modification cassettes in histone biology and beyond. Nature 425, 475–479. doi: 10.1038/nature02017 [DOI] [PubMed] [Google Scholar]
- Fletcher GC, Xue L, Passingham SK, Tolkovsky AM, 2000. Death commitment point is advanced by axotomy in sympathetic neurons. J Cell Biol 150, 741–754. doi: 10.1083/jcb.150.4.741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KG, Fingar DC, 2010. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J. Biol. Chem 285, 14071–14077. doi: 10.1074/jbc.R109.094003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser NW, Lawrence WC, Wroblewska Z, Gilden DH, Koprowski H, 1981. Herpes simplex type 1 DNA in human brain tissue. Proc. Natl. Acad. Sci. U.S.A 78, 6461–6465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geden MJ, Deshmukh M, 2016. Axon degeneration: context defines distinct pathways. Curr. Opin. Neurobiol 39, 108–115. doi: 10.1016/j.conb.2016.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaser R, Kiecolt-Glaser JK, 1997. Chronic stress modulates the virus-specific immune response to latent herpes simplex virus Type 1. Ann. Behav. Med 19, 78–82. doi: 10.1007/BF02883323 [DOI] [PubMed] [Google Scholar]
- Gold MS, Dastmalchi S, Levine JD, 1997. Alpha 2-adrenergic receptor subtypes in rat dorsal root and superior cervical ganglion neurons. Pain 69, 179–190. doi: 10.1016/S0304-3959(96)03218-6 [DOI] [PubMed] [Google Scholar]
- Gordon L, McQuaid S, Cosby SL, 1996. Detection of herpes simplex virus (types 1 and 2) and human herpesvirus 6 DNA in human brain tissue by polymerase chain reaction. Clin Diagn Virol 6, 33–40. [DOI] [PubMed] [Google Scholar]
- Halford WP, Gebhardt BM, Carr DJ, 1996. Mechanisms of herpes simplex virus type 1 reactivation. J. Virol 70, 5051–5060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford WP, Schaffer PA, 2001. ICP0 is required for efficient reactivation of herpes simplex virus type 1 from neuronal latency. J. Virol 75, 3240–3249. doi: 10.1128/JVI.75.7.3240-3249.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M, Ban J-J, Bae J-S, Shin C-Y, Lee DH, Chung JH, 2017. UV irradiation to mouse skin decreases hippocampal neurogenesis and synaptic protein expression via HPA axis activation. Sci. Rep 7, 1–11. doi: 10.1038/s41598-017-15773-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao Y, Frey E, Yoon C, Wong H, Nestorovski D, Holzman LB, Giger RJ, DiAntonio A, Collins C, 2016. An evolutionarily conserved mechanism for cAMP elicited axonal regeneration involves direct activation of the dual leucine zipper kinase DLK. Elife 5, e14048. doi: 10.7554/eLife.14048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Hayderi L, Delvenne P, Rompen E, Senterre JM, Nikkels AF, 2013. Herpes simplex virus reactivation and dental procedures. Clin Oral Investig 17, 1961–1964. doi: 10.1007/s00784-013-0986-3 [DOI] [PubMed] [Google Scholar]
- Hill JM, Garza HH, Helmy MF, Cook SD, Osborne PA, Johnson EM, Thompson HW, Green LC, O’Callaghan RJ, Gebhardt BM, 1997. Nerve growth factor antibody stimulates reactivation of ocular herpes simplex virus type 1 in latently infected rabbits. J Neurovirol 3, 206–211. [DOI] [PubMed] [Google Scholar]
- Hirai S, Kawaguchi A, Suenaga J, Ono M, Cui DF, Ohno S, 2005. Expression of MUK/DLK/ZPK, an activator of the JNK pathway, in the nervous systems of the developing mouse embryo. Gene Expr Patterns 5, 517–523. doi: 10.1016/j.modgep.2004.12.002 [DOI] [PubMed] [Google Scholar]
- Hu G, Huang K, Hu Y, Du G, Xue Z, Zhu X, Fan G, 2016. Single-cell RNA-seq reveals distinct injury responses in different types of DRG sensory neurons. Sci. Rep 6, 1–11. doi: 10.1038/srep31851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukkanen V, Mattila RK, Harila K, Kangas SM, Paavilainen H, Heape AM, Mohr IJ, 2015. An investigation of herpes simplex virus type 1 latency in a novel mouse dorsal root ganglion model suggests a role for ICP34.5 in reactivation. J Gen Virol 96, 2304–2313. doi: 10.1099/vir.0.000138 [DOI] [PubMed] [Google Scholar]
- Ives AM, Bertke AS, 2017. Stress Hormones Epinephrine and Corticosterone Selectively Modulate Herpes Simplex Virus 1 (HSV-1) and HSV-2 Productive Infections in Adult Sympathetic, but Not Sensory, Neurons. J. Virol 91, e00582–17–12. doi: 10.1128/JVI.00582-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- James SH, Whitley RJ, 2010. Treatment of herpes simplex virus infections in pediatric patients: current status and future needs. Clin. Pharmacol. Ther 88, 720–724. doi: 10.1038/clpt.2010.192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen S, Schotta G, Sorensen CS, 2013. Histone H4 Lysine 20 methylation: key player in epigenetic regulation of genomic integrity. Nucleic Acids Res 41, 2797–2806. doi: 10.1093/nar/gkt012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurak I, Hackenberg M, Kim JY, Pesola JM, Everett RD, Preston CM, Wilson AC, Coen DM, 2014. Expression of Herpes Simplex Virus 1 MicroRNAs in Cell Culture Models of Quiescent and Latent Infection. J. Virol 88, 2337–2339. doi: 10.1128/JVI.03486-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur G, Janik J, Isaacson LG, Callahan P, 2007. Estrogen regulation of neurotrophin expression in sympathetic neurons and vascular targets. Brain Res. 1139, 6–14. doi: 10.1016/j.brainres.2006.12.084 [DOI] [PubMed] [Google Scholar]
- Kelkar N, Gupta S, Dickens M, Davis RJ, 2000. Interaction of a Mitogen-Activated Protein Kinase Signaling Module with the Neuronal Protein JIP3. Mol Cell Biol 20, 1030–1043. doi: 10.1128/MCB.20.3.1030-1043.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC, 2012. Transient Reversal of Episome Silencing Precedes VP16-Dependent Transcription during Reactivation of Latent HSV-1 in Neurons. PLoS Pathog 8, e1002540. doi: 10.1371/journal.ppat.1002540.s004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinchington PR, Leger AJS, Guedon J-MG, Hendricks RL, 2012. Herpes simplex virus and varicella zoster virus, the house guests who never leave. Herpesviridae 3, 1–1. doi: 10.1186/2042-4280-3-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knipe DM, Cliffe A, 2008. Chromatin control of herpes simplex virus lytic and latent infection. Nat Rev Micro 6, 211–221. doi: 10.1038/nrmicro1794 [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Wilson AC, Chao MV, Mohr I, 2012. Control of viral latency in neurons by axonal mTOR signaling and the 4E-BP translation repressor. Genes Dev 26, 1527–1532. doi: 10.1101/gad.190157.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole AJ, Annis RP, Deshmukh M, 2013. Mature neurons: equipped for survival. Cell Death Dis 4, e689. doi: 10.1038/cddis.2013.220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MF, Jurak I, Pesola JM, Boissel S, Knipe DM, Coen DM, 2011. Herpes simplex virus 1 microRNAs expressed abundantly during latent infection are not essential for latency in mouse trigeminal ganglia. Virology 417, 239–247. doi: 10.1016/j.virol.2011.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM, Liang Y, Vogel JL, 2010. Control of alpha-herpesvirus IE gene expression by HCF-1 coupled chromatin modification activities. Biochim. Biophys. Acta 1799, 257–265. doi: 10.1016/j.bbagrm.2009.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristie TM, Vogel JL, Sears AE, 1999. Nuclear localization of the C1 factor (host cell factor) in sensory neurons correlates with reactivation of herpes simplex virus from latency. Proc. Natl. Acad. Sci. U.S.A 96, 1229–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubat NJ, Amelio AL, Giordani NV, Bloom DC, 2004a. The Herpes Simplex Virus Type 1 Latency-Associated Transcript (LAT) Enhancer/rcr Is Hyperacetylated during Latency Independently of LAT Transcription. J. Virol 78, 12508–12518. doi: 10.1128/JVI.78.22.12508-12518.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubat NJ, Tran RK, McAnany P, Bloom DC, 2004b. Specific Histone Tail Modification and Not DNA Methylation Is a Determinant of Herpes Simplex Virus Type 1 Latent Gene Expression. J. Virol 78, 1139–1149. doi: 10.1128/JVI.78.3.1139-1149.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushnir AS, Davido DJ, Schaffer PA, 2009. Role of Nuclear Factor Y in Stress-Induced Activation of the Herpes Simplex Virus Type 1 ICP0 Promoter. J. Virol 84, 188–200. doi: 10.1128/JVI.01377-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowski DL, Thompson HW, Bloom DC, 2009. The Polycomb Group Protein Bmi1 Binds to the Herpes Simplex Virus 1 Latent Genome and Maintains Repressive Histone Marks during Latency. J. Virol 83, 8173–8181. doi: 10.1128/JVI.00686-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lallemend F, Ernfors P, 2012. Molecular interactions underlying the specification of sensory neurons. Trends in Neurosciences 35, 373–381. doi: 10.1016/j.tins.2012.03.006 [DOI] [PubMed] [Google Scholar]
- Laycock KA, Lee SF, Brady RH, Pepose JS, 1991. Characterization of a murine model of recurrent herpes simplex viral keratitis induced by ultraviolet B radiation. Invest. Ophthalmol. Vis. Sci 32, 2741–2746. [PubMed] [Google Scholar]
- Lee JS, Raja P, Pan D, Pesola JM, Coen DM, Knipe DM, 2018. CCCTC-Binding Factor Acts as a Heterochromatin Barrier on Herpes Simplex Viral Latent Chromatin and Contributes to Poised Latent Infection. mBio 9, e02372–17–13. doi: 10.1128/mBio.02372-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T-H, Nam JG, Lee HM, Kim B-Y, Kang M-K, Bae WY, Hur DY, Park SK, 2014. Dexamethasone Induces Apoptosis of Nasal Polyp-Derived Tissue Cultures Through JNK and p38 MAPK Activation. Clin Exp Otorhinolaryngol 7, 112–7. doi: 10.3342/ceo.2014.7.2.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA, 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J. Virol 63, 2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Arbuckle JH, Rai G, Jadhav A, Simeonov A, Maloney DJ, Kristie TM, 2013. Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Sci Transl Med 5, 167ra5–167ra5. doi: 10.1126/scitranslmed.3005145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM, 2009. Inhibition of the histone demethylase LSD1 blocks α-herpesvirus lytic replication and reactivation from latency. Nat Med 15, 1312–1317. doi: 10.1038/nm.2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linderman JA, Kobayashi M, Rayannavar V, Fak JJ, Darnell RB, Chao MV, Wilson AC, Mohr I, 2017. Immune Escape via a Transient Gene Expression Program Enables Productive Replication of a Latent Pathogen. Cell Rep 18, 1312–1323. doi: 10.1016/j.celrep.2017.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ma Q, 2011. Generation of somatic sensory neuron diversity and implications on sensory coding. Curr. Opin. Neurobiol 21, 52–60. doi: 10.1016/j.conb.2010.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma JZ, Russell TA, Spelman T, Carbone FR, Tscharke DC, 2014. Lytic Gene Expression Is Frequent in HSV-1 Latent Infection and Correlates with the Engagement of a Cell-Intrinsic Transcriptional Response. PLoS Pathog 10, e1004237. doi: 10.1371/journal.ppat.1004237.s014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahar M, Cavalli V, 2018. Intrinsic mechanisms of neuronal axon regeneration. Nat Rev Neuro Jun;19(6):323–337. doi: 10.1038/s41583-018-0001-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mar FM, Bonni A, Sousa MM, 2014. Cell intrinsic control of axon regeneration. EMBO Rep 15, 254–263. doi: 10.1002/embr.201337723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus A, Lebenthal-Loinger I, Yang IH, Kinchington PR, Goldstein RS, 2015. An In Vitro Model of Latency and Reactivation of Varicella Zoster Virus in Human Stem Cell-Derived Neurons. PLoS Pathog 11, e1004885–22. doi: 10.1371/journal.ppat.1004885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, Lindberg MR, Brennand KJ, Piper JC, Voet T, Cowing-Zitron C, Shumilina S, Lasken RS, Vermeesch JR, Hall IM, Gage FH, 2013. Mosaic copy number variation in human neurons. Science 342, 632–637. doi: 10.1126/science.1243472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McQuillan G, Kruszon-Moran D, Flagg EW, Paulose-Ram R, 2018. Prevalence of Herpes Simplex Virus Type 1 and Type 2 in Persons Aged 14–49: United States, 2015–2016. NCHS Data Brief 1–8. [PubMed] [Google Scholar]
- Messer HGP, Jacobs D, Dhummakupt A, Bloom DC, 2015. Inhibition of H3K27me3-Specific Histone Demethylases JMJD3 and UTX Blocks Reactivation of Herpes Simplex Virus 1 in Trigeminal Ganglion Neurons. J. Virol 89, 3417–3420. doi: 10.1128/JVI.03052-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo A, Mukamel EA, Davis FP, Luo C, Henry GL, Picard S, Urich MA, Nery JR, Sejnowski TJ, Lister R, Eddy SR, Ecker JR, Nathans J, 2015. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 86, 1369–1384. doi: 10.1016/j.neuron.2015.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann DM, Bhattacharjee PS, Giordani NV, Bloom DC, Hill JM, 2007a. In Vivo Changes in the Patterns of Chromatin Structure Associated with the Latent Herpes Simplex Virus Type 1 Genome in Mouse Trigeminal Ganglia Can Be Detected at Early Times after Butyrate Treatment. J. Virol 81, 13248–13253. doi: 10.1128/JVI.01569-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann DM, Bhattacharjee PS, Hill JM, 2007b. Sodium Butyrate: a Chemical Inducer of In Vivo Reactivation of Herpes Simplex Virus Type 1 in the Ocular Mouse Model. J. Virol 81, 6106–6110. doi: 10.1128/JVI.00070-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll MP, Hann W, Shivkumar M, Harman LER, Connor V, Coleman HM, Proença JT, Efstathiou S, 2016. The HSV-1 Latency-Associated Transcript Functions to Repress Latent Phase Lytic Gene Expression and Suppress Virus Reactivation from Latently Infected Neurons. PLoS Pathog 12, e1005539. doi: 10.1371/journal.ppat.1005539.s007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noh K-M, Maze I, Zhao D, Xiang B, Wenderski W, Lewis PW, Shen L, Li H, Allis CD, 2014. ATRX tolerates activity-dependent histone H3 methyl/phos switching to maintain repetitive element silencing in neurons. Proc. Natl. Acad. Sci. U.S.A 112, 201411258–6827. doi: 10.1073/pnas.1411258112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noisakran S, Halford WP, Veress L, Carr DJ, 1998. Role of the hypothalamic pituitary adrenal axis and IL-6 in stress-induced reactivation of latent herpes simplex virus type 1. J. Immunol 160, 5441–5447. [PubMed] [Google Scholar]
- Padgett DA, Sheridan JF, Dorne J, Berntson GG, Candelora J, Glaser R, 1998. Social stress and the reactivation of latent herpes simplex virus type 1. Proc. Natl. Acad. Sci. U.S.A 95, 7231–7235. doi: 10.1073/pnas.95.12.7231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poon DC, Ho YS, Chiu K, Wong HL, Chang RCC, 2015. Sickness: From the focus on cytokines, prostaglandins, and complement factors to the perspectives of neurons. Neurosci Biobehav Rev 57, 30–45. doi: 10.1016/j.neubiorev.2015.07.015 [DOI] [PubMed] [Google Scholar]
- Pourchet A, Modrek AS, Placantonakis DG, Mohr I, Wilson AC, 2017. Modeling HSV-1 Latency in Human Embryonic Stem Cell-Derived Neurons. Pathog 6. doi: 10.3390/pathogens6020024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proenca JT, Coleman HM, Connor V, Winton DJ, Efstathiou S, 2008. A historical analysis of herpes simplex virus promoter activation in vivo reveals distinct populations of latently infected neurones. J Gen Virol.89, 2965–2974. doi: 10.1099/vir.0.2008/005066-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja P, Lee JS, Pan D, Pesola JM, Coen DM, Knipe DM, 2016. A Herpesviral Lytic Protein Regulates the Structure of Latent Viral Chromatin. mBio 7, e00633–16–10. doi: 10.1128/mBio.00633-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymon HK, Thode S, Zhou J, Friedman GC, Pardinas JR, Barrere C, Johnson RM, Sah DW, 1999. Immortalized human dorsal root ganglion cells differentiate into neurons with nociceptive properties. J. Neurosci 19, 5420–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter ER, Dias JK, Gilbert JE II, Atherton SS, 2009. Distribution of Herpes Simplex Virus Type 1 and Varicella Zoster Virus in Ganglia of the Human Head and Neck. J. Infect. Dis 200, 1901–1906. doi: 10.1086/648474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rishal I, Fainzilber M, 2014. Axon-soma communication in neuronal injury. Nat Rev Neurosci 15(1):32–42. doi: 10.1038/nrn3609. [DOI] [PubMed] [Google Scholar]
- Roizman B, Knipe DM, Whitley R, 2013. Herpes simplex viruses, in: Fields Virology. Lippincott Williams & Wilkins, Philadelphia. [Google Scholar]
- Roizman B, Whitley RJ, 2013. An Inquiry into the Molecular Basis of HSV Latency and Reactivation. Annu. Rev. Microbiol 67, 355–374. doi: 10.1146/annurev-micro-092412-155654 [DOI] [PubMed] [Google Scholar]
- Roosterman D, Goerge T, Schneider SW, Bunnett NW, Steinhoff M, 2006. Neuronal control of skin function: the skin as a neuroimmunoendocrine organ. Physiol. Rev 86, 1309–1379. doi: 10.1152/physrev.00026.2005 [DOI] [PubMed] [Google Scholar]
- Rootman DS, Haruta Y, Hill JM, 1990. Reactivation of HSV-1 in primates by transcorneal iontophoresis of adrenergic agents. Invest. Ophthalmol. Vis. Sci 31, 597–600. [PubMed] [Google Scholar]
- Sawtell NM, 1997. Comprehensive quantification of herpes simplex virus latency at the single-cell level. J. Virol 71, 5423–5431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL, 2016a. Herpes simplex virus and the lexicon of latency and reactivation: a call for defining terms and building an integrated collective framework. F1000Res 5, 2038–9. doi: 10.12688/f1000research.8886.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL, 2016b. De Novo Herpes Simplex Virus VP16 Expression Gates a Dynamic Programmatic Transition and Sets the Latent/Lytic Balance during Acute Infection in Trigeminal Ganglia. PLoS Pathog 12, e1005877–27. doi: 10.1371/journal.ppat.1005877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL, 2004. Comparison of Herpes Simplex Virus Reactivation in Ganglia In Vivo and in Explants Demonstrates Quantitative and Qualitative Differences. J. Virol 78, 7784–7794. doi: 10.1128/JVI.78.14.7784-7794.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawtell NM, Thompson RL, 1992. Rapid in vivo reactivation of herpes simplex virus in latently infected murine ganglionic neurons after transient hyperthermia. J. Virol 66, 2150–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sears AE, Hukkanen V, Labow MA, Levine AJ, Roizman B, 1991. Expression of the herpes simplex virus 1 alpha transinducing factor (VP16) does not induce reactivation of latent virus or prevent the establishment of latency in mice. J. Virol 65, 2929–2935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta Ghosh A, Wang B, Pozniak CD, Chen M, Watts RJ, Lewcock JW, 2011. DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J Cell Biol 194, 751–764. doi: 10.1083/jcb.201103153.dv [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimeld C, Easty DL, Hill TJ, 1999. Reactivation of herpes simplex virus type 1 in the mouse trigeminal ganglion: an in vivo study of virus antigen and cytokines. J. Virol 73, 1767–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimeld C, Whiteland JL, Nicholls SM, Easty DL, Hill TJ, 1996. Immune cell infiltration in corneas of mice with recurrent herpes simplex virus disease. J Gen Virol 77 (Pt 5), 977–985. doi: 10.1099/0022-1317-77-5-977 [DOI] [PubMed] [Google Scholar]
- Stefanato CM, Yaar M, Bhawan J, Phillips TJ, Kosmadaki MG, Botchkarev V, Gilchrest BA, 2003. Modulations of nerve growth factor and Bcl-2 in ultraviolet-irradiated human epidermis. J. Cutan. Pathol 30, 351–357. [DOI] [PubMed] [Google Scholar]
- Steiner I, Spivack JG, Deshmane SL, Ace CI, Preston CM, Fraser NW, 1990. A herpes simplex virus type 1 mutant containing a nontransinducing Vmw65 protein establishes latent infection in vivo in the absence of viral replication and reactivates efficiently from explanted trigeminal ganglia. J. Virol 64, 1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JG, Cook ML, 1971. Restriction of herpes simplex virus by macrophages. An analysis of the cell-virus interaction. J. Exp. Med 133, 19–38. doi: 10.1084/jem.133.1.19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens JG, Wagner EK, Devi-Rao GB, Cook ML, Feldman LT, 1987. RNA complementary to a herpesvirus alpha gene mRNA is prominent in latently infected neurons. Science 235, 1056–1059. [DOI] [PubMed] [Google Scholar]
- Stroud H, Su SC, Hrvatin S, Greben AW, Renthal W, Boxer LD, Nagy MA, Hochbaum DR, Kinde B, Gabel HW, Greenberg ME, 2017. Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 171, 1151–1154.e16. doi: 10.1016/j.cell.2017.09.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tedeschi A, Bradke F, 2013. The DLK signalling pathway--a double-edged sword in neural development and regeneration. EMBO Rep 14, 605–614. doi: 10.1038/embor.2013.64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenser RB, 2015. Occurrence of Herpes Simplex Virus Reactivation Suggests a Mechanism of Trigeminal Neuralgia Surgical Efficacy. World Neurosurg 84, 279–282. doi: 10.1016/j.wneu.2015.03.022 [DOI] [PubMed] [Google Scholar]
- Thellman NM, Botting C, Madaj Z, Triezenberg SJ, 2017. An Immortalized Human Dorsal Root Ganglion Cell Line Provides a Novel Context To Study Herpes Simplex Virus 1 Latency and Reactivation. J. Virol 91, e00080–17–18. doi: 10.1128/JVI.00080-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thellman NM, Triezenberg SJ, 2017. Herpes Simplex Virus Establishment, Maintenance, and Reactivation: In Vitro Modeling of Latency. Pathog 6, 28–14. doi: 10.3390/pathogens6030028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Preston CM, Sawtell NM, 2009. De Novo Synthesis of VP16 Coordinates the Exit from HSV Latency In Vivo. PLoS Pathog 5, e1000352. doi: 10.1371/journal.ppat.1000352.g007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM, 2006. Evidence that the Herpes Simplex Virus Type 1 ICP0 Protein Does Not Initiate Reactivation from Latency In Vivo. J. Virol 80, 10919–10930. doi: 10.1128/JVI.01253-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RL, Sawtell NM, 2001. Herpes Simplex Virus Type 1 Latency-Associated Transcript Gene Promotes Neuronal Survival. J. Virol 75, 6660–6675. doi: 10.1128/JVI.75.14.6660-6675.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trousdale MD, Steiner I, Spivack JG, Deshmane SL, Brown SM, MacLean AR, Subak-Sharpe JH, Fraser NW, 1991. In vivo and in vitro reactivation impairment of a herpes simplex virus type 1 latency-associated transcript variant in a rabbit eye model. J. Virol 65, 6989–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR, 2008. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 23, 187. doi: 10.1038/nature07103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valtcheva MV, Copits BA, Davidson S, Sheahan TD, Pullen MY, McCall JG, Dikranian K, Gereau RW, 2016. Surgical extraction of human dorsal root ganglia from organ donors and preparation of primary sensory neuron cultures. Nat Protoc 11, 1877–1888. doi: 10.1038/nprot.2016.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vásquez C, Lewis DL, 2003. The β2-adrenergic receptor specifically sequesters gs but signals through both Gs and Gi/o in rat sympathetic neurons. Neurosci 118, 603–610. doi: 10.1016/S0306-4522(03)00024-1 [DOI] [PubMed] [Google Scholar]
- Vicetti Miguel RD, Sheridan BS, Harvey SAK, Schreiner RS, Hendricks RL, Cherpes TL, 2010. 17- Estradiol Promotion of Herpes Simplex Virus Type 1 Reactivation Is Estrogen Receptor Dependent. J. Virol 84, 565–572. doi: 10.1128/JVI.01374-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q-Y, Zhou C, Johnson KE, Colgrove RC, Coen DM, Knipe DM, 2005. Herpesviral latency-associated transcript gene promotes assembly of heterochromatin on viral lytic-gene promoters in latent infection. Proc Natl Acad Sci U S A 102, 16055–16059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren KG, Brown SM, Wroblewska Z, Gilden D, Koprowski H, Subak-Sharpe J, 1978. Isolation of latent herpes simplex virus from the superior cervical and vagus ganglions of human beings. N. Engl. J. Med 298, 1068–1069. doi: 10.1056/NEJM197805112981907 [DOI] [PubMed] [Google Scholar]
- Washington SD, Edenfield SI, Lieux C, Watson ZL, Taasan SM, Dhummakupt A, Bloom DC, Neumann DM, 2018a. Depletion of the Insulator Protein CTCF Results in Herpes Simplex Virus 1 Reactivation In Vivo. J. Virol 92, e00173–18–15. doi: 10.1128/JVI.00173-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washington SD, Musarrat F, Ertel MK, Backes GL, Neumann DM, 2018b. CTCF Binding Sites in the Herpes Simplex Virus 1 Genome Display Site-Specific CTCF Occupation, Protein Recruitment, and Insulator Function. J. Virol 92, e00156–18–15. doi: 10.1128/JVI.00156-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlow ZW, Kristie TM, 2009. Recruitment of the Transcriptional Coactivator HCF-1 to Viral Immediate-Early Promoters during Initiation of Reactivation from Latency of Herpes Simplex Virus Type 1. J. Virol 83, 9591–9595. doi: 10.1128/JVI.01115-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Johnson EM, 1987. Nerve growth factor deprivation results in the reactivation of latent herpes simplex virus in vitro. J. Virol 61, 2311–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox CL, Smith RL, Freed CR, Johnson EM, 1990. Nerve growth factor-dependence of herpes simplex virus latency in peripheral sympathetic and sensory neurons in vitro. J. Neurosci 10, 1268–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willey DE, Trousdale MD, Nesburn AB, 1984. Reactivation of murine latent HSV infection by epinephrine iontophoresis. Invest. Ophthalmol. Vis. Sci 25, 945–950. [PubMed] [Google Scholar]
- Wilson AC, Mohr I, 2012. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol 20, 1–8. doi: 10.1016/j.tim.2012.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter S, Simboeck E, Fischle W, Zupkovitz G, Dohnal I, Mechtler K, Ammerer G, Seiser C, 2007. 14-3-3 Proteins recognize a histone code at histone H3 and are required for transcriptional activation. EMBO J 27, 88–99. doi: 10.1038/sj.emboj.7601954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman A, Eudy J, Smith L, da Silva LF, Sinani D, Bricker H, Cook E, Doster A, Jones C, 2012. Cellular transcription factors induced in trigeminal ganglia during dexamethasone-induced reactivation from latency stimulate bovine herpesvirus 1 productive infection and certain viral promoters. J. Virol 86, 2459–2473. doi: 10.1128/JVI.06143-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C-C, Wu H-J, Wang C-H, Lin C-H, Hsu S-C, Chen Y-R, Hsiao M, Schuyler SC, Lu FL, Ma N, Lu J, 2015. Akt suppresses DLK for maintaining self-renewal of mouse embryonic stem cells. Cell Cycle 14, 1207–1217. doi: 10.1080/15384101.2015.1014144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan D, Jin Y, 2012. Regulation of DLK-1 Kinase Activity by Calcium-Mediated Dissociation from an Inhibitory Isoform. Neuron 76, 534–548. doi: 10.1016/j.neuron.2012.08.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanez A, Harrell T, Sriranganathan H, Ives A, Bertke A, 2017. Neurotrophic Factors NGF, GDNF and NTN Selectively Modulate HSV1 and HSV2 Lytic Infection and Reactivation in Primary Adult Sensory and Autonomic Neurons. Pathog 6, 5–13. doi: 10.3390/pathogens6010005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JP, Wong CK, Lam CWK, 2000. Role of caspases in dexamethasone-induced apoptosis and activation of c-Jun NH2-terminal kinase and p38 mitogen-activated protein kinase in human eosinophils. Clin Exp Immunol 122, 20–27. doi: 10.1046/j.1365-2249.2000.01344.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Thompson J, Ma F, Eudy J, Jones C, 2017. Effects of the synthetic corticosteroid dexamethasone on bovine herpesvirus 1 productive infection. Virology 505, 71–79. doi: 10.1016/j.virol.2017.02.012 [DOI] [PubMed] [Google Scholar]