

Abstract
Background
Peach (Prunus persica) is an important fruit crop that generally softens rapidly after harvest resulting in a short shelf-life. By contrast, stony hard (SH) peach fruit does not soften and hardly produces ethylene. To explore the candidate genes responsible for the SH phenotype, a high-density genetic map was constructed by restriction-site associated DNA sequencing technology.
Results
In the present study, the linkage map consisted of 1310 single nucleotide polymorphism markers, spanning 454.2 cM, with an average marker distance of 0.347 cM. The single nucleotide polymorphisms were able to anchor eight linkage groups to their corresponding chromosomes. Based on this high-density integrated peach linkage map and two years of fruit phenotyping, two potential quantitative trait loci for the SH trait were identified and positioned on the genetic map. Additionally, Prupe.6G150900.1, a key gene in abscisic acid (ABA) biosynthesis, displayed a differential expression profile identical to the ABA accumulation pattern: mRNA transcripts were maintained at a high level during storage of SH peaches but occurred at low levels in melting fruit.
Conclusion
Thus Prupe.6G150900.1 might play a crucial role in the SH phenotype of peach in which ABA negatively regulates ethylene production. Also, this high-density linkage map of peach will contribute to the mapping of important fruit traits and quantitative trait loci identification.
Electronic supplementary material
The online version of this article (10.1186/s12864-018-4952-y) contains supplementary material, which is available to authorized users.
Keywords: Peach, Stony hard, Restriction-site associated DNA sequencing, Genetic map, Quantitative trait loci, Gene expression
Background
Peach [Prunus persica (L.) Batsch] is a typical climacteric fruit, and it generally softens rapidly after harvest, resulting in a short shelf-life that unfavorably affects its market value [1, 2]. Therefore, peach fruit texture is an important quality in the breeding of fresh market varieties.
To date, peach cultivars have been classified into three flesh texture types, melting (M), no-melting (NM) and stony hard (SH) based on the characteristics of fruit firmness and texture changes during peach ripening and softening [3–5]. Generally, M fruit are characterized by their prominent softening after harvest, NM fruits are characterized by slow softening at the later stages of ripening and never melt, while the SH type does not produce ethylene and barely softens after harvest (both on- and off-tree) [3, 4, 6]. A genetic analysis indicated that the SH genotype is controlled by a single recessive gene [5] and is epistatic to the locus [4].
In general, the softening of climacteric fruit, is related to endo-polygalacturonase enzyme activity, and is induced by ethylene [7–9]. A low level of ethylene production may contribute to the suppression of fruit softening in SH peach cultivars, and this depends on the suppressed transcription of PpACS1 [10]. The suppression of PpACS1 is caused by low indole-3-acetic acid concentrations in SH peaches [11]. PpYUC11 may play an essential role in auxin biosynthesis during peach fruit ripening and is a candidate gene for controlling the SH phenotype in peach [6]. However, Dong et al. [12] showed that ABI4-mediated the transcriptional repression of the ethylene biosynthesis genes ACS4 and ACS8 in Arabidopsis. Li et al. [13] suggested that in ethylene over-producer mutants, abscisic acid (ABA) treatments suppress ACS5 transcripts levels and the ethylene content, indicating that ethylene production is determined by various metabolic pathways.
The advancements in next-generation sequencing (NGS) technologies coupled with continually reducing costs, offers unprecedented conditions for genome-wide marker development and genotyping by sequencing [14]. Restriction-site associated DNA sequencing (RAD-seq) is a foremost NGS technology for high-throughput genotyping [15], in which a reduced representation of the genome produced by the DNA flanking specific a restriction enzyme is sequenced, and the reduced genome is bound to an adapter containing multiplex identifiers to form reduced-representation libraries [15–17]. To date, high-density genetic maps of many species, including eggplant [18], ryegrass [19], barley [20], grape [16], sesame [17], pear [21], apple [22] and cowpea [15], have been constructed using the RAD-seq method. However, RAD-seq technology is barely applied in peach.
To clarify the mechanism responsible for the SH phenotype and improve fruit quality, especially texture by peach breeding, further research is needed. Currently, genetic maps constructed with molecular markers have been constructed to detect the genomic loci or genes related to the traits of interest may provide efficient methods [23]. To explore the candidate gene which controls the SH phenotype, the phenotype-related quantitative trait loci (QTLs) were identified through a high-density single nucleotide polymorphism (SNP) linkage map of the peach genome with the RAD-seq technology and a family of 103 F1 lines phenotypic identification.
Results
Phenotypic identification
In the present study, we identified 42 SH and 49 M fruit trees based on the ethylene production phenotype during storage at room temperature in 2016, and individuals 5, 37, 39, 42, 63, 67, 69, 70, 71, 88, 102 and 103 were unidentified due to less fruits (Fig. 1 and Additional file 1a). In addition, all 42 SH fruit tree and 12 unidentified trees were identified according to ethylene production in 2017. The 42 SH fruit trees identified in 2016 also exhibited the SH phenotype in 2017 (hardly synthesizing ethylene), and trees 5, 37, 70, 88 and 103 were identified as SH phenotype in 2017 according to ethylene production. Trees 63, 67 and 102 were identified as M phenotype, while trees 39, 42, 69 and 71 remained unidentified owing to less fruits in 2017 (Fig. 2). Furthermore, 45 fruit trees were identified as SH phenotype and 47 fruit trees as M phenotype based on the phenotype of firmness in 2017, while 11 fruit trees remained unidentified (39, 42, 57, 59, 69, 71, 73, 76, 78, 93 and 95) owning to less fruits (Fig. 3 and Additional file 1b). The segregation of the SH phenotype in the F1 family (103) of the cross between ‘Yumyeoung’ (YM) (female) and ‘Hujingmilu’ (HJML) (male) presented 1:1 ratio (Table 1) based on both ethylene production and fruit firmness, which indicated that the SH phenotype may be controlled by a single recessive gene. Finally, the phenotypic identifications based on ethylene production in 2016 and 2017 were used for a QTL analysis (Additional file 1c).
Fig. 1.

Ethylene production in F1 offspring during storage at room temperature (2016). Numbers on the x-axis represent individuals; the y-axis represents ethylene production; different colors represent the days after harvest when samples were measured; The letter ‘S’, ‘M’ and ‘N’ represent stony hard phenotype, melting phenotype and unidentified phenotype, respectively. Data are means ± SEs (n = 3)
Fig. 2.

Ethylene production in F1 offspring during storage at room temperature (2017). Numbers on the x-axis represent individuals; the y-axis represents ethylene production; different colors represent the days after harvest when samples were measured; The letter ‘S’, ‘M’ and ‘N’ represent stony hard phenotype, melting phenotype and unidentified phenotype, respectively. Individuals of 16, 80 and 98 as a contrast. Data are means SEs (n = 3)
Fig. 3.

Fruit firmness of F1 offspring during storage at room temperature (2017). Numbers on the x-axis represent individuals; the y-axis represents fruit firmness; different colors represent the days after harvest when samples were measured; The letter ‘S’, ‘M’ and ‘N’ represent stony hard phenotype, melting phenotype and unidentified phenotype, respectively. Data are means ± SEs (n = 3)
Table 1.
Segregation of the SH phenotype in F1 population (103) of the cross between ‘YM’ and ‘HJML’
| Year | based trait | SHa | Mb | Unidentified | Expected rate | χ2c |
|---|---|---|---|---|---|---|
| 2016 | ethylene | 42 | 49 | 12 | 1:1 | 1.650 |
| 2017 | firmness | 45 | 47 | 11 | 1:1 | 1.010 |
aSH, stony hard
bM, melting
cχ2 represents the chi-squared value with the P-value set at 0.05
ABA content determination
There is no significant difference in the early stage of storage between the SH fruit ‘YM’ and the M fruit of ‘HJML’ of the ABA content, but significantly higher in ‘YM’ cultivar than that in ‘HJML’ cultivar during the later storage period (Fig. 4).
Fig. 4.
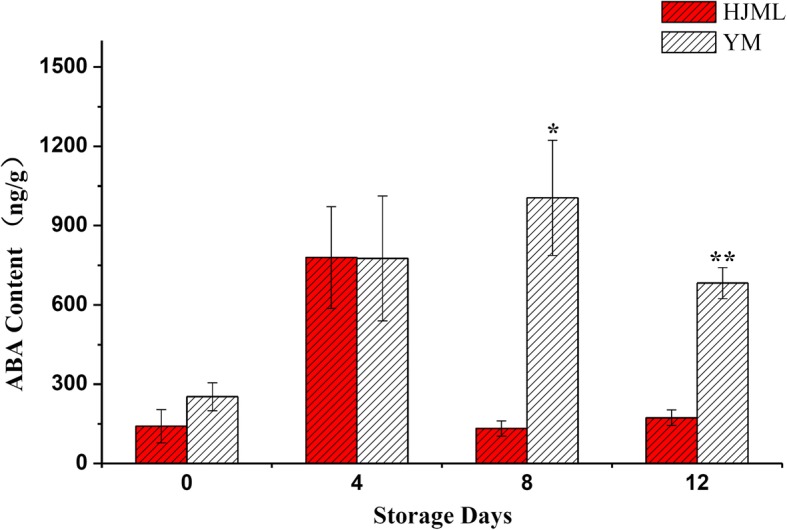
ABA contents of ‘HJML’ and ‘YM’ during storage at room temperature. The x-axis represents the storage days after harvest; the y-axis represents the ABA content. Data are means ± SEs (n = 3). Significant differences (p < 0.05) between means are indicated by the symbol ‘*’ and significant differences (p < 0.01) between means are indicated by the symbol ‘**’
RAD-seq and linkage map construction
After filtering, there were 8550.55 million clean reads, consisting of ∼85.51 Gb, that were used to generate RAD tags (Additional file 2). Thus, the reads ensured that more than 91 and 96% of the nucleotides had a quality values above Q30 (equal to a 0.1% sequencing error) and Q20 (equal to a 1% sequencing error), respectively (Additional file 2). The GC content for both parents and offspring were ∼38.7%. A total of 6150 SNP markers were used for linkage map construction (Additional file 3). As a result, 1310 markers were selected and mapped onto eight different linkage groups (LGs), covering 454.2 cM of the peach genome and having an average distance of 0.347 cM between adjacent markers (Table 2 and Fig. 5). The length of individual LGs ranged from 36.9 cM (LG7) to 83.0 cM (LG3), with inter-locus distances of between 0.18 (LG7) and 0.809 cM (LG5). LG1 was the densest, having 212 SNP loci, while LG5 had the least number of SNP loci (55). A linkage map including genetic distances and loci names, associated with SNP marker positions in different LGs is presented in Fig. 5.
Table 2.
Distribution of mapped markers among eight linkage groups in peach
| Linkage group | Number of markers | Length (cM) | Average distance (cM) |
|---|---|---|---|
| LG1 | 212 | 41.9 | 0.198 |
| LG2 | 129 | 65.3 | 0.506 |
| LG3 | 195 | 83.0 | 0.426 |
| LG4 | 119 | 55.7 | 0.468 |
| LG5 | 55 | 44.5 | 0.809 |
| LG6 | 186 | 74.2 | 0.399 |
| LG7 | 205 | 36.9 | 0.180 |
| LG8 | 209 | 52.7 | 0.252 |
| Total | 1310 | 454.2 | |
| Average | 163.75 | 56.775 | 0.347 |
Fig. 5.

Integrated linkage groups 1 to 8 for ‘YM’ × ‘HJML’. Marker names are shown to the right of each group, and map distances (in cM) are shown on the left. Red symbols represent the position of QTLs
SNP and QTL analyses
Overall, 51,253 polymorphic SNPs were identified between the two parental genotypes and those of their individual offspring (Additional file 4). To characterize the SNPs in peach, we determined the percentages of different SNP types. Among these SNPs, the dominant types were transitions, and the A/G and C/T types represented to 32.1 and 31.9% of the SNPs, respectively. The other four SNP types were the trans-versions A/T, A/C, G/T, and G/C. Their proportions varied from 6.9 to 10.0%, accounting for 36% of all SNPs (Table 3). All of the SNP types present in each individual are summarized in Additional file 5. A High-Density Genetic Map constructed using two years of fruit phenotyping was employed to identify SH phenotype-linked QTL in peach. Two SH phenotypic-related QTLs were found in one linkage group (LG6) using the ICIM-ADD method (Table 4).
Table 3.
Statistics of all SNP types in the parent plants and their offspring
| SNP types | Numbers | Ratio (%) |
|---|---|---|
| A/T | 274, 225 | 10.0 |
| A/G | 879, 057 | 32.1 |
| A/C | 256, 293 | 9.4 |
| G/T | 265, 966 | 9.7 |
| G/C | 188, 305 | 6.9 |
| C/T | 874, 086 | 31.9 |
| Total SNP numbers | 273, 793, 2 |
Table 4.
Analysis of SH phenotypic-related QTLs in peach
| Trait name | Group | Left marker | Right marker | LODa | PVE(%)b |
|---|---|---|---|---|---|
| SH | 6 | Pp06_14932683 | Pp06_15264452 | 9.2038 | 44.19% |
| SH | 6 | Pp06_14742390 | Pp06_12326708 | 2.5097 | 42.29% |
aLOD, likelihood of odds
bPVE, phenotypic variation explained
Functional analysis of candidate genes
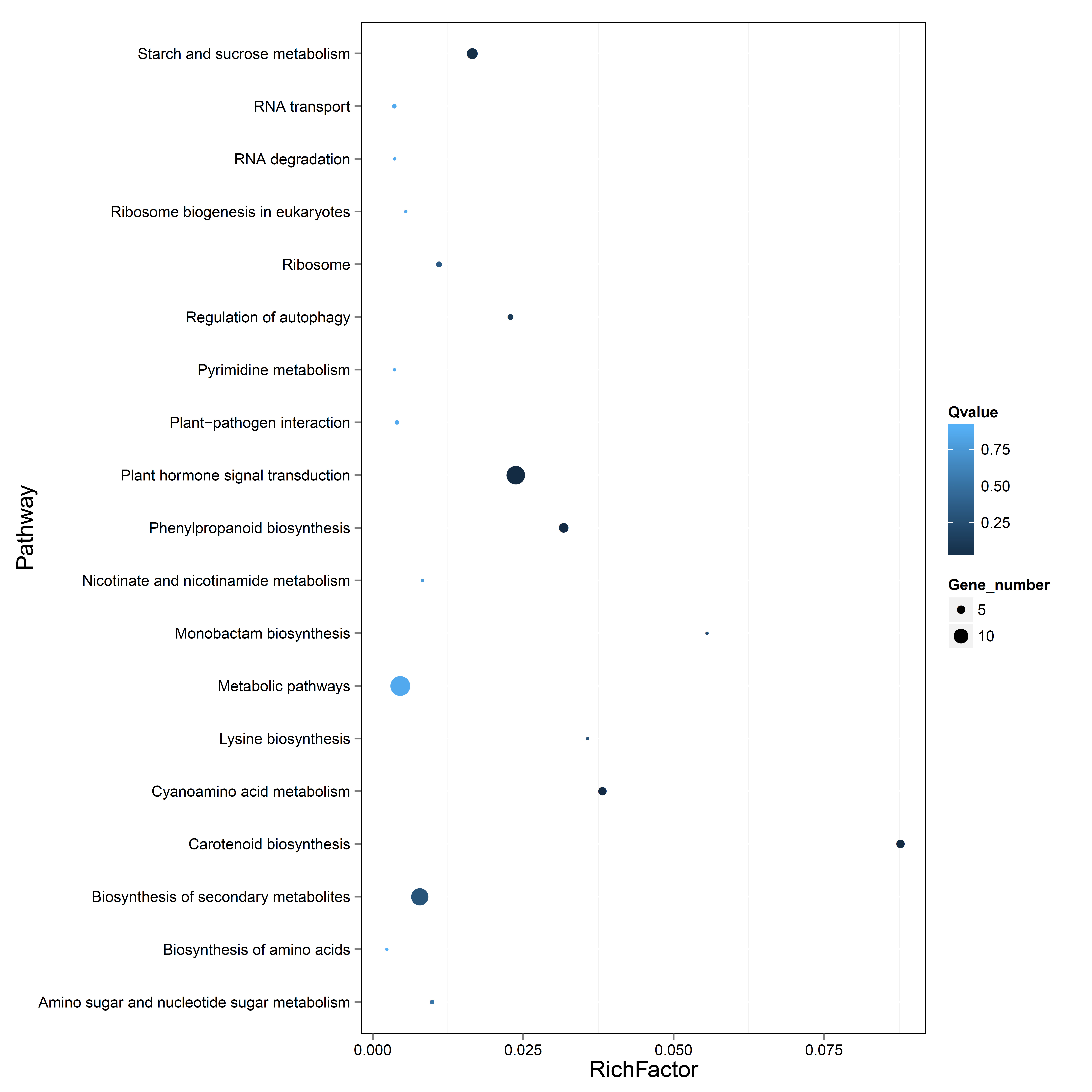
A total of 249 discrete genes were obtained from the two QTL regions (Additional file 6). Of these 80 genes were identified in the Gene Ontology (GO) database and, could be categorized into 28 functional groups, including 14 in biological processes, 9 in cellular components and 5 in molecular functions. The distribution of these eighty genes ranged from 1(in growth) to 48 (in metabolic processes) in the different functional groups. Most of the genes were involved in the cellular process (39), metabolic process (48), cell (42), cell part (42), binding (46) and catalytic activity (39) (Fig. 6). The distribution of the 80 genes in GO Database is list in Additional file 6. Of the 249 candidate genes, 84 were identified in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database, and they were associated with 19 pathways. The pathway with the highest enrichment factor was carotenoid biosynthesis, and a Q-value of < 0.05 was found for plant hormone signal transduction, carotenoid biosynthesis, phenylpropanoid biosynthesis, cyanoamino acid metabolism, starch and sucrose metabolism and regulation of autophagy (Additional file 7). These pathways may be significantly involve in determining the SH phenotype, and the 84 genes identified in the KEGG pathway database are listed in Additional file 6.
Fig. 6.

Histogram showing the gene ontology functional enrichment of differentially expressed genes. Different colors represent distinct functional groups
Expression analysis using real-time quantitative PCR (RT-qPCR)
249 genes were aligned to the Genome Database for Rosaceae (GDR) (https://www.rosaceae.org/node/1) to identify the candidate genes. In total, 11 candidate genes (with P-value < 0.05 in pathway) in peach cultivars with distinct softening characteristics were analyzed using RT-qPCR (Fig. 7). In the present study, the expression of Prupe.6G150900.1 was significantly higher in SH fruit (‘YM’) than that in M fruit (‘HJML’) during storage (Fig. 7), the same phenomenon occurred in other M (‘XH8’) and SH (‘XC’) fruit, which indicated that Prupe.6G150900.1 was expressed higher in SH fruit than in M fruit. The profiles were similar to those of the ABA content in ‘HJML’ and ‘YM’. Likewise, Prupe.6G147600.1 was expressed significantly higher in ‘YM’ than in ‘HJML’ fruit, but in ‘XH8’ and ‘XC’ expressed no significant diversity during later storage stage. Prupe.6G156500.1 was expressed significantly higher in ‘HJM’L fruit than in ‘YM’ fruit (Fig. 7) and also expressed no significant diversity in ‘XH8’ and ‘XC’ cultivars. The other genes in Fig. 7 showed no significant diversity between SH and M-type cultivars.
Fig. 7.

Relative mRNA expression levels of candidate genes during fruit softening. The x-axis represents the storage days after harvest; the y-axis represents relative expression levels of genes. Data are means ± SEs (n = 3). Significant differences (p < 0.05) between means are indicated by the symbol ‘*’ and significant differences (p < 0.01) between means are indicated by the symbol ‘**’
Discussion
SH inheritance in peach
SH phenotype is projected to be used as a genetic resource for breeding to meet fresh market requirements [24]. Additionally, identifying the mechanisms responsible for the SH phenotype is of great importance. The segregation of the SH phenotype in the F1 family (103) of the cross between ‘YM’ and ‘HJML’ in our study was consistent with Mendelian inheritance (1,1ratio). Which suggested that the SH trait is controlled by a single recessive gene. These results are consistent with Yoshida’s research [5].
Construction of a high-density genetic map for peach
Generally, molecular markers and genetic maps are widely used in the genetic mapping of important traits in crops [25]. RAD-seq technology has been employed as an efficient tool for high-density genetic mapping and QTL analyses with the capacity to discover larger numbers of markers in any organism (with or without reference genomes) [15, 26–29]. To date, no RAD-seq-based SNP discover technique has been reported in peach. In the present study, a high-density SNP linkage map in peach was constructed and characterized using the RAD-seq method. In 1994, Chaparro et al. [30] constructed the first linkage map of peach. With the development of molecular marker technology, RFLP, RAPD, AFLP and SSR markers [31–36], especially SSR were widely used to construct linkage maps in peach. Compared with microsatellites and other markers, SNPs are efficient tools for linkage mapping that can be genotyped on a more abundant and much larger scale [21, 37]. The ‘Texas’ × ‘Earlygold’ genetic map is considered the reference map for Prunus, having 562 markers and, covering 517 cM of the peach genome with an average distance of 0.92 cM between adjacent markers (http://www.rosaceae.org/maps) [38]. Recently, two high-density linkage maps were constructed: the Pop-DF map, covering 422 cM of the peach genome and including 1037 SNP markers (0.407 cM/SNP locus), and the Pop-DG map covering 369 cM and including 738 SNPs (0.5 cM/SNP locus) [39]. The map size reported here is 454.2 cM, which is shorter than the previously published maps of 562.3 cM [35] and 1061.8 cM [40] in peach. This might be the result of the limited LGs linkage groups or segregation distortion markers present in our map. The same phenomenon occurred for sesame [17]. This was the first mapping family reported in peach. Additionally, this genetic map had the highest marker density, and fewer distortion markers compared with other published genetic maps in peach [35, 38, 41]. The inter-marker distance of 0.347 cM suggested that it would be favorable for locating sequence scaffolds on the physical peach genome sequence map. Because the whole-genome sequence of peach has been released [42], it could be beneficial for obtaining genes near each marker.
Identification of QTLs related to the SH phenotype in peach
Mapping QTLs in peach is challenging because the peach plant is highly heterozygous with a long growth and breeding cycle. Mapping and QTL have been performed using F1 [39, 41, 43] F2 and BC1 families, as shown at http://www.rosaceae.org/maps. Fruit quality is considered the primary significant selection criteria by peach breeders [44] and, mainly includes color, flavor and texture. Shen et al. [35] mapped the Dominant Blood-Flesh locus, which may control the blood-flesh phenotype of ‘Wu Yue Xian’ peach to the top of LG5. Aromatic and other volatile compounds were mapped onto the Prunus reference map using gas chromatography-mass spectrometry [45, 46]. The NM flesh trait locus may be located in the central region of LG4 with recessive alleles determining the characteristics [47]. The activity of an endo-polygalacturonase gene was associated with M flesh [48]. Recently, Serra et al. [49] identified the qP-MD5 QTL as a key factor for slow M flesh, and qP-MD6 may modulate the maturity date trait. With the development of sequencing and SNP-genotyping platforms, high-resolution linkage maps have been successfully used to locate qualitative and quantitative traits [21, 50]. A total of 32 potential QTLs for 11 traits (5 quantitative and 6 qualitative traits), including length of pedicel, single fruit weight, soluble solid content, transverse diameter, vertical diameter, calyx status, flesh color juice content, number of seeds, skin color, and skin smooth, were identified and positioned in pear [21]. As an important texture type, the SH phenotype is of importance in peach [4, 6, 10, 11]. In this study, the pathway with the highest enrichment factor was carotenoid biosynthesis and plant hormone signal transduction, these indicated that the SH phenotype may much correlation with ABA, and the crosstalk of ethylene and ABA is important for multiple physiological processes [51–53]. These will be of great beneficial in clarifying the mechanism responsible for the SH phenotype.
Identification and expression analyses of candidate genes
Three genes (Prupe.6G150900.1, Prupe.6G147600.1 and Prupe.6G156500.1) were identified as good candidates for controlling the SH trait. Prupe.6G150900.1 was annotated as encoding abscisic-aldehyde oxidase 3, which was thought to catalyze the oxidation of the abscisic aldehyde reaction in the last step of ABA biosynthesis [54]. The expression of Prupe.6G150900.1 in the SH-type cultivar (‘YM’) was significantly higher than that in the M-type cultivar (‘HJML’) during storage. The same expression phenomenon also occurred in the ‘XC’ (SH) and ‘XH8’ (M) cultivars. Furthermore, the ABA content in the SH-type cultivar (‘YM’) was significant high than in M-type cultivar (‘HJML’). Thus the SH phenotype may have a close relationship with ABA. Multiple physiological processes are determined by the crosstalk between ABA and ethylene [51–53]. Particularly, the induction of ethylene biosynthesis can be prevented by ABA treatments [53, 55, 56]. Recent research suggested that ABA negatively regulates ethylene production through the ABI4-mediated transcriptional repression of the ethylene biosynthesis genes ACS4 and ACS8 in Arabidopsis [12]. This indicates that Prupe.6G150900.1 may be an important factor in the inhibition of ethylene production through ABA. In addition Prupe.6G147600.1 was annotate as being related to the transcription factor YABBY, and Prupe.6G156500.1 was annotate as encoding beta-glucosidase (EC3.2.1.21) an important hydrolase that may be related to the softening of peach fruit. In M and NM fruit cultivars, the endo-polygalacturonase activity during peach fruit ripening is responsible for the difference in softening [8, 9]. Ethylene treatment resulted in SH type peach fruit softening rapidly and increasing endo-polygalacturonase enzyme activity and PpPG mRNA expression [57, 58]. And Prupe.6G156500.1 may related to the fruit softening as endo-polygalacturonase for the higher expression in M cultivars and lower expression in SH cultivars. Further studies are needed to reveal the candidate genes’ function in peach.
Conclusion
The use of high-density genetic map will be beneficial for mapping important fruit traits and for QTL identification in peach. Prupe.6G150900.1 may be an important factor involved in the inhibition of the ethylene production through ABA and might be an important candidate gene for controlling the SH phenotype. These results will be useful for further analyses of SH phenotype.
Methods
Materials and DNA extraction
The two parents showed distinct softening characteristics, including fruit firmness and ethylene production. ‘YM’ and ‘HJML’ have significantly different texture phenotypes: ‘YM’ is SH, maintaining fruit firmness and hardly synthesizing ethylene during storage at room temperature, and ‘HJML’ is M, rapidly softening and exhibiting an ethylene production peak during storage at room temperature [59]. The sample family consisted of 103 F1 progeny from a cross between two peach cultivars: the SH-type cultivar ‘YM’ and the M-type cultivar ‘HJML’. The family was hybridized in 2008, These plants were grown at the experimental peach orchard in Nanjing, Jiangsu, China. Young leaves (first few leaves of the apex) were collected and immediately stored in liquid nitrogen and transferred to a − 80 °C freezer in the laboratory. A sample from each individual was ground in liquid nitrogen and total genomic DNA was extracted using a Plant Genomic DNA Kit (TIANGEN, Beijing, China). A 1% agarose gels and a spectrophotometer (Qubit 2.0 Fluorometer, Invitrogen) were used to determine the genomic DNA’s integrity and quality, respectively.
Detection of ethylene production and fruit firmness
Ethylene production was measured by gas chromatography (Agilent 7890A, CA, USA) according to the method followed by Guo et al. [59] in 2016 and 2017, and fruit flesh firmness was measured using the TA-XT. Plus Texture Analyser (Stable Micro Systems Ltd., Godalming, Surrey, UK) according to the method of Guo et al. [59] in 2017. All fruit samples reached commercial maturity, had no diseases and mechanical damage with uniform maturity were randomly collected. And the fruits were stored in a room at at 25 ± 0.5 °C with a relative humidity of 75–85%. Ten fruits are collected each time. Three independent biological replicates were conducted for these measurements.
Determination of the ABA content
The ABA content was measured using ESI-HPLC-MS/MS (Waters, Milford, USA) as described previously [60]. Briefly, 0.6 g of peach flesh was homogenized in liquid nitrogen, transferred to a 20-mL centrifuge tube with 5 mL of isopropanol/hydrochloric acid extraction buffer, and then the reaction solution was vortexed for 30 min at 4 °C. Subsequently, 10 mL dichloromethane was added, and the mixture was vortexed at 4 °C for 30 min. Then, samples were centrifuged at 13000 rpm for 5 min to separate the organic phase, which was subsequently protected from light and dried under nitrogen gas. It was then re-dissolving dissolved 400 μL methanol/methane acid (99.9/0.1, v/v). Then, the solution was sequentially passed through a 0.22 μm filter membrane to measure with HPLC-MS/MS. The HPLC separation used an ACQUITY UPLC R BEH C18, 100 mm × 2.1 mm × 1.7 μm column (Waters) and was eluted with H2O/methanol (98/2, v/v), 0.05% methane acid and 5 mmol/L ammonium acetate (eluent A) and acetonitrile (eluent B) at a flow rate 0.3 mL min− 1. The temperature of the column was maintained at 40 °C and the sample size was 5 μL. The MS methods were as follows: positive, negative ion electrospray ionization, capillary voltage of 3.0 Kv, ionization temperature of 150 °C, cone gas flow of 50 L/Hr, de-solvation temperature of 400 °C, de-solvation gas flow of 800 L/Hr, monitoring mode of MRM. Comparisons of the peak area ratio (analyte/IS) to concentrations were used to construct the calibration curve, and then the ABA content was calculated based on the calibration curve. Three biological replicates were conducted.
RAD library construction and sequencing
Illumina DNA sequencing combined with a RAD strategy was used for the effective identification of SNP markers. The RAD-seq library construction protocol was similar to that described in a set of previously published papers [21, 26] . Briefly, 1 μg genomic DNA from each individual was digested by the restriction enzyme EcoR1(NEB), and the P1 adapter was added with T4 DNA ligase (NEB) for 1 h at 37 °C. Ligation products were pooled and fragmented by a Covaris sonicator. Then fragments between 300 and 500 bp were excised after agarose gel electrophoresis selection and purified using a QIAquick Gel Purification Kit (Qiagen). The purified products were combined with End Repair Mix incubated at 20 °C for 30 min, and then purified again. An end-repaired DNAdA overhang was added by the A-Tailing mix (NEB) at 37 °C for 30 min. The P2 adapter was added to the product for 15 min at 20 °C, and the samples were then purified using a QIAquick Gel Purification Kit (Qiagen). PCR amplification was used to enrich the collected fragments, followed by 2% agarose gel electrophoresis to recover the target fragments. Finally, the library was validated as follows: including: the Agilent 2100 bio analyzer instrument (Agilent DNA 1000 Reagents) was used to determine the average molecule length and RT-qPCR (TaqMan Probe) was used to quantify the libraries. Then, the qualified libraries were amplified on cBot to generate a cluster on the flow cells (PE Cluster Ki,Illumina) and the amplified flow cell were pair-end sequenced in individual lanes of the Illumina HiSeq4000 NGS platform (BGI-Shenzhen, ShenZhen, China).
SNP discovery and genotyping
After filtering and splitting, Illumina raw sequence reads were retained. Briefly, low-quality data were discarded including: reads with adaptors, reads with more than 50% bases whose quality values ≤5, and reads that could not be identified with barcode sequences, which are used to classify reads between samples. Clean reads were mapped against the peach genome sequences from the GDR website (https://www.rosaceae.org/species/prunus_persica/genome_v2.0.a1) with SOAPaligner [61]. Finally, the SNP loci were identified from alignment results using soapsnp software (version 2.23) [62]. Genotypes of offspring individual were based on parental genotypes and markers were tested by chi-squared with the P-value set to 0.01. In addition, the family type was cross pollinators, which is a cross between two heterozygous diploid parents, and its linkage phases were originally unknown [63]. Three segregation types were genotyped, lmxll, nnxnp and hkxhk, with an expected segregation ratio for marker codes lmxll and, nnxnp was being 1:1, and that for lmxll, nnxnp and hkxhk was being 1:2:1. SNP markers with < 10% missing data in the each individual and consistent with the above standards were used for linkage map construction.
Linkage mapping and QTL analysis
Construction of the genetic linkage map was accomplished using JoinMap (version 4.1) [63] with 6150 markers and cross pollinators family type. Initially, LGs were constructed using a LOD threshold of 3.0 to 4.0, Kosambi as the mapping function [64], and regression mapping as mapping algorithm. MapChart software (version 2.2) was used to make the map figures [65]. Mean phenotypic data from all 105 individuals (two parents and 103 F1 progeny) are list in Additional file 1. IciMapping software (version 4.1) was used to calculate the QTLs [66] with the ICIM-ADD method. LOD significance thresholds (P < 0.05) were analyzed by running 1000 permutation tests.
Candidate gene mining in silico and a functional analysis
Mapping-associated markers were used to identify the homologous regions of QTLs on the physical map. Corresponding genes in QTLs were referred to the peach genome from GDR [42]. To identify the main biological functions of corresponding genes in QTLs, these genes were mapped to each node of the GO database (http://www.geneontology.org/) [67], and the pathway enrichment was also analyzed in KEGG (http://www.genome.jp/kegg/pathway.html).
RNA isolation and expression analyses
The SH cultivars ‘YM’ and ‘XC’, and M cultivars ‘HJML’ and ‘XH8’ were used to expression analyses. The fruit samples were same as description from Guo et al. [59]. All fruit samples reached commercial maturity, had no diseases and mechanical damage with uniform maturity were randomly collected. And the fruits were stored in a room at at 25 ± 0.5 °C with a relative humidity of 75–85%. For HJML, YM and XH8 fruit samples were taken at 0, 4, 8 and 12 d postharvest; and for XC at 0, 4, 9 and 12d. The pulp from ten fruit were frozen in liquid nitrogen and stored at − 80 °C until further analysis. Three independent biological replicates were conducted for expression analysis. Total RNA was extracted from peach fruit samples using the RNAprep Pure Plant Kit (Polysaccharides & Polyphenolics-rich) (TIANGEN, Beijing, China), and an ultraviolet spectrophotometer (Eppendorf, Hamburg, Germany) and 1% agarose gels electrophoresis were used to detect RNA integrity and quality, respectively. cDNA was synthesized using the PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa, Dalian, China). The NCBI/Primer-BLAST on-line server was used to design specific primers for each gene. All primer sequences are listed in Additional file 8. The translation elongation factor 2 was used as the internal reference gene as in Tong et al. [68]. RT-qPCR was performed on a 7500 Real Time PCR System (Applied Biosystems, NY, USA) with SYBR®Premix Ex Taq™ (TaKaRa) and gene-specific primers in a volume of 20 μL. PCR conditions were as follows: an initial denaturation at 95 °C for 30 s, and 40 cycles of 95 °C for 5 s and 60 °C for 34 s. The specificity of primer amplifications was checked by a melting curve analysis. The comparative cycle threshold method (ΔΔCt) was used to analyze relative expression level data [69]. Each sample was analyzed in triplicate.
Statistical analyses
Microsoft Excel 2010 was used to calculate standard errors (SEs). Graphs were produced using Origin 8.0 software. Significant differences between means of experimental data and a correlation analysis were determined using SPSS 19.0 software (SPSS, Chicago, IL, USA).
Additional files
Phenotypic identification of the 103 F1 individuals from a cross between two peach cultivars the SH-type cultivar ‘YM’ and the M-type cultivar ‘HJML’. a. Identification of SH phenotype based on the ethylene production in 2016; b. Identification of SH phenotype based on the fruit firmness in 2017; c. Phenotype identification used for QTL analysis. (DOCX 16 kb)
Statistics for the data of RAD-seq in the parent plants and those of their individual offspring. (XLSX 21 kb)
6150 SNP markers identified after filtering used to construct the linkage map in peach. (XLSX 2185 kb)
Analysis of the SNP polymorphic sites in the parent plants and their individual offspring in peach. (XLSX 21652 kb)
Numbers of SNP transitions types in parent plants and their offspring in peach. (DOCX 21 kb)
Discrete genes obtained from QTL regions and the alignment results to GO database and KEGG pathway database. (XLSX 23 kb)
{kind=link}
Scatter plot illustrating pathway rich factor analysis. (JPG 3748 kb)
Specific primers used for RT-qPCR to detect the expression of 11 candidate genes. (DOCX 14 kb)
Acknowledgments
We acknowledge the critical reading of this manuscript from Dr. Zhijun Shen.
Funding
This work was financially supported by the earmarked fund for China Agriculture Research System (CARS-30) and grants from Jiangsu Agriculture Science and Technology Innovation Fund (CX (15)1020), Six Talent Fund of Jiangsu Province (No. NY068) and the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).
Availability of data and materials
The datasets generated and analyzed during the current study are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (http://www.ncbi.nlm.nih.gov/sra/) with the accession number of SRP139715. Additional supporting Tables and Figures are included as Additional files. All plant materials were available from Institute of Pomology, Jiangsu Academy of Agricultural Sciences, P. R. China.
Abbreviations
- M
Melting
- NM
No-melting
- SH
Stony hard
- ABA
Abscisic acid
- NGS
Next-generation sequencing
- RAD-seq
Restriction-site associated DNA sequencing
- QTL
Quantitative trait loci
- LG
Linkage group
- SE
Standard error
- GDR
Genome Database for Rosaceae
- SNP
Single nucleotide polymorphism
- YM
Yumyeoung
- HJML
Hu Jing Mi Lu
- XH8
Xia Hui 8
- XC
Xia Cui
- LOD
Likelihood of odd
- RT-qPCR
real-time quantitative PCR
- GO
Gene Ontology
- KEGG
Kyoto Encyclopedia of Genes and Genomes
Authors’ contributions
ZG, MY and SG conceived and designed the study, SG and SI phenotyped the plants, prepared the DNA, SG and JS performed the RAD sequencing and bioinformatics analysis, RM made the cross and did data analysis, SG and ZG wrote the paper, and MY, RM, ZG revised the paper. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable. No human samples and no animals were used and no field permissions were necessary to collect the plant samples for this study. The authors declared that experimental research works on the plants described in this paper comply with institutional, national and international guidelines.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Mingliang Yu, Email: mly1008@aliyun.com.
Zhihong Gao, Email: gaozhihong@njau.edu.cn.
References
- 1.Karabulut OA, Cohen L, Wiess B, Daus A, Lurie S, Droby S. Control of brown rot and blue mold of peach and nectarine by short hot water brushing and yeast antagonists. Postharvest Biol Tec. 2002;24(2):103–111. doi: 10.1016/S0925-5214(01)00132-6. [DOI] [Google Scholar]
- 2.Yoshioka H, Hayama H, Tatsuki M, Nakamura Y. Cell wall modification during development of mealy texture in the stony-hard peach “Odoroki” treated with propylene. Postharvest Biol Tec. 2010;55(1):1–7. doi: 10.1016/j.postharvbio.2009.08.005. [DOI] [Google Scholar]
- 3.Haji T, Yaegaki H, Yamaguchi M. Changes in ethylene production and flesh firmness of melting, nonmelting and stony hard peaches after harvest. Engei Gakkai Zasshi. 2001;70(4):458–459. doi: 10.2503/jjshs.70.458. [DOI] [Google Scholar]
- 4.Haji T, Yaegaki H, Yamaguchi M. Inheritance and expression of fruit texture melting, non-melting and stony hard in peach. Sci Hortic-amsterdam. 2005;105(2):241–248. doi: 10.1016/j.scienta.2005.01.017. [DOI] [Google Scholar]
- 5.Yoshida M. Genetic studies on the fruit quality of peach varieties III texture and keeping quality. Bull Fruit Tree Res Station. 1976;3:1–16. [Google Scholar]
- 6.Pan L, Zeng W, Niu L, Lu Z, Liu H, Cui G, Zhu Y, Chu J, Li W, Fang W, et al. PpYUC11, a strong candidate gene for the stony hard phenotype in peach (Prunus persica L. Batsch), participates in IAA biosynthesis during fruit ripening. J Exp Bot. 2015;66(22):7031–7044. doi: 10.1093/jxb/erv400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Biale J, Young R. Respiration and ripening in fruits — retrospect and prospect. In: Friend J, Rhodes MJC (eds). Recent advances in the Biochemistry of fruit and vegetables. London: Academic Press; 1981. p. 1–39.
- 8.Callahan AM, Scorza R, Bassett C, Nickerson M, Abeles FB. Deletions in an endopolygalacturonase gene cluster correlate with non-melting flesh texture in peach. Funct Plant Biol. 2004;31(2):159–168. doi: 10.1071/FP03131. [DOI] [PubMed] [Google Scholar]
- 9.Pressey R, Avants JK. Separation and characterization of Endopolygalacturonase and Exopolygalacturonase from peaches. Plant Physiol. 1973;52(3):252. doi: 10.1104/pp.52.3.252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tatsuki M. The involvement of 1-aminocyclopropane-1-carboxylic acid synthase isogene, Pp-ACS1, in peach fruit softening. J Exp Bot. 2006;57(6):1281–1289. doi: 10.1093/jxb/erj097. [DOI] [PubMed] [Google Scholar]
- 11.Tatsuki M, Nakajima N, Fujii H, Shimada T, Nakano M, Hayashi K, Hayama H, Yoshioka H, Nakamura Y. Increased levels of IAA are required for system 2 ethylene synthesis causing fruit softening in peach (Prunus persica L. Batsch) J Exp Bot. 2013;64(4):1049. doi: 10.1093/jxb/ers381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong Z, Yu Y, Li S, Wang J, Tang S. Abscisic acid antagonizes ethylene production through the ABI4-mediated transcriptional repression of ACS4 and ACS8 in Arabidopsis. Mol Plant. 2016;9(1):126–135. doi: 10.1016/j.molp.2015.09.007. [DOI] [PubMed] [Google Scholar]
- 13.Li Z, Zhang L, Yu Y, Quan R, Zhang Z, Zhang H, Huang R. The ethylene response factor AtERF11 that is transcriptionally modulated by the bZIP transcription factor HY5 is a crucial repressor for ethylene biosynthesis in Arabidopsis. Plant J. 2011;68(1):88–99. doi: 10.1111/j.1365-313X.2011.04670.x. [DOI] [PubMed] [Google Scholar]
- 14.Peterson BK, Weber JN, Kay EH, Fisher HS, Hoekstra HE. Double digest RADseq: an inexpensive method for De novo SNP discovery and genotyping in model and non-model species. PLoS One. 2012;7(5):e37135. doi: 10.1371/journal.pone.0037135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pan L, Wang N, Wu Z, Guo R, Yu X, Zheng Y, Xia Q, Gui S, Chen C. A high density genetic map derived from RAD sequencing and its application in QTL analysis of yield-related traits in Vigna unguiculata. Front Plant Sci. 2017;8:1544. doi: 10.3389/fpls.2017.01544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang N, Fang L, Xin H, Wang L, Li S. Construction of a high-density genetic map for grape using next generation restriction-site associated DNA sequencing. BMC Plant Biol. 2012;12(1):148. doi: 10.1186/1471-2229-12-148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu K, Liu H, Yang M, Ye T, Ma H, Wu W, Yang Z, Zhao Y. High-density genetic map construction and QTLs analysis of grain yield-related traits in sesame (Sesamum indicum L.) based on RAD-Seq techonology. BMC Plant Biol. 2014;14(1):1–14. doi: 10.1186/1471-2229-14-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barchi L, Lanteri S, Portis E, Volante A, Pulcini L, Ciriaci T, Acciarri N, Barbierato V, Toppino L, Rotino GL. A RAD tag derived marker based eggplant linkage map and the location of QTLs determining anthocyanin pigmentation. PLoS One. 2012;7(8):e43740. doi: 10.1371/journal.pone.0043740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pfender WF, Saha MC, Johnson EA, Slabaugh MB. Mapping with RAD (restriction-site associated DNA) markers to rapidly identify QTL for stem rust resistance in Lolium perenne. Theor Appl Genet. 2011;122(8):1467–1480. doi: 10.1007/s00122-011-1546-3. [DOI] [PubMed] [Google Scholar]
- 20.Chutimanitsakun Y, Nipper RW, Cuestamarcos A, Cistué L, Corey A, Filichkina T, Johnson EA, Hayes PM. Construction and application for QTL analysis of a restriction site associated DNA (RAD) linkage map in barley. BMC Genomics. 2011;12(1):4. doi: 10.1186/1471-2164-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu J, Li LT, Li M, Khan MA, Li XG, Chen H, Yin H, Zhang SL. High-density genetic linkage map construction and identification of fruit-related QTLs in pear using SNP and SSR markers. J Exp Bot. 2014;65(20):5771–5781. doi: 10.1093/jxb/eru311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun R, Chang Y, Yang F, Wang Y, Li H, Zhao Y, Chen D, Wu T, Zhang X, Han Z. A dense SNP genetic map constructed using restriction site-associated DNA sequencing enables detection of QTLs controlling apple fruit quality. BMC Genomics. 2015;16(1):747. doi: 10.1186/s12864-015-1946-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chapman MA, Pashley CH, Wenzler J, Hvala J, Tang S, Knapp SJ, Burke JM. A genomic scan for selection reveals candidates for genes involved in the evolution of cultivated sunflower (Helianthus annuus) Plant Cell. 2008;20(11):2931. doi: 10.1105/tpc.108.059808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Haji T. Inheritance of flesh texture in peach and effects of ethylene treatment on softening of the stony hard peach. Jarq-Jpn Agr Res Q. 2014;48(1):57–61. doi: 10.6090/jarq.48.57. [DOI] [Google Scholar]
- 25.Zhou X, Xia Y, Ren X, Chen Y, Li H, Huang S, Liao B, Lei Y, Yan L, Jiang H. Construction of a SNP-based genetic linkage map in cultivated peanut based on large scale marker development using next-generation double-digest restriction-site-associated DNA sequencing (ddRADseq) BMC Genomics. 2014;15(1):351. doi: 10.1186/1471-2164-15-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baird NA, Etter PD, Atwood TS, Currey MC, Shiver AL, Lewis ZA, Selker EU, Cresko WA, Johnson EA. Rapid SNP discovery and genetic mapping using sequenced RAD markers. PLoS One. 2008;3(10):e3376. doi: 10.1371/journal.pone.0003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davey JW, Blaxter ML. RADSeq: next-generation population genetics. Brief Funct Genomics. 2010;9(5–6):416–423. doi: 10.1093/bfgp/elq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Raman H, Raman R, Coombes N, Song J, Prangnell R, Bandaranayake C, Tahira R, Sundaramoorthi V, Killian A, Meng J. Genome-wide association analyses reveal complex genetic architecture underlying natural variation for flowering time in canola. Plant Cell Environ. 2016;39(6):1228–1239. doi: 10.1111/pce.12644. [DOI] [PubMed] [Google Scholar]
- 29.Zou J, Hu D, Liu P, Raman H, Liu Z, Liu X, Parkin IAP, Chalhoub B, Meng J. Co-linearity and divergence of the a subgenome of Brassica juncea compared with other Brassica species carrying different a subgenomes. BMC Genomics. 2016;17(1):1–14. doi: 10.1186/s12864-015-2343-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chaparro JX, Werner DJ, O'Malley D, Sederoff RR. Targeted mapping and linkage analysis of morphological isozyme, and RAPD markers in peach. Theor Appl Genet. 1994;87(7):805–815. doi: 10.1007/BF00221132. [DOI] [PubMed] [Google Scholar]
- 31.Blenda AV, Verde I, Georgi LL, Reighard GL, Forrest SD, Muñoz-Torres M, Baird WV, Abbott AG. Construction of a genetic linkage map and identification of molecular markers in peach rootstocks for response to peach tree short life syndrome. Tree Genet Genomes. 2007;3(4):341–350. doi: 10.1007/s11295-006-0074-9. [DOI] [Google Scholar]
- 32.Dirlewanger E, Cosson P, Boudehri K, Renaud C, Capdeville G, Tauzin Y, Laigret F, Moing A. Development of a second-generation genetic linkage map for peach [Prunus persica (L.) Batsch] and characterization of morphological traits affecting flower and fruit. Tree Genet Genomes. 2006;3(1):1–13. doi: 10.1007/s11295-006-0053-1. [DOI] [Google Scholar]
- 33.Dirlewanger E, Pronier V, Parvery C, Rothan C, Guye A, Monet R. Genetic linkage map of peach [Prunus persica (L.) Batsch] using morphological and molecular markers. Theor Appl Genet. 1998;97(5–6):888–895. doi: 10.1007/s001220050969. [DOI] [Google Scholar]
- 34.Rajapakse S, Belthoff LE, He G, Estager AE, Scorza R, Verde I, Ballard RE, Baird WV, Callahan A, Monet R. Genetic linkage mapping in peach using morphological, RFLP and RAPD markers. Theor Appl Genet. 1995;90(3–4):503. doi: 10.1007/BF00221996. [DOI] [PubMed] [Google Scholar]
- 35.Shen Z, Confolent C, Lambert P, Poëssel JL, Quilot-Turion B, Yu M, Ma R, Pascal T. Characterization and genetic mapping of a new blood-flesh trait controlled by the single dominant locus DBF in peach. Tree Genet Genomes. 2013;9(6):1435–1446. doi: 10.1007/s11295-013-0649-1. [DOI] [Google Scholar]
- 36.Yamamoto T, Shimada T, Imai T, Yaegaki H, Haji T, Matsuta N, Yamaguchi M, Hayashi T. Characterization of morphological traits based on a genetic linkage map in peach. Jpn J Breed. 2001;51(4):271–278. [Google Scholar]
- 37.Slate J, Gratten J, Beraldi D, Stapley J, Hale M, Pemberton JM. Gene mapping in the wild with SNPs: guidelines and future directions. Genetica. 2009;136(1):97. doi: 10.1007/s10709-008-9317-z. [DOI] [PubMed] [Google Scholar]
- 38.Joobeur T, Viruel MA, Vicente MCD, Jáuregui B, Ballester J, Dettori MT, Verde I, Truco MJ, Messeguer R, Batlle I. Construction of a saturated linkage map for Prunus using an almond×peach F2 progeny. Theor Appl Genet. 1998;97(7):1034–1041. doi: 10.1007/s001220050988. [DOI] [Google Scholar]
- 39.Martínez-García PJ, Dan EP, Ogundiwin EA, Fass J, Chan HM, Ahmad R, Lurie S, Dandekar A, Gradziel TM, Crisosto CH. High density SNP mapping and QTL analysis for fruit quality characteristics in peach (Prunus persica L.) Tree Genet Genomes. 2013;9(1):19–36. doi: 10.1007/s11295-012-0522-7. [DOI] [Google Scholar]
- 40.Wu J, Shu H, Zhang K, Jiang L, Zhou X, Xin C. Construction and analysis of peach genetic map. Acta Horticulturae Sinica. 2004;31(5):593–597. [Google Scholar]
- 41.Zeballos JL, Abidi W, Giménez R, Monforte AJ, Moreno MÁ, Gogorcena Y. Mapping QTLs associated with fruit quality traits in peach [Prunus persica (L.) Batsch] using SNP maps. Tree Genet Genomes. 2016;12(3):37. doi: 10.1007/s11295-016-0996-9. [DOI] [Google Scholar]
- 42.Verde I, Abbott AG, Scalabrin S, Jung S, Shu S, Marroni F, Zhebentyayeva T, Dettori MT, Grimwood J, Cattonaro F. The high-quality draft genome of peach (Prunus persica) identifies unique patterns of genetic diversity, domestication and genome evolution. Nat Genet. 2013;45(5):487. doi: 10.1038/ng.2586. [DOI] [PubMed] [Google Scholar]
- 43.Ogundiwin EA, Peace CP, Gradziel TM, Dan EP, Bliss FA, Crisosto CH. A fruit quality gene map of Prunus. BMC Genomics. 2009;10(1):587. doi: 10.1186/1471-2164-10-587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Abbott A, Georgi L, Yvergniaux D, Wang Y, Blenda A, Reighard G, Inigo M, Sosinski B. Peach: the model genome for Rosaceae. Acta Hortic. 2002;575(1):145–156. doi: 10.17660/ActaHortic.2002.575.14. [DOI] [Google Scholar]
- 45.Eduardo I, Chietera G, Pirona R, Pacheco I, Troggio M, Banchi E, Bassi D, Rossini L, Vecchietti A, Pozzi C. Genetic dissection of aroma volatile compounds from the essential oil of peach fruit: QTL analysis and identification of candidate genes using dense SNP maps. Tree Genet Genomes. 2013;9(1):189–204. doi: 10.1007/s11295-012-0546-z. [DOI] [Google Scholar]
- 46.Illa E, Eduardo I, Audergon JM, Barale F, Dirlewanger E, Li X, Moing A, Lambert P, Le DL, Gao Z. Saturating the Prunus (stone fruits) genome with candidate genes for fruit quality. Mol Breed. 2011;28(4):667–682. doi: 10.1007/s11032-010-9518-x. [DOI] [Google Scholar]
- 47.Peace CP, Crisosto CH, Gradziel TM. Endopolygalacturonase: a candidate gene for freestone and melting Fleshin peach. Mol Breed. 2005;16(1):21–31. doi: 10.1007/s11032-005-0828-3. [DOI] [Google Scholar]
- 48.Lester DR, Sherman WB, Atwell BJ. Endopolygalacturonase and the melting flesh (M) locus in peach. J Am Soc Hortic Sci. 1996;121(2):231–235. [Google Scholar]
- 49.Serra O, Giné-Bordonaba J, Eduardo I, Bonany J, Echeverria G, Larrigaudière C, Arús P. Genetic analysis of the slow-melting flesh character in peach. Tree Genet Genomes. 2017;13(4):77. doi: 10.1007/s11295-017-1160-x. [DOI] [Google Scholar]
- 50.Frett TJ, Reighard GL, Okie WR, Gasic K. Mapping quantitative trait loci associated with blush in peach [Prunus persica (L.) Batsch] Tree Genet Genomes. 2014;10(2):367–381. doi: 10.1007/s11295-013-0692-y. [DOI] [Google Scholar]
- 51.Beaudoin N, Serizet C, Gosti F, Giraudat J. Interactions between Abscisic acid and ethylene signaling cascades. Plant Cell. 2000;12(7):1103–1115. doi: 10.1105/tpc.12.7.1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ghassemian M, Nambara E, Cutler S, Kawaide H, Kamiya Y, Mccourt P. Regulation of Abscisic acid signaling by the ethylene response pathway in Arabidopsis. Plant Cell. 2000;12(7):1117–1126. doi: 10.1105/tpc.12.7.1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luo X, Chen Z, Gao J, Gong Z. Abscisic acid inhibits root growth in Arabidopsis through ethylene biosynthesis. Plant J. 2014;79(1):44–55. doi: 10.1111/tpj.12534. [DOI] [PubMed] [Google Scholar]
- 54.Koiwai H, Nakaminami K, Seo M, Mitsuhashi W, Toyomasu T, Koshiba T. Tissue-specific localization of an abscisic acid biosynthetic enzyme, AAO3, in Arabidopsis. Plant Physiol. 2004;134(4):1697–1707. doi: 10.1104/pp.103.036970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Woeste KE, Ye C, Kieber JJ. Two Arabidopsis mutants that overproduce ethylene are affected in the posttranscriptional regulation of 1-Aminocyclopropane-1-carboxylic acid synthase. Plant Physiol. 1999;119(2):521–529. doi: 10.1104/pp.119.2.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wright STC. The effect of plant growth regulator treatments on the levels of ethylene emanating from excised turgid and wilted wheat leaves. Planta. 1980;148(4):381–388. doi: 10.1007/BF00388127. [DOI] [PubMed] [Google Scholar]
- 57.Hayama H, Shimada T, Fujii H, Ito A, Kashimura Y. Ethylene-regulation of fruit softening and softening-related genes in peach. J Exp Bot. 2006;57(15):4071–4077. doi: 10.1093/jxb/erl178. [DOI] [PubMed] [Google Scholar]
- 58.Hayama H, Tatsuki M, Ito A, Kashimura Y. Ethylene and fruit softening in the stony hard mutation in peach. Postharvest Biol Tec. 2006;41(1):16–21. doi: 10.1016/j.postharvbio.2006.03.006. [DOI] [Google Scholar]
- 59.Guo S, Song J, Zhang B, Jiang H, Ma R, Yu M. Genome-wide identification and expression analysis of beta-galactosidase family members during fruit softening of peach [Prunus persica (L.) Batsch] Postharvest Biol Tec. 2018;136(Supplement C):111–123. doi: 10.1016/j.postharvbio.2017.10.005. [DOI] [Google Scholar]
- 60.Yin X, He D, Ravi G, Yang P. Physiological and proteomic analyses on artificially aged Brassica napusseed. Front Plant Sci. 2015;6(112):112. doi: 10.3389/fpls.2015.00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li R, Yu C, Li Y, Lam TW, Yiu SM, Kristiansen K, Wang J. SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics. 1966;25(15):1966–1967. doi: 10.1093/bioinformatics/btp336. [DOI] [PubMed] [Google Scholar]
- 62.Li R, Li Y, Fang X, Yang H, Wang J, Kristiansen K, Wang J. SNP detection for massively parallel whole-genome resequencing. Genome Res. 2009;19(6):1124. doi: 10.1101/gr.088013.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Ooijen JW. JoinMap 4. Wageningen: Softw Calc Genet Link maps Exp Popul Kyazma BV; 2006.
- 64.Kosambi DD. The estimation of map distance from recombination values. Ann Hum Genet. 1943;12(1):172–175. [Google Scholar]
- 65.Voorrips RE. MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93(1):77–78. doi: 10.1093/jhered/93.1.77. [DOI] [PubMed] [Google Scholar]
- 66.Wang J, Li H, Zhang L, Meng L. Users’ manual of QTL IciMapping version 3.2. Quant Genet Group, Inst Crop Sci Chinese Acad Agric Sci (CAAS), Beijing 2012, 100081. http://www.sciencedirect.com/science/article/pii/S2214514115000161.
- 67.Harris MA, Clark J, Ireland A, Lomax J, Ashburner M, Foulger R, Eilbeck K, Lewis S, Marshall B, Mungall C, Richter J, Rubin GM, Blake JA, Bult C, Dolan M, Drabkin H, Eppig JT, Hill DP, Ni L, Ringwald M, Balakrishnan R, Cherry JM, Christie KR, Costanzo MC, Dwight SS, Engel S, Fisk DG, Hirschman JE, Hong EL, Nash RS, Sethuraman A, Theesfeld CL, Botstein D, Dolinski K, Feierbach B, Berardini T, Mundodi S, Rhee SY, Apweiler R, Barrell D, Camon E, Dimmer E, Lee V, Chisholm R, Gaudet P, Kibbe W, Kishore R, Schwarz EM, Sternberg P, Gwinn M, Hannick L, Wortman J, Berriman M, Wood V, de la CN, Tonellato P, Jaiswal P, Seigfried T, White R. The gene ontology (GO) database and informatics resource. Nucleic Acids Res. 2004;32:D258–D261. 10.1093/nar/gkh066. [DOI] [PMC free article] [PubMed]
- 68.Tong ZG, Gao ZH, Wang F, Zhou J, Zhang Z. Selection of reliable reference genes for gene expression studies in peach using real-time PCR. BMC Mol Biol. 2009;10(1):1–13. doi: 10.1186/1471-2199-10-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Phenotypic identification of the 103 F1 individuals from a cross between two peach cultivars the SH-type cultivar ‘YM’ and the M-type cultivar ‘HJML’. a. Identification of SH phenotype based on the ethylene production in 2016; b. Identification of SH phenotype based on the fruit firmness in 2017; c. Phenotype identification used for QTL analysis. (DOCX 16 kb)
Statistics for the data of RAD-seq in the parent plants and those of their individual offspring. (XLSX 21 kb)
6150 SNP markers identified after filtering used to construct the linkage map in peach. (XLSX 2185 kb)
Analysis of the SNP polymorphic sites in the parent plants and their individual offspring in peach. (XLSX 21652 kb)
Numbers of SNP transitions types in parent plants and their offspring in peach. (DOCX 21 kb)
Discrete genes obtained from QTL regions and the alignment results to GO database and KEGG pathway database. (XLSX 23 kb)
Scatter plot illustrating pathway rich factor analysis. (JPG 3748 kb)
Specific primers used for RT-qPCR to detect the expression of 11 candidate genes. (DOCX 14 kb)
Data Availability Statement
The datasets generated and analyzed during the current study are available in the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) database (http://www.ncbi.nlm.nih.gov/sra/) with the accession number of SRP139715. Additional supporting Tables and Figures are included as Additional files. All plant materials were available from Institute of Pomology, Jiangsu Academy of Agricultural Sciences, P. R. China.