

Abstract
TNF-α siRNA has shown promising therapeutic benefits in animal models of rheumatoid arthritis. However, there continues to be a need for siRNA delivery systems that have high siRNA encapsulation efficiency and minimum burst release of TNF-α siRNA, and can target inflamed tissues after intravenous administration. Herein we report a novel acid-sensitive sheddable PEGylated solid-lipid nanoparticle formulation of TNF-α-siRNA, AS-TNF-α-siRNA-SLNs, prepared by incorporating lipophilized TNF-α-siRNA into solid-lipid nanoparticles composed of biocompatible lipids such as lecithin and cholesterol. The nanoparticles are approximately 120 nm in diameter, have a high siRNA encapsulation efficiency (> 90%) and a minimum burst release of siRNA (<5%), and increase the deilvery of the siRNA in chronic inflammation sites in mouse models, including in a mouse model with collagen-induced arthritis. Importantly, in a mouse model of collagen antibody-induced arthritis that does not respond to methotrexate therapy, intravenous injection of the AS-TNF-α-siRNA-SLNs significantly reduced paw thickness, bone loss, and histopathological scores. These findings highlight the potential of using this novel siRNA nanoparticle formulation to effectively treat arthritis, potentially in patients who do not respond adequately to methotrexate.
Keywords: CAIA mouse model, acid-sensitive, PEGylation, drug targeting, chronic inflammation, nanoparticles
Graphical Abstract
1. Introduction
Inflammation is an acute, signal-mediated process that occurs in response to harmful stimuli. It involves the infiltration of immune cells and soluble mediators, such as tumor necrosis factor alpha (TNF-α), to the site of inflammation, which is highly elevated in many chronic inflammation-related diseases [1]. Chronic inflammation-related diseases, such as rheumatoid arthritis (RA), may develop in response to failure to resolve acute inflammation [1, 2].
Anti-TNF-α therapies (e.g., Humira) have proven effective in treating arthritis [3–7]. In the past decade, there has been a growing interest in using TNF-α small interfering RNA (siRNA) to selectively reduce the production of the pro-inflammatory TNF-α cytokine to treat arthritis (16–21). Small interfering RNA has been formulated into nanoparticles to address issues related to siRNA’s short half-life, poor extravasation from blood vessels to target tissues, poor cellular uptake, and potential immunogenicity [8, 9]. Data from several studies showed that TNF-α siRNA-loaded nanoparticles or nanocomplexes, prepared with polymers (e.g., chitosan, poly (lactic-co-glycolic) acid (PLGA)) or lipids, are effective against RA in mouse models [10–15]. Various methods and compositions have been used to formulate nanoparticles with high siRNA encapsulation efficiency [15–18]. Previously, we developed acid-sensitive sheddable PEGylated PLGA nanoparticles that increase the distribution and retention of siRNA in chronic inflammation sites in a mouse model [19]. However, the nanoparticles suffer from low encapsulation efficiency and high burst release of siRNA [19]. In fact, high burst release of siRNA (20% or more within two days) is a common problem for siRNA formulations that have high encapsulation efficiencies [16, 20]. Therefore, a need persists for siRNA formulations that maintain high siRNA encapsulation efficiency but with minimum siRNA burst release [21].
In the present paper, we report the development of such a TNF-α siRNA nanoparticle formulation by complexing TNF-α siRNA with a biocompatible cationic lipid and then incorporating the nanocomplexes into solid-lipid nanoparticles prepared using lecithin, cholesterol, and a previously reported acid-sensitive stearic acid-polyethylene glycol (2000) hydrazone conjugate (PHC) [22]. Previously, we have shown that nanoparticles PEGylated with the PHC have increased distribution and retention in chronic inflammation sites in a mouse model, likely due to the relatively lower pH microenvironment in chronic inflammation sites [19, 23]. Herein, we have also evaluated the biodistribution of the new TNF-α siRNA nanoparticles in mouse models of chronic inflammation, including a mouse model of collagen-induced arthritis (CIA), and tested their efficacy in a mouse model of collagen antibody-induced arthritis (CAIA). Both CIA and CAIA models develop clinical features representative of RA in humans, including increased capillary permeability, accumulation of white blood cells, and severe joint damage and bone erosion [24–26]. Importantly, it is known that the CAIA model does not respond to methotrexate treatment [26]. Methotrexate is the first-line therapy for patients with early RA or low disease activity. However, some patients do not respond adequately to methotrexate, and biologics such as anti-TNF-α antibodies are combined with methotrexate (or other disease-modifying antirheumatic drugs) to manage the disease [24, 27].
2. Materials and Methods
2.1 Materials
The polyethylene glycol (2000)-hydrazone-stearic acid (C18) derivative (PHC) and polyethylene glycol (2000)-amide-stearic acid (C18) derivative (PAC) were synthesized following our previously published methods [22]. Cholesterol, chloroform, tetrahydrofuran (THF), Lugol’s solution, Tris-EDTA (TE), sodium dodecyl sulfate, Triton X-100, N, N-dimethyl-9,9-biacridinium dinitrate (Lucigenin), lipopolysaccharides (LPS) from Salmonella enterica serotype enteritidis, MISSION® siRNA Fluorescent Universal Negative Control #1 (Cyanine 5), Amicon Ultra centrifugal filter units Ultra-15 (MWCO 30 kDa) were from Sigma-Aldrich (St. Louis, MO). TopFluor cholesterol and 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP) were from Avanti Polar Lipids (Alabaster, AL). Lecithin was from Alfa Aesar (Ward Hill, MA). BLOCK-iT™ Fluorescent Oligo siRNA (labeled with fluorescein) was from Life Technologies (Grand Island, NY). Negative control siRNA (Medium GC Duplex), Dulbecco’s Modified Eagle Medium (DMEM), FBS, and streptomycin/penicillin were from Invitrogen (Carlsbad, CA). TNF-α siRNA (5′-GUCUCAGCCUCUUCUCAUUCCUGCT-3′) was synthesized by Integrated DNA Technologies (Coralville, IA). Methotrexate was from MP Biomedicals (Santa Ana, CA). Bio-Rad Bradford Protein Assays was from Bio-Rad Laboratories (Hercules, CA).
2.2 Preparation of siRNA-incorporated nanoparticles
The siRNA was first lipophilized by complexing it with DOTAP in a monophase [28]. Briefly, a 100 μl solution of 20 μM siRNA in TE buffer (10 mM Tris–HCl and 1 mM EDTA in water, pH 7.5) was added to 400 μl of RNase-free water. DOTAP in chloroform (1.25 mg in 680 μl) was then added drop-wise to the siRNA solution while stirring. The mixture was sonicated briefly in a water bath sonicator and mixed with 1.36 ml of methanol to form a monophase. After one hour of incubation at room temperature, the siRNA/DOTAP complexes were extracted into chloroform by phase separation. At the siRNA/DOTAP ratio used, more than 99% of the siRNA partitioned into the chloroform phase (by measuring the fluorescence intensity in the water phase and using fluorescently labeled siRNA).
Lecithin (3.2 mg) and cholesterol (1.6 mg), dissolved in chloroform, were added drop-wise to the siRNA/DOTAP complexes in chloroform while stirring. PHC or PAC (2 mg) dissolved in chloroform was then added drop-wise to the siRNA-lipids mixture. The resultant mixture was dried under nitrogen gas and then dissolved in 500 μl of THF, which was then added drop-wise into water while stirring to form nanoprecipitats. The resultant nanoparticle suspension was stirred at room temperature for 6 h to faciliate the evaporation of THF, subjected to ultrafiltation (MWCO, 30 KDa), washed once with water, and reconstituted in diethylpyrocarbonate (DEPC)-treated water (Invitrogen). Nanoparticles prepared with PHC were named AS-siRNA-SLNs, where AS indicates that the nanoparticles were PEGylated with acid-sensitive sheddable PEG(2000) (i.e., PHC) [19, 29]. Nanoparticles prepared with PAC were named AI-siRNA-SLNs, where AI indicates that the nanoparticles were PEGylated with the PAC, which is acid-insensitive. Fluorescently labeled nanoparticles were prepared by using fluorescently labeled siRNA or TopFluor cholesterol (62.5% of total cholesterol) in the preparation.
2.3 Characterization of siRNA-incorporated nanoparticles
The particle size, polydispersity index (PDI), and zeta potential of the siRNA-incorporated nanoparticles were determined using a Malvern Zeta Sizer Nano ZS (Westborough, MA). To determine the encapsulation efficiency of the siRNA in the nanoparticles, nanoparticles were prepared with fluorescein-labeled siRNA to measure the fluorescence intensity of the siRNA in the (ultra)filtrate using a BioTek Synergy HT Multi-Mode Microplate Reader (Winooski, VT, Ex = 485 nm, Em = 528 nm).
2.4 Transmission electron microscopy (TEM)
The morphology of the AS-TNF-α siRNA-SLNs was examined using an FEI Tecnai Transmission Electron Microscope in the Institute for Cellular and Molecular Biology (ICMB) Microscopy and Imaging Facility at The University of Texas at Austin (Austin, TX). Carbon-coated 400-mesh grids were activated for 1–2 min. One drop of the nanoparticle suspension was deposited on the grids and incubated overnight at room temperature before examination.
2.5 In vitro release of siRNA from the nanoparticles
The release of siRNA from the nanoparticles was measured using nanoparticles prepared with fluorescein-labeled siRNA. Briefly, about 9 mg of AS-siRNA-SLNs were suspended in 1 ml PBS (10 mM, pH 7.4) inside a dialysis device (MWCO 50 kDa, Spectrum Laboratories, Rancho Dominguez, CA), which was then placed into 50 ml PBS (10 mM, pH 7.4) and maintained in a shaker incubator (100 rpm, 37°C) (MAQ 5000, MODEL 4350, Thermo Fisher Scientific, Waltham, MA). At given time points (1, 24, 48, 96, 192, 450 and 720 h), the amount of siRNA in the release medium was determined by measuring the fluorescence intensity using a BioTek Synergy HT Multi-Mode Microplate Reader. The percent of siRNA released was calculated using the following equation: % released = 100 X (fluorescence intensity in the release medium/total fluorescence intensity of the encapsulated siRNA).
2.6 Effect of pH on the shedding of PEG from the nanoparticles and in vitro binding/uptake of siRNA-incorporated nanoparticles by macrophages
Murine macrophage J774A.1 cells (American Type Culture Collection, Manassas, VA) were seeded in a 12-well plate (2 × 105 cells/well). To study the effect of the acid-sensitive sheddable PEGylation of the nanoparticles on their uptake and/or binding by the cells, the AS-siRNA-SLNs or AI-siRNA-SLNs were pre-incubated in PBS (200 mM, pH 6.8 or 7.4) for 6 h to facilitate the shedding of PEG before the nanoparticles were added into the cell culture medium. After 50 min of co-incubation, the cells were washed with PBS (10 mM, pH 7.4) and lysed with a lysis solution that contained 2% (v/v) sodium dodecyl sulfate and 1% Triton X-100. The fluorescence intensity in the cell lysate was measured using a plate reader (Ex = 485 nm, Em = 528 nm). Bradford protein assay did not show any significant difference in the total protein concentrations in the lysates among the groups.
The shedding of PEG from the the nanoparticles was tested using an iodide staining method with Lugol’s solution [19]. Briefly, ~4.5 mg of nanoparticles were incubated in PBS (pH 6.8 or 7.4) for 6 h, ultrafiltrated, and then reconstituted in 1 ml of water. One hundred fifty microliters of the nanoparticles in dispersion were added to a solution that contained 950 μl of PBS (pH 7.4, 10 mM) and 68 μl of Lugol’s solution. After 5 min of incubation at room temperature, the absorbance (OD490 nm) was measured using a BioTek Synergy HT MultiMode Microplate Reader.
2.7 TNF-α release from J774A.1 macrophages in culture
J774A.1 cells were seeded in 96-well plates (10,000 cells per well). After 20 h incubation at 37°C, 5% CO2, the culture medium was replaced with serum-free DMEM containing TNF-α siRNA incorporated AS-siRNA-SLNs at a final siRNA concentration of 500 ng/ml. The culture medium was replaced 4 h later with fresh DMEM containing 10% FBS. Nineteen hours later, LPS was added into the cell culture medium to a final concentration of 100 ng/ml. The cell culture medium was harvested after five additional hours of incubation to measure TNF-α concentration using a mouse TNF-α ELISA kit (Thermo Fisher Scientific).
2.8 LPS-induced mouse model of chronic inflammation
All animal studies were conducted in accordance with the U.S. National Research Council Guidelines for the Care and Use of Laboratory Animals. The animal protocol was approved by the Institutional Animal Care and Use Committee at The University of Texas at Austin. Female C57BL/6 mice (6–8 weeks) were from Charles River Laboratories (Wilmington, MA). For imaging, mice were fed with alfalfa-free diet (Harlan, Indianapolis, IN) to minimize unwanted background signals. An LPS-induced mouse model of chronic inflammation was established as previously described [30]. Briefly, LPS was dissolved in sterile PBS (pH 7.4, 10 mM) at a concentration of 1 mg/ml. Mice were injected with 50 μl of the solution into the right hind footpad on day 0. On day 8, chronic inflammation was confirmed using an IVIS® Spectrum (Caliper, Hopkinton, MA) with a bioluminescence imaging system 20 min following intraperitoneal (i.p.) injection of lucigenin (15 mg/kg) (exposure time 60 s, large binning, field B). Lucigenin is known to react with the superoxide produced by macrophages during chronic inflammation [30]. Mice positive for chronic inflammation, as confirmed by IVIS® Spectrum imaging, were randomized into groups. Mice that did not show any significant chronic inflammation were excluded.
2.9 CIA model
Collagen-induced arthritis was induced in 8–12 week-old female DBA/1J mice (Taconic Biosciences, Hudson, NY) with a Hooke CIA Induction Kit following the manufacturer’s instructions (Hooke Laboratories, Lawrence, MA). Chicken type II collagen in an emulsion with Freund’s Complete Adjuvant was used for initial immunization, and chicken type II collagen in an emulsion with Freund’s Incomplete Adjuvant was used for boosting. The emulsions were intradermally injected in the base of the tail. Mice were observed daily for signs of joint inflammation.
2.10 Biodistribution studies
To evaluate the accumulation of the AI -siRNA-SLNs and AS-siRNA-SLNs in LPS-induced inflamed mouse feet, mice were i.v. injected with AI-siRNA-SLNs or AS-siRNA-SLNs (labeled with TopFluor cholesterol, 0.2 mg/kg), and the inflamed foot (i.e., right hind) was imaged using IVIS® Spectrum at 6 h and 24 h after the injection. As controls, mice were i.v. injected with sterile PBS. Mice were euthanized 24 h later to collect the inflamed foot and major organs (i.e., heart, kidneys, liver, spleen, and lung). All samples were then imaged using IVIS® Spectrum (Ex = 495 nm, Em = 507 nm). In another study, CIA mice were i.v. injected with PBS, free siRNA, or AS-siRNA-SLNs. The siRNA was labeled with Cy5 and was given at a dose of 0.5 mg siRNA/kg body weight. Mouse rear legs were imaged using IVIS® Spectrum at 24 h and 48 h after the injection (Ex = 485 nm, Em = 528 nm). All the fluorescence units are photons per second per centimeter square per steradian (p/s/cm2/sr).
2.11 CAIA model and siRNA treatment
CAIA was induced in 8–12 week-old male BALB/c mice (Charles River Laboratories) with an Arthrogen-CIA® 5-Clone Cocktail Kit (Chondrex Inc., Redmond, WA) following the manufacturer’s instructions. Mice were i.p. injected with the cocktail of antibodies on day 0. Three days later, mice were i.p. injected with LPS from Escherichia coli 0111:B4 (Chondrex) to trigger arthritis development. Animals were evaluated every 2 days for arthritis incidence. Paw thickness was measured and evaluated individually on a scale of 0–4, where a score of 4 indicates the most severe inflammation [31]. On days 1, 3, 5, and 7, mice were i.v. injected with AS-TNF-α siRNA-SLNs (TNF-α siRNA, 2 mg/kg) or AS-siRNA-SLNs containing the GC Duplex negative control siRNA (AS-Cont siRNA-SLNs). Other controls include CAIA mice i.v. injected with methotrexate (5 mg/kg) or left untreated. One group of healthy mice did not receive any treatment. Mice were euthanized on day 9.
2.12 Micro-CT analysis
The legs of CAIA mice were assessed at The University of Texas at Austin High-Resolution X-ray CT Facility. After the mice were euthanized, their right legs were immediately collected and stored at −80 °C. The specimens were placed with the implant axis perpendicular to the scanning section, and cross-section images of the specimens were acquired at an isotropic resolution of 14.5 μm using a micro-CT system (NSI scanner, Fein Focus High Power Xradia microXCT 400, Carl Zeiss, Oberkochen, Germany). Scanning parameters were: voltage of 100 kV, a current of 200 μA, no filter, Perkin Elmer detector, 0.25 pF gain, 1 fps, 1×1 binning, no flip, source to object 155.0 mm, source to detector 1316.961 mm, continuous CT scan, no frames averaged, 0 skip frames, 1800 projections, 5 gain calibrations, 5 mm calibration phantom, data range (−3.0, 40.0) (grayscale adjusted from NSI defaults), hardening correction 0.1. Voxel size was 4.5 μm, and total slices were 1790. About 30 slides were used to determine calcaneus bone density using the NIH ImageJ (Bethesda, MD).
2.13 Histopathologic examination
Upon euthanization, the hind legs of CAIA mice were immediately fixed in 10% neutral buffered formalin and transferred to 70% ethanol 24 h later. After decalcification, paraffin embedding, and sectioning, the specimens were stained with hematoxylin and eosin (H&E) or safranin O/fast green. H&E slides were given scores of 0–5 for inflammation according to the following criteria: 0, normal; 1, minimal infiltration of inflammatory cells in the periarticular area; 2, mild infiltration; 3, moderate infiltration; 4, marked infiltration; and 5, severe infiltration [32].
For the safranin O/fast green slides the following criteria was used: 0, normal; 1, minimal-to-mild loss of toluidine blue staining with no obvious chondrocyte loss or collagen disruption; 2, mild loss of toluidine blue staining with focal mild (superficial) chondrocyte loss and/or collagen disruption; 3, moderate loss of toluidine blue staining with multifocal moderate (to middle-zone depth) chondrocyte loss and/or collagen disruption; 4, marked loss of toluidine blue staining with multifocal marked (to deep-zone depth) chondrocyte loss and/or collagen disruption; and 5, severe diffuse loss of toluidine blue staining with multifocal severe (to tidemark depth) chondrocyte loss and/or collagen disruption [33, 34]. Each slide was scored by two independent observers (one blinded) and the average score was reported.
2.14 Statistical analysis
Statistical analyses were completed by performing analysis of variance followed by Fisher’s protected least significant difference procedure. A p value of ≤ 0.05 (two-tail) was considered significant.
3. Results and discussion
3.1 Preparation and in vitro characterization AS-TNF-α siRNA-SLNs
AS-TNF-α siRNA-SLNs were prepared by encapsulating siRNA complexed with DOTAP into solid-lipid nanoparticles comprised of cholesterol, lecithin, and an acid-sensitive stearoyl PEG(2000) hydrazone conjugate (PHC) previously synthesized in our laboratories. As a control, DOTAP-complexed siRNA was also encapsulated into solid-lipid nanoparticles comprised of cholesterol, lecithin, and an acid-insensitive stearoyl PEG(2000) amide conjugate (PAC). The AS-TNF-α siRNA-SLNs were 118 ± 7 nm in diameter, with a polydispersity index of 0.16 ± 0.01 and a zeta potential value of −13.8 ± 5.8 mV. The AS-TNF-α siRNA-SLNs were slightly negatively charged, which is preferred to reduce particle aggregation and recognition by the mononuclear phagocyte system (e.g. liver, spleen) after i.v. injection, thereby reducing the potential toxicity of the nanoparticles [35]. Importantly, the encapsulation efficiency (EE) of the siRNA in the nanoparticles was determined to be 93 ± 2 %. The hydrophobic ion pairing (HIP) technique helps to increase the encapsulation efficiency of the siRNA via complexation with the cationic DOTAP lipid [16]. The complexes were dispersed in THF or ethanol and then added drop wise into an aqueous solution to form nanoparticles using the solvent displacement method. The nanoparticles were comprised of a lipid core, which was wrapped with anionic and neutral lipids (i.e. lecithin and cholesterol). Transmission electron microscopic images show that the AS-TNF-α siRNA-SLNs are spherical in shape (Fig. 1A). Unlike other siRNA nanoparticle formulations that release ~20% of siRNA within two days [16, 20], data from an in vitro siRNA release study showed that there was a minimum burst release of siRNA from our AS-TNF-α siRNA-SLNs, and only about 5% of siRNA was released in a one-month release study (Fig. 1B). The United States Food and Drug Administration (FDA) Nanotechnology Task Force (NTF) recommends no or miminum burst release of the drug from nanoparticles in in vitro evaluation [36].
Fig. 1. Physical characterization of the AS-TNF-α siRNA-SLNs.

(A) Representative TEM images of AS-TNF-α siRNA-SLNs. (B) The in vitro release profile of fluorescently-labeled siRNA from AS-siRNA-SLNs. The diffusion of the siRNA across the dialysis membrane is included to show that the diffusion of siRNA across the membrane is not the rate-limiting step. Data are mean ± S.E.M. (n = 3).
It is noted that a similar composition has been reported to prepare solid-lipid nanoparticles by wrapping a hydrophobic core with lecithin and amphiphilic PEG conjugates [16]. However, the nanoparticles we report herein were made by a simpler method and have favorable in vitro parameters (i.e. a minimum burst release of siRNA vs. high burst release of about 50% of siRNA within 2 days). It is likely that our method of preparation, the composition of the SLNs (i.e. siRNA to DOTAP ratio, the inclusion of cholesterol), and the ratio of siRNA to total lipids all have contributed to the minimum burst release of siRNA from the siRNA-nanoparticles [16]. Moreover, unlike other methods reported to increase siRNA encapsulation in nanoparticles, the method used herein does not require a change in temperature or any chemical modifications [10, 15, 18].
3.2 Confirmation of pH-sensitive sheddale PEGylation of the AS-TNF-α siRNA-SLNs and their functionality in cell culture
To confirm the acid-sensitive PEGylation of the AS-TNF-α siRNA-SLNs, fluorescein-labeled siRNA was used to prepare AS-siRNA-SLNs and AI-siRNA-SLNs (as a control). Preincubation of the AS-siRNA-SLNs in pH 6.8 PBS (200 mM) for 6 h to facilitate the shedding of PEG chains, as compared to preincuabtion in pH 7.4 PBS (200 mM), before incubating with J774A.1 cells significantly increased the amount of siRNA associated with the J774A.1 cells (i.e. uptake and/or binding, Fig. 2A). For the AI-siRNA-SLNs, preincubation in pH 6.8 PBS did not significantly increase the amount of siRNA associated with J774A.1 cells (Fig. 2A), indicating that the AS-siRNA-SLNs and the AI-siRNA-SLNs were PEGylated as intended. The shedding of PEG from the AS-siRNA-SLNs was further confirmed by estimating PEG content with an iodide staining method. As shown in Fig. 2B, after preincubation of the AS-siRNA-SLNs in pH 6.8 PBS for 6 h, the content of PEG in the nanoparticles in dispersion was reduced by about 40%, as compared when the AS-siRNA-SLNs were preincubated in pH 7.4. In contrast, preincubation of the AI-siRNA-SLNs in pH 6.8 for 6 h did not lead to a significant decrease in the PEG content, as compared to incubation at pH 7.4 (Fig. 2B). Taken together, it is concluded that the PEGylation of the AS-siRNA-SLNs was more sensitive to shedding or hydrolysis at a lower pH (i.e. pH 6.8 vs. 7.4), whereas the PEGylation of the AI-siRNA-SLNs was not. Finally, it was observed that in contrast to the AI-siRNA-SLNs, the size of the AS-siRNA-SLNs, after 6 h of incubation in pH 6.8, was increased by around 18%, as compared to incubation in pH 7.4 (data not shown). It is likely that the shedding of PEG from the AS-siRNA-SLNs may have reduced their stability in aqueous dispersion, potentially leading to particle aggregation.
Fig. 2. Confirmation of acid-sensitive sheddable PEGylation of the AS-TNF-α siRNA-SLNs and their functionality.
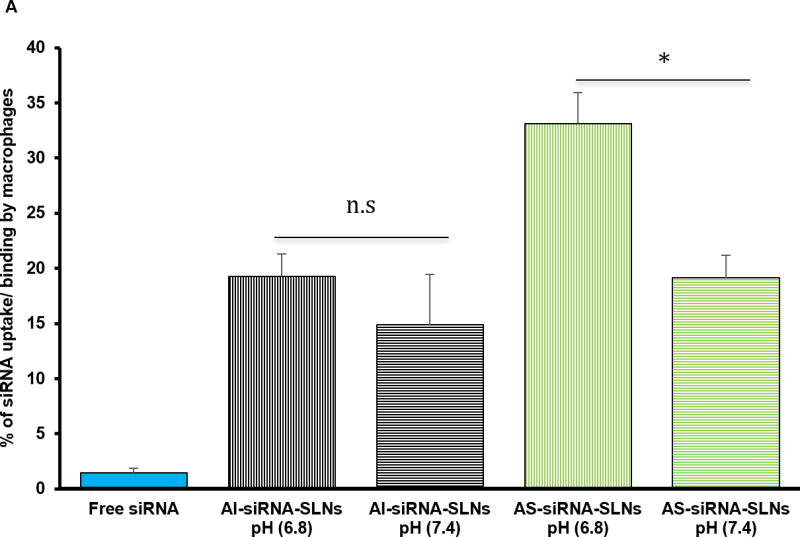
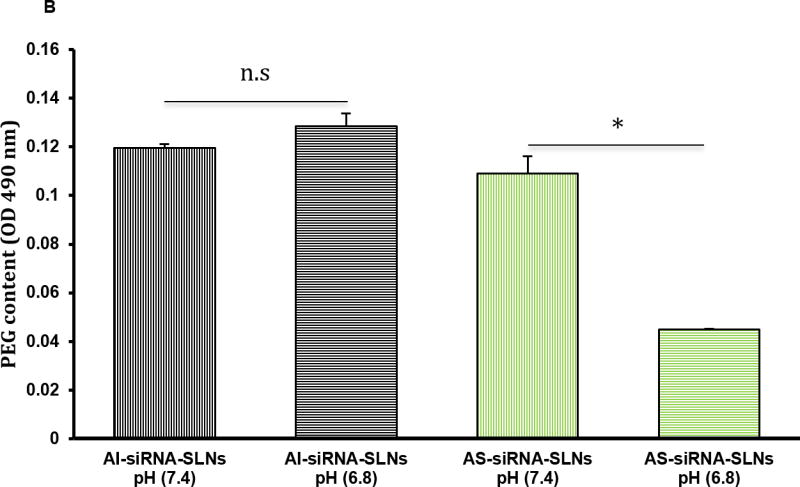
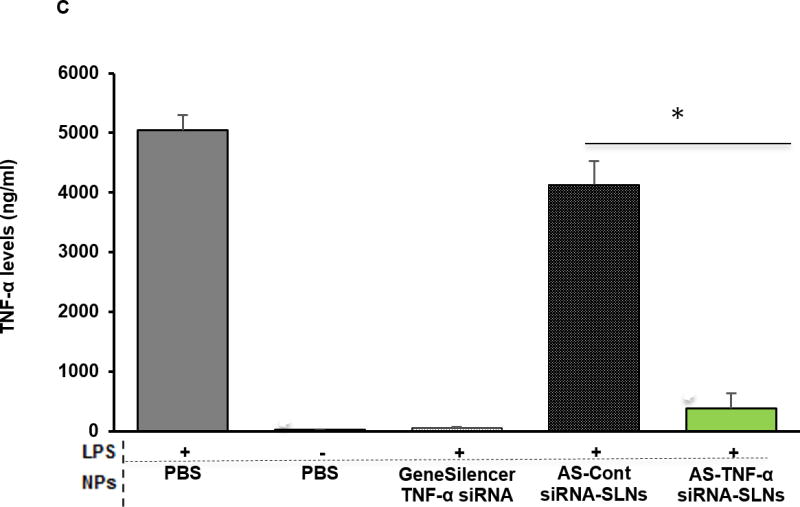
(A) Uptake and/or binding of AS-siRNA-SLNs by J774A.1 macrophages. J774A.1 cells (2 × 105) were seeded. Twenty hours later, the medium was replaced with serum-free DMEM containing fluorescein-labeled AS-siRNA-SLNs or AI-siRNA-SLNs that were pre-incubated at pH 6.8 or pH 7.4 for 6 h. The cells were washed after 50 min of incubation and lysed, and the fluorescence intensity was measured (*, p < 0.05). Data are mean ± S.E.M. (n = 10–12). (B) Levels of PEG on the surface of AI-siRNA-SLNs or AS-siRNA-SLNs after they were pre-incubated in pH 6.8 or pH 7 .4 for 6 h. Shown are OD490 values after samples were reacted with Lugol’s solution (n.s., no significant difference; * p < 0.05). Data are mean ± S.E.M. (n = 3). (C) Evaluation of the function of the TFN-α siRNA in down-regulating TNF-α release. J774A.1 cells (1 × 104) were seeded. Twenty hours later, the medium was replaced with serum-free DMEM containing AS-TNF-α-siRNA-SLNs (siRNA, 50 ng/well). Controls include AS-siRNA-SLNs containing a control siRNA, TNF-α siRNA complexes with the GeneSilencer, or sterile PBS. After 4 h of incubation, the medium was replaced with fresh medium. LPS (100 ng/ml) was added 19 h later, and the cells were incubated for five additional hours. TNF-α level in culture media was measured and divided by the % of cells alive estimated with an MTT assay (* p < 0.05). Data are mean ± S.E.M. (n = 5–10).
To validate the functionality of the TNF-α siRNA in the AS-TNF-α-siRNA-SLNs, AS-TNF-α-siRNA-SLNs were used to inhibit TNF-α cytokine production by LPS-stimulated J774A.1 cells. As controls, LPS-stimulated J774A.1 cells were treated with sterile PBS, siRNA-free AS-SLNs, or AS-siRNA-SLNs containing a control siRNA. As shown in Fig. 2C, only the AS-TNF-α-siRNA-SLNs significantly decreased TNF-α release by J774A.1 cells after LPS stimulation, demonstrating that the siRNA in the AS-TNF-α-siRNA-SLNs remained functional.
3.3 Distribution and accumulation of AS-TNF-α siRNA-SLNs in chronic inflammation sites in mouse models
Surface modification of nanoparticles with PHC generates a hydrophilic and flexible layer to shield nanoparticles, thereby preventing opsonization and reducing the clearance of nanoparticles by the mononuclear phagocyte system [19, 23]. To evaluate the effect of acid-sensitive sheddable PEGylation on the distribution and accumulation of the siRNA-nanoparticles in chronic inflammation sites, the accumulation and biodistribution of the AI -siRNA-SLNs and AS-siRNA-SLNs, both fluorescently labeled with TopFluor-cholesterol, in the inflamed feet of mice with LPS-induced chronic inflammation were evaluated. At 6 and 24 h after i.v. injection, the fluorescence intensity of the inflamed foot was measured using an in vivo imaging system. As shown in Fig. 3A–B, the fluorescence intensity was significantly higher in the inflamed feet in mice i.v. injected with the AS-siRNA-SLNs than in mice injected with the AI-siRNA-SLNs. Ex vivo IVIS® Spectrum imaging also showed that the biodistribution of the AI -siRNA-SLNs and AS-siRNA-SLNs in major organs of the mice are not significantly different, but different in the inflamed feet 24 h after i.v. injection (Fig. 3C). The extravasation through leaky vasculature and subsequent inflammatory cell-mediated sequestration (i.e. ELVIS) phenomenon in inflamed tissues is likely related to the enhanced accumulation and retention of the AS-siRNA-SLNs, as compared to the AI-siRNA-SLNs [19]. As we have previously reported, once the nanoparticles extravasate into inflamed tissues, the low pH environment likely facilitates the shedding of the PEG chains on the surface of the AS-siRNA-SLNs, enabling inflammatory cells, such as macrophages, in the inflamed tissues to readily take up the PEG -shed siRNA-SLNs [19]. For the AI-siRNA-SLNs, they can still extravasate into inflamed tissues, but the PEG chains on them that cannot be readily shed reduce their uptake by macrophages in inflamed tissues [19].
Figure 3. The biodistribution of AI-siRNA-SLNs and AS-siRNA-SLNs in the inflamed feet of mice with LPS-induced chronic inflammation.
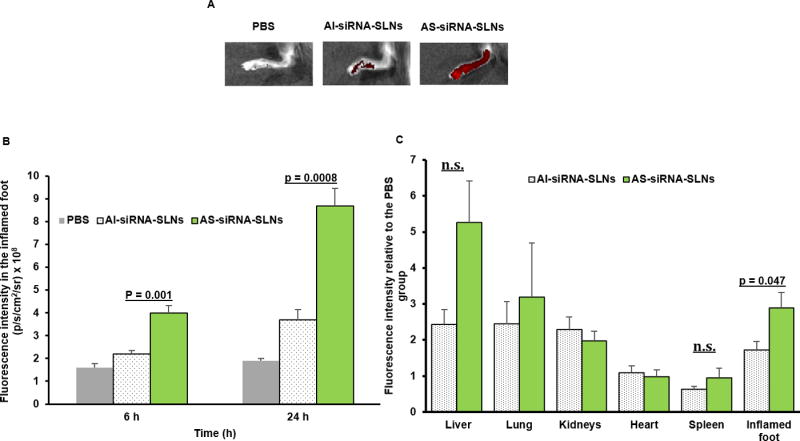
(A) Representative in vivo fluorescence images of inflamed mouse feet at 24 h after i.v. injection of PBS, AI-siRNA-SLNs or AS-siRNA-SLNs. The nanoparticles were labeled with TopFluor cholesterol. (B) Mean fluorescence intensity values of inflamed mouse feet 6 or 24 h after mice were i.v. injected with AI-siRNA-SLNs or AS-siRNA-SLNs. (C) Fluorescence intensity values in major organs and inflamed feet of mice 24 h after they were i.v. injected with AI-siRNA-SLNs or AS-siRNA-SLNs (values in Y-axis were obtained by dividing the fluorescence intensity values of the SLNs by the mean values of the PBS group). In B and C, Data are mean ± S.E.M. (n = 3–5).
To directly evaluate the extent to which our AS-TNF-α-siRNA-SLNs can improve the delivery of TNF-α siRNA into chronic inflammation sites, CIA mice (Fig. 4A) were i.v. injected with fluorescently-labeled siRNA, free or in AS-siRNA-SLNs, and the fluorescence intensity in the inflamed joints/feet of the mice was measured 48 h later. As shown in Fig. 4B–C, the fluorescent signals in the inflamed joints/feet in mice i.v. injected with the AS-siRNA-SLNs were significantly higher than that in mice i.v. injected with free siRNA. It is likely that free siRNA was extensively degraded and renally eliminated [19, 21], but the siRNA in the AS-TNF-α-siRNA-SLNs was protected and accumulated into the inflamed joints/feet while within the SLNs. Taken together, data in the LPS-induced chronic inflammation model and the CIA model demonstrate that our AS-siRNA-SLNs signficantly increased the distribution and retention of siRNAs in chronic inflammation sites in mouse models.
Figure 4. Distribution of siRNA, free or in AS-siRNA-SLNs, in the inflamed feet in CIA mice.

(A) Representative images of healthy feet or inflamed feet in DBA/1 mice with CIA. (B) Representative IVIS® Spectrum images of the left hind legs of CIA mice 48 h after i.v. injection of fluorescently -labeled siRNA, free or in AS-siRNA-SLNs. (C) Mean normalized fluorescence intensity values in inflamed hind legs 48 h after mice were i.v. injected with free siRNA or AS-siRNA-SLNs. Data are mean ± S.E.M. (n = 3–5, both hind legs).
3.4 Therapeutic effect of AS-TNF-α siRNA-SLNs in mice with CAIA
The therapeutic efficacy of the AS-TNF-α-siRNA-SLNs was evaluated in a mouse model of CAIA. The CAIA model was established by injecting mice i.p. with Arthrogen-CIA® 5-Clone Cocktail. The CAIA model has several advantages over the CIA model, including a rapid disease onset, a higher disease rate, applicability in a larger number of mouse strains. Moreover, it is known that the CAIA model does not respond to methotrexate. Methotrexate is the first-line therapy for patients with more than mild RA symptoms [24, 26, 37], but only about half of the patients will benefit from treatment with methotrexate alone [38]. Mice were then i.v. injected with AS-TNF-α-siRNA-SLNs on days 1, 3, 5 and 7. Control mice were left untreated or i.v. injected with AS-siRNA-SLNs containing a control siRNA or with methotrexate. As shown in Fig. 5A, when left untreated, the thickness of the mouse paws increased continuously. Treatment with AS-siRNA-SLNs prepared with a control siRNA or with methotrexate did not significantly affect the thickness of the mouse paws, as compared to when the mice were left untreated. However, treatment with our AS-TNF-α-siRNA-SLNs significantly reduced the paw thickness on days 6 and 8 (Fig. 5A). The clinical scores on day 6 also suggested a significant reduction in inflammation in mice treated with AS-TNF-α-siRNA-SLNs (data not shown). Moreover, micro-CT 3D images of the calcaneus bone of mice shows less roughness in mice treated with the AS-TNF-α-siRNA-SLNs than in mice treated with AS-siRNA-SLNs prepared with a control siRNA or mice treated with methotrexate (Fig. 5B). As expected, CAIA caused bone loss (Fig. 5C). Treatment with AS-TNF-α-siRNA-SLNs, but not with AS-siRNA-SLNs prepared with a control siRNA or with methotrexate, significantly inhibited bone loss (Fig. 5C).
Figure 5. Evaluation of the effect of AS-TNF-α siRNA-SLNs in a mouse model of CAIA.
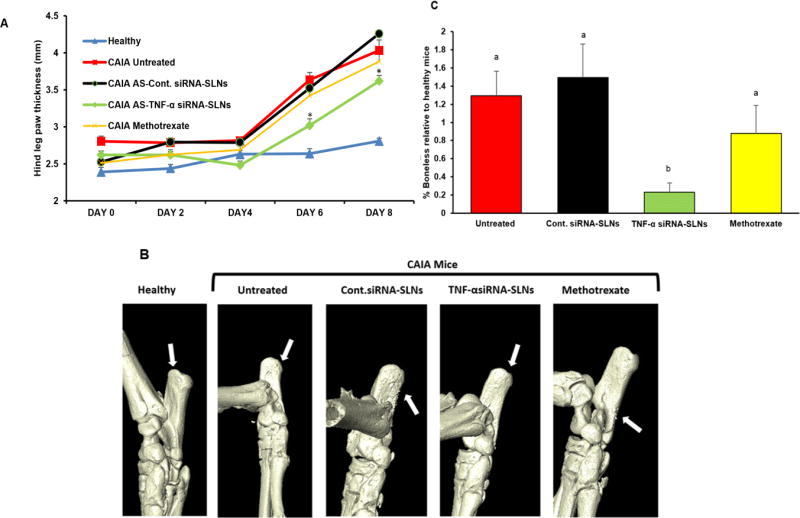
(A) Effect of AS-TNF-α siRNA-SLNs on hind leg paw thickness (* p < 0.05, AS-TNF-α siRNA-SLNs vs. AS-Cont siRNA-SLNs and CAIA untreated on days 6 and 8). (B) Representative 3D X-ray micro-CT reconstructed images of calcaneus bone of the ankle. (C) Relative bone density loss determined using ImageJ (a,b p ≤ 0.05). Data are mean ± S.E.M. (n = 3–5). Note: AS-TNF-α siRNA-SLNs and AS-Cont siRNA-SLNs are indicated as TNF-α siRNA-SLNs and Cont siRNA-SLNs in B–C.
Shown in Fig. 6A and 6B are mouse hind leg ankle joints after H&E and safranin-O staining, respectively. CAIA caused inflammatory cell infiltration in the joints and damage to articular cartilage and bones (Fig. 6A). Treatment with methotrexate or AS-siRNA-SLNs prepared with control siRNA did not show any significant effect. However, CAIA mice treated with AS-TNF-α-siRNA-SLNs showed only minimum inflammatory cell infiltration in the joints, with intact articular cartilage and healthy bones (Fig. 6A). Histopathological evaluation also showed that mice treated with AS-TNF-α-siRNA-SLNs showed significantly lower H&E and cartilage damage scores than untreated mice or mice treated with methotrexate or AS-siRNA-SLNs prepared with a control siRNA (Fig. 6B–C). The CAIA mouse model is known to be an aggressive antibody-dependent RA model, but is not dependent on T cells, which has been used to explain its unresponsiveness to methotrexate [26]. The observed effects of the AS-TNF-α-siRNA-SLNs are likely related to their silencing of TNF-α production by macrophages in the inflamed joints (30). In a previous study, Chia et al. reported that the concentration of TNF-α in hind paws in CAIA mice was below the lowest detection limit of ELISA [32]. Similarly, we could not detect any significant difference in TNF-α concentrations in the inflamed tissues or the serum samples of CAIA mice left untreated or treated with the AS-TNF-α-siRNA-SLNs. It is important to mention that during the study, there was not any significant difference in mouse body weight among all the CAIA groups (data not shown). An i.v. injection of siRNAs, even in nanoparticles, can induce a strong innate immune response triggered by the systemic induction of pro-inflammatory cytokines such as type I interferons [39, 40]. We did not detect any significant difference in pro-inflammatory cytokines (i.e. interferon-α, TNF-α) in the serum samples of CAIA mice treated with AS-TNF-α siRNA-SLNs, CAIA micr and healthy mice upon the euthanization of the mice (data not shown).
Figure 6. Histological evaluation of AS-TNF-α siRNA-SLNs in a mouse model of CAIA.
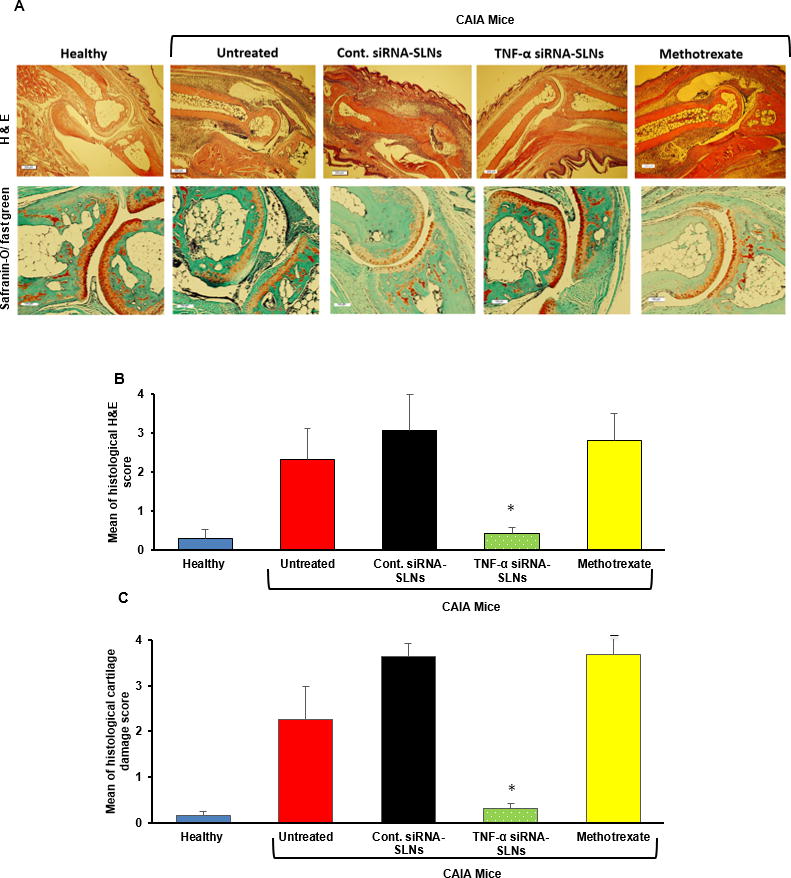
(A) Represent H&E (A) and Safranin-O/fast green images of arthritic joints. (B–C) The average scores of the severity of pathological factors such as synovial hypertrophy, density of resident cells and inflammatory cell infiltrates, matrix degradation and osteolysis after H&E staining (B) or safranin-O/fast green staining (C). In B–C, * p ≤ 0.05, AS-TNF-α siRNA-SLNs v s. other CAIA groups. Data are mean ± S.E.M. (n = 3–5). Note: AS-TNF-α siRNA-SLNs and AS-Cont siRNA-SLNs are indicated as TNF-α siRNA-SLNs and Cont siRNA-SLNs in the figures.
The AS-siRNA-SLNs were prepared with biocompatible materials. Further evaluatons of the efficacy of the AS-TNF-α siRNA-SLNs will be carried out after pharmacokinetics and toxicity tests in a murine model. Nonetheless, our data in Figs. 5–6 clearly demonstrate the potential feasibility of developing our AS-TNF-α siRNA-SLNs as a therapy to treat RA that does not respond adequately to methotrexate. This aspect of the TNF-α siRNA therapy, to our best knowledge, has not been noticed before. Other TNF-α siRNA formulations were tested in the CAIA model previously, but the efficacies of the formulations were not compared with that of methotrexate [34, 41]. There is currently no cure for RA. Current treatments that may control the disease include non-steroidal anti-inflammatory drugs (NSAIDs), corticosteroids, disease-modifying antirheumatic drugs (DMARDs, e.g. methotrexate), and biologic agents (e.g. anti-TNF-α monoclonal antibodies), which are all associated with significant side effects [42]. Biologic agents directly target components of the immune responses, including pro-inflammatory cytokines (e.g. TNF-α) and immune cells (e.g. B & T cells). Biologic agents are often reserved for patients who cannot tolerate or do not responsed adequately to DMARDs such as methotrexate [43, 44]. In RA therapy, when a patient begins to respond inadequately to methotrexate, the patient will be treated with another DMARD or a TNF inhibitor. However, clinical experience showed that biological agents exhibit enhanced efficacy to RA only when they are combined with methotrexate [45]. Based on our data in the CAIA model, there is the possibility that TNF-a siRNA may be effective in patients who respond inadequately to methotrexate. Finally, similar to many biological agents, TNF-α siRNA will likely be effective against other chronic inflammatory diseases as well.
4. Conclusion
In conclusion, we report the development of a TNF-α siRNA nanoparticle formulation that shows promising efficacy against arthritis in a mouse model of CAIA that does not respond to methotrexate. The nanoparticles have a high siRNA encapsulation efficiency (> 90%) and a minimum burst release of siRNA. The nanoparticles increase the delivery of siRNA into chronic inflammation sites.
Acknowledgments
This work was supported in part by the Alfred and Dorothy Mannino Fellowship in Pharmacy at UT Austin (to Z.C) and a grant from the U.S. National Institutes of Health (CA135274 to Z.C.). A.M.A. is a King Abdullah International Medical Research Center (KAIMRC) Scholar and was supported by the KAIMRC Scholarship Program. S.A.V. was supported in part by the Becas-Chile Scholarship from the Government of Chile. H.L.O and S.G.T were supported in part by the University Graduate Continuing Fellowship from The University of Texas at Austin.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jahoor A, Patel R, Bryan A, Do C, Krier J, Watters C, Wahli W, Li G, Williams SC, Rumbaugh KP. Peroxisome proliferator-activated receptors mediate host cell proinflammatory responses to Pseudomonas aeruginosa autoinducer. J Bacteriol. 2008;190:4408–4415. doi: 10.1128/JB.01444-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Choy EH, Panayi GS. Cytokine pathways and joint inflammation in rheumatoid arthritis. N Engl J Med. 2001;344:907–916. doi: 10.1056/NEJM200103223441207. [DOI] [PubMed] [Google Scholar]
- 3.van de Putte LB, Rau R, Breedveld FC, Kalden JR, Malaise MG, van Riel PL, Schattenkirchner M, Emery P, Burmester GR, Zeidler H, Moutsopoulos HM, Beck K, Kupper H. Efficacy and safety of the fully human anti-tumour necrosis factor alpha monoclonal antibody adalimumab (D2E7) in DMARD refractory patients with rheumatoid arthritis: a 12 week, phase II study. Ann Rheum Dis. 2003;62:1168–1177. doi: 10.1136/ard.2003.009563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schiff MH, Burmester GR, Kent JD, Pangan AL, Kupper H, Fitzpatrick SB, Donovan C. Safety analyses of adalimumab (HUMIRA) in global clinical trials and US postmarketing surveillance of patients with rheumatoid arthritis. Ann Rheum Dis. 2006;65:889–894. doi: 10.1136/ard.2005.043166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schreiber S. Certolizumab pegol for the treatment of Crohn’s disease. Therap Adv Gastroenterol. 2011;4:375–389. doi: 10.1177/1756283X11413315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Haens GR. Infliximab (Remicade), a new biological treatment for Crohn’s disease. Ital J Gastroenterol Hepatol. 1999;31:519–520. [PubMed] [Google Scholar]
- 7.Weinblatt ME, Bingham CO, 3rd, Mendelsohn AM, Kim L, Mack M, Lu J, Baker D, Westhovens R. Intravenous golimumab is effective in patients with active rheumatoid arthritis despite methotrexate therapy with responses as early as week 2: results of the phase 3, randomised, multicentre, double-blind, placebo-controlled GO-FURTHER trial. Ann Rheum Dis. 2013;72:381–389. doi: 10.1136/annrheumdis-2012-201411. [DOI] [PubMed] [Google Scholar]
- 8.Gao S, Dagnaes-Hansen F, Nielsen EJB, Wengel J, Besenbacher F, Howard KA, Kjems J. The Effect of Chemical Modification and Nanoparticle Formulation on Stability and Biodistribution of siRNA in Mice. Mol Ther. 2009;17:1225–1233. doi: 10.1038/mt.2009.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Leng Q, Woodle MC, Lu PY, Mixson AJ. ADVANCES IN SYSTEMIC siRNA DELIVERY. Drug Future. 2009;34:721–737. doi: 10.1358/dof.2009.034.09.1413267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Howard KA, Paludan SR, Behlke MA, Besenbacher F, Deleuran B, Kjems J. Chitosan/siRNA nanoparticle-mediated TNF-alpha knockdown in peritoneal macrophages for anti-inflammatory treatment in a murine arthritis model. Mol Ther. 2009;17:162–168. doi: 10.1038/mt.2008.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim SS, Ye C, Kumar P, Chiu I, Subramanya S, Wu H, Shankar P, Manjunath N. Targeted delivery of siRNA to macrophages for anti-inflammatory treatment. Mol Ther. 2010;18:993–1001. doi: 10.1038/mt.2010.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Komano Y, Yagi N, Onoue I, Kaneko K, Miyasaka N, Nanki T. Arthritic joint-targeting small interfering RNA-encapsulated liposome: implication for treatment strategy for rheumatoid arthritis. J Pharmacol Exp Ther. 2012;340:109–113. doi: 10.1124/jpet.111.185884. [DOI] [PubMed] [Google Scholar]
- 13.Presumey J, Salzano G, Courties G, Shires M, Ponchel F, Jorgensen C, Apparailly F, De Rosa G. PLGA microspheres encapsulating siRNA anti-TNFalpha: efficient RNAi-mediated treatment of arthritic joints. Eur J Pharm Biopharm. 2012;82:457–464. doi: 10.1016/j.ejpb.2012.07.021. [DOI] [PubMed] [Google Scholar]
- 14.te Boekhorst BC, Jensen LB, Colombo S, Varkouhi AK, Schiffelers RM, Lammers T, Storm G, Nielsen HM, Strijkers GJ, Foged C, Nicolay K. MRI-assessed therapeutic effects of locally administered PLGA nanoparticles loaded with anti-inflammatory siRNA in a murine arthritis model. J Control Release. 2012;161:772–780. doi: 10.1016/j.jconrel.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 15.Lee SJ, Lee A, Hwang SR, Park JS, Jang J, Huh MS, Jo DG, Yoon SY, Byun Y, Kim SH, Kwon IC, Youn I, Kim K. TNF-alpha gene silencing using polymerized siRNA/thiolated glycol chitosan nanoparticles for rheumatoid arthritis. Mol Ther. 2014;22:397–408. doi: 10.1038/mt.2013.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lobovkina T, Jacobson GB, Gonzalez-Gonzalez E, Hickerson RP, Leake D, Kaspar RL, Contag CH, Zare RN. In vivo sustained release of siRNA from solid lipid nanoparticles. ACS Nano. 2011;5:9977–9983. doi: 10.1021/nn203745n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Choi B, Cui ZK, Kim S, Fan J, Wu BM, Lee M. Glutamine-chitosan modified calcium phosphate nanoparticles for efficient siRNA delivery and osteogenic differentiation. J Mater Chem B Mater Biol Med. 2015;3:6448–6455. doi: 10.1039/C5TB00843C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Z, Li H, Wang K, Guo Q, Li C, Jiang H, Hu Y, Oupicky D, Sun M. Bioreducible Cross-Linked Hyaluronic Acid/Calcium Phosphate Hybrid Nanoparticles for Specific Delivery of siRNA in Melanoma Tumor Therapy. ACS Appl Mater Interfaces. 2017;9:14576–14589. doi: 10.1021/acsami.6b15347. [DOI] [PubMed] [Google Scholar]
- 19.Aldayel AM, Naguib YW, O/’Mary HL, Li X, Niu M, Ruwona TB, Cui Z. Acid-Sensitive Sheddable PEGylated PLGA Nanoparticles Increase the Delivery of TNF-[alpha] siRNA in Chronic Inflammation Sites. Mol Ther Nucleic Acids. 2016;5:e340. doi: 10.1038/mtna.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cun DM, Jensen DK, Maltesen MJ, Bunker M, Whiteside P, Scurr D, Foged C, Nielsen HM. High loading efficiency and sustained release of siRNA encapsulated in PLGA nanoparticles: Quality by design optimization and characterization. European Journal of Pharmaceutics and Biopharmaceutics. 2011;77:26–35. doi: 10.1016/j.ejpb.2010.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Lu Z, Wientjes MG, Au JL. Delivery of siRNA therapeutics: barriers and carriers. AAPS J. 2010;12:492–503. doi: 10.1208/s12248-010-9210-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu SJ, Wonganan P, Lansakara-P DSP, O’Mary HL, Li Y, Cui ZR. The effect of the acid-sensitivity of 4-(N)-stearoyl gemcitabine-loaded micelles on drug resistance caused by RRM1 overexpression. Biomaterials. 2013;34:2327–2339. doi: 10.1016/j.biomaterials.2012.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Mary HL, Aldayel AM, Valdes SA, Naguib YW, Li X, Salvady K, Cui Z. Acid-Sensitive Sheddable PEGylated, Mannose-Modified Nanoparticles Increase the Delivery of Betamethasone to Chronic Inflammation Sites in a Mouse Model. Mol Pharm. 2017;14:1929–1937. doi: 10.1021/acs.molpharmaceut.7b00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peres RS, Liew FY, Talbot J, Carregaro V, Oliveira RD, Almeida SL, Franca RF, Donate PB, Pinto LG, Ferreira FI, Costa DL, Demarque DP, Gouvea DR, Lopes NP, Queiroz RH, Silva JS, Figueiredo F, Alves-Filho JC, Cunha TM, Ferreira SH, Louzada-Junior P, Cunha FQ. Low expression of CD39 on regulatory T cells as a biomarker for resistance to methotrexate therapy in rheumatoid arthritis. Proc Natl Acad Sci U S A. 2015;112:2509–2514. doi: 10.1073/pnas.1424792112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bendele A. Animal models of rheumatoid arthritis. J Musculoskelet Neuronal Interact. 2001;1:377–385. [PubMed] [Google Scholar]
- 26.Lange F, Bajtner E, Rintisch C, Nandakumar KS, Sack U, Holmdahl R. Methotrexate ameliorates T cell dependent autoimmune arthritis and encephalomyelitis but not antibody induced or fibroblast induced arthritis. Ann Rheum Dis. 2005;64:599–605. doi: 10.1136/ard.2004.026120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kinder AJ, Hassell AB, Brand J, Brownfield A, Grove M, Shadforth MF. The treatment of inflammatory arthritis with methotrexate in clinical practice: treatment duration and incidence of adverse drug reactions. Rheumatology (Oxford) 2005;44:61–66. doi: 10.1093/rheumatology/keh512. [DOI] [PubMed] [Google Scholar]
- 28.Cui Z, Mumper RJ. Plasmid DNA-entrapped nanoparticles engineered from microemulsion precursors: in vitro and in vivo evaluation. Bioconjug Chem. 2002;13:1319–1327. doi: 10.1021/bc0255586. [DOI] [PubMed] [Google Scholar]
- 29.Zhu S, Lansakara PD, Li X, Cui Z. Lysosomal delivery of a lipophilic gemcitabine prodrug using novel acid-sensitive micelles improved its antitumor activity. Bioconjug Chem. 2012;23:966–980. doi: 10.1021/bc2005945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tseng JC, Kung AL. In vivo imaging method to distinguish acute and chronic inflammation. J Vis Exp. 2013;78:50690. doi: 10.3791/50690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lu LD, Stump KL, Seavey MM. Novel method of monitoring trace cytokines and activated STAT molecules in the paws of arthritic mice using multiplex bead technology. BMC Immunol. 2010;11:55. doi: 10.1186/1471-2172-11-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chia WT, Chen YW, Cheng LY, Lee HS, Chang DM, Sytwu HK. MMP-9 mRNA as a therapeutic marker in acute and chronic stages of arthritis induced by type II collagen antibody. J Formos Med Assoc. 2008;107:245–252. doi: 10.1016/S0929-6646(08)60143-6. [DOI] [PubMed] [Google Scholar]
- 33.Bendele AM, Chlipala ES, Scherrer J, Frazier J, Sennello G, Rich WJ, Edwards CK., 3rd Combination benefit of treatment with the cytokine inhibitors interleukin-1 receptor antagonist and PEGylated soluble tumor necrosis factor receptor type I in animal models of rheumatoid arthritis. Arthritis Rheum. 2000;43:2648–2659. doi: 10.1002/1529-0131(200012)43:12<2648::AID-ANR4>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 34.Ye C, Bhan AK, Deshpande V, Shankar P, Manjunath N. Silencing TNF-alpha in macrophages and dendritic cells for arthritis treatment. Scand J Rheumatol. 2013;42:266–269. doi: 10.3109/03009742.2013.777779. [DOI] [PubMed] [Google Scholar]
- 35.Frohlich E. The role of surface charge in cellular uptake and cytotoxicity of medical nanoparticles. Int J Nanomedicine. 2012;7:5577–5591. doi: 10.2147/IJN.S36111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tyner K, Sadrieh N. Considerations when submitting nanotherapeutics to FDA/CDER for regulatory review. Methods Mol Biol. 2011;697:17–31. doi: 10.1007/978-1-60327-198-1_3. [DOI] [PubMed] [Google Scholar]
- 37.Khachigian LM. Collagen antibody-induced arthritis. Nat Protoc. 2006;1:2512–2516. doi: 10.1038/nprot.2006.393. [DOI] [PubMed] [Google Scholar]
- 38.Romao VC, Canhao H, Fonseca JE. Old drugs, old problems: where do we stand in prediction of rheumatoid arthritis responsiveness to methotrexate and other synthetic DMARDs? BMC medicine. 2013;11:17. doi: 10.1186/1741-7015-11-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kanasty RL, Whitehead KA, Vegas AJ, Anderson DG. Action and reaction: the biological response to siRNA and its delivery vehicles. Mol Ther. 2012;20:513–524. doi: 10.1038/mt.2011.294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson AL, Linsley PS. Recognizing and avoiding siRNA off-target effects for target identification and therapeutic application. Nat Rev Drug Discov. 2010;9:57–67. doi: 10.1038/nrd3010. [DOI] [PubMed] [Google Scholar]
- 41.Shi Q, Rondon-Cavanzo EP, Dalla Picola IP, Tiera MJ, Zhang X, Dai K, Benabdoune HA, Benderdour M, Fernandes JC. In vivo therapeutic efficacy of TNFalpha silencing by folate-PEG-chitosan-DEAE/siRNA nanoparticles in arthritic mice. Int J Nanomedicine. 2018;13:387–402. doi: 10.2147/IJN.S146942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dinarello CA. Anti-inflammatory Agents: Present and Future. Cell. 2010;140:935–950. doi: 10.1016/j.cell.2010.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.O’Mary H, Del Rincomicronn I, Cui Z. Nanomedicine for Intra-Articular Drug Delivery in Rheumatoid Arthritis. Curr Med Chem. 2016;23:2490–2506. doi: 10.2174/0929867323666160530144445. [DOI] [PubMed] [Google Scholar]
- 44.Prasad LK, O’Mary H, Cui Z. Nanomedicine delivers promising treatments for rheumatoid arthritis. Nanomedicine. 2015;10:2063–2074. doi: 10.2217/nnm.15.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gibofsky A. Rheumatoid arthris: Challenges and opportunities in the evolving treatment landscape. WebMD, Transcript. 2017 https://www.google.com/search?source=hp&ei=N93gWpHSDM-MsQWZ97XoCw&q=Rheumatoid+Arthritis%3A+Challenges+and+Opportunities+in+the+Evolving+Treatment+Landscape&oq=Rheumatoid+Arthritis%3A+Challenges+and+Opportunities+in+the+Evolving+Treatment+Landscape&gs_l=psy-ab.3...1390.1390.0.1742.1.1.0.0.0.0.56.56.1.1.0....0...1c.2.64.psy-ab..0.0.0....0.rvuTthOF3rI.