

Summary
Skeletal muscle stem cell (MuSC) function declines with age and contributes to impaired muscle regeneration in older individuals. Acting through AMPK/p27Kip1, we have identified a pathway regulating the balance between autophagy, apoptosis, and senescence in aged MuSCs. While p27Kip1 is implicated in MuSC aging, its precise role and molecular mechanism have not been elucidated. Age-related MuSC dysfunction was associated with reduced autophagy, increased apoptosis, and hypophosphorylation of AMPK and its downstream target p27Kip1. AMPK activation or ectopic expression of a phosphomimetic p27Kip1 mutant was sufficient to suppress in vitro apoptosis, increase proliferation, and improve in vivo transplantation efficiency of aged MuSCs. Moreover, activation of the AMPK/p27Kip1 pathway reduced markers of cell senescence in aged cells, which was, in part, dependent on p27Kip1 phosphorylation. Thus, the AMPK/p27Kip1 pathway likely regulates the autophagy/apoptosis balance in aged MuSCs and may be a potential target for improving muscle regeneration in older individuals.
Keywords: muscle stem cell, regeneration, autophagy, apoptosis, senescence, AMPK, p27Kip1, aging, caloric restriction, cell transplantation
Graphical Abstract
Highlights
-
•
Aged MuSCs have reduced autophagy associated with apoptotic susceptibility
-
•
AMPK signaling is dysfunctional in the aged MuSC
-
•
AMPK phosphorylates p27Kip1, causing translocation from the nuclei to the cytoplasm
-
•
Cytoplasmic p27Kip1 enhances autophagy and prevents apoptosis and senescence
In this paper, White et al. show aged muscle stem cells have a reduction in AMPK/p27Kip1 signaling, resulting in decreased autophagy and susceptibility to apoptosis or senescence. The rescue of AMPK signaling in aged stem cells returns cellular function and regenerative capacity. These results show AMPK and related downstream mediators are viable targets to enhance aged muscle regeneration.
Introduction
Muscle stem cells (MuSCs) play a vital role in skeletal muscle regeneration (Fry et al., 2014, Moss and Leblond, 1971, von Maltzahn et al., 2013). MuSCs become dysfunctional with advanced age, leading to the inability to successfully regenerate muscle tissue after injury (Blau et al., 2015, Garcia-Prat et al., 2016b, Merlini et al., 2015). Age-associated reductions in MuSC numbers are associated with impairments in muscle regenerative capacity (Brooks et al., 2009, Collins et al., 2007, Conboy et al., 2010, Hikida, 2011, Jang et al., 2011, Shefer et al., 2010). In addition to reductions in satellite cell numbers, age-related impairments in MuSC activation contribute to sarcopenia (Sousa-Victor and Munoz-Canoves, 2016). The contribution of both systemic (Conboy et al., 2005, Sinha et al., 2014) and intracellular (Chakkalakal et al., 2012) factors influence this process; however, the mechanism(s) mediating this phenomenon are currently unclear.
Aged mammalian MuSCs have alterations in intracellular processes including autophagy and apoptosis (Garcia-Prat et al., 2016a, Jejurikar et al., 2006, Krajnak et al., 2006). Autophagy is initiated during periods of negative energy stress and generates substrates for energy production via degradation of cellular organelles and proteins (Mizushima et al., 2004). In the MuSC, autophagy provides initial energetics necessary for proliferation (Tang and Rando, 2014). Inhibition of the autophagy program results in perturbations in MuSC function including alterations in mitochondrial function, ATP production and induction of senescence (Garcia-Prat et al., 2016a, Tang and Rando, 2014). As a result, whole muscle regeneration is compromised when autophagy is genetically reduced in the MuSC (Garcia-Prat et al., 2016a). Aging MuSCs have an inherent reduction in autophagy (Garcia-Prat et al., 2016a), which contributes to the age-related dysfunction. Rescuing autophagy through genetic or pharmacological methods can recover aged MuSC function and rescue muscle regenerative capacity (Garcia-Prat et al., 2016a). Although the initial reports have established the importance of autophagy in MuSC function, upstream regulatory pathways have not been investigated, especially in the context of aging.
When autophagy is unable to meet the energy needs of activated cells, they become susceptible to apoptosis. During cell fate decisions, younger stem cells can initiate autophagy over apoptosis, while aged MuSCs are more likely to undergo apoptosis (Jejurikar et al., 2006). In an unchallenged state, in vitro activation of aged human MuSCs results in increased cell death (Fulle et al., 2013). Similarly, when mouse MuSCs are treated with pro-apoptotic factors such as tumor necrosis factor alpha and actinomycin D, apoptosis is more prevalent in aged MuSCs (Jejurikar et al., 2006). Thus, aged MuSCs have alterations in both apoptosis and autophagy processes critical for muscle regenerative capacity; yet, nodal signaling pathways responsible for such perturbations are currently unknown.
The AMPK signaling pathway has emerged as a potent regulator of autophagy, apoptosis, and proliferation (Liang et al., 2007, Sanli et al., 2014, Sun et al., 2014). In times of energy stress AMPK can promote autophagy directly through phosphorylation of ULK1 (Egan et al., 2011) or indirectly through inhibition of mammalian target of rapamycin (mTOR) complex 1 by phosphorylation of the tuberous sclerosis complex 2 (Garami et al., 2003, Inoki et al., 2003a, Inoki et al., 2003b, Tee et al., 2003, Zhang et al., 2003) and/or through phosphorylation of raptor (Gwinn et al., 2008). Furthermore, AMPK has been shown to regulate apoptosis in part, through phosphorylation of p27Kip1 (CDKN1B) (Liang et al., 2007). In the context of the MuSC, AMPK function is necessary for optimal muscle regeneration (Fu et al., 2015, Theret et al., 2017) however, functional effects of downstream p27Kip1 signaling in the aged MuSC warrants further investigation.
Once thought to simply function as a cyclin inhibitor, p27Kip1 is now recognized as a critical mediator of cell fate during metabolic stress conditions. p27Kip1 is involved in both cell-cycle inhibition and pathways related to autophagy and apoptosis (Liang et al., 2007). In times of cell stress, p27Kip1 can prevent apoptosis by directly inhibiting Cdk2 activation and downstream activity of the pro-apoptotic factor Bax (Gil-Gomez et al., 1998, Hiromura et al., 1999). The function of p27Kip1 is regulated by transcription (Rathbone et al., 2008), phosphorylation (Liang et al., 2002, Liang et al., 2007, Motti et al., 2005), degradation (Carrano et al., 1999, Montagnoli et al., 1999, Pagano et al., 1995), and subcellular location (Liang et al., 2002, Liang et al., 2007, Motti et al., 2005). Liang et al. (2007) reported that AMPK-dependent phosphorylation of p27Kip1 on Thr198 promotes p27Kip1 protein stability, resulting in more autophagy and less apoptosis. In addition, the mTOR-raptor complex can also regulate p27Kip1 phosphorylation and cellular localization through the serum and glucocorticoid-inducible kinase (SGK) (Hong et al., 2008). In aged MuSC, there is less mRNA expression of p27Kip1 (Chakkalakal et al., 2012), yet protein expression is greater in the nuclei where it can serve as cyclin inhibitor (Machida and Booth, 2004) with minimal effect on cell survival. In addition, p27Kip1 expression associates with maintenance of satellite cell populations that proliferate less frequently, but have long-term self-renewal capacity (Chakkalakal et al., 2014). It follows that the functional regulation of p27Kip1 may serve as a key regulatory pathway of both autophagy and apoptosis in MuSCs.
Here, we describe a molecular mechanism controlling apoptosis/autophagy and cell fate decisions involving AMPK signaling to the cyclin inhibitor, p27Kip1 in MuSCs. Furthermore, our results suggest that AMPK/p27Kip1 signaling is a critical regulatory step contributing to the phenotype of aged MuSCs.
Results
Aging Leads to a Reduction in MuSC Autophagy and Increased Apoptosis
We first determined whether aging affects MuSC autophagy and apoptosis in vitro during the initial days (first 48 hr) in culture. To determine the temporal pattern of autophagic flux in MuSCs isolated from young mice, we measured LC3B puncta at 12, 24, and 48 hr in culture. To accumulate and quantify LC3B puncta, 12 hr prior to each time point, we treated cells with chloroquine (10 μM), an inhibitor of autophagosome and lysosomal fusion (Figure S1A). During the first 48 hr in culture, puncta increased steadily in young MuSCs: puncta were abundant after 24 hr in culture and further increased at 48 hr. We next determined MuSC autophagic flux across a time course of physiological aging after 48 hr in culture. LC3B puncta was determined in MuSCs from 3 to 4 months (young), 12 months (middle-aged), 24 months (old), and >28 months (geriatric) mice. Compared with young cells, there was no change in flux in the middle-aged group (Figure 1A). In contrast, puncta formation was less in old MuSCs and even less in geriatric cells. A parallel experiment measuring protein expression of LC3B I and II isoforms showed similar trends. We observed a reduction in both LC3B I and II isoforms in old cells and a further reduction in geriatric cells (Figure 1B). Using the same time course of murine aging, we quantified markers of apoptosis, observing an inverse relationship between autophagy and apoptosis with aging. Protein expression of a marker of apoptosis, cleaved poly(ADP-ribose) polymerase (PARP), was minimal in young and middle-aged cells; however, expression was observed in old cells and was even greater in geriatric cells (Figures 1C and S1C). We also examined apoptosis by analysis of TUNEL and Annexin V staining. Again, compared with young and middle-aged cells, old, and geriatric cells had progressively more TUNEL and Annexin (+) cells (Figures 1D and S1D). In sum, MuSCs from aging mice exhibited progressively less autophagy and increased apoptosis across the lifespan; these trends were especially apparent in the geriatric mice, a state associated with reduced MuSC proliferative capacity (Figure 1E).
Figure 1.

Altered Autophagy and Apoptosis in Aged MuSCs
MuSCs were isolated throughout a time course of aging and plated for 48 hr for autophagy and apoptosis markers.
(A) Upper: representative images of LC3B punta after 10 μM chloroquine treatment in the final 12 hr of culture. Arrowheads indicate LC3B punta. Lower: quantification of relative punta number within age groups. DAPI, blue; LC3B, green; actin, red (N = 3 independent experiments) (A). Scale bar, 5 μm. Representative western blot of LC3B isoforms I and II after 10 μM chloroquine treatment in the final 12 hr of culture (B).
(C) Total and cleaved PARP protein expression in MuSCs after 48 hr in culture.
(D) Upper: representative TUNEL staining during respective ages. DAPI, blue; TUNEL, green. Scale bar, 50 μm. Lower: quantification of percentage TUNEL-positive MuSCs (N = 4 independent experiments). (E). Upper: representative EdU staining during respective ages. DAPI, blue; EdU, pink. Scale bar, 100 μm. Lower: quantification of percentage EdU-positive MuSCs (N = 3 independent experiments).
∗ Signifies different from young; † signifies different than old (p < 0.05). Data are presented as means ± SE.
Geriatric MuSCs Are Preferentially Susceptible to Cell Death during Autophagy Suppression
To determine whether there was a causal relationship between the reduction in autophagy and increase in MuSC apoptosis, we inhibited autophagy via lentivirus containing a short hairpin antisense RNA to Atg5, a key protein in formation of the autophagosome (Hara et al., 2006). The knockdown resulted in an 82% less Atg5 mRNA (Figure S2A) and 87% less Atg5 protein expression (data not shown). The reduction in Atg5 expression after 24 hr of infection resulted in a marked suppression of MuSC autophagy; this was indicated by the lack of LC3B isoform II protein expression (Figure S2B).
Suppression of autophagy did not induce apoptosis in young MuSCs (Figure 2A). In contrast, geriatric MuSCs were especially sensitive to autophagy suppression. Apoptosis increased by nearly 2-fold. Similar effects were observed with measurement of cell proliferation. The inhibition of autophagy by Atg5 knockdown delayed, but did not inhibit young MuSC proliferation throughout 72 hr in culture (Figure 2B). Geriatric MuSCs failed to undergo proliferation under these conditions (Figure 2B). To confirm the induction of apoptosis is associated with cell death, we quantified cell number for each treatment (Figure S2C). A total of 250 cells were plated for both young and geriatric groups with or without Atg5 knockdown. After 72 hr of autophagy inhibition, nearly half the geriatric cells were lost (Figure S2C). Autophagy has been linked to both canonical and non-canonical apoptotic pathways in adult stem cells (Chung and Yu, 2013). To investigate if the observed induction of cell death was through the canonical apoptotic pathways, we replicated the Atg5 knockdown experiments in both young and geriatric cells, with or without a pan-caspase inhibitor (Figure 2C). In both young and geriatric cells, the caspase inhibitor rescued the induction of cell death caused by inhibition of autophagy. Further investigation of the canonical apoptotic pathway showed overexpressed of Bcl2 under conditions of autophagy suppression via shATG5 expression was able to attenuate the onset of apoptosis (Figure S2D). Thus, apoptosis-induction via autophagy withdrawal appears to be dependent on the canonical apoptotic pathway. In morphologic analyses, 48 hr of Atg5 knockdown modestly reduced cell size in the young MuSC. However, the surviving geriatric Atg5 knockdown MuSCs failed to hypertrophy based on quantification of cell size (Figure 2D).
Figure 2.

Aged MuSCs Are Highly Susceptible to Apoptosis during Conditions of Autophagy Impairment
(A) Percentage of TUNEL-positive cells in young and geriatric groups infected with scramble or Atg5 sh lentivirus (N = 3 independent experiments). (B) A 72-hr time course of EdU labeling in young and geriatric MuSCs with or without an sh to Atg5 (N = 3 independent experiments).
(C) Percentage of TUNEL-positive cells in young and geriatric cells infected with Atg5 sh with or without caspase inhibitor Q-VD-OPh (N = 4 independent experiments).
(D) Upper: representative images of young and geriatric MuSCs infected with GFP scramble or shAtg5 lentivirus for 48 hr in culture. Lower: quantification of relative cell size normalized to young control cells (N = 3 independent experiments). Scale bar, 5 μm. Actin, green; nuclei, blue.
∗ Difference among age-matched treatment groups; # different from treatment-matched young cells; † difference between age-matched groups at 48 and 72 hr (p < 0.05). Data are presented as means ± SE.
Restoring AMPK Activity Prevents Cell Death in Aged MuSCs
We next sought a mechanism for the susceptibility of geriatric MuSCs to cell death under autophagy withdrawal. AMPK can regulate both autophagy and apoptosis through phosphorylation of the downstream target, p27Kip1 (Liang et al., 2007). In young MuSCs, phosphorylation of AMPK (Thr172) and p27Kip1 (Thr198) increased during the initial 48 hr in culture (Figure 3A). Comparing across age groups, we observed reduced AMPK/p27Kip1 phosphorylation in old mice; this was further reduced in geriatric mice at 48 hr in culture (Figure 3B). To determine if rescue of AMPK activation would prevent cell death in geriatric MuSCs, we treated cells with the AMP analog, 5-aminoimidazole-4-carboxamide ribonucleotide (AICAR). AICAR was effective at activating AMPK in geriatric MuSCs, as indicated by phosphorylation at Thr172 and increased AMPK-specific phosphorylation of p27Kip1 (Figure S3A). As signified by reductions in TUNEL staining (Figure 3C) and expression of cleaved PARP (Figure 3D), AICAR was sufficient to prevent cell death. To investigate the functional role of AMPK in the MuSC we genetically enhanced or suppressed AMPK activity before transplantation into injured muscle from young severe combined immunodeficiency (SCID) hosts (Figure 3E). Transplantation efficiency was high in young control MuSCs, showing numerous GFP(+) cells 4 days after transplantation (Figure 3F) and eventually incorporated into GFP(+) myofibers after 28 days (Figure 3F). Forced expression of a dominate negative AMPK prior to transplantation in young cells resulted in a roughly 70% reduction in GFP(+) donor cells/myofibers at each respective time point. Geriatric MuSCs had inherently poor transplantation efficiency, as represented by the presence of few GFP(+) cells at 4 days and GFP(+) myofibers at 28 days after transplant (Figure 3G). Prior forced expression of constitutively active AMPK to geriatric cells increased the number of GFP(+) cells and myofibers at 4 and 28 days after transplantation, respectively (Figure 3G). To show further physiological relevance to the AMPK/p27Kip1 pathway, we isolated MuSCs from old mice with or without short-term caloric restriction (CR) (40% CR), an established method to enhance MuSC function in old mice (Cerletti et al., 2012). After 48 hr in culture, phosphorylated and total forms of each protein were measured to determine if CR-mediated effects were associated with activation of the AMPK/p27Kip1 pathway in old MuSCs. MuSCs from CR-treated old mice increased phosphorylation status of AMPK and p27Kip1 compared with old mice fed ad libitum (Figure S3B).
Figure 3.

Restored AMPK Activation Rescues Inherent Apoptotic Susceptibility in Geriatric MuSCs
(A) Phosphorylated and total protein expression of young MuSC AMPK and p27Kip1 throughout 48 hr in culture.
(B) Phosphorylated and total protein expression of AMPK and p27Kip1 at 48 hr in culture across different ages.
(C) TUNEL-positive cells in young, geriatric and geriatric MuSCs treated with AICAR (N = 3 independent experiments). (D). Left: representative blot of total and cleaved PARP; right: quantification of cleaved PARP protein expression normalized to young cells (N = 3 independent experiments).
(E) Young and geriatric MuSCs infected with respective AMPK lentivirus, transplanted into injured SCID muscle and quantified 4 or 28 days later.
(F and G) Upper: young GFP-labeled MuSCs overexpressing GFP control or a dominant-negative (DN) AMPK 4 days; lower: 28 days after transplantation (N = 5 independent experiments) (F). Upper: geriatric GFP-labeled MuSCs overexpressing GFP control or constitutively active (CA) AMPK 4 days; lower: 28 days after transplantation. Quantification includes counts of GFP(+) cells (4d) or myofibers (28d) per muscle (N = 5 independent experiments) (G).
∗ Signifies difference from young cells; # signifies difference from GFP control transplanted cells (p < 0.05). Scale bars, 100 μm. Data are presented as means ± SE.
p27Kip1-Mediated Cell Survival Is Dependent on AMPK-Specific Thr198 Phosphorylation
After establishing the importance of AMPK signaling for regulation of cell survival and proliferation in the aging MuSC, we investigated downstream regulation of p27Kip1 function in the MuSC. Based on previously reported constructs (Liang et al., 2007), we generated lentiviral mutants of p27Kip1, identified as p27Kip1(198A) and p27Kip1(198D). The p27Kip1(198A) mutant cannot be phosphorylated at the AMPK-specific Thr 198, while p27Kip1(198D) serves as a Thr 198 phosphomimetic. We confirmed the potent suppression of AMPK-specific phosphorylation in the p27Kip1(198A) mutant after 24 hr of forced expression (Figure S4A). Next, we showed the anti-apoptotic effects of AICAR were dependent on phosphorylation of p27Kip1. Overexpression of the p27Kip1 mutant p27Kip1(198A), negated the effect of AICAR on MuSC cell death (Figure 4A) and protein expression of cleaved PARP (Figure 4B). To further validate AMPK/p27Kip1 signaling as a cell survival mechanism, we suppressed autophagy in young and geriatric cells with concurrent overexpression of the p27Kip1(198A) or p27Kip1(198D) mutants. In the young MuSC, overexpression of the constitutively active p27Kip1(198D) had minimal effect on cell death; however, overexpression of p27Kip1(198A) resulted in increased cell death (Figure 4C). In geriatric MuSCs, overexpression of p27Kip1(198D) rescued the discernible induction of cell death during Atg5 knockdown, in contrast, the p27Kip1(198A) mutant could not rescue the induction of cell death. Overexpression of the p27Kip1(198D) mutant also prevented cleaved PARP expression in both untreated and ATG5 knockdown geriatric MuSCs (Figure S4B).
Figure 4.

AMPK/p27Kip1 Signaling Mediates Cell Survival in the MuSC
Geriatric MuSCs were cultured for 48 hr treated with or without AICAR and/or overexpression of p27Kip1 mutant-p27Kip1(198A).
(A) Percentage of TUNEL-positive cells with respective treatment (N = 4 independent experiments).
(B) Upper: representative blot of total and cleaved PARP; lower: quantification of protein expression normalized to untreated geriatric cells (N = 3 independent experiments).
(C) Percentage of TUNEL-positive cells during Atg5 knockdown with concurrent overexpression of p27Kip1 mutants p27Kip1(198D) or p27Kip1(198A) (N = 3 independent experiments).
(D) Percentage of TUNEL-positive cells during treatment of compound C to young MuSCs with concurrent overexpression of p27Kip1 mutant p27Kip1(198D) (N = 3 independent experiments).
(E) Geriatric MuSCs treated with or without AICAR and infected with respective p27Kip1 mutant lentivirus. Cells were transplanted into injured young SCID muscle and quantified 4 or 28 days later.
(F) Geriatric MuSCs treated with or without AICAR and overexpression of p27Kip1 mutants analyzed 4 days after transplantation.
(G) Quantification of GFP(+) cells per muscle (N = 6 independent experiments).
(H) Geriatric MuSCs treated with or without AICAR and overexpression of p27Kip1 mutants analyzed 28 days after transplantation.
(I) Quantification of GFP(+) myofibers per muscle (N = 6 independent experiments).
Scale bar, 100 μm. ∗ Signifies difference from young or untreated control cells (p < 0.05). Data are presented as means ± SE.
Next, tested whether the phosphomimetic p27Kip1(198D) mutant could prevent cell death without upstream AMPK activation. We treated young MuSCs with the AMPK inhibitor compound C (10 μM) for 48 hr with or without forced expression of the of p27Kip1(198D) mutant. Compound C was effective in suppressing AMPK phosphorylation and phosphorylation of downstream AMPK target p27Kip1 in young MuSCs 48 hr in culture (Figure S4C). Compound C treatment nearly doubled the incidence of cell death in the young MuSC; this result was blocked by overexpression of p27Kip1(198D) (Figure 4D), further establishing p27Kip1 as a pro survival mechanism. We then tested the role of AMPK/p27Kip1 signaling in transplanted geriatric MuSCs (Figure 4E). Similar to genetic AMPK gain of function, AICAR treatment to geriatric MuSCs resulted in a high number of GFP(+) cells 4 days after transplantation (Figures 4F and 4G). The inability of AMPK to phosphorylate p27Kip1 decreased AICAR-induced enhancement of transplantation efficiency as shown by a vast reduction in GFP(+) cells. Overexpression of the phosphomimetic p27Kip1(198D) was sufficient to improve transplantation capacity independent of AICAR treatment (Figures 4F and 4G). At 28 days after transplantation, AICAR pre-treatment resulted in the sustainable incorporation of geriatric MuSCs as represented by GFP(+) myofibers (Figures 4H and 4I). Concomitant overexpression of p27Kip1(198A) once again reduced AICAR-related effects while overexpression of p27Kip1(198D) showed an AICAR-independent increase in GFP(+) myofibers 28 days after transplantation (Figures 4H and 4I).
Cellular Location of p27Kip1 Dictates Function in the Activating MuSC
The anti-apoptotic capacity of p27Kip1 is dependent on its translocation from the nuclei to the cytoplasm (Liang et al., 2002, Liang et al., 2007, Wu et al., 2006). To investigate whether the same is true in activating MuSCs, we determined the localization pattern of p27Kip1 in quiescent and activated MuSCs. Across all aging groups, p27Kip1 was located in the nuclei in freshly isolated quiescent MuSCs (Figure 5A). After 48 hr in culture, p27Kip1 in young MuSCs was located purely in the cytoplasm; in contrast old MuSCs had roughly equal amount in the nuclei and cytoplasm; in geriatric MuSCs p27Kip1 remaining primarily in nuclei (Figure 5B). We made similar observations with p27Kip1 staining in young and geriatric MuSCs after 48 hr in culture (Figure S5A). Furthermore, we demonstrated the efficacy of AICAR treatment in geriatric MuSCs to translocate p27Kip1 to the cytosol (Figure S5A). We also tested the cellular location of the p27Kip1 mutants. Overexpression of the p27Kip1(198A) mutant was located primarily in the nuclei; in contrast, the AMPK phosphomimetic p27Kip1(198D) mutant was located in the cytoplasm (Figure S5B).
Figure 5.

Cellular Location of p27Kip1 Regulates MuSC Function in Part through the Induction of Autophagy
(A) Upper: p27Kip1 protein expression in both cytoplasmic and nuclear compartments of freshly sorted MuSCs across age groups. Lower: quantification of percentage of cytoplasmic and nuclear expression of p27Kip1 (N = 3 independent experiments).
(B) Upper: p27Kip1 protein expression in both cytoplasmic and nuclear compartments of activated MuSCs (48 hr in culture) across age groups. Lower: quantification of percentage of cytoplasmic and nuclear expression of p27Kip1 (N = 3 independent experiments).
(C) p27Kip1 phosphorylation status-mediated cellular location and autophagy. Left: immunofluorescence staining of LC3B and p27Kip1 expression of young MuSCs overexpressing GPF-labeled WT p27Kip1, p27Kip1(198A), or p27Kip1(198D). All cells were treated with chloroquine for the final 12 hr of culture. LC3B, red; p27Kip1, green; DAPI, blue. Scale bar, 5 μm in (C). Right: quantification of relative change in LC3B puncta across treatment groups (N = 3 independent experiments).
(D) LC3B isoform protein expression in young MuSCs overexpressing WT p27Kip1, p27Kip1(198A), or p27Kip1(198D).
(E) Upper: EdU incorporation in control or p27Kip1(198A) overexpressing young MuSCs. Lower: quantification of percentage of EdU(+) MuSCs in each group (N = 3 independent experiments).
(F) Upper: EdU incorporation in control, p27Kip1(198A) overexpressing or p27Kip1(198D) overexpressing geriatric MuSCs with or without Atg5 knockdown. Lower: quantification of percentage of EdU(+) MuSCs in each group (N = 3 independent experiments).
∗ Signifies difference from young or control cells; † signifies difference from old cells (p < 0.05). Scale bars, 100 μm in (E) and (F). Data are presented as means ± SE.
Next, we determined if the cytoplamic p27Kip1 could induce autophagy as previously reported in non-muscle cell lines (Liang et al., 2007). Overexpression of the p27Kip1(198D) in young activated MuSCs increased LC3B puncta by roughly 50% (Figure 5C); and increased the ratio of LC3B II:I by 2-fold (Figure 5D). As indicated by immunofluorescence or protein analysis, overexpression of the p27Kip1(198A) mutant did not affect MuSC autophagy (Figures 5C and 5D). The ability to translocate p27Kip1 out of the nuclei appeared critical for cell proliferation. Young MuSCs overexpressing p27Kip1(198A) had a marked reduction in EdU labeling (Figure 5E). In contrast, overexpression of the p27Kip1(198D) mutant rescued proliferation in geriatric MuSCs. This effect was dependent on the induction of autophagy, as Atg5 knockdown negated the proliferative effects on p27Kip1(198D) overexpressing geriatric MuSCs (Figure 5F). These data suggest p27Kip1 induces cell survival in an autophagy-independent fashion, while its proliferative effects are autophagy dependent.
AMPK Activation through p27Kip1 Regulates Cellular Senescence
Loss of autophagy in satellite cells of young mice results in the induction of senescence markers (Garcia-Prat et al., 2016a). We sought to determine if restoring AMPK signaling activity would suppress senescence markers. Beta-galactosidase (β-gal), an established senescence marker (Dimri et al., 1995), was greater in old MuSCs and even greater in geriatric cells (Figure S6). AICAR treatment in geriatric cells suppressed β-gal staining (Figure 6A). Overexpression of the p27Kip1(198A) mutant attenuated AICAR-induced suppression of β-gal staining (Figure 6A). Similar effects were observed when quantifying protein expression of phosphorylated S6 Kinase (Figure 6B). mRNA expression of senescence genes p16INK4a and p21CIP1 increased in geriatric MuSCs (Figure 6C). AICAR treatment prevented p16INK4a and p21CIP1 expression while overexpression of p27Kip1(198A) attenuated this effect. Thus, the AMPK/p27Kip1 pathway appears to regulate MuSC cellular senescence in addition to the autophagy and apoptotic processes.
Figure 6.

The AMPK/p27Kip1 Pathway Regulates Cellular Senescence
(A) Upper: β-gal staining in geriatric MuSCs treated with AICAR and simultaneous overexpression of p27Kip1(198A). Arrows represent β-gal-positive cells. Lower: percentage of β-gal-positive cells in each group (N = 3 independent experiments).
(B) Upper: representative western blot of phosphorylated and total S6 kinase protein expression. Lower: quantification of phospho/total S6 ratio normalized to geriatric control cells (N = 4 independent experiments). (C) mRNA expression of p21CIP1 and p16INK4a in young and geriatric MuSCs treated with AICAR with or without p27Kip1(198A) overexpression (N = 4 independent experiments).
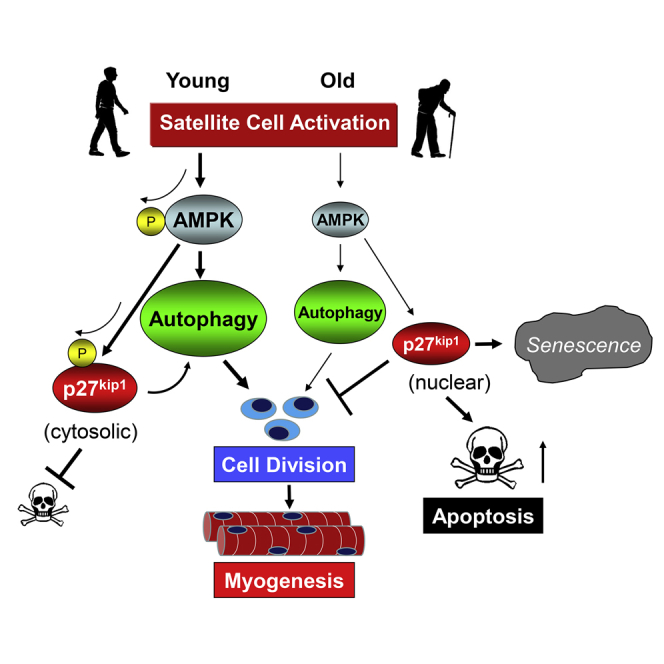
(D) Summary figure illustrating role of AMPK/p27Kip1 signaling on autophagy, proliferation, senescence, and apoptosis.
∗ Signifies difference from untreated geriatric control cells; † signifies difference from AICAR-treated geriatric cells; # signifies difference from young cells; ˆ signifies difference from untreated geriatric control cells (p < 0.05). Scale bars, 5 μm. Data are presented as means ± SE.
Discussion
Stem cell transition from quiescence to cell-cycle entry is a critical transformation. As cells proceed to the “blast” phenotype, in order to facilitate energy production and morphological changes, there is cytoplasmic hypertrophy (Rodgers et al., 2014). Cellular anabolism comes with high energy demands. As these processes require energy in excess of the minimal energy stores of the activating stem cell, this is a period of relative energy deficit and nutrient stress. Until energy demands are met or adequate survival measures are in place, the cell is susceptible to apoptosis. Our data support the role of AMPK/p27Kip1 signaling in ensuring cell survival through prevention of apoptosis and enhancing the autophagy program during early activation (Figure 6D). Defects in this pathway, through aging or genetic manipulation, lead stem cells to default to apoptosis or senescence (Garcia-Prat et al., 2016b).
Cell fate is determined by a delicate balance between energy availability and survival mechanisms. The interplay between autophagy and apoptosis is becoming increasing apparent across many cell types (Ravanan et al., 2017). In the MuSC, autophagy is critical for activation and proliferation, acting as a temporary energy reserve to fuel the initial stages of cell proliferation (Tang and Rando, 2014). Aging MuSCs have a reduction in autophagy while rescuing the aged-related defect has a rejuvenating effect, improving function and suppressing senescence (Garcia-Prat et al., 2016a). Aged MuSCs are more susceptibility to cell death when challenged with cellular stress (Jejurikar et al., 2006). We observed an age-dependent reduction in autophagy during the initial days of in vitro activation, associated with reduced proliferation and increased apoptosis. Furthermore, advanced age leaves the MuSC extremely susceptible to apoptosis under conditions of autophagy suppression. This is in contrast to what happens in young MuSCs during autophagy suppression (Tang and Rando, 2014), suggesting the possible presence of age-related perturbations in key survival mechanisms.
The AMPK pathway promotes cell survival during times of nutrient stress. Although AMPK targets numerus proteins and cellular pathways (Sanli et al., 2014), downstream signaling to the multifunctional p27Kip1 can induce autophagy; increase proliferation; and increase cell survival (Liang et al., 2007, Sun et al., 2014). We hypothesized and subsequently observed this pathway to be relevant in activating MuSCs. In activated young MuSCs, there was greater phosphorylation of AMPK and its downstream target p27Kip1 yet, in activated old and geriatric cells, there was less phosphorylation of these proteins. This observation is consistent with the idea that aging reduces AMPK sensitivity in muscle and likely throughout all organs and tissues (Salminen and Kaarniranta, 2012). Activation of the AMPK pathway by AICAR prevented apoptosis in aged cells; conversely, AMPK inhibition in young cells resulted in greater apoptosis. The lack of AMPK signaling could explain the differential age effects of apoptosis under autophagy inhibition. In the young MuSC, AMPK activation suppresses apoptosis and results in succeeding energy production, while the aged MuSC has a reduction in AMPK activity and the added stress of suppressed autophagy defaults the cell to apoptosis.
We observed the anti-apoptotic capacity of AMPK is dependent on phosphorylation and subsequent cytoplasmic translocation of p27Kip1. Machida and Booth (2004) reported aged MuSCs to have a greater expression of nuclear p27Kip1; we confirmed these findings. However, our data suggest cellular location of p27Kip1 is dynamic, shifting from the nucleus to the cytoplasm as the cell undergoes activation. Failure to translocate p27Kip1 during activation is one mechanism of age-related cellular dysfunction. Maintaining p27Kip1 in the nuclei not only inhibits cell-cycle progression, it also leaves the cell susceptible to apoptosis. In fact, inhibition of the cytoplasmic translocation of p27Kip1 is a popular pharmacological target to encouraging cell death in various cancer cell line (Hiromura et al., 1999, Liang et al., 2002, Motti et al., 2005, Wu et al., 2006). Our current work shows the potency of this pathway and its dysfunction with age in the MuSC.
Our current work and that of others (Garcia-Prat et al., 2016a) show advanced age drives senescence in muscle MuSCs. Considering the potency of the AMPK/p27Kip1 pathway in revitalizing cellular function in aged MuSCs, we strove to determine its regulatory role in senescence. AMPK activation was effective in blocking the induction of senescence markers. However, inability of AMPK to phosphorylate p27Kip1 negated these effects. Autophagy is a negative regulator of MuSC senescence (Garcia-Prat et al., 2016a). The translocation of p27Kip1 from the nuclei to the cytoplasm could play a role in preventing senescence by relieving the breaks on the cell cycle and promoting autophagy.
In sum, we observed a reduction in autophagy and more apoptosis in activating aged MuSCs when compared with their younger counterparts. This effect was due, in part by inactivation of the AMPK/p27Kip1 pathway. Reactivation of AMPK and/or downstream p27Kip1 translocation restored proliferation and cell survival in aged MuSCs. We observed functional differences in cytoplasmic and nuclear p27Kip1, the former enhancing cell survival and autophagy and the later promoting quiescence, apoptosis, and senescence. Thus, dysfunctional signaling of the AMPK/p27Kip1 pathway may be a central mechanism for age-induced loss of MuSCs, subsequent decline in regenerative function and perhaps a mechanism of and a viable target for enhancing regeneration in aged muscle.
Experimental Procedures
Animals
C57BL/6J (wild-type [WT]) mice were used at 3–4, 12, 24, or >28 months of age. All aged mice were obtained from the GSK aging colony (Jackson Laboratory) and NIA rodent colony. All animal care followed the guidelines and was approved by the Institutional Animal Care and Use Committees at the Duke Medical Center.
Lentivirus Infection
Lentivirus for GFP, sh-Atg5 and scrambled sh were purchased from Dharmacon (GE Lifesciences) with titer 108 transduction units (TU)/mL. Lentivirus for WT p27Kip1 and p27Kip1 mutant vectors, originally described in Liang et al. (2007) were cloned and amplified at the Duke Viral Vector Core with titer 109 TU/mL. For in vitro experiments lentivirus would be added shortly after plating and left overnight before replaced with fresh media.
Senescence-Associated β-Gal Activity
Senescence-associated (SA) β-gal activity was detected in MuSCs using the senescence β-Galactosidase Staining Kit (Cell Signaling Technology), according to manufacturer's instructions. SA β-gal+ cells were quantified as percentage of the total number of cells analyzed.
Isolation of Muscle Satellite Cells
Satellite cells were isolated by methods described previously (Bareja et al., 2014). In brief, dissected muscle was digested in gentleMACS C tubes (Miltenyi Biotec). subjected to dissociation using a GentleMACS dissociator (Miltenyi Biotec). After digestion, samples were filtered, spun, re-suspended, and transferred to a 5-mL fluorescence-activated cell sorting (FACS) tube for staining. Cells were stained with the following monoclonal antibodies—CD11b (1:100), CD31 (1:100), CD45 (1:100), Ter119, and Sca1 (all conjugated to APC, eBioscience), CD34 (1:50), phycoerythrin (BD Biosciences), and ITGA7 (FITC, R&D Scientific). Following a 45-min-long incubation on ice, the cell suspension was spun down and incubated with anti-APC magnetic beads (1:10, Miltenyi Biotec) for 15 min on ice. All cells bound to APC-conjugated antibodies were separated from the original suspension using the manual MACS cell separation protocol (Miltenyi Biotec). Mouse skeletal muscle precursors were isolated by FACS using CD34 and ITGA7 as positive selection markers while CD31, CD45, CD11b, SCA1, and Ter119 were used as negative selection markers (for further details, see Supplemental Experimental Procedures).
Protein and mRNA Expression
RNA isolation, cDNA synthesis and mRNA expression was performed as described previously (White et al., 2014). Primer sequences are as follows: p21cip1 F- CCTGGTGATGTCCGACCTG, R- CCATGAGCGCATCGCAATC, p16INK4a F- CGCAGGTTCTTGGTCACTGT, R- TGTTCACGAAAGCCAGAGCG. Atg5 F- TGTGCTTCGAGATGTGTGGTT, R- ACCAACGTCAAATAGCTGACTC. Protein isolation and western blotting was performed as described previously (White et al., 2012). Primary antibodies were as follows; tubulin, PARP, AMPK, p AMPK (Thr172), S6, pS6 (Ser240/244), LC3B (Cell Signaling Technologies), p27Kip1 (BD Biosciences), p p27Kip1 (thr198) (Thermo Fisher Scientific), Histone H1 (Santa Cruz) (see Table S1).
In Vitro Immunofluorescence and Quantification
MuSCs were fixed with 4% paraformaldehyde for 15 min, washed with PBS and permeabilized with TBS-T (0.1% Triton X- [pH 8.0] TBS) for another 15 min. The cells were then blocked in 2% normal goat serum for 20 min. This was followed by incubation with respective antibodies diluted in blocking solution for 1 hr. An Alexa Fluor 488 secondary antibody (Invitrogen, cat. no. A-21121) was used for detection. The cells were counterstained with DAPI (Cell Signaling, cat. no. 4083) and Phalloidin (Cell Signaling Technology, cat. no. 8953) to visualize nuclei and actin, respectively. Images were taken using fluorescent microscopes (Zeiss). Total cell number and the percentage of cells expressing a particular fluorophore were quantified using the automated Cellomics ArrayScan. ImageJ/FIJI was used to measure densitometry for cytoplasmic/nuclear localization and quantification of cell size.
Muscle Injury
Skeletal muscle injury was performed by injection of barium chloride (BaCl2) into the tibialis anterior (TA) muscle as described previously (White et al., 2009). Thirty microliters of 1.2% BaCl2 (Sigma) was injected into the TA muscle.
Proliferation Assay
Quantification of cell proliferation was assayed with Click-iT EdU Alexa Fluor 594 Imaging Kit (Thermo Fisher Scientific) according to manufacturer's instructions. Quantification of EdU-positive cells was performed on the automated Cellomics ArrayScan.
Cell Apoptosis
Quantification of cellular apoptosis was assayed with TUNEL and Annexin V/PI staining. TUNEL Click-iT Plus TUNEL Assay for In Situ Apoptosis Detection, Alexa Fluor 488 dye according to manufacturer's instructions. Quantification of TUNEL-positive cells was performed on the automated Cellomics ArrayScan.
Satellite Cell Engraftment
Transplantation methods were performed as described previously (Garcia-Prat et al., 2016a, Sousa-Victor et al., 2014). In brief, FACS isolated satellite cells from young or geriatric C57BL/6 mice were plated and treated with AICAR and/or infected with the respective lentivirus overnight. The next day the media was briefly replaced and cells were collected, re-suspended in 20% fetal bovine serum F10 medium and injected into muscles previously injured with BaCl2 the day before. Recipient mice were young (10–12 weeks) SCID mice. For each mouse, 10,000 cells were injected into the TA muscle. Transplantation experiments were analyzed 4 or 28 days post transplantation.
In Vivo Muscle Immunofluorescence
Immediately post dissection the whole muscle was incubated in 20% sucrose (w/v) for 12 hr then cut at the mid-belly and placed in OCT for later use. Sections (12 μm) were cut on a cryostat and dried at room temperature for 10 min. Sections were then blocked for 0.5 hr with 10% goat serum, followed by a 2-hr incubation with an anti-GFP antibody conjugated with Alex Flour 488 (Thermo Fisher Scientific, A-21311). After staining, slides were washed with PBS and mounted with mounting medium containing DAPI (Vector Laboratories, cat. no. H-1200). For quantification, six images (20×) were taken per TA muscle and GFP(+) cells or myofibers were counted and totaled for each muscle.
Cell Culture Reagents
To quantify LC3B flux MuSCs were treated with 10 μM chloroquine (Sigma) for 12 hr. To inhibit AMPK activity MuSCs were treated with 10 μM compound C (Sigma) throughout time in culture. To induce AMPK activity MuSCs were treated with 250 μM AICAR (Sigma) for the duration of time in culture. For transplantation experiments, MuSCs were treated with AICAR overnight, typically during lentiviral infection and transplanted the following day.
Cell Fractionation
Cytosolic and nuclear fractionation was performed with NE-PER Nuclear and Cytoplasmic Extraction Reagent (Thermo Scientific, no. 78833) according to manufacturer's instructions. Approximately 1 × 105 cells were plated for each sample to adequately quantify protein expression in nuclear and cytoplasmic compartments.
Statistical Analysis
Data were analyzed by one-way ANOVA using Tukey's post-hoc analysis or two-tailed Student's t test. All data are reported as means ± SEM. Statistical analysis was performed using GraphPad Prism 6 software.
Author Contributions
J.W. conceived, designed, and executed the experiments. A.B., W.K., K.H., and A.R. conceived and designed the experiments. J.W., A.B., W.K., and K.H. wrote the manuscript. M.C. executed experiments.
Acknowledgments
The authors gratefully acknowledge the Duke's Cancer Center Flow Cytometry, Functional Genomics and Viral Vector Cores, for support on this project. J.P.W. was supported by funds from GlaxoSmithKline, Duke Aging Center/Pepper Center grant P30 (AG028716) and K01 (AG056664).
Published: July 19, 2018
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, six figures, and one table and can be found with this article online at https://doi.org/10.1016/j.stemcr.2018.06.014.
Supplemental Information
References
- Bareja A., Holt J.A., Luo G., Chang C., Lin J., Hinken A.C., Freudenberg J.M., Kraus W.E., Evans W.J., Billin A.N. Human and mouse skeletal muscle stem cells: convergent and divergent mechanisms of myogenesis. PLoS One. 2014;9:e90398. doi: 10.1371/journal.pone.0090398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blau H.M., Cosgrove B.D., Ho A.T. The central role of muscle stem cells in regenerative failure with aging. Nat. Med. 2015;21:854–862. doi: 10.1038/nm.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks N.E., Schuenke M.D., Hikida R.S. No change in skeletal muscle satellite cells in young and aging rat soleus muscle. J. Physiol. Sci. 2009;59:465–471. doi: 10.1007/s12576-009-0058-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrano A.C., Eytan E., Hershko A., Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat. Cell Biol. 1999;1:193–199. doi: 10.1038/12013. [DOI] [PubMed] [Google Scholar]
- Cerletti M., Jang Y.C., Finley L.W., Haigis M.C., Wagers A.J. Short-term calorie restriction enhances skeletal muscle stem cell function. Cell Stem Cell. 2012;10:515–519. doi: 10.1016/j.stem.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakkalakal J.V., Christensen J., Xiang W., Tierney M.T., Boscolo F.S., Sacco A., Brack A.S. Early forming label-retaining muscle stem cells require p27kip1 for maintenance of the primitive state. Development. 2014;141:1649–1659. doi: 10.1242/dev.100842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakkalakal J.V., Jones K.M., Basson M.A., Brack A.S. The aged niche disrupts muscle stem cell quiescence. Nature. 2012;490:355–360. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung K.M., Yu S.W. Interplay between autophagy and programmed cell death in mammalian neural stem cells. BMB Rep. 2013;46:383–390. doi: 10.5483/BMBRep.2013.46.8.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins C.A., Zammit P.S., Ruiz A.P., Morgan J.E., Partridge T.A. A population of myogenic stem cells that survives skeletal muscle aging. Stem Cells. 2007;25:885–894. doi: 10.1634/stemcells.2006-0372. [DOI] [PubMed] [Google Scholar]
- Conboy I.M., Conboy M.J., Wagers A.J., Girma E.R., Weissman I.L., Rando T.A. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature. 2005;433:760–764. doi: 10.1038/nature03260. [DOI] [PubMed] [Google Scholar]
- Conboy M.J., Cerletti M., Wagers A.J., Conboy I.M. Immuno-analysis and FACS sorting of adult muscle fiber-associated stem/precursor cells. Methods Mol. Biol. 2010;621:165–173. doi: 10.1007/978-1-60761-063-2_11. [DOI] [PubMed] [Google Scholar]
- Dimri G.P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E.E., Linskens M., Rubelj I., Pereira-Smith O. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc. Natl. Acad. Sci. USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry C.S., Lee J.D., Mula J., Kirby T.J., Jackson J.R., Liu F., Yang L., Mendias C.L., Dupont-Versteegden E.E., McCarthy J.J. Inducible depletion of satellite cells in adult, sedentary mice impairs muscle regenerative capacity without affecting sarcopenia. Nat. Med. 2014;21:76–80. doi: 10.1038/nm.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X., Zhu M.J., Dodson M.V., Du M. AMP-activated protein kinase stimulates Warburg-like glycolysis and activation of satellite cells during muscle regeneration. J. Biol. Chem. 2015;290:26445–26456. doi: 10.1074/jbc.M115.665232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulle S., Sancilio S., Mancinelli R., Gatta V., Di Pietro R. Dual role of the caspase enzymes in satellite cells from aged and young subjects. Cell Death Dis. 2013;4:e955. doi: 10.1038/cddis.2013.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garami A., Zwartkruis F.J., Nobukuni T., Joaquin M., Roccio M., Stocker H., Kozma S.C., Hafen E., Bos J.L., Thomas G. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell. 2003;11:1457–1466. doi: 10.1016/s1097-2765(03)00220-x. [DOI] [PubMed] [Google Scholar]
- Garcia-Prat L., Martinez-Vicente M., Perdiguero E., Ortet L., Rodriguez-Ubreva J., Rebollo E., Ruiz-Bonilla V., Gutarra S., Ballestar E., Serrano A.L. Autophagy maintains stemness by preventing senescence. Nature. 2016;529:37–42. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- Garcia-Prat L., Munoz-Canoves P., Martinez-Vicente M. Dysfunctional autophagy is a driver of muscle stem cell functional decline with aging. Autophagy. 2016;12:612–613. doi: 10.1080/15548627.2016.1143211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gil-Gomez G., Berns A., Brady H.J. A link between cell cycle and cell death: Bax and Bcl-2 modulate Cdk2 activation during thymocyte apoptosis. EMBO J. 1998;17:7209–7218. doi: 10.1093/emboj/17.24.7209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gwinn D.M., Shackelford D.B., Egan D.F., Mihaylova M.M., Mery A., Vasquez D.S., Turk B.E., Shaw R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara T., Nakamura K., Matsui M., Yamamoto A., Nakahara Y., Suzuki-Migishima R., Yokoyama M., Mishima K., Saito I., Okano H. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–889. doi: 10.1038/nature04724. [DOI] [PubMed] [Google Scholar]
- Hikida R.S. Aging changes in satellite cells and their functions. Curr. Aging Sci. 2011;4:279–297. doi: 10.2174/1874609811104030279. [DOI] [PubMed] [Google Scholar]
- Hiromura K., Pippin J.W., Fero M.L., Roberts J.M., Shankland S.J. Modulation of apoptosis by the cyclin-dependent kinase inhibitor p27(Kip1) J. Clin. Invest. 1999;103:597–604. doi: 10.1172/JCI5461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F., Larrea M.D., Doughty C., Kwiatkowski D.J., Squillace R., Slingerland J.M. mTOR-raptor binds and activates SGK1 to regulate p27 phosphorylation. Mol. Cell. 2008;30:701–711. doi: 10.1016/j.molcel.2008.04.027. [DOI] [PubMed] [Google Scholar]
- Inoki K., Li Y., Xu T., Guan K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17:1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K., Zhu T., Guan K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115:577–590. doi: 10.1016/s0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- Jang Y.C., Sinha M., Cerletti M., Dall'Osso C., Wagers A.J. Skeletal muscle stem cells: effects of aging and metabolism on muscle regenerative function. Cold Spring Harb. Symp. Quant. Biol. 2011;76:101–111. doi: 10.1101/sqb.2011.76.010652. [DOI] [PubMed] [Google Scholar]
- Jejurikar S.S., Henkelman E.A., Cederna P.S., Marcelo C.L., Urbanchek M.G., Kuzon W.M., Jr. Aging increases the susceptibility of skeletal muscle derived satellite cells to apoptosis. Exp. Gerontol. 2006;41:828–836. doi: 10.1016/j.exger.2006.06.053. [DOI] [PubMed] [Google Scholar]
- Krajnak K., Waugh S., Miller R., Baker B., Geronilla K., Alway S.E., Cutlip R.G. Proapoptotic factor Bax is increased in satellite cells in the tibialis anterior muscles of old rats. Muscle Nerve. 2006;34:720–730. doi: 10.1002/mus.20656. [DOI] [PubMed] [Google Scholar]
- Liang J., Shao S.H., Xu Z.X., Hennessy B., Ding Z., Larrea M., Kondo S., Dumont D.J., Gutterman J.U., Walker C.L. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat. Cell Biol. 2007;9:218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- Liang J., Zubovitz J., Petrocelli T., Kotchetkov R., Connor M.K., Han K., Lee J.H., Ciarallo S., Catzavelos C., Beniston R. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 2002;8:1153–1160. doi: 10.1038/nm761. [DOI] [PubMed] [Google Scholar]
- Machida S., Booth F.W. Increased nuclear proteins in muscle satellite cells in aged animals as compared to young growing animals. Exp. Gerontol. 2004;39:1521–1525. doi: 10.1016/j.exger.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Merlini L., Bonaldo P., Marzetti E. Editorial: pathophysiological mechanisms of sarcopenia in aging and in muscular dystrophy: a translational approach. Front. Aging Neurosci. 2015;7:153. doi: 10.3389/fnagi.2015.00153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N., Yamamoto A., Matsui M., Yoshimori T., Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol. Biol. Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montagnoli A., Fiore F., Eytan E., Carrano A.C., Draetta G.F., Hershko A., Pagano M. Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev. 1999;13:1181–1189. doi: 10.1101/gad.13.9.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss F.P., Leblond C.P. Satellite cells as the source of nuclei in muscles of growing rats. Anat. Rec. 1971;170:421–435. doi: 10.1002/ar.1091700405. [DOI] [PubMed] [Google Scholar]
- Motti M.L., Califano D., Troncone G., De Marco C., Migliaccio I., Palmieri E., Pezzullo L., Palombini L., Fusco A., Viglietto G. Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/AKT pathway: regulation of p27kip1 expression and localization. Am. J. Pathol. 2005;166:737–749. doi: 10.1016/S0002-9440(10)62295-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano M., Tam S.W., Theodoras A.M., Beer-Romero P., Del Sal G., Chau V., Yew P.R., Draetta G.F., Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- Rathbone C.R., Booth F.W., Lees S.J. FoxO3a preferentially induces p27Kip1 expression while impairing muscle precursor cell-cycle progression. Muscle Nerve. 2008;37:84–89. doi: 10.1002/mus.20897. [DOI] [PubMed] [Google Scholar]
- Ravanan P., Srikumar I.F., Talwar P. Autophagy: the spotlight for cellular stress responses. Life Sci. 2017;188:53–67. doi: 10.1016/j.lfs.2017.08.029. [DOI] [PubMed] [Google Scholar]
- Rodgers J.T., King K.Y., Brett J.O., Cromie M.J., Charville G.W., Maguire K.K., Brunson C., Mastey N., Liu L., Tsai C.R. mTORC1 controls the adaptive transition of quiescent stem cells from G0 to G(Alert) Nature. 2014;510:393–396. doi: 10.1038/nature13255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salminen A., Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res. Rev. 2012;11:230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- Sanli T., Steinberg G.R., Singh G., Tsakiridis T. AMP-activated protein kinase (AMPK) beyond metabolism: a novel genomic stress sensor participating in the DNA damage response pathway. Cancer Biol. Ther. 2014;15:156–169. doi: 10.4161/cbt.26726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shefer G., Rauner G., Yablonka-Reuveni Z., Benayahu D. Reduced satellite cell numbers and myogenic capacity in aging can be alleviated by endurance exercise. PLoS One. 2010;5:e13307. doi: 10.1371/journal.pone.0013307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha M., Jang Y.C., Oh J., Khong D., Wu E.Y., Manohar R., Miller C., Regalado S.G., Loffredo F.S., Pancoast J.R. Restoring systemic GDF11 levels reverses age-related dysfunction in mouse skeletal muscle. Science. 2014;344:649–652. doi: 10.1126/science.1251152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa-Victor P., Gutarra S., Garcia-Prat L., Rodriguez-Ubreva J., Ortet L., Ruiz-Bonilla V., Jardi M., Ballestar E., Gonzalez S., Serrano A.L. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature. 2014;506:316–321. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- Sousa-Victor P., Munoz-Canoves P. Regenerative decline of stem cells in sarcopenia. Mol. Aspects Med. 2016;50:109–117. doi: 10.1016/j.mam.2016.02.002. [DOI] [PubMed] [Google Scholar]
- Sun X., Momen A., Wu J., Noyan H., Li R., von Harsdorf R., Husain M. p27 protein protects metabolically stressed cardiomyocytes from apoptosis by promoting autophagy. J. Biol. Chem. 2014;289:16924–16935. doi: 10.1074/jbc.M113.542795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang A.H., Rando T.A. Induction of autophagy supports the bioenergetic demands of quiescent muscle stem cell activation. EMBO J. 2014;33:2782–2797. doi: 10.15252/embj.201488278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tee A.R., Manning B.D., Roux P.P., Cantley L.C., Blenis J. Tuberous sclerosis complex gene products, Tuberin and Hamartin, control mTOR signaling by acting as a GTPase-activating protein complex toward Rheb. Curr. Biol. 2003;13:1259–1268. doi: 10.1016/s0960-9822(03)00506-2. [DOI] [PubMed] [Google Scholar]
- Theret M., Gsaier L., Schaffer B., Juban G., Ben Larbi S., Weiss-Gayet M., Bultot L., Collodet C., Foretz M., Desplanches D. AMPKalpha1-LDH pathway regulates muscle stem cell self-renewal by controlling metabolic homeostasis. EMBO J. 2017;36:1946–1962. doi: 10.15252/embj.201695273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Maltzahn J., Jones A.E., Parks R.J., Rudnicki M.A. Pax7 is critical for the normal function of satellite cells in adult skeletal muscle. Proc. Natl. Acad. Sci. USA. 2013;110:16474–16479. doi: 10.1073/pnas.1307680110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J.P., Baltgalvis K.A., Sato S., Wilson L.B., Carson J.A. Effect of nandrolone decanoate administration on recovery from bupivacaine-induced muscle injury. J. Appl. Physiol. 2009;107:1420–1430. doi: 10.1152/japplphysiol.00668.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J.P., Gao S., Puppa M.J., Sato S., Welle S.L., Carson J.A. Testosterone regulation of Akt/mTORC1/FoxO3a signaling in skeletal muscle. Mol. Cell Endocrinol. 2012;365:174–186. doi: 10.1016/j.mce.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White J.P., Wrann C.D., Rao R.R., Nair S.K., Jedrychowski M.P., You J.S., Martinez-Redondo V., Gygi S.P., Ruas J.L., Hornberger T.A. G protein-coupled receptor 56 regulates mechanical overload-induced muscle hypertrophy. Proc. Natl. Acad. Sci. USA. 2014;111:15756–15761. doi: 10.1073/pnas.1417898111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu F.Y., Wang S.E., Sanders M.E., Shin I., Rojo F., Baselga J., Arteaga C.L. Reduction of cytosolic p27(Kip1) inhibits cancer cell motility, survival, and tumorigenicity. Cancer Res. 2006;66:2162–2172. doi: 10.1158/0008-5472.CAN-05-3304. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Gao X., Saucedo L.J., Ru B., Edgar B.A., Pan D. Rheb is a direct target of the tuberous sclerosis tumour suppressor proteins. Nat. Cell Biol. 2003;5:578–581. doi: 10.1038/ncb999. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.