

Abstract
Previous work from our laboratories utilized a novel skin taping method and mass spectrometry-based proteomics to discover clinical biomarkers of skin conditions; these included atopic dermatitis, Staphylococcus aureus colonization, and eczema herpeticum. While suitable for discovery purposes, semi-quantitative proteomics is generally time-consuming and expensive. Furthermore, depending on the method used, discovery-based proteomics can result in high variation and inadequate sensitivity to detect low abundant peptides. Therefore, we strove to develop a rapid, sensitive, and reproducible method to quantitate disease-related proteins from skin tapings. We utilized isotopically-labeled peptides and tandem mass spectrometry to obtain absolute quantitation values on 14 peptides from 7 proteins; these proteins had shown previous importance in skin disease. The method demonstrated good reproducibility, dynamic range, and linearity (R2 > 0.993) when n=3 standards were analyzed across 0.05-2.5 pmol. The method was used to determine if differences exist between skin proteins in a small group of atopic versus non-atopic individuals (n=12). While only minimal differences were found, peptides were detected in all samples and exhibited good correlation between peptides for 5 of the 7 proteins (R2 = 0.71 – 0.98). This method can be applied to larger cohorts to further establish the relationships of these proteins to skin disease.
Keywords: Skin tapings, mass spectrometry, proteomics, atopic dermatitis, quantitation, isotope dilution
1.0 Introduction
Previous work from our laboratory has resulted in the development of a semi-quantitative mass spectrometry-based strategy to study skin diseases such as atopic dermatitis and eczema herpeticum. The strategy uses non-invasively obtained skin taping samples; a collection method that is appropriate for all age groups, including infants and children. In general, a total of 10 tapings from a single sampling results in confident identification of over 100 proteins. These include proteins that have previously been determined to be present in the cornified and granular layers of the skin[1].
When applied to a study on atopic dermatitis (AD), the semi-quantitative strategy successfully revealed novel proteins involved in the pathogenesis of AD[2]. Specifically, it was found that there was a lower expression of skin barrier proteins and enzymes in individuals with AD. These include filaggrin 2, corneodesmosin, and transglutaminase 3. It was further shown that patients colonized with methicillin-resistant Staphylococcus aureus (S. aureus) had greater amounts of epidermal fatty acid-binding protein (FABP)[3]; it was hypothesized that this might perpetuate the inflammatory response through eicosanoid signaling. While suitable for discovery based experiments, the semi-quantitative approach was deemed inappropriate for larger scale clinical studies. This was attributed to several factors including the time consuming and expensive nature of the approach and the concentration of protein required for technical replicates. Finally, relatively high variance in peptide abundances was observed during discovery experiments; this was considered to be largely due to matrix effects. The quantitative analysis of skin proteins has numerous additional applications, including cancer, psoriasis, and photodamage[4-7]. Therefore, the current study aimed to develop a method that overcame these challenges to produce a rapid, sensitive, and validated method to quantitate skin proteins.
Isotope dilution is a commonly used strategy for obtaining absolute concentrations (eg. Pg/ml) of molecules in complex matrices. The strategy has been applied in multiple examples of peptide quantitation[8-10]. Briefly, a synthetic, isotope-labeled peptide is added during sample preparation and acts as an internal standard; tandem mass spectrometry is used to resolve and quantitate both the endogenous and labeled peptides[11]. The known amount of the labeled peptide is used to determine the concentration of endogenous peptide in the sample. This strategy is sometimes referred to as isotope dilution.
In the current study, we developed an isotope dilution technique to quantitatively measure 14 peptides from 7 proteins; these proteins were previously shown to be related to skin disease[2, 3, 12]. Overall, the 17 min method exhibited good linearity for all peptides and good reproducibility when standards were spiked into control samples. When applied to a small study evaluating the effect of location on skin proteins, it was determined that only two peptides were consistently different between patients with atopic dermatitis. However, 5 of 7 proteins had good correlation between the two peptides used to quantitate that protein, with R2 values greater than 0.71.
2.0 Experimental
2.1 Sample Collection & Storage
Six atopic and six non-atopic subjects were consented under National Jewish Health IRB protocol # HS 1962, NJ209. Tapings were obtained for the following locations: bicep, antecubital fossae (anterior elbow), forearm, abdomen, back, thigh, and behind the knee. Sample collection procedures have been described elsewhere in detail [2]. Briefly, 20 standard D-Squame Skin Sampling Discs (CuDerm, Dallas, TX) were applied sequentially to the same location and placed adhesive side up in its own 20 ml borosilicate scintillation vial (Wheaton, Fisher Scientific, Pittsburgh, PA) and frozen in a -80°C freezer. Participants were advised to not to use soap, lotion, or perfume on the day of collection. In addition, forearm samples were obtained from one non-atopic individual for the purpose of method and assay development; this included the generation of linearity curves for all labeled standards.
2.2 Reagents
Ammonium bicarbonate (ABC), sodium dodecyl sulfate (SDS), LC/MS grade acetonitrile and formic acid were obtained from Fisher Scientific. LC grade water was obtained from Burdick and Jackson. HALT protease inhibitor 100× was obtained from Pierce. Dithiothreitol (DTT) and Iodoacetamide (IAA) were obtained from Bio-rad. Trypsin was obtained from Promega. Labeled peptides were custom synthesized from Sigma-Aldrich and New England Peptides. Since the peptides used for quantitation were from a tryptic digest, the C-terminus of the peptide was labeled with U-13C6, U-15N4 arginine (+10 da shift) or a U-13C6, U-15N4 lysine (+8 da shift). Individual peptides were reconstituted in either 0.1% formic acid and 30% acetonitrile or 5% ammonium hydroxide based on the solubility of the individual peptide. A mixture of all of the labeled peptides was then prepared at a concentration of 250pmol/ul in 0.1% formic acid in 30% acetonitrile. Peptide digestion tubes were then prepared by pipetting 20ul (5pmol total of each labeled peptide) of the combined mix into a 1.5 ml low retention microfuge tube and frozen at -70°C until ready for use.
2.3 Protein extraction
Discs (i.e. tape strips) were removed from the freezer and protein was harvested as follows: 500μl of extraction buffer (1% SDS, 50mM ABC, 10mM DTT, 1× HALT) was placed in the well of a 6 well cell culture plate and discs were placed in to the well with the sticky side down using forceps and incubated for 1 minute. While holding with forceps, the entire sticky side of the disc was scraped with a methanol cleaned cell scraper; as much sample was recovered as possible. The extracted disc was discarded. This process was repeated for up to 10 discs. The extract from 10 discs was then transferred to a 1.5 ml low retention microfuge tube and stored on ice. The volume was measured and the extraction well was washed with enough extraction buffer to bring the total volume of protein extract to 500ul. This process was performed separately for each disc obtained at each location.
Following extraction from the disc, the samples were vortexed briefly and centrifuged to collect all liquid. Each sample was sonicated 3 times for 2 sec each using a probe sonicator at a frequency of 25%. The samples were stored on ice for ∼1 min after each sonication. Between each sonication the probe was washed by sonication in Contrad, followed by rinsing with 70% EtOH, then sonication with Millipore water. The sonicated extracts were then centrifuged at 14,000 RPM for 10 min at 4°C and the supernatant was placed in a new microfuge tube.
Detergent was removed from the samples prior to protein digestion using a Pierce Detergent Removal Spin Column; the standard Pierce protocol was used with 50 mM ABC as wash buffer. The protein content was then determined using a Bradford protein assay with bovine serum albumin as the standard.
2.4 Protein digestion
Following initial processing, including detergent removal, a volume equivalent to 100ug of total protein was added to a 1.5ml centrifuge tube containing 5pM of each isotope labeled peptide. Samples were dried in a speedvac prior to alkylation and digestion. A volume of 52.5 ul of 50% trifluoroethanol in 50mM ABC and 0.2mM DTT was added to the sample; tubes were vortexed for 1 min to denature and reduce the proteins. The sample was then heated for 45 minutes at 65°C. 10ul of 4mM iodoacetamide was added and the sample was incubated in the dark at room temperature for 1 hour to alkylate cysteine residues. 2.5ul of 4mM DTT was added and the sample was incubated in the dark at room temperature for 1 hour to remove excess iodoacetamide. 3.3 ug of trypsin in 400ul of 25mM ABC was added and the sample was digested for 17 hours at 37°C. Following digestion, trypsin was deactivated with 2ul of neat formic acid; the sample was placed into a speedvac until dry. Digested protein samples were frozen at -80°C until analysis. On the day of analysis the samples were removed from the freezer and reconstituted in 100ul of 0.1% ammonium hydroxide in 3% acetonitrile. The samples were analyzed within 24 hours of reconstitution.
2.5 Quadrupole time-of-flight mass spectrometry and database searching for method development
For initial experiments designed to select peptides for inclusion in the assay, digested peptides were analyzed using liquid chromatography mass spectrometry (LC/MS) as described previously[13, 14]. Briefly, tryptic peptides were analyzed using an Agilent Technologies 1100 series nanoflow HPLC-chip/MS system coupled to an Agilent 6520 Quadrupole Time-of-Flight (QTOF) mass spectrometer. Separation of peptides was accomplished using a Protein ID high capacity chip (0.075 mm × 150 mm 300Å C18 analytical column with a 160 nL enrichment column). A total of 1 ug total protein was injected onto the column with the following run conditions: nano pump flow rate = 450 nL/min; capillary pump flow rate = 4 μL/min; drying gas temperature = 300°C; gas flow rate = 4 L/min and; capillary voltage = 1800V-2100V. The analytical separation employed a combination of Buffer A (3% Acetonitrile, 97% water, 0.1% formic acid) and Buffer B (90% Acetonitrile, 10% water, 0.1% formic acid). The linear gradient employed was from 3%-36% Buffer B in 33 minutes; a linear gradient to 80% Buffer B at 35 minutes; holding at 80% Buffer B until 40 minutes and an 8-minute post-run equilibration in 3% Buffer A.
Data analyses were performed using the Spectrum Mill MS Proteomics Workbench software suite (Rev A.03) (Agilent Technologies, Santa Clara, CA). Data were searched against the Swiss-Prot database (UniProt Release 14). Search parameters included setting the species selection to “human”; the missed cleavage allowance to “2” and; fixed modifications to include carbamidomethylation and oxidized methionine. Search criteria for data acquired on the Q-TOF allowed for 20 ppm precursor and 50 ppm product ion mass tolerances with a minimum matched peak intensity of 50%. Protein identifications were filtered based on the following criteria; protein score ≥ 11, individual peptide scores ≥ 7, and Scored Percent Intensity (SPI) ≥ 70%. Matching spectra were confirmed manually. Note that protein identification was only used for the purpose of identifying spectra that corresponded to specific peptides of interest. For a more complete catalog of proteins detected in skin tapings, please see [2].
2.6 Selection of MRM transitions
Proteins were selected for inclusion based on known biological relevance and differential analysis results from previous studies [2, 3]. Table 1 details the proteins and specific peptides used; briefly these were filaggrin I and II, corneodesmosin, fatty acid binding protein, serpin B3, transglutaminase 3, and alpha enolase. Agilent Spectrum Mill MRM selector was used to determine which tryptic peptides to use for the MRM transitions; Skyline was used to verify the fragment ions chosen for the MRM method. 2 peptides per protein and 2 transitions per peptide were monitored for each targeted protein.
Table 1.
Proteins and peptides selection for inclusion in quantitative assay. Proteins were chosen based on results from previous studies and are listed based on retention time. SpectrumMill MRM and Skyline were used to choose initial peptides and transitions. Precursor ions (Prec Ion), fragmentation voltage (CEV), Product Ions, and Ion fragments are shown for each peptide. Transitions for endogenous peptides are highlighted in grey, labeled peptides are shown in white.
| Peptide, protein name, accession # | RT | Prec Ion | CE (V) | Product Ion | Ion fragment |
|---|---|---|---|---|---|
| LAQAYYESTR Filaggrin I P20930 | 5.14 | 601.3 | 16.8 | 889.4 | y7 |
| 655.3 | y5 | ||||
| LAQAYYESTR* Filaggrin I P20930 (AQUA) | 606.3 | 16.8 | 899.4 | y7 | |
| 665.3 | y5 | ||||
| IILQPCGSK (alk) Corneodesmosin Q15517 | 6.44 | 508.3 | 13.5 | 789.4 | y7 |
| 548.2 | y5 | ||||
| IILQPCGSK* (alk) Corneodesmosin Q15517 (AQUA) | 512.3 | 13.5 | 797.4 | y7 | |
| 556.3 | y5 | ||||
| TTQFSCTLGEK (alk) Fatty Acid Binding Protein Q01469 | 6.52 | 636.3 | 18.1 | 941.4 | y8 |
| 794.4 | y7 | ||||
| TTQFSCTLGEK* (alk) Fatty Acid Binding Protein Q01469 (AQUA) | 640.3 | 18.1 | 949.5 | y8 | |
| 802.4 | y7 | ||||
| TNSILFYGR Serpin B3 P29508 | 9.65 | 535.8 | 14.5 | 855.5 | y7 |
| 655.4 | y5 | ||||
| TNSILFYGR* Serpin B3 P29508 (AQUA) | 540.8 | 14.5 | 865.5 | y7 | |
| 665.4 | y5 | ||||
| ITWLYDNTTGK Transglutaminase 3 Q08188 | 10.09 | 656.3 | 18.8 | 911.4 | y8 |
| 798.4 | y7 | ||||
| ITWLYDNTTGK* Transglutaminase 3 Q08188 (AQUA) | 660.3 | 18.8 | 919.5 | y8 | |
| 806.4 | y7 | ||||
| ELGVGIALR Fatty Acid Binding Protein Q01469 | 10.49 | 464.3 | 11.9 | 685.4 | y7 |
| 529.3 | y6 | ||||
| ELGVGIALR* Fatty Acid Binding Protein Q01469 (AQUA) | 469.3 | 11.9 | 695.4 | y7 | |
| 539.4 | y6 | ||||
| FYQTSVESVDFANAPEESR Serpin B3 P29508 | 10.7 | 726.0 | 21.3 | 802.4 | y7 |
| 617.3 | y5 | ||||
| FYQTSVESVDFANAPEESR* Serpin B3 P29508 (AQUA) | 729.3 | 21.3 | 812.4 | y7 | |
| 627.3 | y5 | ||||
| GSPGVPSFAAGPPISEGK Corneodesmosin Q15517 | 10.88 | 827.9 | 25 | 755.9 | y16++ |
| 629.3 | y13++ | ||||
| GSPGVPSFAAGPPISEGK* Corneodesmosin Q15517 (AQUA) | 831.9 | 25 | 759.9 | y16++ | |
| 633.3 | y13++ | ||||
| GNPTVEVDLFTSK Alpha enolase P06733 | 11.38 | 703.9 | 20.5 | 938.5 | y8 |
| 618.3 | y11++ | ||||
| GNPTVEVDLFTSK* Alpha enolase P06733 (AQUA) | 707.9 | 20.5 | 946.5 | y8 | |
| 622.3 | y11++ | ||||
| SQGVFQCGPASVIGVR (alk) Transglutaminase 3 Q08188 | 11.78 | 831.4 | 25.1 | 1015.5 | y10 |
| 855.5 | y9 | ||||
| SQGVFQCGPASVIGVR* (alk) Transglutaminase 3 Q08188 (AQUA) | 836.4 | 25.1 | 1025.5 | y10 | |
| 865.5 | y9 | ||||
| AAVPSGASTGIYEALELR Alpha enolase P06733 | 13.56 | 903.0 | 27.7 | 1164.6 | y10 |
| 1063.6 | y9 | ||||
| AAVPSGASTGIYEALELR* Alpha enolase P06733 (AQUA) | 908.0 | 27.7 | 1174.6 | y10 | |
| 1073.6 | y9 | ||||
| SVVTVIDVFYK Filaggrin 2 Q5D862 | 14.9 | 635.4 | 16.8 | 984.5 | y8 |
| 784.4 | y6 | ||||
| SVVTVIDVFYK* Filaggrin 2 Q5D862 AQUA (AQUA) | 639.4 | 16.8 | 992.6 | y8 | |
| 792.4 | y6 | ||||
| NPDDPDMVDVFMDHLDIDHNK Filaggrin I P20930 | 15.43 | 621.3 | 17.6 | 799.9 | y13++ |
| 741.4 | y6 | ||||
| NPDDPDMVDVFMDHLDIDHNK*Filaggrin I P20930 (AQUA) | 623.3 | 17.6 | 803.9 | y13++ | |
| 749.4 | y6 | ||||
| NPDDPDTVDVIMHMLDR Filaggrin 2 Q5D862 | 16.19 | 661.6 | 27.4 | 771.4 | y13++ |
| 623.6 | y16+++ | ||||
| NPDDPDTVDVIMHMLDR* Filaggrin 2 Q5D862 (AQUA) | 665.0 | 27.4 | 776.4 | y13++ | |
| 627.0 | y16+++ |
2.7 Labeled peptides
For each peptide selected for MRM quantitation, a labeled peptide was synthesized. For each labeled peptide, the C-terminus was labeled with either a U-13C6, U-15N4 arginine or a U-13C6, U-15N2 lysine. The “heavy” labeled arginine resulted in a +10 Da shift in mass; the heavy labeled lysine resulted in a +8 Da mass shift. Labeled peptides were added to extracted samples at a concentration of 5pmol per 100ug of total protein digested; this was performed immediately before reduction, alkylation, and digestion. A total of 14 peptides were synthesized.
2.8 Linearity in response of labeled peptides
To demonstrate linearity and dynamic range of the labeled peptides across a range of concentrations, 6 @ 100ug aliquots of pooled protein harvested from skin tapings was spiked will all 14 labeled peptides at concentrations of 0.5, 1.25, 2.5, 5, 12.5, and 25 pmol per 100ug of protein. The samples were then digested and analyzed as described.
2.9 Quantitative LC/MS/MS using triple quadrupole (QQQ)
Liquid chromatography was carried out using an Agilent 1200 series HPLC equipped with a quaternary pump (pump “A”), a binary pump (pump “B”), an autosampler with thermostat, and a column heating compartment with switching valve (Agilent Technologies, Palo Alto, CA). Buffer A was 0.1% formic acid in HPLC water and Buffer B was 0.1% formic acid in 90:10 acetonitrile:H2O. Gradient conditions for pump A were as follows: 3% B from 0 - 1 minute, 100% B from 1.01 - 10 minutes, 3% B from 10.01 – 17 minutes. Gradient conditions for pump B were as follows: 5% B from 0 - 1 minutes, 5-30% B from 1.01 – 15 minutes, 80% B from 15.01-16 minutes, and 100%B from 16.01-17 minutes. 20uL of the digested skin was injected onto the enrichment column for concentration/purification followed by backflushing and separation on the analytical column using the gradient conditions described above. The column switching valve was switched to redirect the flow from pump B to backflush the enrichment column onto the analytical column 1 minute after injection. Flow rates were 1 ml/min for pump A and 0.15 ml/min for pump B. The enrichment column was an SB-C8 2.1 × 12.5mm 5uM guard cartridge (Agilent Technologies, Palo Alto, CA). The analytical column was an Agilent 300SB-C18 column (1.0 × 150 mm) with a 3.5 uM particle size operated at 45C.
Detection of digested skin peptides was accomplished using an Agilent 6410 triple quadrupole mass spectrometer (QQQ) coupled to a positive electrospray ionization source. The QQQ mass spectrometer was tuned and calibrated using Agilent G1969-85000 calibration and tuning mix (Agilent Technologies). Heated (325°C) drying gas flowing at 9 L/min, with a nebulizer pressure of 30 PSIG, was used for droplet desolvation. Spray was induced with a capillary voltage of 4000V. A fragmentor voltage of 130V was used for all transitions, and collision energy was determined using Skyline (Table 1).
2.10 Data analysis
Quantitative analysis of all peptides was accomplished using MassHunter quantitative analysis software (Agilent). The peak area of both the light and heavy peptide were obtained and the concentration of each peptide was calculated using the following formula: Area_endogenous/Area_labeled*5pM=pM peptide/100ug of skin protein.
2.11 Method accuracy, precision, limits of detection (LOD) and limits of quantitation (LOQ)
Linearity curves for the labeled peptide standards were used to determine the recovery of each labeled peptide in a matrix of 100ug of digested skin protein. Analysis was conducted in triplicate; recovery and relative standard deviation (RSD) were determined for each peptide. LOD and LOQ were also calculated using a digested skin sample spiked with 0.5pmol/100ug of each AQUA peptide run in triplicate (Supplemental Material 1). The LOD was calculated by multiplying the standard deviation by 3 (3σ) and the LOQ was calculated by multiplying the standard deviation by 10 (10σ).
2.12 Clinical sample analysis
The method was applied to the clinical pilot study described above. Samples from each of the 7 locations was digested as described and then analyzed in triplicate using the LC/MS/MS method described. The results for each peptide in each sample were determined using the formula: Area_endogenous/Area_labeled*5pM=pM peptide/100ug of skin protein. The average of the 3 injections was used for the final concentration.
2.13 Statistics
The mean and standard error for the triplicate analyses were determined for each peptide and for the normal non-atopic and atopic subjects separately. An unpaired t-test was performed against both phenotypes on each peptide for each location. Statistical significance determined using the Holm-Sidak method, with alpha = 0.05. Computations assumed that all peptide values were sampled from populations with the same scatter (SD). A two-way ANOVA was conducted for each peptide to determine if differences existed across sampling locations. Linear regression analysis was conducted to determine if the two peptides had similar responses across locations for each protein. All statistics were performed using GraphPad Prism v. 6.0.
3.0 Results and Discussion
3.1 Peptide selection from QTOF data
Using previous results, 15 candidate proteins were considered for inclusion in the assay [2, 3]. To verify the presence of candidate proteins, a set of fresh skin taping samples was obtained from a non-atopic volunteer. The sample was prepared as described and analyzed using an LC/Q-TOF to verify the presence of candidate proteins using Agilent Spectrum Mill as previously described [2, 3]. Spectrum Mill MRM selector was used to identify potential target peptides and MS/MS transitions from the Q-TOF data; Skyline was used as further verification of the MS/MS spectra. Skyline was used to build and export an MRM method compatible with Agilent Mass Hunter Acquisition software. A 10 ug digest was analyzed on the QQQ and compared with the Q-TOF data (Figure 1). Based on these results, a final set of 7 proteins and 14 peptides was selected for inclusion in the assay: alpha enolase, corneodesmosin, FABP, filaggrin 1 and 2, serpin B3, and transglutaminase 3. Additional criteria that were considered include the following: the presence of potentially modified amino acids such as cysteines, oxidation sites, number of amino acids, and potential for missed trypsin cleavages.
Figure. 1.
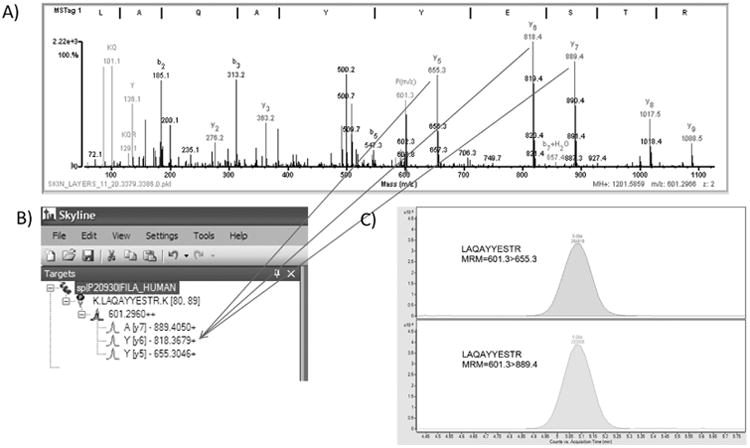
QTOF data was used to determine peptides and transitions. 1A: Product MS/MS spectrum for peptide LAQAYYESTR with precursor m/a 601.2966. Data was generated on a QTOF mass spectrometer as described in the methods. Data was searched against a SpectrumMill database as previously described 2,3. 1B: Skyline was used to generate predicted peptides from a theoretical trypsin digest and the predicted y and b peptide ion fragments. Arrows indicate the experimentally determined fragments in the Q-TOF data. 1C: An extracted ion chromatogram is shown for two transitions, 601.3 > 655.3 and 601.3 > 889.4, generated for peptide LAQAYYESTR. Data was generated on the QQQ mass spectrometer.
3.2 Linearity of labeled peptide standards
It is impossible to create a blank matrix for skin taping samples that are completely free of the target peptides being analyzed; therefore, linearity and dynamic range were assessed by spiking labeled peptides into extracted and pooled skin taping samples. The samples were digested and analyzed as described in the methods. Good resolution of the labeled and endogenous peptides was achieved at the MS-level (Figure 2A). There was no overlap of isotopes for any of the labeled:endogenous peptide pairs included in the assay. Figure 2B shows the peak areas for both labeled and endogenous peptides; peptide concentration was determined based on this value for each of the peptide pairs.
Figure. 2.
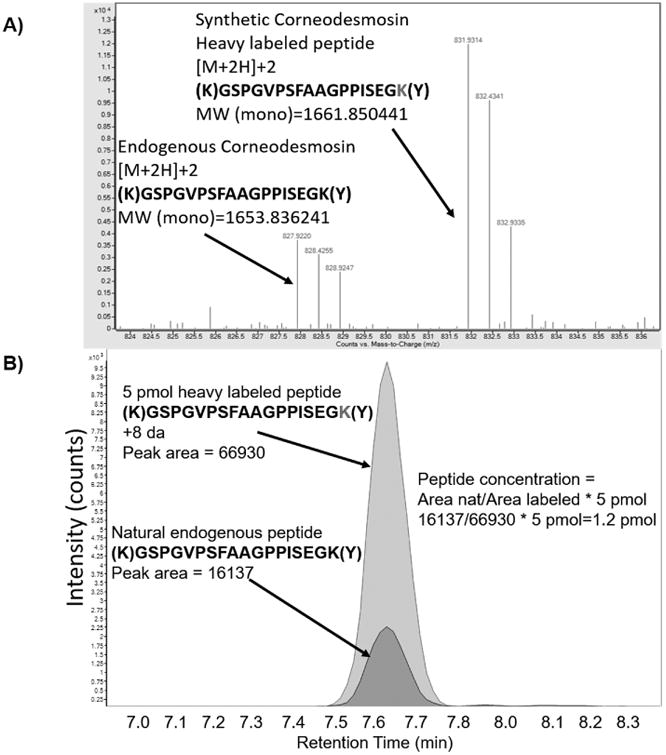
Resolution of labeled:endogenous peptide pairs in MS-mode and determination of endogenous peptide concentration. 2A) Full scan MS spectrum (QTOF) of endogenous (left side of spectrum) and labeled (right side of spectrum) peptide. Data was collected using a QTOF mass spectrometer. 2B) Overlaid extracted ion chromatograms of labeled and endogenous peptides. Peak area was determined using MassHunter software (Agilent) and peptide concentration was determined using the following formula: Area_endogenous/Area_labeled*5pM=pM peptide/100ug of skin protein.
Six aliquots of pooled protein were used for linearity experiments. Each aliquot was comprised of 100 ug pooled protein; this was spiked with all 14 labeled peptides at concentrations of 0.5, 1.25, 2.5, 5, 12.5, and 25 pmol. Figure 3 shows a representative calibration curve for both peptides associated with one protein, Filaggrin I. The R2 values for these peptides, LAQAYYESTR (Figure 3A) and NPDDPDMVDVFMDHLDIDHNK (Figure 3B), were 0.993 and 0.997 respectively.
Figure. 3.
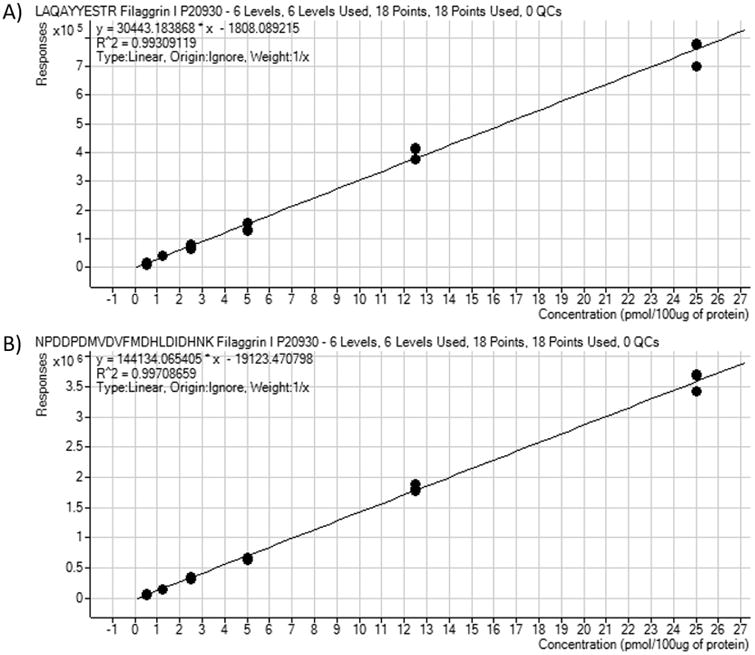
Calibration curves for labeled peptides of one representative protein, Filaggrin I. Pooled protein samples were spiked with 6 concentrations of labeled peptides prior to digestion. Samples were analyzed on the QQQ mass spectrometer as described. Curves were generated as described in the methods. Calibration curve for peptide LAQAYYESTR is shown in 3A. Calibration curve for peptide NPDDPDMVDVFMDHLDIDHNK is shown in 3B.
Calibration curves for all peptides are shown in Supplemental Material 1. R2 values for all labeled peptides ranged from 0.99363 to 0.99942. Overall, the chromatography method demonstrated good-to-excellent separation of peptides (Figure 4). The total length of QQQ analysis time was 17 minutes.
Figure. 4.
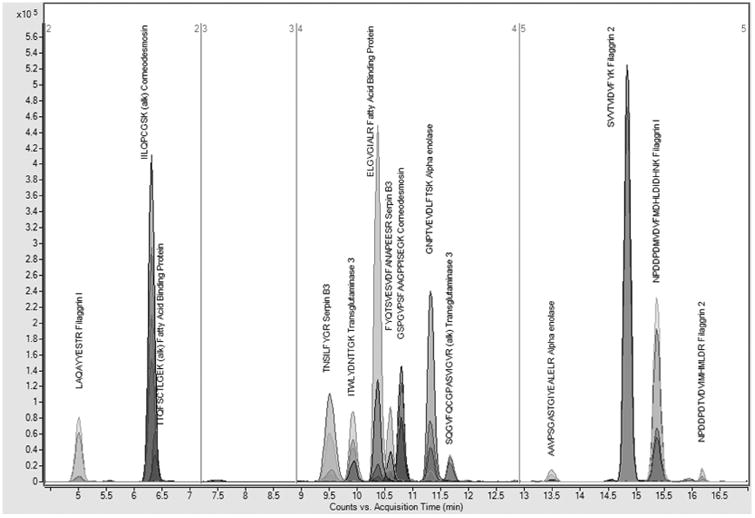
Extracted ion chromatograms of all MRM transitions of all peptides. Good-to-excellent separation of peptides is demonstrated using a 16.5 minute liquid chromatography gradient.
3.3 Precision, accuracy, LOD and LOQ of LC/MS/MS method
The majority of the labeled peptides demonstrated very good linearity, reproducibility and accuracy (Table 2) with the exception of NPDDPDTVDVIMHMLDR (Table 2 and Supplemental Material 1). The majority of labeled peptides had recoveries between 89.3 – 120.6% with relative standard deviations (RSDs) between 0.5 – 21.4 (Table 2). LODs ranged from 0.032-0.449pmol/100ug protein and LOQs ranged from 0.106-1.496pmol/100ug protein (Supplemental Material 1).
Table 2. Recovery and RSD values for each of the 14 labeled peptides.
Aliquots of 100 ug pooled sample were spiked with 0.5-25 pmol of labeled peptides. Samples were analyzed in triplicate using an LC/QQQ mass spectrometer system. Data was analyzed as described in methods.
| Protein | Filaggrin 1 | Corneodesmosin | FABP | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide | LAQ..TR | NPD..NK | IIL..SK | GSP.. GK | TTQ..EK | ELG..LR | ||||||
| pmol/100ug protein | Rec. | RSD | Rec. | RSD | Rec. | RSD | Rec. | RSD | Rec. | RSD | Rec. | RSD |
| 0.5 | 101.3 | 21.8 | 115.8 | 4.2 | 110.2 | 1.9 | 114.3 | 8.7 | 119.6 | 6.4 | 111.8 | 2.5 |
| 1.25 | 108.5 | 3.0 | 91.1 | 5.0 | 93.5 | 4.8 | 98.2 | 4.3 | 94.2 | 4.9 | 93.8 | 2.2 |
| 2.5 | 94.3 | 10.0 | 97.4 | 3.8 | 96.4 | 1.1 | 92.6 | 6.8 | 92.6 | 2.9 | 94.2 | 2.3 |
| 5 | 91.1 | 11.0 | 92.6 | 2.7 | 98.0 | 1.8 | 94.4 | 3.3 | 91.3 | 2.4 | 96.8 | 3.5 |
| 12.5 | 105.8 | 5.3 | 102.5 | 3.0 | 102.1 | 2.2 | 97.9 | 3.4 | 99.9 | 3.0 | 104.3 | 1.9 |
| 25 | 99.0 | 6.1 | 100.6 | 4.3 | 99.8 | 1.6 | 102.7 | 2.9 | 102.4 | 1.6 | 99.2 | 2.8 |
| Protein | Serpin B3 | Transglutaminase 3 | Alpha Enolase | Filaggrin 2 | ||||||||||||
| Peptide | TNS..GR | FYQ..SR | ITW..GK | SQG..VR | GNP..SK | AAV..LR | SVV..YK | NPD..DR | ||||||||
| pmol/100ug protein | Rec. | RSD | Rec. | RSD | Rec. | RSD | Rec. | RSD | Rec. | RSD | Rec. | RSD | Rec. | RSD | Rec. | RSD |
| 0.5 | 120.6 | 6.0 | 119.9 | 8.7 | 115.7 | 2.7 | 115.0 | 25.3 | 118.2 | 11.6 | 100.1 | 8.6 | 107.2 | 3.3 | ND | NC |
| 1.25 | 89.3 | 3.9 | 93.0 | 14.5 | 95.3 | 1.4 | 91.9 | 5.4 | 89.9 | 5.8 | 102.1 | 12.3 | 95.8 | 2.1 | ND | NC |
| 2.5 | 91.2 | 3.2 | 92.1 | 3.6 | 93.7 | 9.6 | 96.7 | 6.8 | 92.6 | 5.3 | 102.5 | 7.5 | 96.2 | 0.8 | 126.3 | 63.1 |
| 5 | 94.3 | 4.1 | 91.3 | 2.3 | 92.0 | 3.3 | 96.7 | 4.0 | 97.1 | 3.8 | 94.1 | 4.4 | 98.6 | 0.5 | 101.2 | 21.4 |
| 12.5 | 105.0 | 1.5 | 102.5 | 0.8 | 102.2 | 1.6 | 97.5 | 2.7 | 101.5 | 3.1 | 100.8 | 5.0 | 102.9 | 2.0 | 120.7 | 12.6 |
| 25 | 99.7 | 1.8 | 101.2 | 3.7 | 101.1 | 3.4 | 102.4 | 4.1 | 100.7 | 3.7 | 100.4 | 3.0 | 99.3 | 1.3 | 89.1 | 11.5 |
3.4 Reproducibility of protein digestion
Because variation may occur during processing, especially in complex matrices, the reproducibility of sample preparation was determined. To account for this, we used a constant 100ug of protein for each sample and a 30:1 protein/trypsin ratio in addition to a typical digestion time (17 hours). We then used inter-day precision to demonstrate that the digest was reproducible. For these experiments, a pooled sample was split into 3 aliquots, spiked with standards, and digested separately over 3 days. Digested samples were analyzed using LC/QQQ in a single batch. Overall, 12 of the 14 peptides were detected in the pooled samples (Table 3). Variability ranged from 1.90% RSD for corneodesmosin to 25.31% for filaggrin I (Table 3). We suspect that the higher variation was due to differences in protein digestion efficiency. However, because our previous experience and others[15, 16] demonstrates that optimal protein digestion times can vary per protein in complex matrices, we did not conduct individual protein digestion time course experiments.
Table 3. Reproducibility of protein digestion.
A single pooled skin sample was split into 3 aliquots, spiked with standards, and digested; each aliquot was digested on a separate day. Digested protein samples were analyzed in triplicate using an LC/QQQ in a single batch. Triplicate values were averaged and intra-day RSD was determined for each of the 3 days. An interday RSD was determined for each peptide using all values. Units are in pmol/100ug protein.
| Protein | Filaggrin 1 | Corneodesmosin | FABP | Serpin B3 | TGase | Alpha Enolase | Filaggrin 2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Peptide | LAQ..TR | NPD..NK | IIL..SK | GSP..GK | TTQ..EK | ELG..LR* | TNS..GR | FYQ..SR | ITW..GK | SQG..VR | GNP..SK* | AAV..LR | SVV..YK | NPD..DR* | |
| Day 1 | AVG | 1.99 | 0.98 | 0.63 | 1.06 | ND | 0.06 | 0.97 | 1.33 | 1.12 | 2.62 | 0.11 | ND | 7.21 | 0.03 |
| RSD | 10.67 | 17.00 | 6.07 | 10.02 | NC | 16.44 | 9.09 | 12.08 | 7.14 | 21.43 | 22.85 | NC | 6.61 | 29.09 | |
| Day 2 | AVG | 2.29 | 1.07 | 0.63 | 1.00 | ND | 0.06 | 1.10 | 1.42 | 1.34 | 2.30 | 0.11 | ND | 8.09 | 0.04 |
| RSD | 6.49 | 22.43 | 3.01 | 11.78 | NC | 18.70 | 8.83 | 8.16 | 5.38 | 12.24 | 10.97 | NC | 2.29 | 17.49 | |
| Day 3 | AVG | 3.20 | 1.30 | 0.65 | 1.05 | ND | 0.07 | 1.28 | 1.80 | 1.33 | 2.67 | 0.13 | ND | 8.29 | 0.03 |
| RSD | 13.52 | 13.07 | 3.16 | 11.56 | NC | 14.75 | 8.12 | 11.82 | 11.25 | 14.52 | 16.67 | NC | 3.46 | 27.32 | |
| Inter-Day | AVG | 2.49 | 1.12 | 0.64 | 1.04 | ND | 0.07 | 1.12 | 1.52 | 1.26 | 2.53 | 0.12 | ND | 7.86 | 0.03 |
| RSD | 25.31 | 14.59 | 1.90 | 2.86 | NC | 8.69 | 13.59 | 16.60 | 9.71 | 7.91 | 8.99 | NC | 7.33 | 17.77 | |
Peptide concentrations were below the lowest calibration level of 0.5pmol/100ug protein.
ND=Not Detected; NC=Not Calculated.
3.5 Assay performance in pilot study
The assay was applied to a small pilot study aimed at determining if differences exist based on sampling location and disease state. A total of 12 participants were sampled in the following locations: bicep, antecubital fossae, forearm, abdomen, back, thigh, and behind the knee. Six of the participants had been diagnosed with atopic dermatitis; an additional six participants with no atopic dermatitis were included. Participants with atopic dermatitis were sampled at non-inflamed regions. Samples were analyzed in technical triplicates. Endogenous peptide concentration was determined as ug peptide/ug total protein as described in methods.
3.6 Comparison of endogenous protein levels
Statistical analyses were conducted to compare peptide and protein abundances across locations and between atopic and non-atopic individuals. To achieve this, peptide abundances were compared following normalization to total ion signal or with no normalization. Regardless of the normalization method used, few significant differences were found between atopic and non-atopic individuals. As shown in Table 4, peptide SVVTVIDVFYK from filaggrin II was found to be significantly different between atopic and non-atopic individuals in the abdomen, anterior elbow bicep, and knee locations. Only the anterior elbow location was different when the Filaggrin II NPDDPDTVDVIMHMLDR peptide was measured. filaggrin I peptide LAQAYYESTR was different when the back, forearm, or thigh were sampled. Conversely, no differences were noted for any of the serpin B3, alpha enolase, corneodesmosin, transglutaminase, or FABP proteins. While not significant, peptides sampled from the back location were consistently higher compared to other locations (Supplemental Material 2).
Table 4. Comparison of peptide abundances across locations and disease state.
T-tests were used to compare atopic vs non-atopic individuals at each location; no normalization was used. P-values < 0.05 are shown in bold font. Two-way ANOVAs were used to determine interactions between disease and sampling location. Values for “location” and “AD vs Normal” are also shown.
| Peptide | Protein | Abdomen | Elbow | Back | Bicep | Forearm | Knee | Thigh | Interaction | Location | AD v N |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AAV…ELR | Alpha enolase | 0.9750 | 0.9910 | 0.9287 | 0.9995 | 0.9774 | 0.9805 | 0.9873 | 0.0793 | 0.0024 | 0.9230 |
| GNP…TSK | Alpha enolase P | 0.9701 | 0.9816 | 0.9057 | 0.9516 | 0.9702 | 0.9854 | 0.9747 | 0.3278 | 0.0112 | 0.2447 |
| GSP…EGK | Corneodesmosin | 0.9340 | 0.9070 | 0.9908 | 0.9680 | 0.9230 | 0.8444 | 0.9336 | 0.6397 | 0.7333 | 0.6465 |
| IIL…GSK | Corneodesmosin | 0.9529 | 0.9506 | 0.9782 | 0.9791 | 0.9886 | 0.9233 | 0.9880 | 0.7649 | 0.9355 | 0.4832 |
| ELG…ALR | FABP | 0.9986 | 0.9927 | 0.8235 | 0.9929 | 0.9898 | 0.9756 | 0.9957 | 0.4833 | 0.3456 | 0.3572 |
| TTQ…GEK | FABP | 0.9976 | 0.9989 | 0.9382 | 0.9927 | 0.9929 | 0.9902 | 0.9965 | 0.4028 | 0.4004 | 0.3288 |
| NPD…LDR | Filaggrin 2 | 0.9168 | 0.0026 | 0.3261 | 0.0735 | 0.8530 | 0.2305 | 0.9745 | 0.0753 | 0.0016 | 0.1092 |
| SVV…FYK | Filaggrin 2 | 0.0002 | 2.68E-09 | 0.9995 | 0.0004 | 0.3091 | 7.93E-05 | 0.4983 | 0.5211 | 0.0001 | 0.0002 |
| LAQ…STR | Filaggrin I | 0.9570 | 0.6292 | 5.24E-06 | 0.4432 | 0.0014 | 0.4570 | 0.0010 | 0.3873 | 0.5214 | 0.4558 |
| NPD…HNK | Filaggrin I | 0.8484 | 0.8786 | 0.3742 | 0.6406 | 0.8286 | 0.8173 | 0.9355 | 0.5097 | 0.0054 | 0.7598 |
| FYQ…ESR | Serpin B3 | 0.8962 | 0.9744 | 0.7890 | 0.8945 | 0.9370 | 0.8952 | 0.9677 | 0.4227 | 0.0064 | 0.1474 |
| TN…YGR | Serpin B3 | 0.7831 | 0.9234 | 0.7912 | 0.9871 | 0.9778 | 0.9280 | 0.9671 | 0.2872 | 0.0417 | 0.3254 |
| ITW…TGK | Tgase 3 | 0.4336 | 0.7821 | 0.9296 | 0.5315 | 0.8218 | 0.5803 | 0.9644 | 0.6456 | 0.0069 | 0.0255 |
| SQG…GVR | Tgase 3 | 0.4618 | 0.7513 | 0.9267 | 0.5681 | 0.8628 | 0.6236 | 0.9870 | 0.7534 | 0.0133 | 0.0347 |
Conversely, when a two-way ANOVA was used to compare samples, the majority of peptides were found to be different between locations (Table 4, “Location” column). Relatively few differences were found when disease status was considered (Table 4 “Disease status” column). However, when both location and disease status were considered, no significant differences were found (Table 4, “Interaction” column).
3.7 Regression analysis
In quantitative proteomics, one challenge lies in determining the relationship between the quantitation of a single peptide and absolute protein amounts. In the current study, two peptides were used to determine the overall quantity of each protein. Linear regression analysis was used to determine if the peptides had similar responses. While peptides from alpha enolase (R2 = 0.9644), FABP (R2 = 0.9883), and serpin B3 (R2 = 0.8199) had relatively good correlation, peptides from corneodesmosin (R2 = 0.716) and transglutaminase 3 (R2 = 0.7426) had only moderately good correlation. Filaggrin 1 (R2 = 0.5693) and filaggrin 2 (R2 = 0.4776) peptides were poorly correlated. Possible explanations for this include post-translational modifications (eg oxidation of methionine), remaining secondary structure, or other factors that affect protein digest efficiency of some peptides more than others. In addition, some loss could occur with longer peptide sequences during the detergent removal step and/or re-solubilization of the labeled standard. Graphs and equations for regression analysis can be found in Supplemental Material.
3.8 Application to Clinical Research
There are currently no comparable techniques to analyze epidermal proteins. This technique is well tolerated by the patient and can conceivably be used to assess any skin condition. Skin tape strips can be obtained in less than one minute, the lab analysis can be completed in less than 3 days, and the technique results in quantitative measurement of proteins of interest. Although we examined 7 key epidermal proteins involved in skin barrier for the current study, any epidermal protein (>150) as reported by Broccardo et. al. can be quantitated. While transcriptome analysis has been performed on skin tapings, transcriptomics is far more tedious and complex to perform compared to the technique described herein[17]. The simplicity of this skin tape strip technique makes it ideal for clinical application.
4.0 Conclusion
We have demonstrated the utility of an isotope dilution technique for the quantitative analysis of 14 peptides from 7 proteins: alpha enolase, corneodesmosin, FABP, filaggrin 1 and 2, serpin B3, and transglutaminase 3. These proteins have been previously shown to be related to skin disease[2, 3, 12]. Overall, LC/QQQ method exhibited good linearity for all peptides and acceptable reproducibility when standards were spiked into control samples. In addition, 5 of 7 proteins had good correlation between the two peptides used to quantitate that protein, with R2 values greater than 0.71. Analytical challenges, including solubility and digestion efficiency of individual peptides, may have contributed to the poor correlation between the two filaggrin proteins. When applied to a small clinical study, it was determined that peptides from filaggrin 2 were consistently different between patients with atopic dermatitis and normal patients. Importantly, all peptides were found consistently in all patient samples. This method is suitable for quantitating these 14 peptides in skin taping samples. It can easily be adopted to non-skin samples such as cells or tissue.
Supplementary Material
Highlights.
We present an LC/MS/MS method to quantitate 7 proteins from skin tapings.
Isotopically-labeled peptides were used to obtain absolute quantitation values
Good reproducibility, dynamic range, and linearity were achieved
Atopic versus non-atopic individuals (n=12) were compared for differences in proteins
Peptides were detected in all samples and in general exhibited good correlation
Acknowledgments
Supported by NIH/NCATS Colorado CTSA Grant Number UL1 TR001082 and NCRR 1S10OD010366 (to Dr. Reisdorph). Contents are the authors' sole responsibility and do not necessarily represent official NIH views. We thank Trudi Madigan who provided excellent nursing support in obtaining the skin tape strips.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–340. doi: 10.1038/nrm1619. [DOI] [PubMed] [Google Scholar]
- 2.Broccardo CJ, Mahaffey SB, Strand M, Reisdorph NA, Leung DY. Peeling off the layers: skin taping and a novel proteomics approach to study atopic dermatitis. J Allergy Clin Immunol. 2009;124:1113–1115 e1111-1111. doi: 10.1016/j.jaci.2009.07.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Broccardo CJ, Mahaffey S, Schwarz J, Wruck L, David G, Schlievert PM, Reisdorph NA, Leung DY. Comparative proteomic profiling of patients with atopic dermatitis based on history of eczema herpeticum infection and Staphylococcus aureus colonization. J Allergy Clin Immunol. 2011;127:186–193. 193 e181–111. doi: 10.1016/j.jaci.2010.10.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Song J, Zhang H, Wang Z, Xu W, Zhong L, Cao J, Yang J, Tian Y, Yu D, Ji J, Cao J, Zhang S. The Role of FABP5 in Radiation-Induced Human Skin Fibrosis. Radiat Res. 2017 doi: 10.1667/RR14901.1. [DOI] [PubMed] [Google Scholar]
- 5.Voegeli R, Monneuse JM, Schoop R, Summers B, Rawlings AV. The effect of photodamage on the female Caucasian facial stratum corneum corneome using mass spectrometry-based proteomics. Int J Cosmet Sci. 2017;39:637–652. doi: 10.1111/ics.12426. [DOI] [PubMed] [Google Scholar]
- 6.Guo Z, Hu Q, Tian J, Yan L, Jing C, Xie HQ, Bao W, Rice RH, Zhao B, Jiang G. Proteomic profiling reveals candidate markers for arsenic-induced skin keratosis. Environ Pollut. 2016;218:34–38. doi: 10.1016/j.envpol.2016.08.035. [DOI] [PubMed] [Google Scholar]
- 7.Williamson JC, Scheipers P, Schwammle V, Zibert JR, Beck HC, Jensen ON. A proteomics approach to the identification of biomarkers for psoriasis utilising keratome biopsy. J Proteomics. 2013;94:176–185. doi: 10.1016/j.jprot.2013.09.010. [DOI] [PubMed] [Google Scholar]
- 8.Vidova V, Spacil Z. A review on mass spectrometry-based quantitative proteomics: Targeted and data independent acquisition. Anal Chim Acta. 2017;964:7–23. doi: 10.1016/j.aca.2017.01.059. [DOI] [PubMed] [Google Scholar]
- 9.Shi T, Song E, Nie S, Rodland KD, Liu T, Qian WJ, Smith RD. Advances in targeted proteomics and applications to biomedical research. Proteomics. 2016;16:2160–2182. doi: 10.1002/pmic.201500449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mermelekas G, Vlahou A, Zoidakis J. SRM/MRM targeted proteomics as a tool for biomarker validation and absolute quantification in human urine. Expert Rev Mol Diagn. 2015;15:1441–1454. doi: 10.1586/14737159.2015.1093937. [DOI] [PubMed] [Google Scholar]
- 11.Liebler DC, Zimmerman LJ. Targeted quantitation of proteins by mass spectrometry. Biochemistry. 2013;52:3797–3806. doi: 10.1021/bi400110b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Simon M, Tazi-Ahnini R, Jonca N, Caubet C, Cork MJ, Serre G. Alterations in the desquamation-related proteolytic cleavage of corneodesmosin and other corneodesmosomal proteins in psoriatic lesional epidermis. Br J Dermatol. 2008;159:77–85. doi: 10.1111/j.1365-2133.2008.08578.x. [DOI] [PubMed] [Google Scholar]
- 13.Legg KM, Powell R, Reisdorph N, Reisdorph R, Danielson PB. Discovery of highly specific protein markers for the identification of biological stains. Electrophoresis. 2014;35:3069–3078. doi: 10.1002/elps.201400125. [DOI] [PubMed] [Google Scholar]
- 14.Legg KM, Powell R, Reisdorph N, Reisdorph R, Danielson PB. Verification of protein biomarker specificity for the identification of biological stains by quadrupole time-of-flight mass spectrometry. Electrophoresis. 2016 doi: 10.1002/elps.201600352. [DOI] [PubMed] [Google Scholar]
- 15.Kuhn E, Whiteaker JR, Mani DR, Jackson AM, Zhao L, Pope ME, Smith D, Rivera KD, Anderson NL, Skates SJ, Pearson TW, Paulovich AG, Carr SA. Interlaboratory evaluation of automated, multiplexed peptide immunoaffinity enrichment coupled to multiple reaction monitoring mass spectrometry for quantifying proteins in plasma. Mol Cell Proteomics. 2012;11:M111 013854. doi: 10.1074/mcp.M111.013854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Proc JL, Kuzyk MA, Hardie DB, Yang J, Smith DS, Jackson AM, Parker CE, Borchers CH. A quantitative study of the effects of chaotropic agents, surfactants, and solvents on the digestion efficiency of human plasma proteins by trypsin. J Proteome Res. 2010;9:5422–5437. doi: 10.1021/pr100656u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dyjack N, Goleva E, Rios C, Kim BE, Bin L, Taylor P, Bronchick C, Hall CF, Richers BN, Seibold MA, Leung DY. Minimally invasive skin tape strip RNA-seq identifies novel characteristics of type 2-high atopic dermatitis disease endotype. J Allergy Clin Immunol. 2018 doi: 10.1016/j.jaci.2017.10.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.