

Summary
During development, neurons undergo apoptosis if they do not receive adequate trophic support from tissues they innervate or when detrimental factors activate the p75 neurotrophin receptor (p75NTR) at their axon ends. Trophic factor deprivation (TFD) or activation of p75NTR in distal axons results in a retrograde degenerative signal. However, the nature of this signal and the regulation of its transport are poorly understood. Here, we identify p75NTR intracellular domain (ICD) and histone deacetylase 1 (HDAC1) as part of a retrograde proapoptotic signal generated in response to TFD or ligand binding to p75NTR in sympathetic neurons. We report an unconventional function of HDAC1 in retrograde transport of a degenerative signal and its constitutive presence in sympathetic axons. HDAC1 deacetylates dynactin subunit p150Glued, which enhances its interaction with dynein. These findings define p75NTR ICD as a retrograde degenerative signal and reveal p150Glued deacetylation as a unique mechanism regulating axonal transport.
Keywords: P75NTR, HDAC1, Axonal Transport, p150Glued, Dynein, Neurotrophin, Dynactin, Sympathetic, BDNF, NGF
Graphical Abstract
eTOC blurb:
Retrograde degenerative signaling is suggested to regulate neuronal survival and degeneration. Pathak et al. identify a retrograde degenerative signal involving the intracellular domain of p75 neurotrophin receptor and histone deacetylase, HDAC1, in sympathetic neurons. HDAC1 regulates retrograde axonal transport by deacetylating dynactin subunit p150Glued, enhancing its binding to dynein.
Introduction
Neuronal apoptosis is necessary for vertebrate development and establishment of proper neural circuitry. However, abnormal apoptosis is the basis for many neuropathologies, highlighting the need to define the mechanisms by which it is regulated in order to develop new therapies. The balance between neuronal survival and degeneration in the peripheral nervous system is mostly governed by the availability of neurotrophic factors, including the neurotrophins. The neurotrophins mediate survival and differentiation through binding to the Trk family of tyrosine kinase receptors (Deinhardt and Chao, 2014). All neurotrophins can also bind to the p75 neurotrophin receptor (p75NTR), a member of the TNF receptor superfamily, which may elicit distinct effects, including the induction of apoptosis and axonal degeneration, depending on the cellular context (Ceni et al., 2014; Kraemer et al., 2014; Vicario et al., 2015)
Neurotrophins are produced by the target tissues innervated by developing neurons, thus these factors act locally on axon ends and generate signals that must be conveyed back to the cell body. The mechanisms of retrograde survival signaling have been extensively studied, primarily in sympathetic and sensory neurons that depend on nerve growth factor (NGF) for survival(Harrington and Ginty, 2013; Scott-Solomon and Kuruvilla; Wu et al., 2009). Loss of trophic factor input leads to a reduction in pro-survival signals reaching the soma. However, there is growing evidence that trophic factor deprivation (TFD) also results in pro-apoptotic signaling that starts at the periphery and is sent back to the cell body (Ghosh et al., 2011; Mok et al., 2009; Simon et al., 2016). When NGF was selectively withdrawn from distal axons of sympathetic neurons, there was an increase in phosphorylation of c-Jun in the cell bodies. This increase was blocked when the axons were treated with the microtubule disrupting agent colchicine, suggesting the existence of a retrograde transport-dependent apoptotic mechanism (Mok et al., 2009). Blocking axonal activation of the stress-induced dual leucine zipper kinase (DLK) or c-Jun N-terminal kinase (JNK), also prevented sensory neuron apoptosis following TFD (Simon et al., 2016). However, the molecular components of this retrograde signaling are yet to be defined.
Activation of p75NTR has also been implicated in apoptosis following TFD; for example, sympathetic neurons from p75−/− mice are resistant to NGF withdrawal (Majdan et al., 2001; Nikoletopoulou et al., 2010). How the receptor contributed to induction of apoptosis was not determined. However, it is notable that direct activation of p75NTR by ligand binding at distal axons can trigger neuronal apoptosis (Tauris et al., 2011; Taylor et al., 2012; Yano et al., 2009), suggesting that there may be a shared mechanism with TFD. Many of p75NTR’s downstream signals, including the activation of cell death pathways, require regulated proteolysis of the receptor by α-secretase and the γ-secretase complex to release the intracellular domain (ICD) (Skeldal et al., 2012). Therefore, we hypothesized that the p75NTR ICD may be a key component of the retrograde degenerative signal following TFD or p75NTR activation by ligand binding.
Retrograde axonal transport is driven by the microtubule minus-end directed motor protein dynein, which consists of a dimer of heavy chains and additional intermediate and light chains. Dynein associates with the activator complex dynactin, which is composed of more than 20 proteins, including the largest subunit p150Glued. A dimer of p150Glued binds to the dynein intermediate chain through its coiled-coil domain (CC1) and is required for processivity of the complex (Karki and Holzbaur, 1995; King and Schroer, 2000; McKenney et al., 2014). Formation of this complex and how it is regulated is still an active area of investigation.
Surprisingly, several histone deacetylases (HDACs) have recently been shown to influence axonal transport. While best characterized for their role in regulating gene expression, several HDACs can shuttle out of the nucleus and modify other targets (Cho and Cavalli, 2014). HDAC1 was recently shown to translocate from the nucleus into the axons of cortical and hippocampal neurons following TNF and glutamate treatment, where it associated with kinesins. This association disrupted mitochondrial transport, which led to axon degeneration (Kim et al., 2010).
Here we reveal a role for HDAC1 in retrograde degenerative signaling through regulating the formation of the dynactin-dynein complex. We further identify the ICD of p75NTR as a retrograde pro-apoptotic signal triggered by p75NTR activation due to ligand binding or trophic factor deprivation in sympathetic neurons.
Results:
Retrograde apoptotic signaling requires localized cleavage of p75NTR
To investigate the mechanisms of retrograde degenerative signaling, sympathetic neurons from rat superior cervical ganglia were cultured in microfluidics chambers, which allow separation of the cell bodies from the distal axons (Fig 1A), thus mimicking the different environmental milieu for distal axons and cell bodies in vivo. Since NGF binds to a complex of p75NTR and TrkA (Hempstead et al., 1991), which would complicate the analysis of activating p75NTR alone, NGF was removed and neurons were kept alive in KCl. To selectively stimulate p75NTR, brain-derived neurotrophic factor (BDNF) was used, since it has been implicated as an endogenous pro-apoptotic ligand for p75NTR in this system (Deppmann et al., 2008). BDNF or NGF was then added to the distal axons or cell bodies, as indicated. There was a significant increase in cell death in response to BDNF treatment of axons (Fig 1B), even if the cell bodies received limited trophic support from 2 ng/ml NGF. These results support the hypothesis that p75NTR activation in axons can trigger a retrograde degenerative signal.
Figure 1. Retrograde apoptotic signaling by BDNF in sympathetic neurons.

A. Schematic representation of microfluidics chamber culture system and representative image of sympathetic neurons immunostained with TUJ1 (green). Scale bar 100um.
B. Activation of p75NTR in distal axons results in apoptosis at the cell soma. After the axons reached the distal sides, NGF was removed and the cell bodies (CB) and distal axons (DAx) were treated with 12.5 mM KCl or NGF, with or without 200 ng/ml BDNF for 48 hrs, as indicated. The bar on the extreme left of the graph represents neurons maintained in 20 ng/ml NGF on the cell bodies and distal axons while for the extreme right bar, axons were treated with 200 ng/ml BDNF and cell bodies were given 2 ng/ml NGF. After 48 hrs of treatment, the neurons were fixed, stained for TUJ1 and DAPI, and pyknotic nuclei were quantified as a measure of apoptosis. Depicted are the means ± SEM for n=5; **, p < 0.01; ***, p < 0.001; 2-way ANOVA with a Tukey’s multiple comparisons test.
Proteolytic cleavage of p75NTR by α- and γ-secretase releases its intracellular domain (ICD) (Fig 2A) as well as a number of associated signaling components, which traffic to the nucleus to mediate apoptosis (Kraemer et al., 2014). Since activation of p75NTR on the distal axons induced cell death back in the soma, we hypothesized that the receptor was locally cleaved in the axons and this proteolysis was required to generate the retrograde apoptotic signal. To test this hypothesis, we added BDNF to the distal axons along with the γ-secretase inhibitor DAPT and compared the subsequent cell death to that induced in the presence of DAPT exclusively on the cell somas. When present in the distal axons, DAPT completely abrogated apoptosis. However, when the γ-secretase inhibitor was applied only to the cell somas and BDNF to the distal axons, there was no protective effect (Fig 2B). These data suggest that activation of p75NTR induces local receptor proteolysis, which is necessary for retrograde apoptotic signaling (Fig 2B and Fig S1A&B).
Figure 2. P75NTR cleavage in distal axons is necessary for apoptosis.
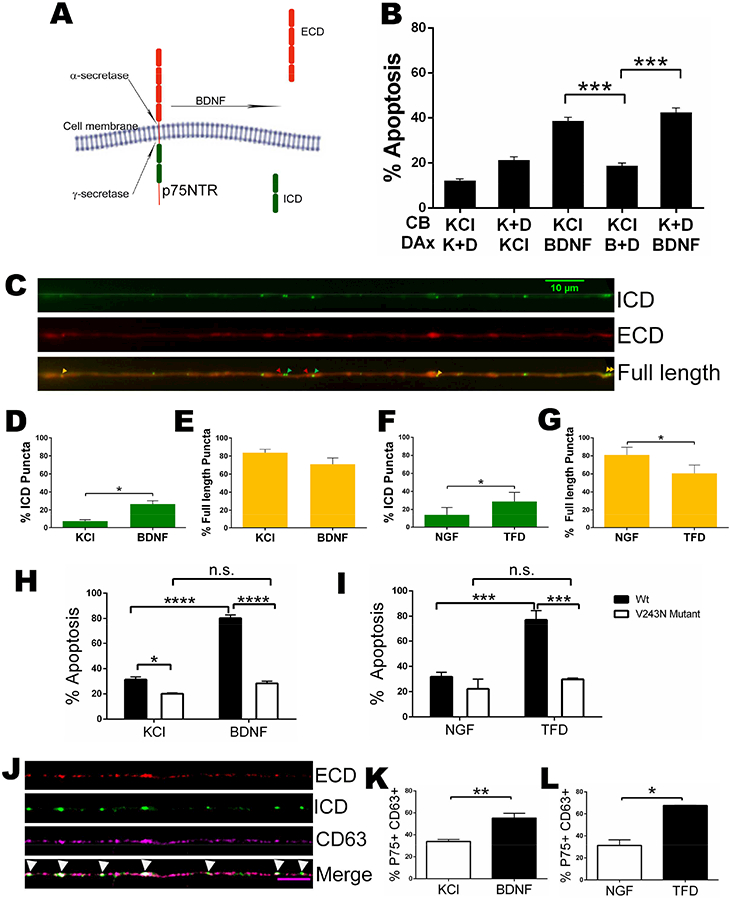
A. Schematic showing the cleavage of p75NTR
B. Inhibition of axonal γ-secretase prevents p75NTR-mediated apoptosis. Sympathetic neurons were cultured in microfluidics chambers and cell bodies (CB) and distal axons (DAx) were treated with 12.5 mM KCl (K) or 200 ng/ml BDNF (B) +/− the γ-secretase inhibitor, 250 nM DAPT (D), for 48 hours as indicated. The bars depict the means ± SEM for n=3–5; ***, p < 0.001, student’s t-test with Welch correction among indicated groups. See also figure S1
C. Representative image of mCherry-p75NTR-GFP in sympathetic neurons showing full length p75NTR (yellow arrowheads), the released ECD (red arrowheads) and the liberated ICD (green arrowheads) in axons. See also figure S1
D and E. Quantification of the number of p75NTR ICD only (green) and full length p75NTR (yellow) puncta in axons after treatment with KCl (control) or BDNF for 2 hrs. Depicted are the means ± SEM from at least 3 different experiments (KCl, n= 266 and BDNF, n=255 events); *, p < 0.05, student’s t-test.
F and G. Quantification of the number of p75NTR ICD only (green) and full length p75NTR (yellow) puncta under control conditions and after trophic factor deprivation (TFD) for 14 hours. Depicted are the means ± SEM from 3 different experiments (NGF, n= 212 and TFD, n= 383 events); *, p < 0.05, student’s t-test.
H and I. Expression of cleavage resistant V243N mutant p75NTR prevents neuronal apoptosis induced by BDNF or trophic factor deprivation (TFD). Sympathetic neurons were electroporated with either wildtype or V243N mutant mCherry-p75NTR-GFP and cultured in NGF. The neurons were then treated with KCl +/− 200 ng/ml BDNF, maintained in 20ng/ml NGF or NGF was removed as indicated for 48 hours. The bars depict the means ± SEM for n=3–5; *, p< 0.05, ***, p < 0.001, *** *, p < 0.0001 2-way ANOVA with a Sidak’s and Tukey’s multiple comparisons test among indicated groups. See also figure S1
J. Visualization of p75NTR in CD63 labelled MVBs. Neurons electroporated with mCherry-p75NTR-GFP and stained for endogenous CD63 (magenta). P75NTR-ECD shown in red and P75NTR-ICD in green. Scale bar: 5 um.
K and L. Quantification of P75NTR and CD63 double labelled puncta after the neurons were treated with KCl +/− 200 ng/ml BDNF for 2 hours (K), or maintained in 20ng/ml NGF or NGF was removed for 14 hours (L). The bars depict the means ± SEM for n=3–5; *, p< 0.05; **, p < 0.01, student’s t-test with Welch correction among indicated groups.
To directly assess axonal p75NTR cleavage in response to a pro-apoptotic ligand, we electroporated a dual-tagged p75NTR reporter construct into the neurons, which has mCherry on the N-terminus and GFP on the C-terminus. The expression of the construct was detected throughout the neuron, including in the axons (Fig S1C). This mCherry-p75NTR-GFP reporter was previously utilized to investigate the localization of the cleaved receptor products in astrocytes and can differentiate between uncleaved, full length p75NTR (yellow), cleaved p75NTR ICD (green) and extracellular domain (ECD) (red) (Schachtrup et al., 2015). After electroporation, the neurons were allowed to recover in NGF, then switched to KCl with or without BDNF for 2 hrs and the number of mCherry, GFP or overlapping puncta in the axons was quantified. The mCherry signal was found to be very weak, making quantification difficult; therefore, to avoid underestimating the amount of ECD, we preincubated live neurons with an antibody to the ECD that does not interfere with ligand binding. This antibody was tagged with ATTO-550 to enhance the mCherry signal of ECD (from here on referred to as mCherry). Puncta positive for both GFP and mCherry (yellow, Fig 2C) were considered to be full length receptor, although we cannot rule out the possibility that p75NTR was cleaved and both the ICD and ECD remained localized too close to be resolved. Puncta that were only GFP+ or mCherry+ were considered as independent ICD and ECD, respectively. BDNF significantly increased the number of GFP+ only (ICD) puncta, suggesting p75NTR cleavage within the axons (Fig 2D). The majority of puncta were labeled with both mCherry and GFP and BDNF treatment did not induce a significant change in the amount of full length receptor, suggesting only a small fraction of p75NTR undergoes cleavage after BDNF treatment (Fig 2E). Similar results were reported using western blot to detect p75NTR cleavage, only a fraction of the receptor is cleaved and the majority remains full length (Jung et al., 2003; Kanning et al., 2003; Kenchappa et al., 2006). We also detected a very small percentage of mCherry+ only puncta, reflecting the ECD, which was not altered by BDNF (data not shown). We interpret the low number of ECD fragments as a majority being lost due to most of the ECD having been released by γ-secretase at the plasma membrane.
Since trophic factor deprivation also initiates a retrograde degenerative signal (Ghosh et al., 2011; Mok et al., 2009; Simon et al., 2016), we investigated the possibility that NGF withdrawal could stimulate local p75NTR proteolysis. NGF was withdrawn and the axons were live imaged after 14 hours, which we reasoned would be long enough to induce p75NTR cleavage, but prior to the neurons committing to apoptosis (Deckwerth and Johnson, 1993). There was a significant decrease in full length receptor after NGF withdrawal and a corresponding increase in the fraction of GFP+ only puncta (Fig 2F&G), indicating that TFD can induce the cleavage of p75NTR in axons.
Although treatment of the axons with DAPT blocked neuronal death (Fig. 2B), DAPT inhibits the cleavage of all γ-secretase substrates. Therefore, we investigated whether cleavage of p75NTR specifically was necessary for the apoptotic signal. We generated a Val243Asn (V243N) mutation in the extracellular, juxtamembrane region of the mCherry-p75NTR-GFP construct, which introduces a glycosylation site that prevents the receptor from undergoing proteolysis and blocks p75NTR-mediated apoptosis (Underwood et al., 2008). We confirmed that the V243N mutant did not undergo proteolysis (Fig S1D&E). Importantly, when this construct was expressed in the neurons, it prevented cell death induced by BDNF or by NGF withdrawal (Fig 2H&I). Since both α- and γ-secretase cleavage were inhibited by the expression of the V243N construct, these results do not distinguish between the ICD, produced by γ-secretase, and the carboxy-terminal fragment produced by α-secretase as the essential death signal. However, these findings indicate that cleavage specifically of p75NTR is required for apoptotic signaling generated by BDNF binding to p75NTR or by NGF withdrawal.
The cleavage of p75NTR in axons raised the question as to how the ICD would be transported back to the soma. It was recently reported that p75NTR is present in CD63+ multi vesicular bodies (MVBs) in sympathetic neuron cell bodies after BDNF treatment (Escudero et al., 2014) and MVBs have been implicated in retrograde NGF-TrkA signaling (Ye et al., 2018). Therefore, we hypothesized that p75NTR ICD may also be transported in MVBs. In neurons electroporated with the mCherry-p75NTR-GFP reporter, we found a significant increase in the colocalization of GFP signal with CD63+ immunostaining in the axons following BDNF stimulation or NGF deprivation (Fig 2J-L). These results suggest MVBs transport the p75NTR apoptotic signal from axons to the neuronal soma.
Axonal HDAC1 is required for retrograde degenerative signaling
Recent studies have revealed a role for several HDACs in regulating axonal transport (Cho and Cavalli, 2014). Therefore, we hypothesized that HDACs may also be involved in retrograde degenerative signaling. Addition of the general class I HDAC inhibitor sodium butyrate selectively to distal axons along with BDNF blocked neuronal death (Fig S2A). Since sodium butyrate is rather nonselective, we tested the effects of MS275, which preferentially inhibits HDAC1 (Bertrand, 2010; Hu et al., 2003). Treatment of the axons with MS275 completely prevented p75NTR-mediated retrograde apoptotic signaling (Fig 3A). Importantly, addition of MS275 to the cell bodies had no effect on the apoptosis induced by BDNF in the distal axons, indicating that nuclear HDAC1 activity is not required for apoptosis. To confirm the specificity for HDAC1, we knocked down the deacetylase with a shRNA expressing lentivirus (Fig 3B) that selectively silences HDAC1 without affecting the other HDACs (Kim et al., 2010). After confirming the knockdown (Fig 3B), we found that the apoptosis induced by axonal BDNF was completely prevented (Fig 3C), further supporting an essential role for HDAC1 in this retrograde degenerative signaling.
Figure 3. Axonal HDAC1 is required for p75NTR-mediated retrograde apoptotic signaling.
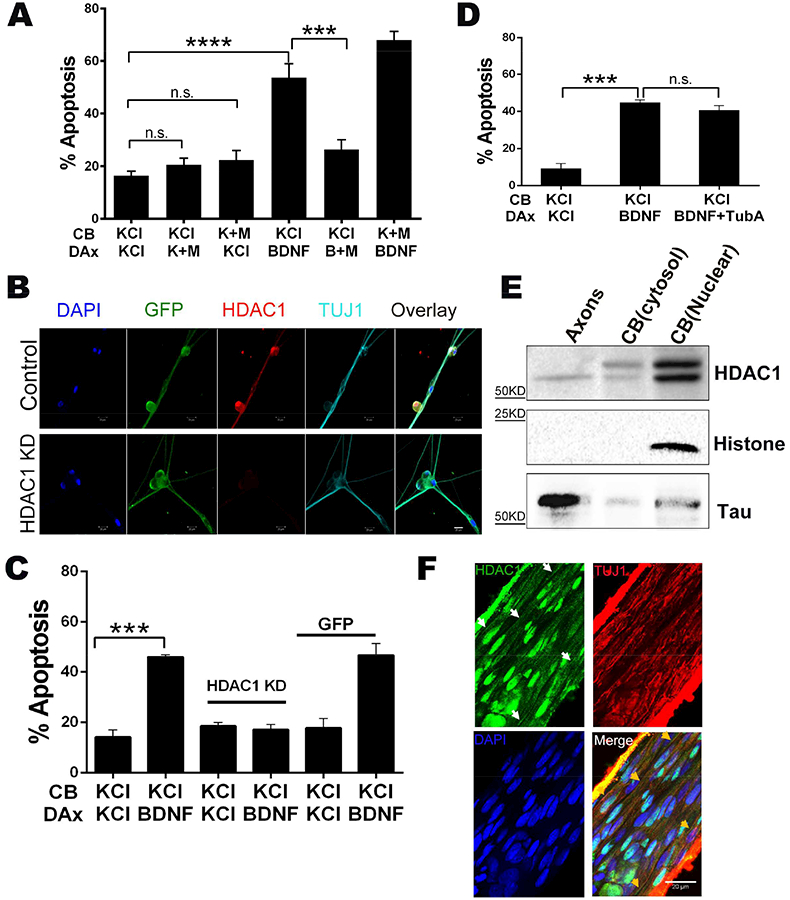
A. Inhibition of axonal HDAC1 blocks p75NTR-mediated neuronal apoptosis. Sympathetic neurons were cultured in microfluidic chambers and cell bodies (CB) and distal axons (DAx) were treated with 12.5 mM KCl or 200 ng/ml BDNF +/− 5 μm MS275 (M) for 48 hrs, as indicated. Depicted are the means ± SEM for n=3–4; ***, p < 0.001; ****, p < 0.0001; 2-way ANOVA with a Tukey’s multiple comparisons test.
B. Representative images of sympathetic neurons infected with a lenti virus expressing HDAC1 knock-down shRNA and co-expressing GFP (green) or a GFP expressing control lenti virus. The neurons were immunostained for HDAC1 (red) and TUJ1 (Teal). DAPI was used to label the nuclei. Scale bar 20 μm
C. Silencing HDAC1 prevents p75NTR-mediated neuronal apoptosis. Sympathetic neurons with HDAC1 knocked-down or expressing a GFP control were treated on their cell bodies (CB) and distal axons (DAx) with 12.5 mM KCl or 200 ng/ml BDNF, for 48 hours as indicated. Depicted are the means ± SEM for n=3–4; ***, p < 0.001, 2-way ANOVA with a Tukey’s multiple comparisons test.
D. Inhibition of HDAC6 did not affect p75NTR-induced apoptosis. Neurons were treated on their cell bodies (CB) and distal axons (DAx) with 12.5 mM KCl, 200 ng/ml BDNF or 200 ng/ml BDNF+ 5 μm Tubastatin A (TubA), as indicated. Depicted are the means ± SEM for n=3–4; ***, p < 0.001; n.s., not significant; 2-way ANOVA with a Tukey’s multiple comparisons test.
E. Axons and cell bodies were collected from sympathetic neurons and lysates were western blotted for HDAC1 and Histone H3 or Tau, to confirm the purity of nuclear and axonal fractions, respectively.
F. HDAC1 is constitutively expressed in sympathetic axons in vivo. Nerve fibers descending proximal to the superior cervical ganglia were isolated from P4–5 rats, fixed and immunostained for HDAC1 and TUJ1. White arrows indicate the HDAC1 stained axons and yellow arrow heads indicate the same axons showing TUJ1 co-staining.
Since HDAC6 is a well-established regulator of axonal transport through deacetylation of tubulin (d’Ydewalle et al., 2011), we also evaluated the effects of Tubastatin A, an HDAC6 selective inhibitor. Addition of Tubastatin A to the distal axons did not affect BDNF-induced apoptosis, indicating that HDAC6 activity is not required (Fig 3D).
Class I HDACs, including HDAC1, primarily localize to the nucleus (Cho and Cavalli, 2014; Haberland et al., 2009); however, it was recently reported that HDAC1 can translocate into axons following injury (Kim et al., 2010). Since the sympathetic neurons used here were not subject to any insult prior to p75NTR activation, our results suggested that HDAC1 is constitutively present in these axons. By immunostaining, we analyzed HDAC1 expression in the presence of NGF, which promotes robust survival of these neurons, and found HDAC1 expression in the axons as well as the nucleus (Fig 3B). The specificity of the staining was confirmed by HDAC1 knockdown. We also isolated pure axons from the neurons, fractionated the cell bodies into cytosolic and nuclear fractions, and then the 3 compartments were immunoblotted for HDAC1. A single band of 66 kD for HDAC1 was detected in the axons whereas a doublet was present in the cytosolic and nuclear fractions (Fig 3E). This doublet is consistent with the phosphorylation of HDAC1 that was recently reported to maintain its localization in the nucleus (Zhu et al., 2017). Finally, to confirm that HDAC1 is present in sympathetic axons in vivo and not just in cultured neurons, we isolated the descending nerve proximal to the superior cervical ganglia from P4–5 rats and immunostained for the deacetylase. HDAC1 was detectable in axon fibers of the nerve (as well as in glial nuclei), indicating that it is constitutively expressed in peripheral axons (Fig. 3F). The specificity of the signal in the nerve fibers was confirmed by staining brain tissue, where only nuclei were detected (Fig S2B).
HDAC1 is required for retrograde axonal transport of the p75NTR intracellular domain
As a first step in retrograde trafficking, p75NTR is internalized through a mechanism requiring the GTPase dynamin (Deinhardt et al., 2007; Hibbert et al., 2006), which can be blocked by the inhibitor Dynasore (Kirchhausen et al., 2008; Macia et al., 2006; Escudero et al, 2014). We also observed p75NTR internalization following BDNF treatment (Fig 4A&B), which was necessary for retrograde apoptotic signaling, since addition of Dynasore along with BDNF in the distal axons prevented neuronal cell death (Fig 4C). However, there was no significant effect of MS275 on receptor internalization (Fig 4A&B), indicating that HDAC1 activity is not required for p75NTR endocytosis.
Figure 4. HDAC1 does not affect p75NTR internalization, which is necessary for neuronal apoptosis.
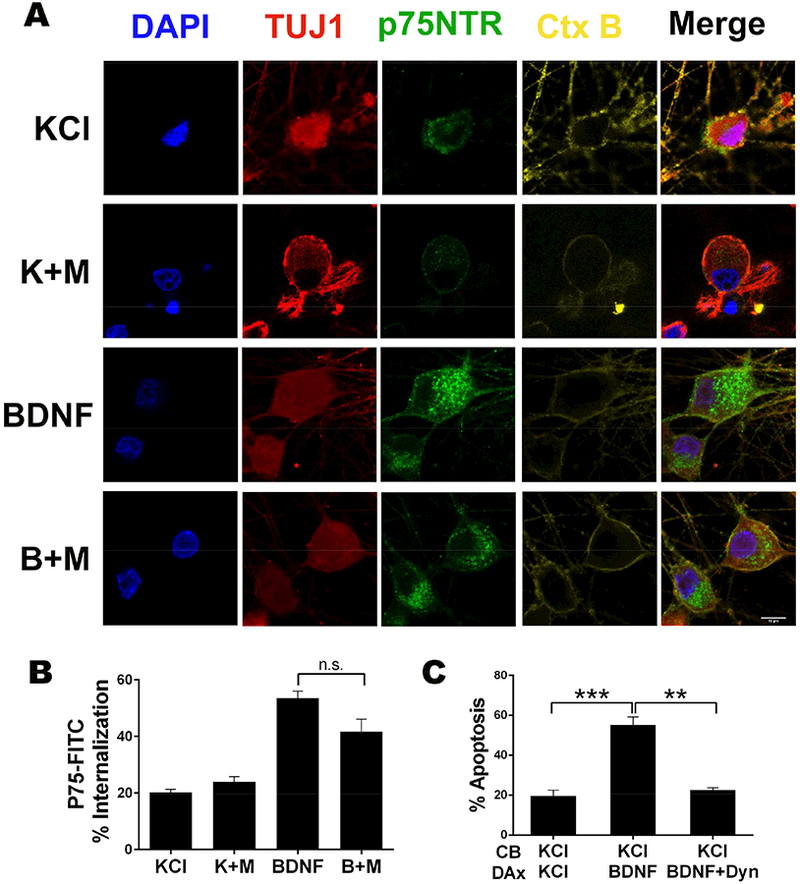
A. Representative images of p75NTR immuno-endocytosis in the presence or absence of BDNF +/− MS275. Sympathetic neurons were incubated with an antibody to p75NTR ECD conjugated to FITC at 4°C then treated for 4 hrs with 12.5 mM KCl or 200 ng/ml BDNF +/− 5 μm MS275 (M) at 37°C, as indicated, then fixed and immunostained for TUJ1 (red). P75NTR is shown in green and nuclei were labeled with DAPI (blue). Cholera toxin B (Ctx B) was used to define the plasma membrane (yellow). Scale bar 10 μm.
B. Quantification of p75NTR internalization following BDNF (B) treatment +/− MS275 (M). The relative amount of internalized fluorescence corresponds to the intracellular fluorescence normalized to the cell surface associated fluorescence in complete z-stacks of confocal images. Shown are the average internalized fluorescence ± SEM for n=3 (70 neurons); n.s. not significant, student’s t-test with Welch correction.
C. Internalization of p75NTR is necessary for neuronal apoptosis. Neurons were cultured in microfluidics chambers and cell bodies (CB) and distal axons (DAx) were treated with 12.5 mM KCl or 200 ng/ml BDNF +/− 80 μm Dynasore (Dyn) for 48 hrs, as indicated. Depicted are the means ± SEM for n=4; **, p < 0.01; ***, p < 0.001; 2-way ANOVA with Tukey’s multiple comparisons test.
Since inhibition of HDAC1 did not affect p75NTR internalization, we hypothesized that the deacetylase was required for retrograde trafficking of the receptor. To investigate p75NTR axonal transport we used the mCherry-p75-GFP reporter, which allowed us to track both the full length receptor and the liberated ICD. The neurons were treated with BDNF in the presence or absence of MS275 for 2 hrs and live imaged to assess the movement of the cleaved ICD and the full length p75NTR (Fig 5A&B). The ATTO-550 labeled antibody to the ECD of p75NTR was included to enhance detection of the ECD fragment. Following BDNF treatment, there was a significant increase in the number of ICD-only, GFP+ particles moving retrogradely; however, this increase was completely blocked by addition of MS275, indicating that HDAC1 activity is required for the BDNF-induced retrograde movement (Fig 5C). BDNF also increased the instantaneous velocity of the GFP+ puncta moving in a retrograde direction; however this was not notably affected by inhibition of HDAC1 (Fig S3).
Figure 5. HDAC1 is required for retrograde axonal transport of p75NTR intracellular domain.
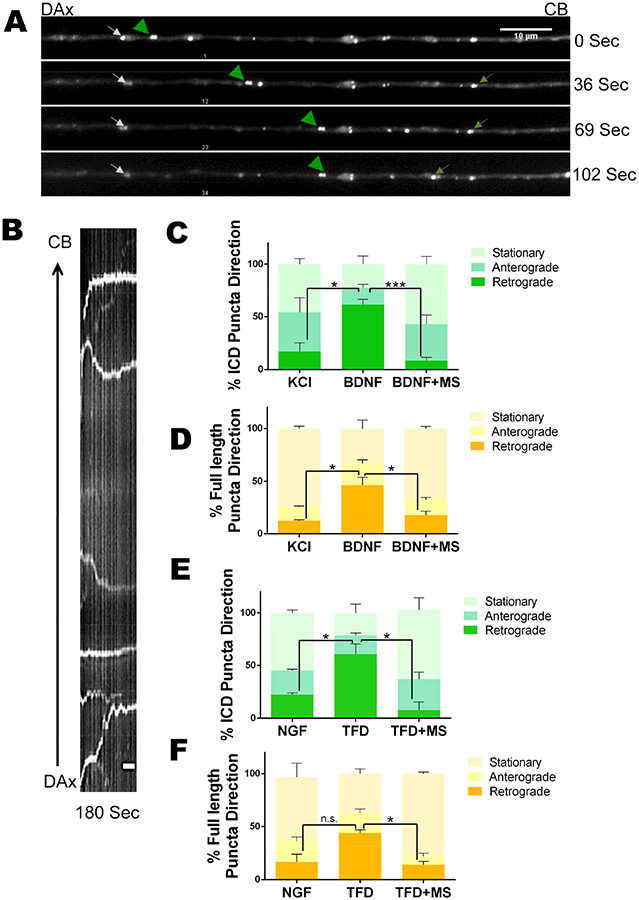
A. Representative images of an axon from a neuron treated with BDNF. Frames were collected from a time-lapse movie showing p75NTR ICD particles moving in the retrograde (green arrowheads) or anterograde (green arrows) direction or stationary (white arrows). Scale bar:10 μm.
B. Kymograph generated from time-lapse images displaying the trajectory of p75NTR ICD. Scale bar: 1 μm.
C-F. Quantification of moving and stationary p75NTR ICD and full length receptor and directionality of traffic under the indicated conditions. P75NTR ICD is displayed by green stacked bar graphs while the movement of full length p75NTR is shown by bar graphs in different shades of yellow. Data pooled from n=3; student’s t-test with Welch correction revealed that for retrograde movement of the ICD: KCl vs BNDF, p<0.05; BDNF vs BDNF+MS, p<0.001; NGF vs TFD, p<0.05; TFD vs TFD+MS, p<0.05; and for the full length p75NTR: KCl vs BDNF, p,0.05; BDNF vs BDNF+MS, p,0.05; NGF vs TFD, n.s.; TFD vs TFD+MS, p<0.01. See also Figure S2.
Interestingly, the percentage of full length p75NTR, or co-localized ECD and ICD, particles moving retrogradely also increased after BDNF treatment and MS275 prevented this increase (Fig 5D). There was also an increase in the instantaneous velocity of full length p75NTR moving in the retrograde direction after BDNF treatment, but MS275 had little effect. Although we cannot rule out the possibility that full length particles containing both the ECD and the ICD contribute to the subsequent cell death, the fact that inhibiting receptor cleavage in the distal axons prevented apoptosis (Fig 2B) suggests that it is the retrograde transport of the liberated ICD that is the determining factor for the fate of the neuron.
Based on the requirement for p75NTR in the apoptosis of sympathetic neurons following NGF withdrawal (Majdan et al., 2001; Nikoletopoulou et al., 2010) and the induction of p75NTR cleavage by TFD (Fig 2F&G), we hypothesized that loss of NGF signaling would also induce retrograde trafficking of the p75NTR ICD and this transport would also require HDAC1 activity. We found a significant increase in the number of p75NTR ICD, GFP+ particles moving in the retrograde direction following NGF withdrawal and this retrograde transport was blocked by HDAC1 inhibition (Fig 5E). Retrograde transport of full length p75NTR, or closely localized ICD and ECD, was also increased following NGF removal and this was also blocked by MS275 (Fig 5F). Similar to p75NTR activation by BDNF binding, the instantaneous velocity of the retrogradely moving p75 was increased after NGF withdrawal, but there was no marked effect of MS275. Taken together, the live imaging data indicate that HDAC1 activity is required for retrograde transport of the p75NTR ICD, which mediates a degeneration signal.
HDAC1 deacetylates the p150Glued subunit of dynactin
To determine the relevant substrates of HDAC1 in the axons, neurons were grown on transwell inserts in the presence of NGF and MS275 to maximize the acetylation of HDAC1 substrates, and axons were collected. Trypsin digested axonal proteins were immunoprecipitated with anti-acetyl lysine and analyzed by mass spectrometry. A total of 100 axonal proteins potentially harboring lysine-acetylation were identified; among these, one candidate that caught our attention was dynactin1/p150Glued. The p150Glued subunit is the major polypeptide of the dynactin complex and is necessary for the processivity of the dynein motor (King and Schroer, 2000; McKenney et al., 2014). We validated the lysine-acetylation site on p150Glued by manual inspection of the spectra (Fig 6A). Interestingly, this acetylated lysine (K230) of p150Glued lies in coiled coil region 1 (CC1), the domain responsible for binding the dynein intermediate chain (DIC) (King et al., 2003; Karki and Holzbaur, 1995). This region is highly conserved across species from flies through humans, emphasizing its importance (Fig 6B). As shown in figure 6 (C&D), inhibition of HDAC1 in HEK293 cells increased the acetylation of p150Glued while over expression of HDAC1 decreased the level of acetylation. These results support the hypothesis that HDAC1 promotes deacetylation of p150Glued.
Figure 6. HDAC1 deacetylates p150Glued at lysine 230.
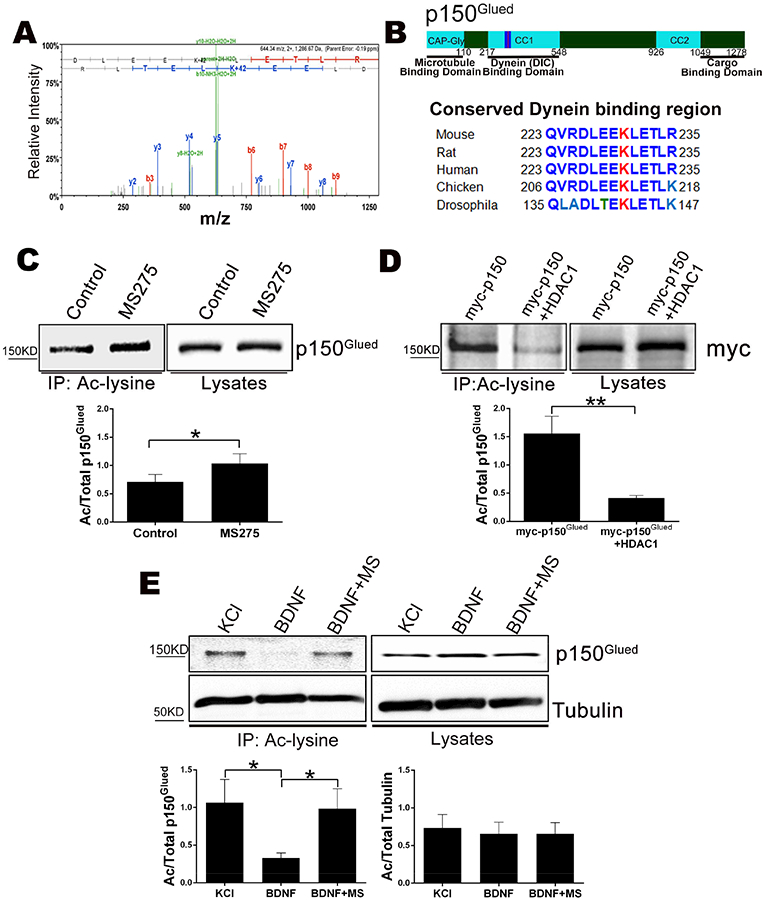
A. MS/MS spectrum of acetylated p150Glued tryptic peptide spanning lysine 230. Annotated product ions highlighted in red and blue correspond to b- and y- type ions, respectively. Neutral loss ions from the peptide precursor or product ions are highlighted in green. The observed product ions confirmed acetylation at K230 as indicated by a 42 Da mass shift.
B. (Top) Schematic of p150Glued. (Bottom) Comparison of protein sequences from the DIC binding region of CC1 around K230 from the indicated species.
C. Inhibition of HDAC1 increases the acetylation of p150Glued. (Top) HEK293 cells were treated +/− MS275 for 48 hrs, fractionated and cytosolic lysates were immunoprecipitated with anti-acetyl lysine and western blotted for p150Glued. (Bottom) Bar graph showing the average ratio of acetylated over total p150Glued ± SEM for n=7; *, p < 0.05, student’s t-test.
D. Over expression of HDAC1 reduces the amount of acetylated p150Glued. (Top) HEK293 cells were transfected with Myc-p150Glued with or without HDAC1-Flag. The cytosolic lysates were immunoprecipitated with anti-acetyl lysine and western blotted with an antibody to Myc. (Bottom) The average ratio of acetylated over total p150Glued ± SEM for n=5; **, p < 0.01, student’s t-test.
E. Activation of p75NTR reduces the level of acetylated p150Glued. Sympathetic neurons were treated with 12.5 mM KCl or 200 ng/ml BDNF +/− 5 μm MS275 (MS) for 48 hrs. The neurons were fractionated and the cytosolic lysates were immunoprecipitated with anti-acetyl lysine and western blotted for p150Glued or alpha-Tubulin. (Top) Representative western blot image. (Bottom) The average ratio of acetylated over total p150Glued or tubulin ± SEM for n=6–7; *, p < 0.05, student’s t-test.
Since HDAC1 activity was required for the retrograde degeneration signal activated by p75NTR (Fig 3), we hypothesized that BDNF would promote HDAC1-dependent deacetylation of p150Glued in sympathetic neurons. We found a significant decrease in the level of acetylated p150Glued following BDNF treatment and the reduction could be completely prevented by MS275 (Fig 6E), suggesting that p75NTR activation results in p150Glued deacetylation by HDAC1. We also evaluated acetylated tubulin, since HDAC5 and 6 can deacetylate microtubules to regulate axonal transport (Cho and Cavalli, 2014); however, there was no change in the expression or acetylation levels of tubulin following BDNF treatment, indicating that activation of p75NTR does not affect these other HDACs.
Deacetylation of p150Glued enhances its interaction with dynein
Retrograde signaling typically depends on active transport of the signaling components by the dynein-dynactin complex. To determine if the retrograde degenerative signal induced by p75NTR activation was dependent on dynein, we tested the inhibitor ciliobrevin A, which blocks the ATPase activity of dynein without affecting kinesins (Firestone et al., 2012). When distal axons were treated with ciliobrevin A, there was a significant reduction in BDNF induced apoptosis (Fig 7A), indicating that the retrograde signal generated by p75NTR was dynein dependent.
Figure 7. De-acetylation of p150Glued increases its interaction with dynein.
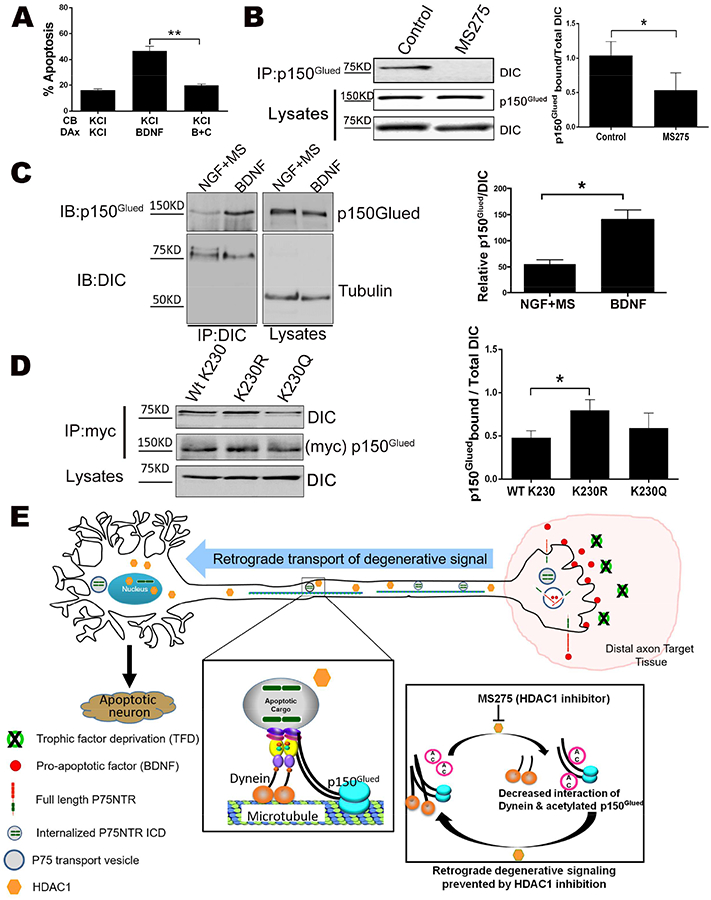
A. Inhibition of dynein blocks p75NTR retrograde apoptotic signaling. Sympathetic neurons were cultured in microfluidic chambers and cell bodies (CB) and distal axons (DAx) were treated with 12.5 mM KCl (K) or 200 ng/ml BDNF, as indicated. During the last 8 hrs, 25 μm ciliobrevin A together with BDNF was applied to the distal axons (B+C). The neurons were then fixed and stained for TUJ1 and DAPI to quantify pyknotic nuclei. The bars depict the means ± SEM for n=3; **, p < 0.01, 2-way ANOVA with Tukey’s multiple comparisons test.
B. Inhibition of HDAC1 reduces the interaction of p150Glued with dynein. (Left) HEK293 cells were treated with MS275 or left untreated (control), then fractionated and the cytosolic lysates immunoprecipitated with anti-p150Glued, then western blotted for dynein intermediate chain (DIC). Also shown are the lysates blotted for p150Glued and DIC. (Right) The average ratio of DIC pulled down to total DIC in the lysates ± SEM for n=3; *, p < 0.05, student’s t-test.
C. Activation of p75NTR enhances the interaction of p150Glued with dynein. (Left) Neurons were treated with 200 ng/ml BDNF or 20 ng/ml NGF + 5 μM MS275 for 48 hrs. The neurons were then lysed, and immunoprecipitated with DIC and western blotted for p150Glued or DIC. (Right) The average ratio of p150Glued pulled down with DIC to total DIC in the lysates ± SEM for n=3; *, p < 0.05, student’s t-test.
D. Mutation of K230 to R in p150Glued increases its interaction with dynein. (Left) HEK293 cells were transfected with Myc-p150Glued wild type, K230R or K230Q mutant. The cells were then fractionated and the cytosolic lysates immunoprecipitated with anti-Myc and western blotted for DIC or p150Glued, as indicated. Also shown are the lysates blotted for DIC. (Right) The average ratio of DIC pulled down to total DIC in the lysates + SEM for n=6; *, p < 0.05, student’s t-test.
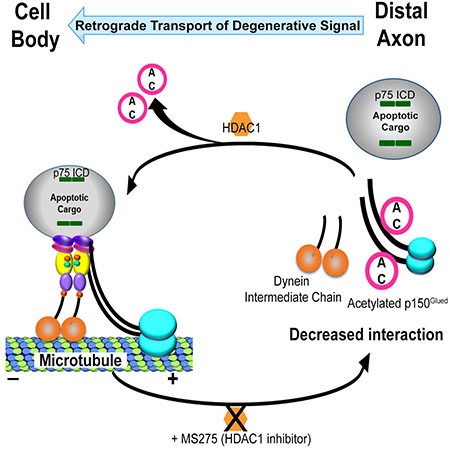
E. Summary model. Axonal HDAC1 is required for retrograde apoptotic signaling by activation of p75NTR or trophic factor deprivation. HDAC1 enhances p75ICD transport in axons by deacetylating p150Glued, which facilitates its binding to the dynein intermediate chain.
Since the lysine (K230) on p150Glued that we found acetylated in the presence of MS275 lies in the CC1 domain, which interacts with the DIC, we hypothesized that deacetylation of p150Glued by HDAC1 affects its interaction with dynein. In HEK293 cells treated with MS275, there was a significant decrease in the association of p150Glued with DIC relative to untreated cells, suggesting that the acetylation reduces their interaction (Fig 7B). Similarly we treated the neurons with BDNF, to stimulate deacetylation of p150Glued, or NGF + MS275, to maximize the acetylation. In the presence of BDNF there was a significant increase in the coimmunoprecipitation of p150Glued and DIC compared to NGF + MS275, supporting a role for p75NTR in regulating this interaction (Fig 7C).
To specifically address the influence of K230 acetylation on p150Glued binding to dynein, we mutated K230 either to arginine (R), which cannot be acetylated, or glutamine (Q), which can mimic an acetylated lysine (Fujimoto et al., 2012). Wild type p150Glued or the mutants were transfected into HEK293 cells and their ability to pull down with DIC was quantified (Fig 7D). The p150Glued-dynein interaction was not significantly altered by the K230Q mutation, which we suggest indicated that glutamine does not adequately mimic acetyl-lysine in this system, as reported earlier for the protein Ku (Fujimoto et al., 2012). However, the K230R mutant associated with dynein significantly better than the wild type p150Glued, supporting the notion that deacetylation of K230 increases the formation of the p150Glued-dynein complex, which is required for retrograde apoptotic signaling.
Discussion
In this study, we identify an HDAC1-dependent signal downstream of the p75NTR as critical for the regulation of retrograde, pro-apoptotic signals generated in response to p75NTR activation by ligand binding or TFD in distal axons (Fig 7E). The involvement of p75NTR in retrograde pro-apoptotic signaling following TFD helps to explain previous findings that sensory and sympathetic neurons in trkA−/− mice could be rescued by simultaneous deletion of p75ntr (Majdan et al., 2001; Nikoletopoulou et al., 2010). The loss of TrkA mimics TFD during development; therefore, based on our results, elimination of p75NTR would reduce proapoptotic signaling. Notably, neurons engineered to express TrkA underwent apoptosis in the absence of NGF. The cell death correlated with p75NTR cleavage and could be prevented by a γ-secretase inhibitor, suggesting that p75NTR cleavage contributed to the apoptosis (Nikoletopoulou et al., 2010). The mechanism by which loss of TrkA signaling induces p75NTR cleavage and retrograde signaling is not known. However, withdrawal of NGF activates c-Jun N-terminal kinase (JNK) (Xia et al., 1995), which can stimulate the cleavage of p75NTR, in part, through up regulation and activation of α-secretase (Kenchappa et al., 2010). It is possible that local activation of JNK causes release of the p75NTR ICD, which then forms a retrograde signaling complex.
Direct activation of p75NTR by ligand binding has also been shown to induce retrograde degenerative signaling. For example, pro-neurotrophin-3 applied exclusively to distal axons of sympathetic neurons induced apoptosis via p75NTR (Yano et al., 2009) and in vivo activation of p75NTR was suggested to contribute to the normal developmental pruning of motor neurons (Taylor et al., 2012). The nature of the retrograde apoptotic signal was not investigated in those studies; however, our results indicate that activation of p75NTR exclusively in axons induces retrograde transport of the p75NTR ICD, which promotes neuronal apoptosis. Indeed, expression of p75NTR ICD alone in sympathetic neurons (including both cell bodies and axons), leads to their death (Kenchappa et al., 2006) and mice engineered to ectopically express the p75NTR ICD in neurons exhibited extensive, global neuron loss (Majdan et al., 1997).
Although apoptosis induced by NGF withdrawal or direct activation of p75NTR both involve cleavage of p75NTR and retrograde transport of the ICD, there are distinct differences between the mechanisms by which apoptosis is ultimately induced. For example, both p75NTR activation by ligand binding and TFD stimulate phosphorylation of c-Jun by JNK; however, c-Jun is only necessary for apoptosis induced by TFD (Palmada et al., 2002). Certainly, multiple signals are generated by these two processes that influence the fate of the cell; nevertheless, both include a pro-apoptotic signal involving JNK and the proteolytic cleavage of p75NTR.
The proteolysis of p75NTR by γ-secretase was reported to occur in endosomes following internalization in PC12 cells (Urra et al., 2007). Therefore, we expect that the p75NTR ICD is generated on endosomes within the axon, although this remains to be determined. In contrast, cleavage of many substrates by α-secretase occurs at the plasma membrane (Murphy, 2008), thus the shedding of p75NTR’s ECD by α-secretase may occur at the axon surface, which would explain why we detected so little free ECD in the axons. Interestingly, p75NTR has been detected in multi vesicular bodies (MVBs) following endocytosis (Butowt and Bartheld, 2009; Escudero et al., 2014). In neurons of the avian isthmo-optic nucleus, which innervate the retina, p75NTR activation by NGF on axon ends in the eye resulted in apoptosis (von Bartheld et al., 1994). This retrograde p75NTR-mediated cell death correlated with accumulation of NGF in the neuronal soma in MVBs (Butowt and Bartheld, 2009), suggesting that retrograde transport of p75NTR (and possibly separated ECD and ICD) occurs in MVBs. The increase in colocalization of p75NTR ICD and CD63 we observed in axons following BDNF treatment or TFD suggests that the retrograde degenerative signal is transported in MVBs, although we cannot rule out the involvement of other vesicles.
How the freed ICD would remain associated with a vesicle is not clear, since this fragment would be released following receptor cleavage. However, it is notable that there is a palmitoylation site in the juxtamembrane region of the ICD that is required for cell death signaling (Underwood et al., 2008). The addition of this lipid may allow the ICD to associate with a vesicle for transport. An analogous mechanism was recently reported for Dual leucine-zipper kinase (DLK): Following axonal injury of sensory neurons, DLK was palmitoylated, which facilitated its recruitment to vesicles for retrograde transport (Holland et al., 2016).
Our findings also reveal that axonal HDAC1 is required for pro-apoptotic retrograde signaling. Although HDAC1 has been considered exclusively nuclear, it was recently reported that this deacetylase can translocate out of the nucleus into the axons of hippocampal and cortical neurons following exposure to toxic insults, such as TNF and glutamate (Kim et al., 2010). In these axons, HDAC1 promoted degeneration by disrupting mitochondrial transport through binding to the kinesins KIF2a and KIF5. In contrast, we found HDAC1 is constitutively present in sympathetic axons, even under strong pro-survival signaling. The constitutive presence of HDAC1 in sympathetic axons may reflect a general difference between peripheral and central axons; however, there must be some mechanism in the peripheral axons to prevent the deacetylase from disrupting mitochondrial transport. In the central nervous system, HDAC1 may promote degeneration by two mechanisms: disrupting mitochondrial transport and enhancing retrograde degenerative signaling. Since most cargo bind both kinesins and dynein (Maday et al., 2014), sequestering the anterograde motors KIF2a and KIF5 may result in a predominance of dynein associated with some cargo, leading to primarily retrograde transport, although this remains to be investigated.
In an effort to understand the role of HDAC1 in regulating p75ICD transport, we identified the dynactin subunit p150Glued as a potential substrate. P150Glued is the largest subunit of the 1 MDa dynactin complex, which is essential for virtually all aspects of dynein function (Carter et al., 2016; King and Schroer, 2000; Schroer, 2004; Urnavicius et al., 2015). In its N-terminus, p150Glued contains a cytoskeleton-associated protein Gly-rich (CAP-Gly) domain, which binds microtubules, followed by two coiled-coil domains, the first of which (CC1) binds directly to the DIC. Our results indicate that p150Glued can be acetylated at K230, in the CC1 region, and deacetylation of K230 increased its association with DIC.
The deacetylation of p150Glued was enhanced following BDNF treatment and this could be prevented by inhibition of HDAC1. The mechanism by which p75NTR induces HDAC1-dependent deacetylation of p150Glued is not known; however, an association between HDAC1 and the p75NTR interacting protein SC1 has previously been reported (Chittka et al., 2004). SC1 is a transcription factor that associates with the ICD of p75NTR and translocate to the nucleus upon neurotrophin binding to p75NTR (Chittka et al., 2004). Therefore, activation of the receptor may lead to recruitment of HDAC1 through association with SC1.
It is interesting to consider the clinical implications of these results. Notably, the ICD of p75NTR was previously identified in association with dynein in the axons of mice harboring the G93A mutation in superoxide dismutase 1 (SOD1) that is associated with a form of amyotrophic lateral sclerosis, but not wild type mice (Perlson et al., 2009, 2010). Inhibition of this retrograde signaling rescued cultured motor neurons expressing the mutant SOD1. Hence, axonal transport of both pro-survival and pro-apoptotic signals needs to be considered in designing therapeutic strategies for neurodegenerative conditions.
Star methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bruce D Carter (bruce.carter@vanderbilt.edu). Requests will be handled according to Vanderbilt University and/ or NIH policy regarding MTA and related matters.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Rats and mice
All animal experiments were approved by the Institutional Animal Care and Use Committee at Vanderbilt University. Wild type, untimed pregnant Sprague Dawley (SD) and CD1 rats and mice were purchased mice from Charles River Laboratory. Pregnant rats and mice were housed in 12-hour day and night cycle environment with ad libitum availability of chow diet and water. Both male and female postnatal day 2–3 old rats and mice were used in all the experiments, other than HDAC1 immunostaining in the nerve where postnatal day 4–5 old rat was used.
Cell culture
Primary sympathetic neurons from superior cervical ganglia were cultured in UltraCULTURE medium (Lonza) supplemented with 3% fetal bovine serum (Invitrogen), 20 ng/ml nerve growth factor (Harlan), 2 mM L-glutamine (Invitrogen), 100 units/ml penicillin (Invitrogen), and 100 ug/ml streptomycin (Invitrogen) or as indicated.
HEK293 cells were cultured in high glucose DMEM with 10% fetal bovine serum, 100U/ml penicillin and 100 μ/ml streptomycin.
Method Details
Cell culture and Treatments
Sympathetic neurons were isolated from superior cervical ganglia, which were dissected from male and female postnatal day 2–3 rats or mice (murine neurons were used for the HDAC1 knockdown experiment) and dissociated with 0.08% trypsin (Worthington) and 0.3% collagenase (Sigma). Dissociated cells were then plated on poly-d-lysine (MP Biomedicals) and laminin (Invitrogen) coated surface.
For mass cultures, neurons were cultured in UltraCULTURE medium (Lonza) supplemented with 3% fetal bovine serum (Invitrogen), 20 ng/ml nerve growth factor (Harlan), 2 mM L-glutamine (Invitrogen), 100 units/ml penicillin (Invitrogen), and 100 ug/ml streptomycin (Invitrogen) or as indicated. 24 hours after plating, the neurons were treated with 10 uM cytosine arabinofuranoside (AraC) (Sigma) for 24 hrs to inhibit the proliferation of non-neuronal cells. Following 2 days of exposure, AraC was removed and after 12–24 hrs, the neurons were treated at the indicated concentrations with the reagents as described in respective figure legends. For all experiments involving NGF removal and treatment with BDNF or KCl, the neurons were rinsed at least 3 times with media lacking NGF, containing 12.5 mM KCl and 100 ng/ml of anti-NGF with at least 10 min incubation each in between the rinses prior to addition of the indicated reagents. For NGF withdrawal condition, KCl was not included in the media used for washing or treatments.
For neuron cultures in microfluidic devices (from Xona Microfluidics or generated in-house by Deyu Li laboratory, Vanderbilt University School of Engineering), the chambers were sterilized in plasma cleaner (PDC-32G from Harrick Plasma) and assembled on coverslips, then coated with poly D lysine and laminin. Dissociated neurons were plated on one side (proximal) of the devices in ultraCULTURE media with 20 ng/ml NGF and axons were allowed to grow and cross into the other side (distal). To direct the growth of axons more towards the distal side, media with 50 ng/ml NGF was added in the distal compartment for initial two days. We confirmed that there was no diffusion of soluble material between the chambers using a fluorescent dye (Cy3). After the axons reached the distal side, the neurons were kept alive by maintaining the cell bodies in media containing 20 ng/ml or 12.5 mM KCl and distal axons were treated for 48 hours as indicated in figure legends. For infection with HDAC1 shRNA lenti virus, mouse neurons were cultured in microfluidic devices in NGF for 2 days and then incubated with lentivirus expressing HDAC1 shRNA (3.7 × 107 transduction units/ml), GFP control (7.55 × 107 transduction units/ml). After 48 hours of infection, the axons and cell bodies were treated with 200 ng/ml BDNF, 12.5 mM KCl or 20 ng/ml NGF as indicated in figure legends. After the treatments, neurons were fixed in 4% PFA for immunostaining and counting apoptotic nuclei.
The mCherry-p75NTR-GFP construct was kindly provided by Katerina Akassoglou (Gladstone Institute of Neurological Disease, UCSF). The mCherry-p75NTR-GFP reporter construct was expressed in primary sympathetic neurons by electroporation immediately after dissociation of sympathetic neurons using rat neuron nucleofactor kit (Amaxa Cat# VPG-1003) on nucleofactor II device from Amaxa according to manufacturer’s instructions. More than 1 million neurons isolated from approximately 20 ganglia were pelleted at 200g for 4 minutes and resuspended in 100 ul of rat neuron nucleofection solution prepared according to manufacturer’s instruction. The cell suspension was immediately transferred to Amaxa transfection cuvette containing 20ug of plasmid in small volume of (10–15ul) and zapped on program settings G-013 (designated on the instrument for “rat” neuron DRG). Atleast 1 ml of Ultraculture media with 50 ng/ml NGF was immediately added to the electroporated neurons and the contents were transferred to a 15 ml tube and incubated at 37C in a water bath incubator for at least 20 minutes to promote recovery prior to plating the neurons.
HEK293 cells were cultured in high glucose DMEM with 10% fetal bovine serum and penicillin/streptomycin (100 U/ml, 100 μg/ml, respectively) and transfected using Lipofectamine 2000 (Invitrogen), according to the manufacturer’s instructions.
For analysis of p75NTR receptor cleavage, mCherry-p75NTR-GFP wildtype (mCh-p75WTGFP), V243D mutant (mCh-p75VD-GFP) and V243N mutant (mCh-p75-GFP) constructs were transfected in HEK293 cells. Cells were then pretreated with 10uM ZLLLH for 1 hour (all). For TAPI treatment, cells were pretreated with 10uM TAPI for one hour concurrently with ZLLLH and for PMA treatment, cells were treated with 1uM PMA for 1 hour. After respective treatments, cells were harvested and lysates were prepared for Western blotting.
Isolation of axons in culture
To get pure axon fractions without contamination of cell bodies, dissociated neurons were plated on the transwell cell culture inserts with 8 um pore size. Cultured neurons were maintained in 20 ng/ml NGF for 3–4 days. Following the indicated treatment, the axons and cell bodies were separately harvested by scraping the membrane with a cell scrapper. Membrane facing the well contains the axons and the opposite side has cell bodies (Zheng et al., 2001).
Site directed mutagenesis of DNA constructs
To generate non cleavable mCherry-p75NTR-GFP construct, nucleotide changes encoding V243D mutation were introduced into wildtype mCherry-p75NTR-GFP construct (Schachtrup C et al, 2015) by site-directed mutagenesis using PfuUltra High-Fidelity DNA polymerase (Agilent Technologies). This V243D mutation was further changed to D243N by site directed mutagenesis PCR, to finally generate the cleavage resistant mCherry-p75NTR-GFP construct with V243N mutation.
To generate the constitutively acetylated mimic construct of p150Glued (K230Q) and nonacetylable p150Glued (K230R), lysine (K) residue of wildtype p150Glued construct at position 230 was changed to glutamine (Q) and arginine (R) respectively by site directed mutagenesis of myc-tagged p150Glued (Chevalier-Larsen ES et al, 2008). The accuracy of nucleotides in all the wild type constructs and the changes introduced in the mutant constructs were confirmed by sequencing the wildtype and mutant constructs.
Immunostaining
Primary sympathetic neurons in culture were infected with lentivirus expressing HDAC1 shRNA and/or GFP (control) for more than 48 hrs, then fixed with 4% PFA, permeabilized with 0.1% Triton X-100 containing 0.1% sodium citrate, blocked with 5% BSA or 10% goat serum in PBS, and incubated with antibodies for HDAC1 (1:100) and TUJ1 (1:1000) in PBS containing 0.05% Triton X-100, followed by incubation with fluorescently tagged secondary antibody. Nuclei were visualized by DAPI, and images were acquired with a confocal laser imaging system (LSM 710; Carl Zeiss MicroImaging, Inc.).
Superior cervical ganglia and brain were collected from post-natal day 4–5 rat pups after cardiac perfusion with 4% PFA and embedded in paraffin wax for tissue sectioning. Antigen retrieval of sections was done in citrate buffer (10 mM, pH 6.0). Sections were blocked with 5% BSA and incubated with antibodies for HDAC1 (1:100, Santa cruz biotechnology) and TUJ1 (1:1000, Covance) in PBS containing 0.05% Triton X-100, followed by incubation with fluorescently tagged secondary antibody. Nuclei were visualized by DAPI, and images were acquired with a confocal laser imaging system (LSM 880; Carl Zeiss MicroImaging, Inc.).
For CD63 immunostaining, wildtype mCherry-p75NTR-GFP electroporated neurons were fixed with cold 3% PFA and 4% sucrose and blocked with 5% Fish gelatin in PBS containing 0.1% TritonX 100. Then incubated with CD63 (1:100, Santa cruz biotechnology) antibody followed by incubation with fluorescently tagged secondary antibody. Nuclei were visualized by DAPI, and images were acquired with a confocal laser imaging system (LSM 880; Carl Zeiss MicroImaging, Inc.). Colocalization of P75NTR and CD63 in the axons was counted blindly and 30–40 axons were scored per condition in each experiment.
Neuronal Death analysis
Following the indicated treatments, sympathetic neurons were fixed with 4% paraformaldehyde and immuno-stained with anti-TUJ1 (1:1000, Covance) primary antibody, Alexa Fluor 488 secondary antibody (1:1000, Invitrogen) and DAPI as nuclear marker. The neurons were scored blindly as apoptotic or non-apoptotic on the basis of the appearance of the nucleus, apoptotic nuclei being condensed or fragmented and much brighter compared to healthy neurons. At least 75–100 TUJ1-positive neurons with their axons crossing to the distal chambers were counted per condition in all experiments in microfluidic devices and mass cultures.
Immuno endocytosis of P75NTR
Primary neurons were grown in NGF containing media for 3 days, NGF was then removed and replaced with DMEM containing 1 mg/ml BSA and 12.5 mM KCl for serum deprivation. After 1 hour, anti-p75ECD antibody tagged with FITC (1:50, Santa cruz biotechnology) was added for 1 hour at 4°C. Neurons were then left in KCl or treat ed with 200 ng/ml BDNF +/− 5μM MS275 for 4 hrs at 37°C, rinsed and incubated at 4°C with Chole ra toxin B (1:500, Invitrogen) for 20 min to label the cell surface. Thereafter, the neurons were fixed in 4% paraformaldehyde and processed for TUJ1 immunostaining as described above. The labelled cells were examined and imaged by using a Zeiss LSM 710 confocal microscope with a Plan apochromat 63×/1.4 oil differential interference contrast (DIC) objective. FIJI was used to quantify the cell-associated fluorescence. The total cellular fluorescence was calculated after subtracting the non-specific fluorescence from images of untreated cells obtained with the same illumination and exposure conditions. Internalized p75NTR ECD was determined as the ratio of the internalized receptor (intracellular fluorescence) versus the cell surface-associated receptor (cell-surface-associated fluorescence) and expressed as a percentage of internalized fluorescence (intracellular p75NTR), considering 100% to be the total cell-associated fluorescence.
Live imaging of neurons and kymograph generation
Sympathetic neurons were electroporated with mCherry-p75NTR-GFP construct and plated on glass bottom dishes. The neurons were first plated in UltraCULTURE media with 50 ng/ml NGF which was changed to media with 20 ng/ml NGF next day. After 2 days in culture, NGF was removed and the neurons were incubated with a p75NTR antibody (Anti-p75NTR (extracellular)-ATTO-550, Alomone labs) for 30 min in phenol red free DMEM with HEPES, BSA (1 mg/ml) and KCl (12.5 mM) on ice. The antibody was then rinsed off and the neurons were treated with or without 200 ng/ml BDNF +/− 5 μM MS275 for 2 hrs.
High-resolution wide-field fluorescence images were acquired on a Nikon Eclipse Ti equipped with a Nikon 100× Plan Apo 1.45 numerical aperture (NA) oil objective and a Nikon DS-Qi2 CMOS camera. Multichannel time lapse images were acquired at 3 sec intervals for a total of 3 min and processed using Nikon Imaging software to generate the kymographs. The cell body is at the top of the kymograph, all sloping (moving) pathways were counted to be moving particles while the horizontal pathways were considered to be stationary. All the kymographs were blindly analyzed to quantify the overall direction of movement. Approximately 100 moving particles were analyzed from at least 8 different neurons in each condition for each experiment.
Analysis of p75NTR transport velocity in the axons
Time lapse image analysis was carried out using FIJI. X-Y coordinates of tracks, distance travelled, velocity were registered from about 40 axons in each experiment using Manual Tracking plugin (Schindelin et al., 2012). Tracks with average velocity < 0.2 um/sec were filtered out. Instantaneous velocities were calculated as the distance a puncta travelled between two consecutive frames, divided by frame interval (3 sec). Velocity distribution is presented for 0.2 μm/sec bins.
P75NTR receptor cleavage analysis in axons
Primary sympathetic neurons from rats were grown in microfluidic devices and infected with lentivirus with GFP tag on the C-terminal of p75 (provided by Eran Perlson, Tel Aviv University). Upon p75NTR overexpression, distal axons were treated with 200 ng/ml BDNF or BDNF+ 250nM DAPT for 2 hours and cell bodies were left in 12.5 mM KCl. Distal axons in BDNF+DAPT treatment group were pretreated with 250nM DAPT alone for 1 hour before BDNF+DAPT treatment. Live neurons were stained with ATTO-550 tagged antibody against p75NTR-ECD and then fixed with 4%PFA. Thereafter, fixed neurons, were stained with GFP and imaged on LSM880 with Airyscan mode. To quantify p75NTR receptor cleavage in axons, green puncta were counted for cleaved p75NTR-ICD and yellow puncta for full length p75NTR receptor.
LC-Tandem MS/MS
Neurons were cultured in trans well cell culture inserts, as described above, to collect the axons. After 2–3 days in 20 ng/ml NGF, 5 μM MS275 was added for 24 hrs to maximize the amount of acetylated HDAC1 substrate. Axon were collected without contamination from cell bodies. Lysates were prepared in 6M urea in 100mM Tris buffer (pH 8.0) and protein concentration was determined.
To prepare tryptic peptides for enrichment, axon lysate (4.7 mg) was diluted 2-fold with trifluoroethanol (TFE). The samples were then reduced by addition of 15 μl of 0.5M TCEP for 1 hr at room temperature, and alkylated with 30 μl of 500 mM iodoacetamide for 30 min in the dark at room temperature. Samples were diluted 10-fold with 100 mM Tris HCl, pH 8.0 and digested overnight at 37°C with proteomics-grade tr ypsin (Sigma) at a ratio of 1:60 enzyme to protein. The resulting peptides were then desalted by solid-phase extraction (Sep-pak C18 cartridges, Waters Corporation). Digested samples were first acidified with Trifluroacetic acid (TFA), diluted 2-fold with 0.1% TFA, and loaded onto the Sep-pak SPE material. After sample loading, the cartridges were washed with 0.1% TFA, and eluted with acetonitrile containing 0.1% TFA. Eight sequential 1 ml elutions were performed, increasing from 10% up to 80% acetonitrile, and eluates were reduced to dryness via vacuum centrifugation.
Acetylated peptide were enriched essentially as described earlier with minor modifications. Protein-A conjugated agarose beads immobilized with anti-acetyl lysine antibodies (ICP0388; ImmunoChem Pharmaceuticals Inc., Burnaby, British Columbia, Canada) were added at a ratio of 30 μl beads per 2 mg soluble protein resuspended in 1 ml final of NETN buffer (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, 0.5% NP40) and the mixture was incubated with rotation at 4°C overnight. The beads were washed fo ur times with 1 ml of NETN buffer and three times with ETN (50 mM Tris-HCl [pH 8.0], 100 mM NaCl, 1 mM EDTA). The bound peptides were eluted from the beads by washing three times with 100 μl of 0.1% TFA. The eluates were combined and dried in a SpeedVac.
LC-MS-MS analysis of the peptides was performed using a LTQ-Orbitrap Velos mass spectrometer (Thermo Scientific) equipped with a nanospray source and an Eksigent NanoLC and AS1 Autosampler. The peptides were loaded onto a self-packed biphasic C18/SCX MudPIT column using a Helium-pressurized cell (pressure bomb). The MudPIT column consisted of 360 μm × 150 μm i.d. fused silica, fritted with a filter-end fitting (IDEX Health & Science), and packed with 5cm of Luna SCX material (5 μm bead, Phenomenex) and 4 cm of Jupiter C18 material (5 μm bead, Phenomenex). After sample loading, the MudPIT column was connected using an M-520 microfilter union (IDEX Health & Science) to an analytical column (360 μm × 100 μm i.d.), equipped with a laser-pulled emitter tip. The analytical column was packed with 20cm Jupiter C18 material (3 μm bead, Phenomenex. Using the Eksigent NanoLC and Autosampler, MudPIT analysis was performed with an 8-step salt pulse gradient (0 mM, 50 mM, 100 mM, 150 mM, 200 mM, 300 mM, 500 mM, and 1 M ammonium acetate). Peptides were eluted from the analytical column after each salt pulse with a 105 min reverse gradient (2–40% acetonitrile, 0.1% formic) for the first 7 salt pulses, and a 2–95% acetonitrile for the last salt pulse. Gradient-eluted peptides were introduced into the mass spectrometer via nanoelectrospray ionization. Data were collected using a 17-scan event, data-dependent method. Full scan (m/z 350–2000) were acquired with the Orbitrap as the mass analyzer (resolution 60,000), and the 16 most abundant ions in each MS scan were selected for fragmentation in the LTQ Velos. An isolation width of 2 m/z, an activation time of 10 ms, and 35% normalized collision energy, a maximum injection time of 100 ms and an AGC target of 1×104 were used to generate MS/MS spectra. Dynamic exclusion was enabled, using a repeat count of 1, repeat duration of 10 sec, and an exclusion duration of 30sec.
For identification of acetylated peptides, raw data were extracted using ScanSifter and searched with SEQUEST (Thermo Fisher Scientific) against a Rattus norvegicus subset database created from the Uniprot KB protein database (www.uniprot.org). The protein database was a concatenated forward and reversed (decoy) database. Searches were configured to use variable modification of +57.0214 on Cys (carbamidomethylation), +15.9949 on Met (oxidation), and +42.0.056 on Lys (acetylation). Search results were assembled using Scaffold 3.0 (Proteome Software), where data were filtered to a protein and peptide threshold of 2% and 0.5% FDR respectively. Acetylated peptides of interest were validated via manual interrogation of the raw tandem mass spectra.
Immunoprecipitation and western blotting:
For p75NTR western blot, whole cell lysates from mCherry-p75NTR-GFP (wildtype or mutant) overexpressing Hek 293 cells were prepared in NP-40 lysis buffer (25 mM Tris (pH 7.4), 137 mM NaCl, 2.7 mM KCl, 1% Nonidet P-40, and 10% glycerol) supplemented with a Complete Mini EDTA-free protease inhibitor mixture tablet (Roche) and a PhosStop phosphatase inhibitor mixture tablet (Roche). Cell lysates were subjected to SDS-PAGE and western blot analysis using antibody against p75NTR ICD (1:1000).
For HDAC1 immunoblotting, primary neurons were cultured in cell culture inserts and axons were lysed in NP-40 lysis buffer (25 mM Tris (pH 7.4), 137 mM NaCl, 2.7 mM KCl, 1% Nonidet P-40, and 10% glycerol) supplemented with a Complete Mini EDTA-free protease inhibitor mixture tablet (Roche) and a PhosStop phosphatase inhibitor mixture tablet (Roche). Cell lysates were subjected to SDS-PAGE and Western blot analysis using antibodies against HDAC1 (1:2000, Millipore), Histone H3 (1:3000, Abcam), Tau (1:1000, Cell signaling technology).
For immunoprecipitation, neurons were grown in mass cultures as described above and treated as indicated in figure legends. After treatment, neurons were collected and suspended in Buffer A (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT and Complete Mini EDTA-free protease inhibitor mixture), then lysed in glass homogenizer. The nuclei were pelleted by centrifugation at 750×g for 5 min, then the supernatant was collected and centrifuged again to remove any remaining nuclei. The supernatant was considered the cytosolic fraction and was treated with a detergent solution to solubilize cytosolic proteins in RIPA buffer (150 mM NaCl, 50 mM Tris (pH 7.5), 1% NP-40, 0.5% deoxycholate, and Complete Mini EDTA-free protease inhibitor mixture). Transfected HEK293 cells were processed similarly to make cytosolic fractions and analyzed by immunoprecipitation and western blotting with the indicated antibodies.
The cytosolic solutions were immunoprecipitated with anti-acetyl lysine agarose beads (25 ul beads per mg of protein), anti-dynein intermediate chain antibody (1ul per 100 ug protein), anti-p150Glued antibody (1ul per 100 ug protein) or anti-myc antibody (1:1000) as indicated in figure legends. Western blotting was done using following antibodies as indicated: anti- p150Glued antibody (1:1000), anti-myc (1: 1000), anti-tubulin (1:1000) or anti-dynein intermediate chain antibody (1: 1000).
Quantification and Statistical Analysis
Statistical analyses
Number of experiments and sample sizes were as indicated in the figure legends. Data were collected randomly. Counts for analyzing neuronal apoptosis, p75NTR cleavage in distal axons and p75NTR co-localization with CD63 was done in a blinded manner such that the investigator was not aware of the treatment while counting. All graphs and statistical analyses were done using GraphPad Prism software. Student’s t tests were performed assuming Gaussian distribution as indicated. One-way or two-way ANOVA analyses were performed when more than two groups were compared. Statistical analyses were based on at least 3 independent experiments, and described in the figure legends. All error bars represent the standard error of the mean (s.e.m).
Supplementary Material
Highlights:
The intracellular domain (ICD) of p75NTR is a retrograde degenerative signal
Trophic factor deprivation induces regulated proteolysis of p75NTR in axons
HDAC1 is constitutively expressed in peripheral axons to regulate P75ICD transport
HDAC1 mediated p150Glued deacetylation promotes its interaction with dynein
Acknowledgements:
The authors thank members of the Carter lab for helpful discussions, Vanderbilt Mass Spectrometry Research Center Proteomics Core Facility for data acquisition on a LTQ Orbitrap Velos mass spectrometer funded by the NIH (S10RR027714). We acknowledge help from Michael Mercier for construct characterization. We are also grateful to the Cell Imaging Shared Resource at Vanderbilt University (supported by NIH grants CA68485, DK20593, DK58404, DK59637 and EY08126). This work was supported by a grant from Knights Templar Eye Foundation to AP and grants from the NIH (R01 NS038220 and R01 NS102365 to BDC, R35 NS097976 to KA, R01 EY024373 to SF and R37 NS42925 to PC). The authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests: The authors declare they have no competing interests.
References:
- von Bartheld CS, Kinoshita Y, Prevette D, Yin Q-W, Oppenheim RW, and Bothwell M (1994). Positive and negative effects of neurotrophins on the isthmo-optic nucleus in chick embryos. Neuron 12, 639–654. [DOI] [PubMed] [Google Scholar]
- Bertrand P (2010). Inside HDAC with HDAC inhibitors. Eur. J. Med. Chem 45, 2095–2116. [DOI] [PubMed] [Google Scholar]
- Butowt R, and Bartheld CS von (2009). Fates of Neurotrophins after Retrograde Axonal Transport: Phosphorylation of p75NTR Is a Sorting Signal for Delayed Degradation. J. Neurosci 29, 10715–10729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AP, Diamant AG, and Urnavicius L (2016). How dynein and dynactin transport cargos: a structural perspective. Curr. Opin. Struct. Biol 37, 62–70. [DOI] [PubMed] [Google Scholar]
- Ceni C, Unsain N, Zeinieh MP, and Barker PA (2014). Neurotrophins in the regulation of cellular survival and death. Handb. Exp. Pharmacol 220, 193–221. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen ES, Wallace KE, Pennise CR, and Holzbaur ELF (2008). Lysosomal proliferation and distal degeneration in motor neurons expressing the G59S mutation in the p150Glued subunit of dynactin. Hum. Mol. Genet 17, 1946–1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittka A, Arevalo JC, Rodriguez-Guzman M, Pérez P, Chao MV, and Sendtner M (2004). The p75NTR-interacting protein SC1 inhibits cell cycle progression by transcriptional repression of cyclin E. J. Cell Biol 164, 985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho Y, and Cavalli V (2014). HDAC signaling in neuronal development and axon regeneration. Curr. Opin. Neurobiol 27, 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deckwerth TL, and Johnson EM (1993). Temporal analysis of events associated with programmed cell death (apoptosis) of sympathetic neurons deprived of nerve growth factor. J. Cell Biol 123, 1207–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deinhardt K, and Chao MV (2014). Trk Receptors In Neurotrophic Factors, Lewin GR, and Carter BD, eds. (Springer; Berlin Heidelberg: ), pp. 103–119. [Google Scholar]
- Deinhardt K, Reversi A, Berninghausen O, Hopkins CR, and Schiavo G (2007). Neurotrophins Redirect p75NTR from a Clathrin-Independent to a Clathrin-Dependent Endocytic Pathway Coupled to Axonal Transport. Traffic 8, 1736–1749. [DOI] [PubMed] [Google Scholar]
- Deppmann CD, Mihalas S, Sharma N, Lonze BE, Niebur E, and Ginty DD (2008). A Model for Neuronal Competition During Development. Science 320, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emiliani S, Fischle W, Van Lint C, Al-Abed Y, and Verdin E (1998). Characterization of a human RPD3 ortholog, HDAC3. Proc. Natl. Acad. Sci. U. S. A 95, 2795–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escudero CA, Lazo OM, Galleguillos C, Parraguez JI, Lopez-Verrilli MA, Cabeza C, Leon L, Saeed U, Retamal C, Gonzalez A, et al. (2014). The p75 neurotrophin receptor evades the endolysosomal route in neuronal cells, favouring multivesicular bodies specialised for exosomal release. J. Cell Sci 127, 1966–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Firestone AJ, Weinger JS, Maldonado M, Barlan K, Langston LD, O’Donnell M, Gelfand VI, Kapoor TM, and Chen JK (2012). Small-molecule inhibitors of the AAA+ ATPase motor cytoplasmic dynein. Nature 484, 125–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto H, Higuchi M, Koike M, Ode H, Pinak M, Bunta JK, Nemoto T, Sakudoh T, Honda N, Maekawa H, et al. (2012). A possible overestimation of the effect of acetylation on lysine residues in KQ mutant analysis. J. Comput. Chem 33, 239–246. [DOI] [PubMed] [Google Scholar]
- Ghosh AS, Wang B, Pozniak CD, Chen M, Watts RJ, and Lewcock JW (2011). DLK induces developmental neuronal degeneration via selective regulation of proapoptotic JNK activity. J. Cell Biol 194, 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haberland M, Montgomery RL, and Olson EN (2009). The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet 10, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington AW, and Ginty DD (2013). Long-distance retrograde neurotrophic factor signalling in neurons. Nat. Rev. Neurosci 14, 177–187. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, and Chao MV (1991). High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature 350, 678–683. [DOI] [PubMed] [Google Scholar]
- Hibbert AP, Kramer BMR, Miller FD, and Kaplan DR (2006). The localization, trafficking and retrograde transport of BDNF bound to p75NTR in sympathetic neurons. Mol. Cell. Neurosci 32, 387–402. [DOI] [PubMed] [Google Scholar]
- Holland SM, Collura KM, Ketschek A, Noma K, Ferguson TA, Jin Y, Gallo G, and Thomas GM (2016). Palmitoylation controls DLK localization, interactions and activity to ensure effective axonal injury signaling. Proc. Natl. Acad. Sci 113, 763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu E, Dul E, Sung C-M, Chen Z, Kirkpatrick R, Zhang G-F, Johanson K, Liu R, Lago A, Hofmann G, et al. (2003). Identification of Novel Isoform-Selective Inhibitors within Class I Histone Deacetylases. J. Pharmacol. Exp. Ther 307, 720–728. [DOI] [PubMed] [Google Scholar]
- Jung K-M, Tan S, Landman N, Petrova K, Murray S, Lewis R, Kim PK, Kim DS, Ryu SH, Chao MV, et al. (2003). Regulated Intramembrane Proteolysis of the p75 Neurotrophin Receptor Modulates Its Association with the TrkA Receptor. J. Biol. Chem 278, 42161–42169. [DOI] [PubMed] [Google Scholar]
- Kanning KC, Hudson M, Amieux PS, Wiley JC, Bothwell M, and Schecterson LC (2003). Proteolytic Processing of the p75 Neurotrophin Receptor and Two Homologs Generates C-Terminal Fragments with Signaling Capability. J. Neurosci 23, 5425–5436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki S, and Holzbaur ELF (1995). Affinity Chromatography Demonstrates a Direct Binding between Cytoplasmic Dynein and the Dynactin Complex. J. Biol. Chem 270, 28806–28811. [DOI] [PubMed] [Google Scholar]
- Kenchappa RS, Zampieri N, Chao MV, Barker PA, Teng HK, Hempstead BL, and Carter BD (2006). Ligand-Dependent Cleavage of the P75 Neurotrophin Receptor Is Necessary for NRIF Nuclear Translocation and Apoptosis in Sympathetic Neurons. Neuron 50, 219–232. [DOI] [PubMed] [Google Scholar]
- Kim JY, Shen S, Dietz K, He Y, Howell O, Reynolds R, and Casaccia P (2010). HDAC1 nuclear export induced by pathological conditions is essential for the onset of axonal damage. Nat. Neurosci 13, 180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SJ, and Schroer TA (2000). Dynactin increases the processivity of the cytoplasmic dynein motor. Nat. Cell Biol 2, 20–24. [DOI] [PubMed] [Google Scholar]
- King SJ, Brown CL, Maier KC, Quintyne NJ, and Schroer TA (2003). Analysis of the Dynein-Dynactin Interaction In Vitro and In Vivo. Mol. Biol. Cell 14, 5089–5097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhausen T, Macia E, and Pelish HE (2008). Use of Dynasore, the Small Molecule Inhibitor of Dynamin, in the Regulation of Endocytosis In Methods in Enzymology, D. CJ and William AH Balch E, ed. (Academic Press; ), pp. 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer BR, Yoon SO, and Carter BD (2014). The biological functions and signaling mechanisms of the p75 neurotrophin receptor. Handb. Exp. Pharmacol 220, 121–164. [DOI] [PubMed] [Google Scholar]
- Macia E, Ehrlich M, Massol R, Boucrot E, Brunner C, and Kirchhausen T (2006). Dynasore, a Cell-Permeable Inhibitor of Dynamin. Dev. Cell 10, 839–850. [DOI] [PubMed] [Google Scholar]
- Maday S, Twelvetrees AE, Moughamian AJ, and Holzbaur ELF (2014). Axonal Transport: Cargo-Specific Mechanisms of Motility and Regulation. Neuron 84, 292–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdan M, Lachance C, Gloster A, Aloyz R, Zeindler C, Bamji S, Bhakar A, Belliveau D, Fawcett J, Miller FD, et al. (1997). Transgenic Mice Expressing the Intracellular Domain of the p75 Neurotrophin Receptor Undergo Neuronal Apoptosis. J. Neurosci 17, 6988–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdan M, Walsh GS, Aloyz R, and Miller FD (2001). TrkA mediates developmental sympathetic neuron survival in vivo by silencing an ongoing p75NTR-mediated death signal. J. Cell Biol 155, 1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenney RJ, Huynh W, Tanenbaum ME, Bhabha G, and Vale RD (2014). Activation of cytoplasmic dynein motility by dynactin-cargo adapter complexes. Science 345, 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mobley BC, Kwon M, Kraemer BR, Hickman FE, Qiao J, Chung DH, and Carter BD (2015). Expression of MYCN in Multipotent Sympathoadrenal Progenitors Induces Proliferation and Neural Differentiation, but Is Not Sufficient for Tumorigenesis. PLOS ONE 10, e0133897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mok S-A, Lund K, and Campenot RB (2009). A retrograde apoptotic signal originating in NGF-deprived distal axons of rat sympathetic neurons in compartmented cultures. Cell Res. 19, 546–560. [DOI] [PubMed] [Google Scholar]
- Murphy G (2008). The ADAMs: signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 8, 932–941. [DOI] [PubMed] [Google Scholar]
- Nikoletopoulou V, Lickert H, Frade JM, Rencurel C, Giallonardo P, Zhang L, Bibel M, and Barde Y-A (2010). Neurotrophin receptors TrkA and TrkC cause neuronal death whereas TrkB does not. Nature 467, 59–63. [DOI] [PubMed] [Google Scholar]
- Palmada M, Kanwal S, Rutkoski NJ, Gustafson-Brown C, Johnson RS, Wisdom R, and Carter BD (2002). c-jun is essential for sympathetic neuronal death induced by NGF withdrawal but not by p75 activation. J. Cell Biol 158, 453–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Jeong G-B, Ross JL, Dixit R, Wallace KE, Kalb RG, and Holzbaur ELF (2009). A Switch in Retrograde Signaling from Survival to Stress in Rapid-Onset Neurodegeneration. J. Neurosci 29, 9903–9917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlson E, Maday S, Fu M, Moughamian AJ, and Holzbaur ELF (2010). Retrograde axonal transport: pathways to cell death? Trends Neurosci. 33, 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schachtrup C, Ryu JK, Mammadzada K, Khan AS, Carlton PM, Perez A, Christian F, Le Moan N, Vagena E, Baeza-Raja B, et al. (2015). Nuclear pore complex remodeling by p75NTR cleavage controls TGF-β signaling and astrocyte functions. Nat. Neurosci 18, 1077–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. (2012). Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroer TA (2004). Dynactin. Annu. Rev. Cell Dev. Biol 20, 759–779. [DOI] [PubMed] [Google Scholar]
- Scott-Solomon E, and Kuruvilla R Mechanisms of neurotrophin trafficking via Trk receptors. Mol. Cell. Neurosci [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon DJ, Pitts J, Hertz NT, Yang J, Yamagishi Y, Olsen O, Tešić Mark M, Molina H, and Tessier-Lavigne M (2016). Axon Degeneration Gated by Retrograde Activation of Somatic Pro-apoptotic Signaling. Cell 164, 1031–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skeldal S, Sykes AM, Glerup S, Matusica D, Palstra N, Autio H, Boskovic Z, Madsen P, Castrén E, Nykjaer A, et al. (2012). Mapping of the Interaction Site between Sortilin and the p75 Neurotrophin Receptor Reveals a Regulatory Role for the Sortilin Intracellular Domain in p75 Neurotrophin Receptor Shedding and Apoptosis. J. Biol. Chem 287, 43798–43809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor AR, Gifondorwa DJ, Robinson MB, Strupe JL, Prevette D, Johnson JE, Hempstead B, Oppenheim RW, and Milligan CE (2012). Motoneuron programmed cell death in response to proBDNF. Dev. Neurobiol 72, 699–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Underwood CK, Reid K, May LM, Bartlett PF, and Coulson EJ (2008). Palmitoylation of the C-terminal fragment of p75NTR regulates death signaling and is required for subsequent cleavage by γ-secretase. Mol. Cell. Neurosci 37, 346–358. [DOI] [PubMed] [Google Scholar]
- Urnavicius L, Zhang K, Diamant AG, Motz C, Schlager MA, Yu M, Patel NA, Robinson CV, and Carter AP (2015). The structure of the dynactin complex and its interaction with dynein. Science 347, 1441–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urra S, Escudero CA, Ramos P, Lisbona F, Allende E, Covarrubias P, Parraguez JI, Zampieri N, Chao MV, Annaert W, et al. (2007). TrkA Receptor Activation by Nerve Growth Factor Induces Shedding of the p75 Neurotrophin Receptor Followed by Endosomal γ-Secretase-mediated Release of the p75 Intracellular Domain. J. Biol. Chem 282, 7606–7615. [DOI] [PubMed] [Google Scholar]
- Vicario A, Kisiswa L, Tann JY, Kelly CE, and Ibáñez CF (2015). Neuron-type-specific signaling by the p75NTR death receptor is regulated by differential proteolytic cleavage. J. Cell Sci 128, 1507–1517. [DOI] [PubMed] [Google Scholar]
- Wu C, Cui B, He L, Chen L, and Mobley WC (2009). The coming of age of axonal neurotrophin signaling endosomes. J. Proteomics 72, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, and Greenberg ME (1995). Opposing Effects of ERK and JNK-p38 MAP Kinases on Apoptosis. Science 270, 1326–1331. [DOI] [PubMed] [Google Scholar]
- Yano H, Torkin R, Martin LA, Chao MV, and Teng KK (2009). Proneurotrophin-3 Is a Neuronal Apoptotic Ligand: Evidence for Retrograde-Directed Cell Killing. J. Neurosci 29, 14790–14802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- d’Ydewalle C, Krishnan J, Chiheb DM, Van Damme P, Irobi J, Kozikowski AP, Berghe PV, Timmerman V, Robberecht W, and Van Den Bosch L (2011). HDAC6 inhibitors reverse axonal loss in a mouse model of mutant HSPB1-induced Charcot-Marie-Tooth disease. Nat. Med 17, 968–974. [DOI] [PubMed] [Google Scholar]
- Ye M, Lehigh KM, and Ginty DD (2018). Multivesicular bodies mediate long-range retrograde NGF-TrkA signaling. eLife 7, e33012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J-Q, Kelly TK, Chang B, Ryazantsev S, Rajasekaran AK, Martin KC, and Twiss JL (2001). A Functional Role for Intra-Axonal Protein Synthesis during Axonal Regeneration from Adult Sensory Neurons. J. Neurosci 21, 9291–9303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Vidaurre OG, Adula KP, Kezunovic N, Wentling M, Huntley GW, and Casaccia P (2017). Subcellular Distribution of HDAC1 in Neurotoxic Conditions Is Dependent on Serine Phosphorylation. J. Neurosci 37, 7547–7559. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.