

Abstract
Adaptations and responses to stress conditions are fundamental processes that all cells must accomplish to maintain or restore cellular homeostasis. Cells have a plethora of response pathways to mitigate the effect of different environmental stressors. The transcriptional regulators transcription factor EB (TFEB) and transcription factor binding to IGHM enhancer 3 (TFE3) play a key role in the control of these stress pathways. Therefore, understanding their regulation under different stress conditions is of great interest. Here, using a range of human and murine cells, we show that TFEB and TFE3 are activated upon induction of acute oxidative stress by sodium arsenite via an mTOR complex 1 (mTORC1)-independent process. We found that the mechanism of arsenite-stimulated TFEB and TFE3 activation instead involves protein phosphatase 2A (PP2A)-mediated dephosphorylation at Ser-211 and Ser-321, respectively. Depletion of either the catalytic (PPP2CA+B) or regulatory (PPP2R2A/B55α) subunits of PP2A, as well as PP2A inactivation with the specific inhibitor okadaic acid, abolished TFEB and TFE3 activation in response to sodium arsenite. Conversely, PP2A activation by ceramide or the sphingosine-like compound FTY720 was sufficient to induce TFE3 nuclear translocation. MS analysis revealed that PP2A dephosphorylates TFEB at several residues, including Ser-109, Ser-114, Ser-122, and Ser-211, thus facilitating TFEB activation. Overall, this work identifies a critical mechanism that activates TFEB and TFE3 without turning off mTORC1 activity. We propose that this mechanism may enable some cell types such as immune or cancer cells that require simultaneous TFEB/TFE3 and mTORC1 signaling to survive and achieve robust cell growth in stressful environments.
Keywords: transcription factor, phosphatase, protein phosphatase 2 (PP2A), oxidative stress, nuclear translocation, mTORC1, okadaic acid, sodium arsenite, transcription factor binding to IGHM enhancer 3, transcription factor EB
Introduction
Eukaryotic cells must respond to fluctuations in a variety of environmental conditions. When those fluctuations exceed a particular threshold, cells activate different stress signaling pathways with the goal of preventing damage and restoring homeostasis.
TFEB2 and TFE3 have recently emerged as critical mediators of the cellular response to stress (1). TFEB and TFE3 belong to the MITF/TFE family of basic helix-loop-helix/leucine zipper transcription factors and share the ability to bind CLEAR motifs located in the promoter region of a variety of lysosomal and autophagic genes (2–4). The role of TFEB and TFE3 in nutrient sensing and energy homeostasis is well established. Under nutrient-rich conditions, TFEB and TFE3 are recruited to the lysosomal surface via direct interaction with the small GTPases Rags (4, 5). Once on the lysosomal surface, TFEB and TFE3 are phosphorylated by the Ser/Thr kinase mTORC1 at several residues (6–8). mTORC1-dependent phosphorylation of TFEB at Ser-211 or TFE3 at Ser-321 is particularly relevant because it promotes binding of TFEB and TFE3 to 14-3-3, leading to their retention in the cytosol (6, 7). Amino acids or serum deprivation causes mTORC1 inactivation, resulting in the dissociation of the TFEB and TFE3/14-3-3 complexes and the rapid accumulation of these transcription factors in the nucleus (6, 7). Once in the nucleus, TFEB and TFE3 induce lysosomal biogenesis and autophagy, and stimulate expression of metabolic regulators, thus helping cells to adapt and survive nutrient withdrawal (3, 4).
Several recent reports have shown that TFEB and TFE3 respond not only to changes in nutrient levels but also to a wide variety of internal and external stressors, including mitochondrial damage (9), accumulation of unfolded proteins in the ER (10), pathogens (11–14), and physical exercise (15, 16). Interestingly, many of these stressors do not cause an obvious mTORC1 inactivation, suggesting that additional, mTORC1-independent mechanisms to promote TFEB and TFE3 activation might exist.
Another open question is the identity of the phosphatase in charge of dephosphorylating key regulatory residues, such as TFEB-S211 and TFE3-S321. Recent evidence suggests that PPP3/calcineurin promotes TFEB-S211 dephosphorylation in response to starvation and some types of oxidative stress (17, 18). However, calcineurin inhibition does not completely prevent TFEB and TFE3 activation in response to ER stress or lipopolysaccharide treatment (10, 14), suggesting that other phosphatases may be involved in TFEB and TFE3 regulation. Also, it has recently been shown that TFEB activation by digoxin is independent of the activity of PPP3/calcineurin, further supporting the existence of uncharacterized TFEB/TFE3 phosphatases (19).
In this study, we assessed the mechanism of TFEB and TFE3 activation in response to sodium arsenite. Arsenic is a potent toxicant and carcinogen. Epidemiological studies have reported an association between arsenic exposure and increased risks of various cancers as well as noncancerous diseases, including diabetes, hypertension, cardiovascular and respiratory diseases, and neurotoxicity. Arsenic perturbs mitochondrial membrane integrity and potential, leading to the release of reactive oxygen species (ROS) from mitochondria and causing DNA damage, impaired protein degradation, ER stress, autophagic cell death, and inflammation (20–24). We found that incubation of different cell types with sodium arsenite resulted in decreased phosphorylation of TFEB-S211 and TFE3-S321 and consequent TFEB and TFE3 nuclear accumulation. TFEB and TFE3 activation occurred despite the robust increase in mTORC1 activity observed under oxidative stress conditions, suggesting an mTORC1-independent process. Importantly, catalytic inhibition or depletion of either the catalytic or regulatory subunits of the PP2A phosphatase was sufficient to prevent TFEB-S211 and TFE3-S321 dephosphorylation in response to arsenite. Mass spectrometry analysis revealed that PP2A dephosphorylates several residues in TFEB, including Ser-109, Ser-114, Ser-122, and Ser-211. Therefore, we propose that the activation of the PP2A phosphatase toward TFEB and TFE3 represents a novel mechanism to induce the cellular stress response without turning off mTOR activity, allowing cells to fine-tune growth and survival.
Results
TFE3 and TFEB are activated in response to sodium arsenite in an mTORC1-independent manner
Cellular adaptation and response to stress under adverse conditions is the key to survival. To gain insight into the cellular machinery involved in TFE3 and TFEB regulation, we investigated the mechanism of TFEB and TFE3 activation under oxidative stress. Treatment of ARPE19 cells with sodium arsenite (NaAsO2), a ROS inducer, caused significant accumulation of endogenous TFE3 into the nucleus (Fig. 1A). Quantification of several independent experiments demonstrated that TFE3 accumulated in the nucleus in ∼75% (76.4 ± 2.9%) of cells after NaAsO2 treatment, whereas only 5% (4.8 ± 0.2%) of cells showed TFE3 nuclear staining in control conditions (Fig. 1B). We have previously shown that dephosphorylation of TFE3-S321 is required for dissociation of the TFE3/14-3-3 complex and subsequent TFE3 nuclear translocation (4, 10, 14). Accordingly, TFE3-S321 dephosphorylation was already observed after 30 min incubation of ARPE19 cells with NaAsO2, and the effect was more robust after a 2-h treatment (Fig. 1, C and D). TFE3 dephosphorylation was also assessed by changes in TFE3 electrophoretic mobility (Fig. 1D, lower panel). Moreover, TFE3 activation upon NaAsO2 treatment was observed in various cell types, including mouse embryonic fibroblasts (MEFs), HeLa, and murine macrophage Raw 264.7 (Fig. S1, A–C). Incubation in starvation medium without serum and amino acids (EBSS), or treatment with the mTOR catalytic inhibitor Torin-1, was used as a positive control for efficient TFE3-S321 dephosphorylation (Fig. 1D and Fig. S1, A–C).
Figure 1.
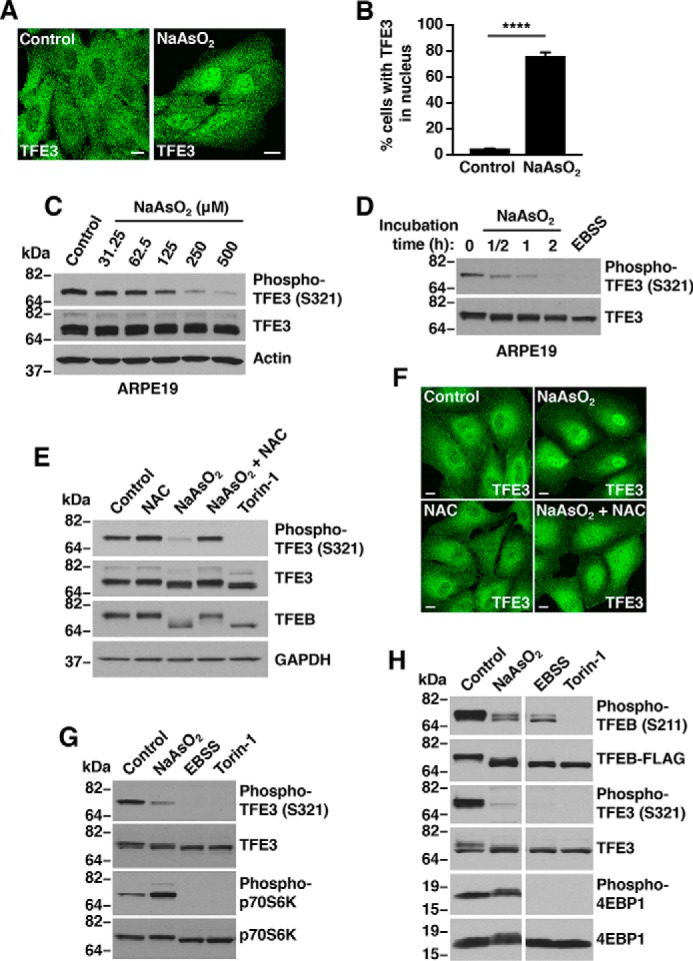
Sodium arsenite activates TFE3 and TFEB independent of mTORC1 activity. A, ARPE19 cells treated with either vehicle or 250 μm NaAsO2 for 1 h were fixed, permeabilized, and immunostained with antibodies against TFE3. Scale bar, 10 μm. B, quantification of cells with TFE3 in nucleus in vehicle- or NaAsO2-treated ARPE19 cells as shown in A. Vehicle, n = 810; NaAsO2, n = 846 from three independent experiments. Error bars denote S.D. p value calculated using two-tailed t test, (****) p < 0.0001. C, immunoblot analysis of protein lysates from ARPE19 cells treated with the indicated concentrations of NaAsO2 for 1 h. D, immunoblot analysis of protein lysates from ARPE19 cells treated with either 250 μm NaAsO2 for the indicated times or EBSS for 1 h. E, immunoblot analysis of protein lysates from ARPE19 cells treated with either 250 μm NaAsO2 in the presence of 15 mm NAC or 250 nm Torin-1 for 1 h. F, ARPE19 cells treated with either vehicle or 250 μm NaAsO2 in the presence of 15 mm NAC for 1 h were fixed, permeabilized, and immunostained with antibodies against TFE3. Scale bar, 10 μm. G, immunoblot analysis of protein lysates from ARPE19 cells and treated with either 250 μm NaAsO2, 250 nm Torin-1 or EBSS for 1 h. H, immunoblot analysis of protein lysates from HeLa cells expressing TFEB-FLAG and treated as indicated in G. Samples were run on the same gel (irrelevant lanes were spliced out; see raw data). Immunoblots are representative of three independent experiments.
Arsenite affects different cellular pathways causing not only oxidative stress but also mitochondria dysfunction, impaired protein degradation, DNA damage, and ER stress (Fig. S2). To determine whether TFE3 and TFEB were activated by oxidative stress or by alteration in cellular compartments, we assessed the effect of the antioxidant N-acetyl-l-cysteine (NAC), a nonselective oxygen-free radical scavenger that confers protection to cells against ROS (25). Pretreatment of ARPE19 cells with NAC prevented TFE3 and TFEB dephosphorylation (Fig. 1E), as well as TFE3 nuclear accumulation (Fig. 1F) in NaAsO2-treated cells, confirming that NaAsO2 induces TFE3 and TFEB activation via ROS generation.
Next, we assessed whether the activation of TFE3 observed in NaAsO2-treated cells was due to reduced mTORC1 activity. ARPE19 and HeLa (CF7) cells were treated with NaAsO2, Torin-1, or starved, and mTORC1 activity was measured by monitoring the phosphorylation status of p70S6 kinase and 4EBP1, two downstream effectors of mTORC1. As expected, starvation and Torin-1 resulted in strong mTORC1 inactivation (Fig. 1, G and H). In contrast, dephosphorylation of TFE3-S321 and TFEB-S211 in NaAsO2-treated cells occurred despite the strongly increased mTORC1 activity observed under these conditions (Fig. 1, G and H). Altogether, our results indicate that NaAsO2 treatment induces TFE3 and TFEB activation and that this process does not require mTORC1 inactivation.
Okadaic acid prevents arsenite-induced TFE3 and TFEB activation
It has been recently suggested that activation of the lysosomal TRPML1 channel by nutrient deprivation and certain ROS-generating conditions promotes release of lysosomal Ca2+, thus resulting in activation of the phosphatase PPP3/calcineurin and concomitant TFEB dephosphorylation (17, 18). Given these observations, we sought to investigate whether PPP3/calcineurin and TRPML1 regulate TFE3 and TFEB activation in response to oxidative stress induced by NaAsO2. As shown in Fig. 2A, pretreatment of ARPE19 cells with FK506, a potent immunosuppressive drug commonly used to inhibit PPP3/calcineurin activity (26), did not noticeably alter NaAsO2-dependent dephosphorylation of endogenous TFE3 (Fig. 2A). Moreover, depletion of PPP3 catalytic subunits (PPP3CA+B) did not prevent TFE3 dephosphorylation in NaAsO2-treated cells (Fig. S3). Likewise, the degree of TFE3-S321 dephosphorylation following NaAsO2 treatment was comparable between TRPML1-deficient human fibroblasts (from mucolipidosis type IV patients; MLIV) and fibroblasts from healthy individuals (Fig. 2B). These results indicate that neither PPP3/calcineurin nor TRPML1 are required for TFE3 and TFEB activation in NaAsO2-treated cells and suggest that other factors might participate in the process.
Figure 2.
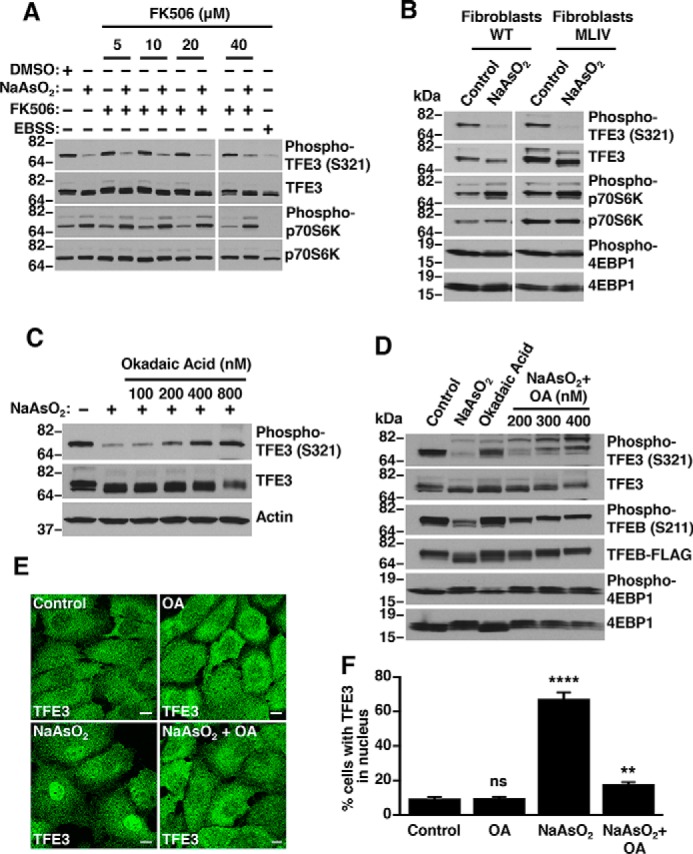
Sodium arsenite-induced TFE3 and TFEB activation is inhibited by okadaic acid. A, immunoblot analysis of protein lysates from ARPE19 cells treated with 250 μm NaAsO2 in the presence of the indicated concentration of Calcineurin inhibitor FK506 for 1 h. B, immunoblot analysis of protein lysates from a MLIV patient and unrelated nondiseased fibroblasts treated with 250 μm NaAsO2 for 1 h. C and D, immunoblot analysis of protein lysates from ARPE19 cells and HeLa (CF7) cells treated with 250 μm NaAsO2 in the presence of the indicated concentration of okadaic acid for 1 h. E, ARPE19 cells treated with either vehicle or 250 μm NaAsO2 for 1 h in the presence of 400 nm okadaic acid were fixed, permeabilized, and immunostained with antibodies against TFE3. Scale bar, 10 μm. F, quantification of cells with TFE3 in nucleus in either vehicle or NaAsO2 and okadaic acid-treated ARPE19 cells as shown in E. Vehicle, n = 517; OA, n = 556 NaAsO2, n = 524; and NaAsO2 + OA, n = 548 from three independent experiments. Error bars denote S.D. p value was calculated using one-way ANOVA, (**) p < 0.01, (****) p < 0.0001. Immunoblots are representative of three independent experiments.
To evaluate this possibility, we looked at other protein phosphatases, which may be involved in TFE3 and TFEB activation during oxidative stress. Okadaic acid (OA) is a potent pharmacological inhibitor of multiple protein serine/threonine phosphatases (27). OA is widely used to study the function of protein phosphatase 2A (PP2A) and protein phosphatase 1 (PPP1), which are the most abundant and ubiquitously expressed phosphatases in cells (27). As seen in Fig. 2, C and D, pretreatment of ARPE19 and HeLa (CF7) cells with OA blocked NaAsO2-induced dephosphorylation of TFE3-S321 and TFEB-S211. Furthermore, whereas the subcellular distribution of endogenous TFE3 in OA-treated ARPE19 cells was similar to that in the control conditions, pretreatment with OA significantly diminished TFE3 nuclear accumulation induced by NaAsO2 (Fig. 2, E and F). Consequently, our results suggest that PP2A and/or PPP1 participate in TFE3 and TFEB activation upon NaAsO2 treatment.
PP2A but not PPP1 dephosphorylates TFE3 at Ser-321
PP2A functions as a trimeric complex formed by a catalytic C subunit (PPP2C), a scaffold subunit (PPP2R1), and a regulatory subunit (PPP2R2), which confers specificity of PP2A for selective substrates (28, 29) (Fig. S4). In addition, two PP2A catalytic isoforms (PPP2CA and PPP2CB) have been described. Interestingly, we found that depletion of either the PP2A catalytic or scaffolding subunits significantly inhibited the dephosphorylation of TFE3 at Ser-321 (Fig. 3, A and B). In contrast, knockdown of the PPP1 catalytic subunits did not have an effect, despite the efficient depletion of PPP1 catalytic subunits PPP1CA and PPP1CB (Fig. 3, A and B).
Figure 3.
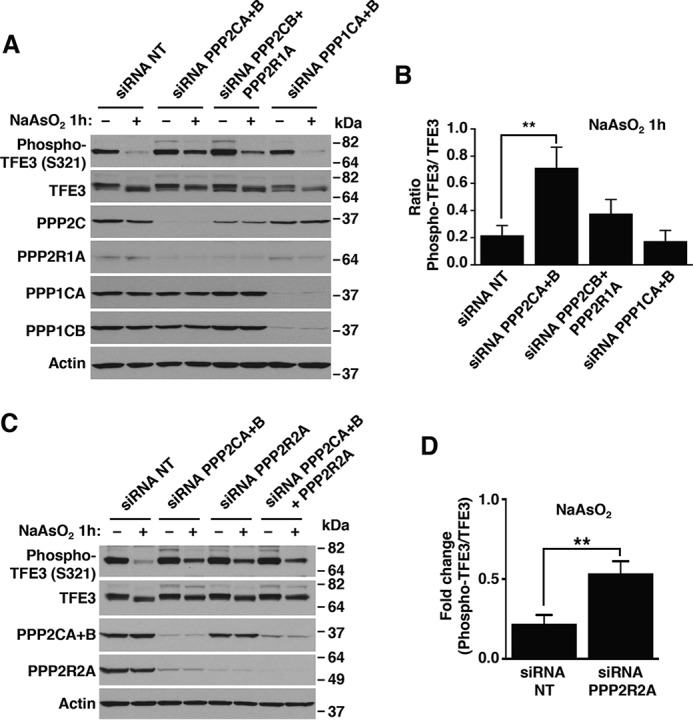
TFE3 is a substrate of protein phosphatase 2A but not protein phosphatase 1A. A, immunoblot analysis of protein lysates from ARPE19 cells depleted of either PPP2A or PPP1A and treated with 250 μm NaAsO2 for 1 h. B, quantification of phospho-TFE3/TFE3 ratios from ARPE19 cells depleted of either PPP2A or PPP1A and treated with 250 μm NaAsO2 for 1 h, as shown in A. Quantified results are fold-change of phospho-TFE3/TFE3 from its corresponding vehicle-treated cells. Data are presented as mean ± S.D. using one-way ANOVA, **, p < 0.01, from three independent experiments. C, immunoblot analysis of protein lysates from ARPE19 cells depleted of either PPP2A catalytic and regulatory subunits and treated with 250 μm NaAsO2 for 1 h. D, quantification of phospho-TFE3/TFE3 ratios from ARPE19 cells depleted of the PPP2A regulatory subunit and treated with 250 μm NaAsO2 for 1 h, as shown in C. Quantified results are fold-change of phospho-TFE3/TFE3 from its corresponding vehicle-treated cells. Data are presented as mean ± S.D. using two-tailed t test; **, p < 0.01, from three independent experiments. Immunoblots are representative of three independent experiments.
The subcellular location and substrate specificity of PP2A is determined by its ability to interact with specific PPP2R2 regulatory subunits to form a heterotrimeric holoenzyme. In humans, at least 26 different PPP2R2 subtypes have been identified, implying the existence of different PP2A holoenzymes with distinctive biological functions. The regulatory subunit PPP2R2A (also known as B55α) has recently been implicated in the cellular response to ROS (30, 31). Interestingly, the depletion of PP2A catalytic subunits led to a reduction in the protein levels of all the components of the PP2A complex. This observation is in agreement with previous reports suggesting unstability and rapid degradation of PP2A subunits (32–34). Accordingly, we found that depletion of PPP2R2A, alone or in combination with the catalytic subunits PPP2CA+B, significantly reduced TFE3-S321 dephosphorylation following NaAsO2 treatment (Fig. 3, C and D). These results suggest that the regulatory subunit PPP2R2A is required for PP2A activity toward TFE3-S321.
Next we assessed whether PP2A activation was sufficient to induce TFE3 dephosphorylation. The immunosuppressant FTY720, a fungal metabolite structurally similar to sphingosine, is a known activator of PP2A (35). In vivo, FTY720 is phosphorylated by the enzyme sphingosine kinase 2 to form FTY720-phosphate, which resembles sphingosine 1-phosphate (36). FTY720 and its phosphorylated form have been used to activate PP2A both in vivo and in vitro (37). As seen in Fig. 4A, treatment of ARPE19 cells with FTY720 resulted in a concentration-dependent dephosphorylation of TFE3-S321, suggesting that PP2A activation is sufficient to induce TFE3 activation. As control, we measured the levels of phospho-p70S6K to confirm that FTY720 was not causing mTORC1 inactivation (Fig. 3A). Ceramide is another well-known activator of PP2A. Ceramide directly binds the PP2A inhibitor I2PP2A/SET, relieving PP2A from SET and increasing PP2A activity (38, 39). Notably, treatment of ARPE19 cells with a cell-permeable ceramide analog resulted in TFE3 nuclear accumulation (Fig. 4B) and concomitant TFE3-S321 dephosphorylation (Fig. 4C). Altogether, our results suggest that PP2A, but not PPP1 or PPP3, is required for TFE3 activation in response to NaAsO2.
Figure 4.
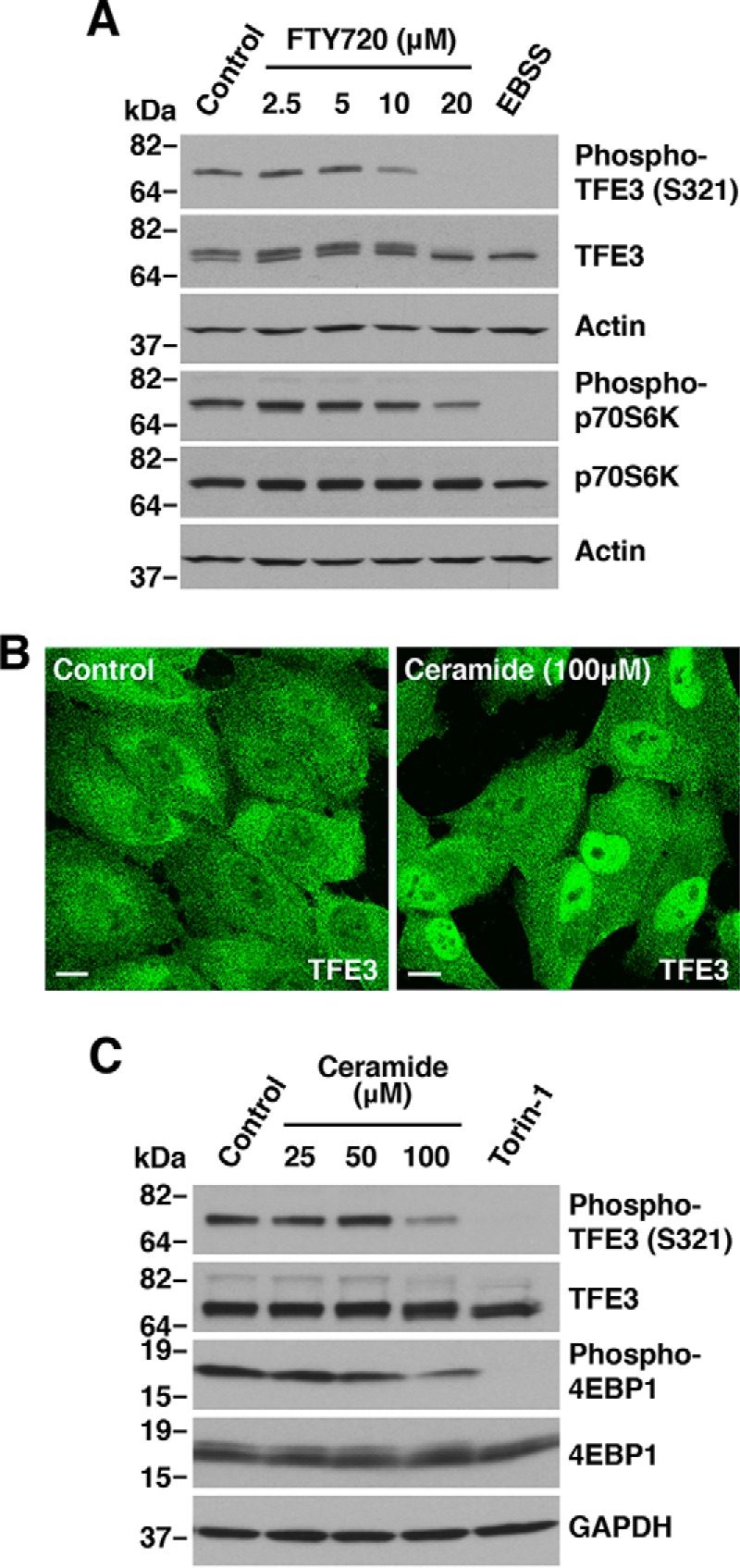
Protein phosphatase 2A dephosphorylation of TFE3 and TFEB. A, immunoblot analysis of protein lysates from ARPE19 cells treated with the indicated concentrations of FTY720 for 1 h. B, ARPE19 cells treated with vehicle or 100 μm C2-ceramide for 2 h were fixed, permeabilized, and immunostained with antibodies against TFE3. Scale bar, 10 μm. C, immunoblot analysis of protein lysates from ARPE19 cells treated with the indicated concentrations of C2-ceramide or 250 nm Torin-1 for 2 h. Immunoblots are representative of three independent experiments.
PP2A dephosphorylates TFE3 and TFEB in vitro
To further investigate the role of PP2A in the regulation of TFE3 and TFEB, we used an in vitro phosphatase assay in which purified substrates TFEB-FLAG and TFE3-FLAG were incubated with PP2A from cell lysates or with the recombinant purified enzyme (Fig. S5). In agreement with our results showing that pharmacological and genetic inhibition of PP2A affect the activation of TFE3 and TFEB under oxidative stress, we found that OA addition to the dephosphorylation reaction (Fig. 5A), or cell treatment with OA prior to the assay (Fig. 5B), greatly delayed the kinetic of TFEB-S211 dephosphorylation. Moreover, depletion of either PP2A catalytic subunits (PPP2AC+B) (Fig. 5C) or the regulatory subunit (PPP2R2A), alone or in combination with the catalytic subunits, markedly decreased TFEB-S211 dephosphorylation (Fig. 5D). Efficient depletion of PPP2C and PPP2R2A subunits was assessed by immunoblot (Fig. 5E). Finally, purified recombinant PP2A catalytic subunit A in combination with scaffold subunit A was sufficient to dephosphorylate both TFEB-S211 and TFE3-S321 (Fig. 5, F and G). These results further support the contribution of PP2A to TFE3 and TFEB activation in NaAsO2-induced oxidative stress.
Figure 5.
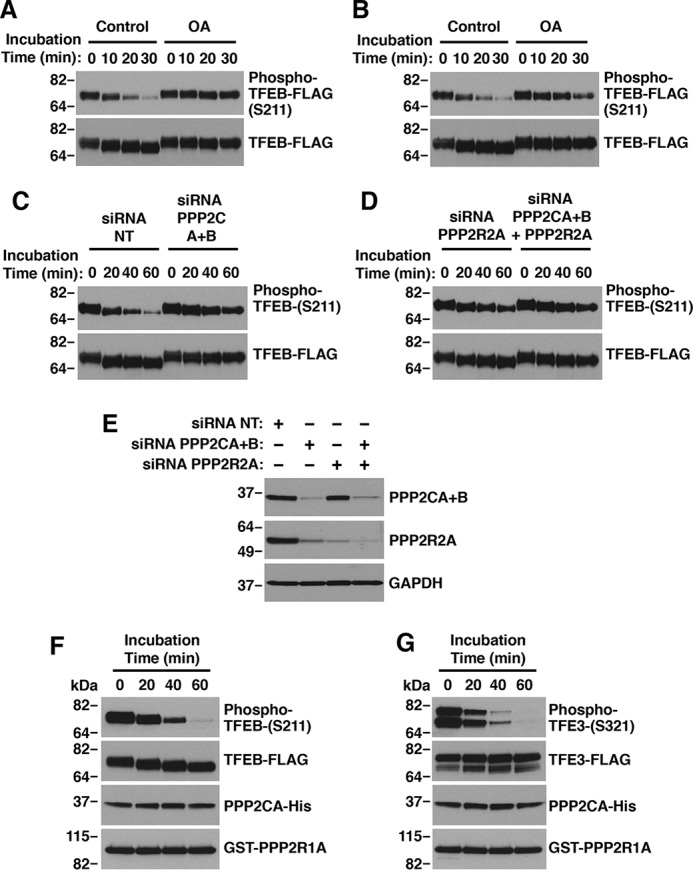
Protein phosphatase 2A dephosphorylates TFEB and TFE3 in vitro. A, in vitro dephosphorylation of TFEB with ARPE19 cell lysates in the presence of 400 nm okadaic acid. B, in vitro dephosphorylation of TFEB with ARPE19 cell lysates treated with 400 nm okadaic acid for 1 h. C and D, in vitro dephosphorylation of TFEB with ARPE19 cell lysates depleted of PP2A subunits. E, immunoblot analysis of protein lysates from ARPE19 cells depleted of either PPP2A catalytic and regulatory subunits. F and G, in vitro dephosphorylation of TFEB (G) or TFE3 (H) with recombinant purified PP2A subunits.
PP2A dephosphorylates TFEB at several residues
Our in vitro phosphatase assays revealed significant changes in TFEB and TFE3 electrophoretic mobility following incubation with recombinant PP2A, suggesting that this phosphatase likely targets several residues. To address this possibility in more detail, we analyzed phosphorylation changes in TFEB in response to NaAsO2 treatment by mass spectrometry. For this, U-2 OS cells expressing recombinant TFEB-FLAG were incubated with DMSO, NaAsO2 or Torin-1. Immunoisolation of TFEB-FLAG followed by MS analysis allowed the identification of several phosphopeptides; their relative abundance was evaluated based on the areas under curve of their corresponding chromatographic peaks (Fig. 6A). As expected, the abundance of peptides containing phosphorylated Ser-211 were noticeably reduced in cells treated with NaAsO2 or Torin-1 compared with DMSO-treated cells (Fig. 6A). Interestingly, we also found decreased abundance of phosphopeptides containing Ser-109, Ser-114, and Ser-122 both under oxidative stress and mTORC1 inactivation conditions (Fig. 6A). To further corroborate the participation of PP2A in the regulation of TFEB-S211, S109, -S114, and -S122 phosphorylation, we analyzed phosphorylation changes in these particular residues in HeLa (CF7) cells treated with NaAsO2 in the presence or absence of OA. Similar to our results in U-2 OS cells, we observed a significant reduction in the phosphorylation levels of TFEB-S211, -S109, -S114, and -S122 in HeLa (CF7) cells treated with either NaAsO2 or Torin-1 (Fig. 6, B–D). More importantly, inhibition of PP2A by OA prevented the dephosphorylation of these residues in response to NaAsO2 (Fig. 6, B–D). Altogether, these results indicate that PP2A dephosphorylates TFEB on several residues including Ser-109, Ser-114, Ser-122, and Ser-211 under oxidative stress conditions.
Figure 6.
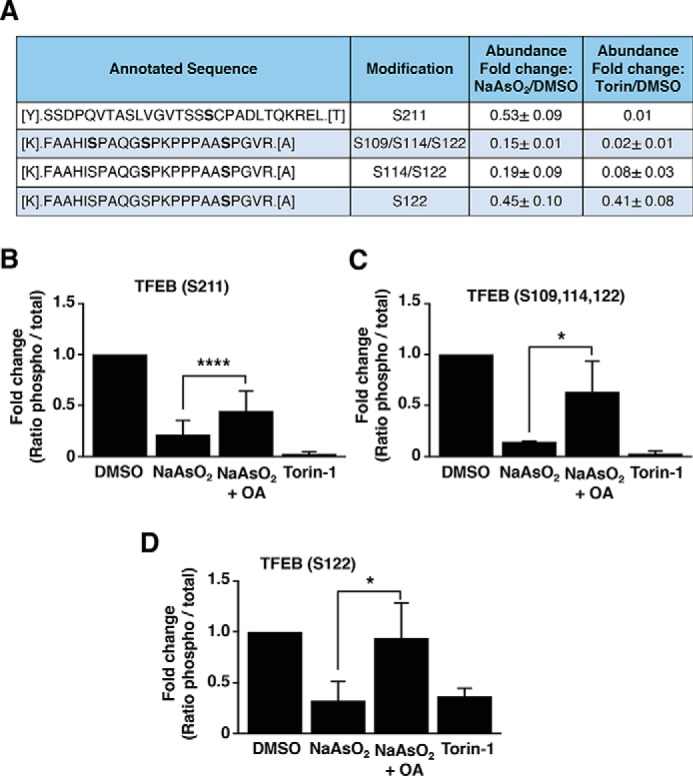
Protein phosphatase 2A dephosphorylates TFEB at different residues. A, mass spectrometry analysis of the phosphorylation status of TFEB-FLAG from U-2 OS cells treated with either vehicle, 150 μm NaAsO2 or 250 nm Torin-1 for 2 h. The relative abundances of the indicated phosphopeptides were compared based on the areas under curve of their corresponding chromatographic peaks and are shown as fold-change from its corresponding vehicle-treated cells. Data are presented as mean ± S.D. from two independent experiments. B–D, mass spectrometry analysis of the phosphorylation status of TFEB-FLAG in Ser-211 (B), Ser-109, -114, and -122 (C), and Ser-122 (D) from HeLa (CF7) cells treated with either vehicle or 300 μm NaAsO2 in the presence or absence of 400 nm okadaic acid or 250 nm Torin-1 for 1.5 h. The relative abundances of the indicated phosphopeptides were compared based on the areas under curve of their corresponding chromatographic peaks and are shown as fold-change from its corresponding vehicle-treated cells. Data are presented as mean ± S.D. using one-way ANOVA, (*) p < 0.05 and (****) p < 0.0001, from three independent experiments.
Dephosphorylation of TFEB at Ser-109, Ser-114, and Ser-122 facilitate its activation in response to oxidative stress
Based on the results of MS analysis, we proceeded to further investigate whether Ser-109, Ser-114, and Ser-122 might participate in the regulation of TFEB activation. To this end, we assessed the functional consequences of mutating these three serine residues to alanine (S109A,S114A,S122A) or aspartic acid (S109D,S114D,S122D) in the context of oxidative stress induced by NaAsO2. As seen in Fig. 7, TFEB-WT and the TFEB-S109A,S114A,S122A mutant behaved similarly and both were efficiently dephosphorylated at Ser-211 and translocated to the nucleus in response to NaAsO2 (Fig. 7, A and B). In contrast, accumulation of TFEB-S109D,S114D,S122D in the nucleus in response to NaAsO2 was significantly reduced (Fig. 7B), suggesting that the dephosphorylation of these residues is required for efficient TFEB activation. It is important to note that mutation of Ser-109, Ser-114, and Ser-122 to aspartic acid did not prevent Ser-211 dephosphorylation (Fig. 7A), indicating that these residues are likely not involved in the formation of the TFEB/14-3-3 complex and might regulate TFEB nuclear translocation by a yet unknown mechanism. Finally, we analyzed the behavior of the TFEB-S109A,S114A,S122A and TFEB-S109D,S114D,S122D mutants in response to starvation. Although we found a reduced number of cells with TFEB-S109D,S114D,S122D in the nucleus compared with TFEB-WT, the differences did not reach statistical significance, suggesting that dephosphorylation of Ser-109, Ser-114, and Ser-122 may play a minor role under a nutrient-depleted condition (Fig. 7, A and B). Moreover, PP2A depletion did not prevent TFE3-S321 dephosphorylation following starvation (Fig. S6), further suggesting that the mechanism of activation of TFEB and TFE3 may differ depending on the type of stress.
Figure 7.
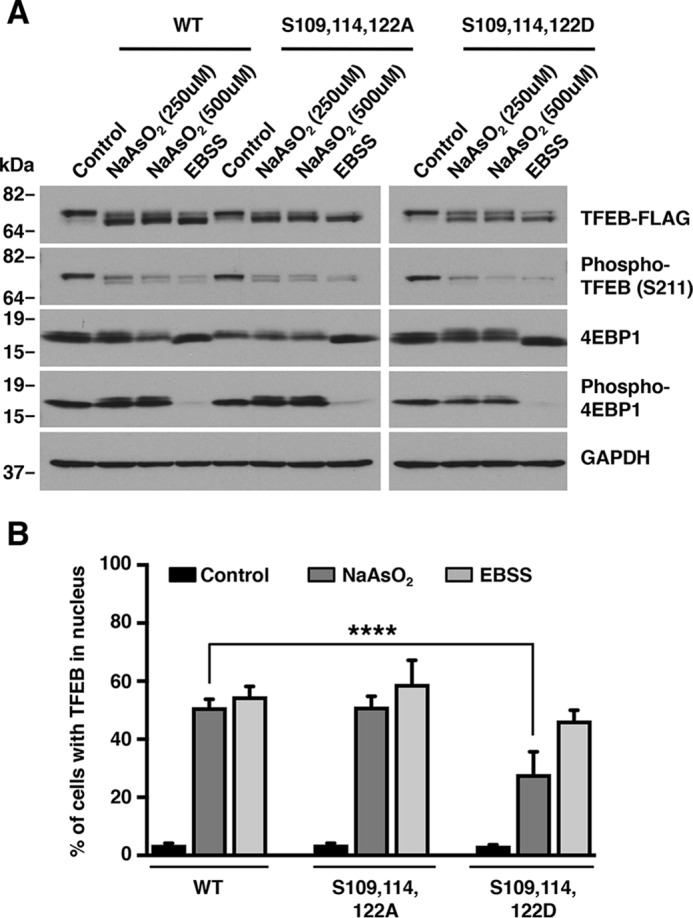
Dephosphorylation of TFEB at Ser-109, Ser-114, and Ser-122 facilitate its activation in response to oxidative stress. A, immunoblot analysis of protein lysates from ARPE19 cells expressing TFEB-WT or the indicated TFEB mutants and treated with either 250 μm NaAsO2 or EBSS for 1 h. Data are representative of at least three independent experiments. B, quantification of cells with TFEB in nucleus in ARPE19 cells expressing TFEB-WT or the indicated TFEB mutants and treated with either 250 μm NaAsO2 or EBSS for 1 h. TFEB-WT (vehicle, n = 654, NaAsO2, n = 687, and EBSS, n = 747), TFEB-S109A,S114A,S122A (vehicle, n = 643, NaAsO2, n = 671, and EBSS n = 669), and TFEB-S109D,S114D,S122D (vehicle, n = 651, NaAsO2, n = 671, and EBSS, n = 658) from three independent experiments are shown. Error bars denote S.D. p value was calculated using one-way ANOVA, (****) p < 0.0001.
Discussion
Phosphorylation/dephosphorylation events play a major role in TFEB/TFE3 regulation. The serine/threonine kinase mTOR is the best-characterized modulator of TFEB/TFE3 activation. Under nutrient-rich conditions, mTORC1 phosphorylates TFEB and TFE3 on critical residues, promoting binding of these transcription factors to 14-3-3 and their consequent retention in the cytosol. Conversely, inactivation of mTORC1 by nutrient withdraw or Torin-1 causes TFEB/TFE3 nuclear translocation (6–8). Recently, additional kinases have been proposed to contribute to TFEB/TFE3 sequestration in the cytosol. These include ERK2 (3), MAP4K3 (40), GSK3 (41), and AKT (42). In most cases, the mechanism by which these kinases prevent TFEB/TFE3 nuclear translocation is unknown, although it has been suggested that MAP4K3 and GSK3 may facilitate TFEB/TFE3 recruitment to the lysosomal surface, thus facilitating mTORC1-mediated phosphorylation.
To be active, TFEB and TFE3 must dissociate from 14-3-3 and this requires dephosphorylation of TFEB-S211 and TFE3-S321. Therefore, identifying the phosphatases involved in TFEB-S211 and TFE3-S321 dephosphorylation is critical for understanding TFEB/TFE3 regulation. The phosphatase PPP3/calcineurin has been shown to play an important role in TFEB dephosphorylation. Under starvation conditions, mTORC1 inactivation along with PPP3/calcineurin activation promote TFEB activation (17). PPP3/calcineurin activity is regulated by intracellular calcium levels (43). It has been proposed that lysosomal calcium release via TRPML1 may result in local calcineurin activation and consequent TFEB transport to the nucleus (17, 18).
It is becoming evident that TFEB and TFE3 are activated in response to multiple types of intracellular stress, including pathogen infection, inflammation, ER stress, and mitocondrial damage. In many of these cases, TFEB/TFE3 activation does not seem to require mTORC1 inactivation; however, TFEB-S211 and TFE3-S321 need to be dephosphorylated, suggesting that activation of phosphatases plays a critical role in this context.
In this study we have identified a novel role for the phosphatase PP2A in TFEB/TFE3 activation in response to oxidative stress. Treatment of different cell types with sodium arsenite induced oxidative stress, resulting in TFEB-S211 and TFE3-S321 dephosphorylation and TFEB/TFE3 nuclear translocation. Importantly, the activity of mTORC1 was enhanced by sodium arsenite, indicating that mTORC1 inactivation does not account for the reduced TFEB-S211 and TFE3-S321 phosphorylation observed under these conditions. Moreover, depletion of PPP3/cacineurin or TRPML1 did not affect TFEB/TFE3 transport to the nucleus, further suggesting the presence of a novel mechanism of TFEB/TFE3 activation. Notably, depletion of either the catalytic (PPP2CA+B) or regulatory (PPPR2A/B55α) subunits of PP2A, as well as PP2A inactivation with okadaic acid, was sufficient to prevent TFEB/TFE3 activation in response to sodium arsenite. Conversely, PP2A activation by ceramide or FTY720 caused TFEB/TFE3 nuclear translocation. Activation of TFEB/TFE3 was assessed by different methods including changes in cellular distribution (nucleus versus cytosol), changes in electrophoretic mobility, and the use of phospho-specific antibodies against TFEB-S211 and TFE3-S321. The ability of PP2A to specifically dephosphorylate TFEB-S211 and TFE3-S321 was also confirmed by in vitro phosphatase assays.
Mass spectometry analysis using recombinant TFEB revealed that sodium arsenite not only induced reduced TFEB-S211 phosphorylation but also caused dephosphorylation of additional residues, including Ser-109, Ser-114, and Ser-122. Furthermore, dephosphorylation of Ser-109, Ser-114, Ser-122, and Ser-211 was prevented by okadaic acid, indicating that PP2A dephosphorylates multiple TFEB residues in sodium arsenite-treated cells. One possibility is that PP2A-mediated dephosphorylation of Ser-109, Ser-114, and Ser-122 makes Ser-211 more accessible to other phosphatases (e.g. PPP3/calcineurin). However, the fact that the TFEB-S109A,S114A,S122A mutant does not exhibit reduced Ser-211 phosphorylation argues against this possibility. Therefore, it appears that distinct protein phosphatases may target the same key phosphorylation sites on TFEB (for example, Ser-211 can be dephosphorylated by PP2A and PPP3/calcineurin).
Our data also identified a novel role of Ser-109, Ser-114, and Ser-122 in TFEB activation. Phosphomimetic mutation of these residues to aspartic acid (TFEB-S109D,S114D,S122D) significantly decreased TFEB nuclear translocation in response to sodium arsenite. These results are in agreement with a recent study showing that mutation of Ser-122 to alanine appears to enhance the effects of the S211A mutation (44).
One important question is to determine whether PP2A might contribute to TFEB/TFE3 activation under nutrient withdrawn conditions. Several reports have shown that inactivation of mTOR results in a robust increase in PP2A/PPP2R2A activity (45–47). Moreover, stimulation of PP2A/PPP2R2A following amino acid starvation is a critical regulatory step to allow ULK1 dephosphorylation and activation (31). On the other hand, our data showed that PP2A depletion did not prevent TFEB/TFE3 activation in starved cells, suggesting that mTOR inactivation or other phosphatases, such as PPP3/calcineurin, may play a predominant role under nutrient deprivation conditions. Alternatively, the fraction of TFEB/TFE3 that remains phosphorylated due to PP2A depletion might be preferentially targeted by the E3 ubiquitin ligase STUB1 and sent to the proteasome for degradation (48), thus preventing its detection; however, simultaneous treatment with starvation medium and proteasome inhibitors did not result in a significant accumulation of phosphorylated TFEB/TFE3 (data not shown), further suggesting that PP2A is not essential for TFEB/TFE3 activation in response to starvation. Nonetheless, modulation of Ser-109, Ser-114, and Ser-122 might contribute to starvation-induced TFEB/TFE3 activation. In support of this, our mass spectrometry experiments reveled that Torin-1 treatment caused a dramatic reduction in Ser-109, Ser-114, and Ser-122 phosphorylation levels, suggesting that these residues are phosphorylated by mTOR and, thereby, likely dephosphorylated under starvation conditions. In addition, activation of the TFEB-S109D,S114D,S122D mutant was reduced in response to low nutrient levels, although the reduction did not reach statistical significance under our experimental conditions. Therefore, our data suggest that there is a number of critical phosphorylation sites in TFEB/TFE3 that are regulated by distinct protein phosphatases and kinases depending on the nature/duration/strength of the stress.
In summary, our study identifies a novel mechanism to promote TFEB/TFE3 activation by stimulating the protein phosphatase PP2A without suppressing mTOR. We propose that this mechanism may be critical in particular cell types, such as immune or cancer cells, which require simultaneous mTORC1 and TFEB/TFE3 signaling as a way to achieve robust cell growth and survival.
Experimental procedures
Cell line cultures and treatments
ARPE-19 cells (CRL-2302, American Type Culture Collection) were grown at 37 °C in a 1:1 mixture of Dulbecco's modified Eagle's medium and Ham's F-12 media supplemented with 10% fetal bovine serum (Invitrogen), 2 mm GlutamaxTM, 100 units/ml of penicillin, and 100 μg/ml of streptomycin (Gibco) in a humidified 5% CO2 atmosphere. MEF WT (CRL-2977, American Type Culture Collection), HeLa cells (CCL-2, American Type Culture Collection), U-2 OS cells (HTB-96 American Type Culture Collection), HeLa (CF7) cells stably expressing TFEB-FLAG (previously described in Martina et al. (6), skin fibroblasts from a MLIV patient (clone GM02527, Coriell Cell Repositories, Coriell Institute for Medical Research, Camden, NJ), and unrelated nondiseased skin fibroblasts (clone MCH065, Repository for Mutant Human Cell Strains of Montreal Children's Hospital, Montreal, Canada) were grown in Dulbecco's modified Eagle's medium supplemented with fetal bovine serum, GlutamaxTM and antibiotics as indicated were used for ARPE-19 cells media. TFEB/TFE3 double KO MEF cells were generated as described by Martina et al. (10). For transient expression, ARPE19 cells were nucleofected using Cell Line Nucleofector® Kit V (Lonza) following the manufacturer's recommendations. Cells were analyzed 24 h post-nucleofection. For drug treatment experiments, cells were incubated for the indicated times at 37 °C in medium containing one of the following reagents: DMSO (Sigma), 250 nm Torin-1 (TOCRIS), 5–40 μm FK-506, 100–800 nm okadaic acid (Cell Signaling Technology), 2.5–20 μm FTY720 (Cayman Chemical), 15 mm N-acetyl-l-cysteine (Sigma), or 31.25–500 μm NaAsO2 (sodium arsenite, Santa Cruz Biotechnology). For starvation experiments cells were washed three times in Hank's balanced salt solution (Invitrogen) and incubated for 1–4 h at 37 °C in EBSS (Sigma).
Antibodies
The following mouse monoclonal antibodies were used: clone Ab5 to actin, clone 29 to TOM20 (BD Transduction Laboratories, 612656 and 612278), clone M2 to FLAG (Sigma, F3165), clone 1D6 to PPP2C (Millipore, 05-421), clone E-9 to PPP1CA, clone C-5 to PPP1CB (Santa Cruz Biotechnology, sc-7482 and sc-373782), clone H4A3 to Lamp1 (Developmental Studies Hybridoma Bank, H4A3-s), and clone 6C5 to GAPDH (Ambion, AM4300). The following polyclonal antibodies were also used: anti-phospho-p70 S6 kinase, anti-p70 S6 kinase, anti-phospho-4E-BP1, anti-4E-BP1, anti-14-3-3, anti-PPP2R2A, anti-PPP2AC, and anti-GST were from Cell Signaling Technology (9205, 2708, 9451, 9644, 8312, 2290, 2259, and 2625, respectively), anti-BiP and anti-PPP3C were from Abcam (ab21685 and ab71149, respectively), anti-giantin (Covance, PRB-114C), anti-TFE3 (Sigma, HPA023881), goat anti-TIA-1 (Santa Cruz Biotechnology, sc-1751), anti-PPP2R1A (GeneTex, GTX102206), and phospho-TFE3-S321 were as previously described by Martina et al. (10). Horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG were acquired from Cell Signaling Technology (7076 and 7074).
Production of anti-phospho-TFEB (Ser-211) antibody
For antibody production, the synthesis and purification of a phospho-specific TFEB peptide (VGVTSS-pS-CPADLTQ; amino acids 205–218), and a nonphosphorylated peptide counterpart (VGVTSSSCPADLTQ) as well as the production of polyclonal antisera was performed by YenZym Antibodies (South San Francisco, CA). Two New Zealand White rabbits were immunized with the phosphopeptide following a 90-day immunization protocol. The antisera were further purified by affinity chromatography against the same phosphopeptide used for immunization. The purified antibody was then affinity-absorbed against the nonphosphorylated peptide counterpart, to separate the phosphopeptide-specific antibody from the cross-reactive population. The specificity of anti-phospho-TFEB antibody was examined by immunoblotting.
Recombinant DNA plasmid
TFE3-FLAG expression vector was generated by cloning the full-length encoding sequence of human TFE3 obtained by RT-PCR amplification from ARPE-19 cells total RNA followed by in-frame cloning into HindIII-BamHI sites of p3×FLAG-CMV-14 (Sigma) with a triple FLAG tag fused to the carboxyl termini of TFE3. Amino acid substitutions in TFEB were made using the QuikChange Lightning site-directed mutagenesis kit (Agilent Technologies) according to the manufacturer's instructions.
Immunofluorescence confocal microscopy
For immunofluorescence, cells grown on coverslips were washed with PBS and fixed with 4% formaldehyde for 15 min at room temperature or with methanol/acetone (1:1, v/v) for 10 min at −20 °C. For monitoring nuclear localization of TFE3, cells were permeabilized in PBS containing 0.2% Triton X-100 for 10 min at room temperature. Cells were then incubated with the indicated primary antibodies in PBS containing 10% fetal bovine serum and 0.1% (w/v) saponin for 1 h at room temperature, followed by incubation with the corresponding secondary antibodies conjugated to Alexa Fluor-488 (Invitrogen, A-11008). After staining, the coverslips were mounted onto glass slides with Fluoromount-G (Southern Biotech, 0100–0101). Images were acquired on a Zeiss LSM 510 confocal system with a ×63 NA 1.4 oil immersion objective using 488-nm laser excitation (Carl Zeiss).
Immunoprecipitation, electrophoresis, and immunoblotting
Cells washed with ice-cold PBS were lysed in lysis buffer containing 25 mm Hepes-KOH, pH 7.4, 150 mm NaCl, 5 mm EDTA, and 1% Triton X-100 (w/v) and supplemented with protease and phosphatase inhibitor mixtures (Roche Applied Science). Cell lysates were incubated on ice for 30 min and then passed 10 times through a 25-gauge needle. Cell lysates were centrifuged at 16,000 × g for 10 min at 4 °C. For immunoprecipitation, the soluble fractions were incubated with 20 μl of anti-FLAG M2 affinity gel beads (Sigma) for 4 h at 4 °C. The immunoprecipitates were collected, washed three times with lysis buffer, and proteins were eluted with Laemmli sample buffer.
Samples were analyzed by SDS-PAGE (4–20% gradient gels, Life Technologies) under reducing conditions and transferred to nitrocellulose. Membranes were immunoblotted using the indicated antibodies. Horseradish peroxidase-conjugated anti-mouse, anti-rabbit IgG, or anti-rat IgG (Cell Signaling Technology) were used at a dilution of 1:8000. Horseradish peroxidase-chemiluminescence was developed using Western Lightning Chemiluminescence Reagent Plus (PerkinElmer Life Sciences). The exposed films were scanned, and the protein band intensities quantified using ImageJ software (NIH) and Photoshop CS4 software was used to produce the figures.
RNA interference (RNAi)
Knockdown of the indicated genes was achieved by transfection of siRNA duplexes. In brief, cells grown in six-well plates were transfected with RNAiMAX transfection reagent (Thermo Fisher Scientific) and 100 nm ON-TARGETplus nontargeting pool siRNA duplexes or ON-TARGETplus smart pool siRNA duplexes targeted against PPP2CA, PPP2CB, PPP2R1A, PPP2R2A, PPP1CA, PPP1CB, PPP3CA, and PPP3CB (GE Dharmacon). Treated cells were analyzed 72 h after transfection.
In vitro dephosphorylation assay
For the preparation of phosphatase substrates (TFEB-FLAG and TFE3-FLAG), CF7 HeLa or ARPE19 cells overexpressing TFEB-FLAG or TFE3-FLAG, respectively, were grown in two 15-cm dishes each for 24 h. Cells were lysed in phosphatase assay buffer (50 mm Hepes, pH 7.5, 100 mm NaCl, 2 mm MgCl2, 1 mm DTT, and 0.5% Nonidet P-40) supplemented with a protease and phosphatase inhibitor mixture (Roche Applied Science). Cell lysates were incubated on ice for 30 min and then were passed 10 times through a 25-gauge needle. Cell lysates were centrifuged at 16,000 × g for 15 min at 4 °C. The soluble fractions were incubated with 50 μl of anti-FLAG M2 affinity gel beads (Sigma) overnight at 4 °C. The immunoprecipitated fractions were washed 5 times and the affinity purified substrates were eluted in 150 μl of phosphatase assay buffer containing 400 μg/ml of 3×FLAG peptide (Sigma). To ensure the stability of the phosphatase substrates in reactions that use recombinant phosphatase, a protective lysate was obtained from TFEB/TFE3 double KO MEF cells as described by Wong et al. (31).
For the phosphatase assay using cell lysates, purified substrates (TFEB-FLAG or TFE3-FLAG) were incubated with 10 μg of lysate proteins in a final volume of 15 μl at 30 °C for the indicated times. For the phosphatase assay using recombinant phosphatase, substrates were incubated with 10 μg of phosphatase activity-depleted protective lysate proteins and 25 nm PPP2CA-His + GST-PPP2R1A (BPS Bioscience) in a final volume of 15 μl at 30 °C for the indicated times.
Mass spectrometry
Immunoprecipitated TFEB-FLAG from CF7 HeLa cells treated with the indicated reagents were sequentially reduced with tris (2-carboxyethyl)phosphine hydrochloride and alkylated with chloroacetamide (both from Sigma). Proteins were then digested with trypsin or chymotrypsin (Promega). The resulting peptide mixtures were analyzed with an Orbitrap Fusion Lumos (Thermo Fisher Scientific) equipped with a Dionex Ultimate 3000 nanoLC system. Peptide IDs and phosphorylation sites were assigned with Mascot version 2.5 (Matrix Science). The confidence of phosphorylation site localization is assessed with ptmRS node in Proteome Discoverer 2.2 platform (Thermo Fisher Scientific). The phosphopeptides were filtered out at 1% false discovery rate and their relative abundances were compared based on the areas under curve of their corresponding chromatographic peaks.
Statistical analysis
Data were processed in Excel (Microsoft Corporation) then Prism (GraphPad Software) to generate curve and bar charts and perform statistical analyses. Student's t test or one-way ANOVA and pairwise post tests were performed for each dependent variable, as specified in each figure legend. All data are presented as mean ± S.D. p < 0.05 was considered statistically significant (*), p < 0.01 very significant (**), p < 0.001 extremely significant (***), and p < 0.0001 extremely significant (****). p > 0.05 was considered not significant (n.s.).
Author contributions
J. A. M. data curation; J. A. M. and R. P. formal analysis; J. A. M. and R. P. investigation; J. A. M. methodology; J. A. M. and R. P. writing-original draft; R. P. conceptualization; R. P. supervision; R. P. funding acquisition.
Supplementary Material
Acknowledgments
We thank Dr. Yong Chen and Dr. Marjan Gucek, NHLBI Proteomics Core Facility, for help to perform, and interpret the MS analysis.
This work was supported by the Intramural Research Program of the National Institutes of Health, NHLBI. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6.
- TFEB
- transcription factor EB
- TFE3
- transcription factor binding to IGHM enhancer 3
- mTORC1
- mechanistic target of rapamycin (serine/threonine kinase) complex 1
- ARPE-19
- retinal pigmented epithelium cell line
- EBSS
- Earle's balanced salt solution
- NaAsO2
- sodium arsenite
- OA
- okadaic acid
- NAC
- N-acetyl-l-cysteine
- PPP2CA
- protein phosphatase 2A catalytic subunit α
- PPP2CB
- protein phosphatase 2A catalytic subunit β
- PPP2R1A
- protein phosphatase 2A scaffold subunit α
- PPP2R2A
- protein phosphatase 2A regulatory subunit B55α
- TRPML1
- mucolipin 1
- ROS
- reactive oxygen species
- ER
- endoplasmic reticulum
- MEF
- mouse embryonic fibroblast
- MLIV
- mucolipidosis type IV.
References
- 1. Raben N., and Puertollano R. (2016) TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu. Rev. Cell Dev. Biol. 32, 255–278 10.1146/annurev-cellbio-111315-125407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sardiello M., Palmieri M., di Ronza A., Medina D. L., Valenza M., Gennarino V. A., Di Malta C., Donaudy F., Embrione V., Polishchuk R. S., Banfi S., Parenti G., Cattaneo E., and Ballabio A. (2009) A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 10.1126/science.1174447 [DOI] [PubMed] [Google Scholar]
- 3. Settembre C., Di Malta C., Polito V. A., Garcia Arencibia M., Vetrini F., Erdin S., Erdin S. U., Huynh T., Medina D., Colella P., Sardiello M., Rubinsztein D. C., and Ballabio A. (2011) TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 10.1126/science.1204592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martina J. A., Diab H. I., Lishu L., Jeong A. L., Patange S., Raben N., and Puertollano R. (2014) The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal. 7, ra9 10.1126/scisignal.2004754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Martina J. A., and Puertollano R. (2013) Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 200, 475–491 10.1083/jcb.201209135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Martina J. A., Chen Y., Gucek M., and Puertollano R. (2012) MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 10.4161/auto.19653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Roczniak-Ferguson A., Petit C. S., Froehlich F., Qian S., Ky J., Angarola B., Walther T. C., and Ferguson S. M. (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal 5, ra42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Settembre C., Zoncu R., Medina D. L., Vetrini F., Erdin S., Erdin S., Huynh T., Ferron M., Karsenty G., Vellard M. C., Facchinetti V., Sabatini D. M., and Ballabio A. (2012) A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 10.1038/emboj.2012.32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nezich C. L., Wang C., Fogel A. I., and Youle R. J. (2015) MiT/TFE transcription factors are activated during mitophagy downstream of Parkin and Atg5. J. Cell Biol. 210, 435–450 10.1083/jcb.201501002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Martina J. A., Diab H. I., Brady O. A., and Puertollano R. (2016) TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 35, 479–495 10.15252/embj.201593428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Campbell G. R., Rawat P., Bruckman R. S., and Spector S. A. (2015) Human immunodeficiency virus type 1 Nef inhibits autophagy through transcription factor EB sequestration. PLoS Pathog. 11, e1005018 10.1371/journal.ppat.1005018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gray M. A., Choy C. H., Dayam R. M., Ospina-Escobar E., Somerville A., Xiao X., Ferguson S. M., and Botelho R. J. (2016) Phagocytosis enhances lysosomal and bactericidal properties by activating the transcription factor TFEB. Curr. Biol. 26, 1955–1964 10.1016/j.cub.2016.05.070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ouimet M., Koster S., Sakowski E., Ramkhelawon B., van Solingen C., Oldebeken S., Karunakaran D., Portal-Celhay C., Sheedy F. J., Ray T. D., Cecchini K., Zamore P. D., Rayner K. J., Marcel Y. L., Philips J. A., and Moore K. J. (2016) Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat. Immunol. 17, 677–686 10.1038/ni.3434,10.1038/nrm.2016.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pastore N., Brady O. A., Diab H. I., Martina J. A., Sun L., Huynh T., Lim J. A., Zare H., Raben N., Ballabio A., and Puertollano R. (2016) TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 12, 1240–1258 10.1080/15548627.2016.1179405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mansueto G., Armani A., Viscomi C., D'Orsi L., De Cegli R., Polishchuk E. V., Lamperti C., Di Meo I., Romanello V., Marchet S., Saha P. K., Zong H., Blaauw B., Solagna F., Tezze C., et al. (2017) Transcription factor EB controls metabolic flexibility during exercise. Cell Metab. 25, 182–196 10.1016/j.cmet.2016.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Pastore N., Vainshtein A., Klisch T. J., Armani A., Huynh T., Herz N. J., Polishchuk E. V., Sandri M., and Ballabio A. (2017) TFE3 regulates whole-body energy metabolism in cooperation with TFEB. EMBO Mol. Med. 9, 605–621 10.15252/emmm.201607204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Medina D. L., Di Paola S., Peluso I., Armani A., De Stefani D., Venditti R., Montefusco S., Scotto-Rosato A., Prezioso C., Forrester A., Settembre C., Wang W., Gao Q., Xu H., Sandri M., Rizzuto R., De Matteis M. A., and Ballabio A. (2015) Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 10.1038/ncb3114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhang X., Cheng X., Yu L., Yang J., Calvo R., Patnaik S., Hu X., Gao Q., Yang M., Lawas M., Delling M., Marugan J., Ferrer M., and Xu H. (2016) MCOLN1 is a ROS sensor in lysosomes that regulates autophagy. Nat. Commun. 7, 12109 10.1038/ncomms12109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang C., Niederstrasser H., Douglas P. M., Lin R., Jaramillo J., Li Y., Oswald N. W., Zhou A., McMillan E. A., Mendiratta S., Wang Z., Zhao T., Lin Z., Luo M., Huang G., et al. (2017) Small-molecule TFEB pathway agonists that ameliorate metabolic syndrome in mice and extend C. elegans lifespan. Nat. Commun. 8, 2270 10.1038/s41467-017-02332-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gong X., Ivanov V. N., Davidson M. M., and Hei T. K. (2015) Tetramethylpyrazine (TMP) protects against sodium arsenite-induced nephrotoxicity by suppressing ROS production, mitochondrial dysfunction, pro-inflammatory signaling pathways and programed cell death. Arch. Toxicol. 89, 1057–1070 10.1007/s00204-014-1302-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelly S. M., Vanslyke J. K., and Musil L. S. (2007) Regulation of ubiquitin-proteasome system mediated degradation by cytosolic stress. Mol. Biol. Cell 18, 4279–4291 10.1091/mbc.e07-05-0487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun H., Yang Y., Shao H., Sun W., Gu M., Wang H., Jiang L., Qu L., Sun D., and Gao Y. (2017) Sodium arsenite-induced learning and memory impairment is associated with endoplasmic reticulum stress-mediated apoptosis in rat hippocampus. Front. Mol. Neurosci. 10, 286 10.3389/fnmol.2017.00286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng C. Y., Lam S. K., Li Y. Y., and Ho J. C. (2015) Arsenic trioxide-induced cytotoxicity in small cell lung cancer via altered redox homeostasis and mitochondrial integrity. Int. J. Oncol. 46, 1067–1078 [DOI] [PubMed] [Google Scholar]
- 24. Zhu X. X., Yao X. F., Jiang L. P., Geng C. Y., Zhong L. F., Yang G., Zheng B. L., and Sun X. C. (2014) Sodium arsenite induces ROS-dependent autophagic cell death in pancreatic beta-cells. Food Chem. Toxicol. 70, 144–150 10.1016/j.fct.2014.05.006 [DOI] [PubMed] [Google Scholar]
- 25. Supinski G. S., Stofan D., Ciufo R., and DiMarco A. (1997) N-Acetylcysteine administration alters the response to inspiratory loading in oxygen-supplemented rats. J. Appl. Physiol. (1985) 82, 1119–1125 10.1152/jappl.1997.82.4.1119 [DOI] [PubMed] [Google Scholar]
- 26. Klee C. B., Draetta G. F., and Hubbard M. J. (1988) Calcineurin. Adv. Enzymol. Relat. Areas Mol. Biol. 61, 149–200 [DOI] [PubMed] [Google Scholar]
- 27. Swingle M., Ni L., and Honkanen R. E. (2007) Small-molecule inhibitors of Ser/Thr protein phosphatases: specificity, use and common forms of abuse. Methods Mol. Biol. 365, 23–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mumby M. C., and Walter G. (1993) Protein serine/threonine phosphatases: structure, regulation, and functions in cell growth. Physiol. Rev. 73, 673–699 10.1152/physrev.1993.73.4.673 [DOI] [PubMed] [Google Scholar]
- 29. Virshup D. M., and Shenolikar S. (2009) From promiscuity to precision: protein phosphatases get a makeover. Mol. Cell 33, 537–545 10.1016/j.molcel.2009.02.015 [DOI] [PubMed] [Google Scholar]
- 30. Reid M. A., Wang W. I., Rosales K. R., Welliver M. X., Pan M., and Kong M. (2013) The B55α subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol. Cell 50, 200–211 10.1016/j.molcel.2013.02.008 [DOI] [PubMed] [Google Scholar]
- 31. Wong P. M., Feng Y., Wang J., Shi R., and Jiang X. (2015) Regulation of autophagy by coordinated action of mTORC1 and protein phosphatase 2A. Nat. Commun. 6, 8048 10.1038/ncomms9048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Silverstein A. M., Barrow C. A., Davis A. J., and Mumby M. C. (2002) Actions of PP2A on the MAP kinase pathway and apoptosis are mediated by distinct regulatory subunits. Proc. Natl. Acad. Sci. U.S.A. 99, 4221–4226 10.1073/pnas.072071699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strack S., Cribbs J. T., and Gomez L. (2004) Critical role for protein phosphatase 2A heterotrimers in mammalian cell survival. J. Biol. Chem. 279, 47732–47739 10.1074/jbc.M408015200 [DOI] [PubMed] [Google Scholar]
- 34. Sablina A. A., Chen W., Arroyo J. D., Corral L., Hector M., Bulmer S. E., DeCaprio J. A., and Hahn W. C. (2007) The tumor suppressor PP2A Aβ regulates the RalA GTPase. Cell 129, 969–982 10.1016/j.cell.2007.03.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Matsuoka Y., Nagahara Y., Ikekita M., and Shinomiya T. (2003) A novel immunosuppressive agent FTY720 induced Akt dephosphorylation in leukemia cells. Br. J. Pharmacol. 138, 1303–1312 10.1038/sj.bjp.0705182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chun J., and Hartung H. P. (2010) Mechanism of action of oral fingolimod (FTY720) in multiple sclerosis. Clin. Neuropharmacol. 33, 91–101 10.1097/WNF.0b013e3181cbf825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rahman M. M., Prünte L., Lebender L. F., Patel B. S., Gelissen I., Hansbro P. M., Morris J. C., Clark A. R., Verrills N. M., and Ammit A. J. (2016) The phosphorylated form of FTY720 activates PP2A, represses inflammation and is devoid of S1P agonism in A549 lung epithelial cells. Sci. Rep. 6, 37297 10.1038/srep37297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chalfant C. E., Kishikawa K., Mumby M. C., Kamibayashi C., Bielawska A., and Hannun Y. A. (1999) Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A: activation is stereospecific and regulated by phosphatidic acid. J. Biol. Chem. 274, 20313–20317 10.1074/jbc.274.29.20313 [DOI] [PubMed] [Google Scholar]
- 39. Saddoughi S. A., Gencer S., Peterson Y. K., Ward K. E., Mukhopadhyay A., Oaks J., Bielawski J., Szulc Z. M., Thomas R. J., Selvam S. P., Senkal C. E., Garrett-Mayer E., De Palma R. M., Fedarovich D., Liu A., et al. (2013) Sphingosine analogue drug FTY720 targets I2PP2A/SET and mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO Mol. Med. 5, 105–121 10.1002/emmm.201201283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hsu C. L., Lee E. X., Gordon K. L., Paz E. A., Shen W. C., Ohnishi K., Meisenhelder J., Hunter T., and La Spada A. R. (2018) MAP4K3 mediates amino acid-dependent regulation of autophagy via phosphorylation of TFEB. Nat. Commun. 9, 942 10.1038/s41467-018-03340-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li Y., Xu M., Ding X., Yan C., Song Z., Chen L., Huang X., Wang X., Jian Y., Tang G., Tang C., Di Y., Mu S., Liu X., Liu K., Li T., Wang Y., Miao L., Guo W., Hao X., and Yang C. (2016) Protein kinase C controls lysosome biogenesis independently of mTORC1. Nat. Cell Biol. 18, 1065–1077 10.1038/ncb3407 [DOI] [PubMed] [Google Scholar]
- 42. Palmieri M., Pal R., Nelvagal H. R., Lotfi P., Stinnett G. R., Seymour M. L., Chaudhury A., Bajaj L., Bondar V. V., Bremner L., Saleem U., Tse D. Y., Sanagasetti D., Wu S. M., Neilson J. R., et al. (2017) mTORC1-independent TFEB activation via Akt inhibition promotes cellularclearance in neurodegenerative storage diseases. Nat. Commun 8, 14338 10.1038/ncomms14338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hogan P. G., and Li H. (2005) Calcineurin. Curr. Biol. 15, R442–R443 10.1016/j.cub.2005.06.006 [DOI] [PubMed] [Google Scholar]
- 44. Vega-Rubin-de-Celis S., Peña-Llopis S., Konda M., and Brugarolas J. (2017) Multistep regulation of TFEB by MTORC1. Autophagy 13, 464–472 10.1080/15548627.2016.1271514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cianfanelli V., Fuoco C., Lorente M., Salazar M., Quondamatteo F., Gherardini P. F., De Zio D., Nazio F., Antonioli M., D'Orazio M., Skobo T., Bordi M., Rohde M., Dalla Valle L., Helmer-Citterich M., et al. (2015) AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol. 17, 20–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Di Conza G., Trusso Cafarello S., Loroch S., Mennerich D., Deschoemaeker S., Di Matteo M., Ehling M., Gevaert K., Prenen H., Zahedi R. P., Sickmann A., Kietzmann T., Moretti F., and Mazzone M. (2017) The mTOR and PP2A pathways regulate PHD2 phosphorylation to fine-tune HIF1α levels and colorectal cancer cell survival under hypoxia. Cell Rep. 18, 1699–1712 10.1016/j.celrep.2017.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hartley D., and Cooper G. M. (2002) Role of mTOR in the degradation of IRS-1: regulation of PP2A activity. J. Cell. Biochem. 85, 304–314 10.1002/jcb.10135 [DOI] [PubMed] [Google Scholar]
- 48. Sha Y., Rao L., Settembre C., Ballabio A., and Eissa N. T. (2017) STUB1 regulates TFEB-induced autophagy-lysosome pathway. EMBO J. 36, 2544–2552 10.15252/embj.201796699 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.