

Abstract
Protoheme (hereafter referred to as heme) is an essential cellular cofactor and signaling molecule that is also potentially cytotoxic. To mitigate heme toxicity, heme synthesis and degradation are tightly coupled to heme utilization in order to limit the intracellular concentration of “free” heme. Such a model, however, would suggest that a readily accessible steady-state, bioavailable labile heme (LH) pool is not required for supporting heme-dependent processes. Using the yeast Saccharomyces cerevisiae as a model and fluorescent heme sensors, site-specific heme chelators, and molecular genetic approaches, we found here that 1) yeast cells preferentially use LH in heme-depleted conditions; 2) sequestration of cytosolic LH suppresses heme signaling; and 3) lead (Pb2+) stress contributes to a decrease in total heme, but an increase in LH, which correlates with increased heme signaling. We also observed that the proteasome is involved in the regulation of the LH pool and that loss of proteasomal activity sensitizes cells to Pb2+ effects on heme homeostasis. Overall, these findings suggest an important role for LH in supporting heme-dependent functions in yeast physiology.
Keywords: heme, biosensor, metal homeostasis, metal, heme oxygenase, stress, xenobiotic, yeast, cell stress, heavy metals, heme, heme sensor, lead, Hap1, labile heme pool
Introduction
Protoheme (iron protoporphyrin IX or heme b) is an essential cofactor and signaling molecule that is also potentially cytotoxic (1–4). The molecules and mechanisms cells employ to utilize protoheme, while mitigating its inherent toxicity, are complex and poorly understood, especially during stress (1, 2). Cells manage protoheme, which is hereafter referred to as heme, by controlling both its total concentration and bioavailability (1, 2). Heme concentration is primarily governed by the relative rates of heme synthesis and degradation, which are well-understood processes (1, 2). Indeed, all of the enzymes involved in eukaryotic heme synthesis and degradation have been structurally characterized to atomic resolution, and the mechanisms of action have been largely delineated (1–4).
On the other hand, the factors that control heme bioavailability are poorly understood (1, 2). Bioavailable heme can be operationally defined as a pool of chelatable, kinetically labile heme that can readily exchange between biomolecules and is accessible for heme-dependent processes. The identity of factors that buffer and traffic labile heme (LH)3 and the mechanisms employed to mobilize it for utilization are largely unknown (1, 2).
To mitigate heme toxicity, it is generally thought that heme is made on demand (i.e. heme synthesis and utilization are tightly coupled), and heme in excess of that required for metabolism is degraded (4–6). As a consequence, it is unclear what role, if any, steady-state LH plays in supporting heme-dependent functions. Using genetically encoded ratiometric fluorescent heme sensors to probe LH and heme homeostatic mechanisms, we have previously demonstrated the existence of a highly dynamic steady-state pool of LH in the cytosol, spanning 20–40 nm, and identified new heme trafficking factors (e.g. glyceraldehyde-3-phosphate dehydrogenase (GAPDH)) and signaling molecules that mobilize LH (e.g. nitric oxide (NO)) (7). However, the physiological importance of LH remained unclear.
In the current work, using Saccharomyces cerevisiae as a model eukaryote and heme sensors and chelating agents, we have established the functional importance of LH in regulating heme signaling, demonstrated that LH is preferentially consumed when cells become heme depleted, and discovered that certain xenobiotics (e.g. lead ions (Pb2+)) can, rather paradoxically, both deplete total cellular heme, primarily through its inhibition of heme synthesis, and increase labile bioavailable heme. We further find that the proteasome is involved in the regulation of labile heme and its response to Pb2+ stress. In total, our results establish a functional role for LH, indicate that total and labile heme pools can be decoupled in response to certain stressors, and provide evidence for heme-based signaling in response to heavy metal stress.
Results
Sequestering labile heme impacts heme signaling
We first sought to determine whether cytosolic LH serves a functional role in heme signaling. Toward this end, we developed an approach to sequester LH and probe its effects on the heme-regulated transcription factor Hap1 (8–11). To chelate cytosolic LH, we induced the expression of a high-affinity hemoprotein, cytochrome b562 (Cyt b562) (12–16), using a galactose (GAL)-inducible promoter (pGAL) (17). Cyt b562 binds ferric heme at pH 7.0 with a dissociation constant (KDIII) of 10 nm (16). Given the reduction potential (EM) of heme-Cyt b562, +200 mV versus NHE at pH 7.0 (18), and the EM of free heme, −38 mV versus NHE (19, 20), one can estimate the ferrous heme dissociation constant (KDII) to be ∼1 pm by completing a thermodynamic cycle (19).
First, we confirmed that Cyt b562 overexpression could sequester heme by measuring changes in LH with the genetically encoded ratiometric fluorescent heme sensor, HS1-M7A (7). HS1 is a tridomain fusion protein consisting of a heme-binding domain, the His/Met coordinating 4-α-helical bundle Cyt b562, fused to a pair of fluorescent proteins, enhanced GFP (eGFP) and mKATE2, that exhibit heme-sensitive and -insensitive fluorescence, respectively (7) (Fig. 1A). Heme binding to the Cyt b562 domain results in the quenching of eGFP fluorescence via resonance energy transfer but has little effect on mKATE2 fluorescence (7). Thus, the ratio of eGFP fluorescence (excitation 488 nm, emission 510 nm) to mKATE2 fluorescence (excitation 588 nm, emission 620 nm) provides a readout of cellular heme independently of sensor concentration, with the eGFP/mKATE2 ratio inversely correlating with heme binding to the sensor (7). The high affinity of HS1 for ferric and ferrous heme, KDIII = 10 nm and KDII < 1 nm at pH 7.0, renders it fully saturated with heme in WT cells (7). On the other hand, a variant of HS1, HS1-M7A, in which the heme-coordinating Met ligand is mutated to Ala, exhibits ferrous and ferric affinities of KDII = 25 nm and KDIII = 2 μm at pH 7.0 and is 20–50% bound in the yeast cytosol (7). The observed sensor eGFP/mKATE2 fluorescence ratio (Rexpt) can be used to determine the fractional heme saturation of the sensor and the concentration of LH, assuming a 1:1 heme/sensor binding model and previously established sensor calibration protocols that involve determining the sensor ratio when the sensor is 100% (Rmax) and 0% (Rmin) bound to heme (see “Experimental procedures”) (7). Notably, heme binding to the sensor is reversible, and expression of the heme sensor in cells does not itself perturb heme homeostasis or otherwise affect viability (7).
Figure 1.
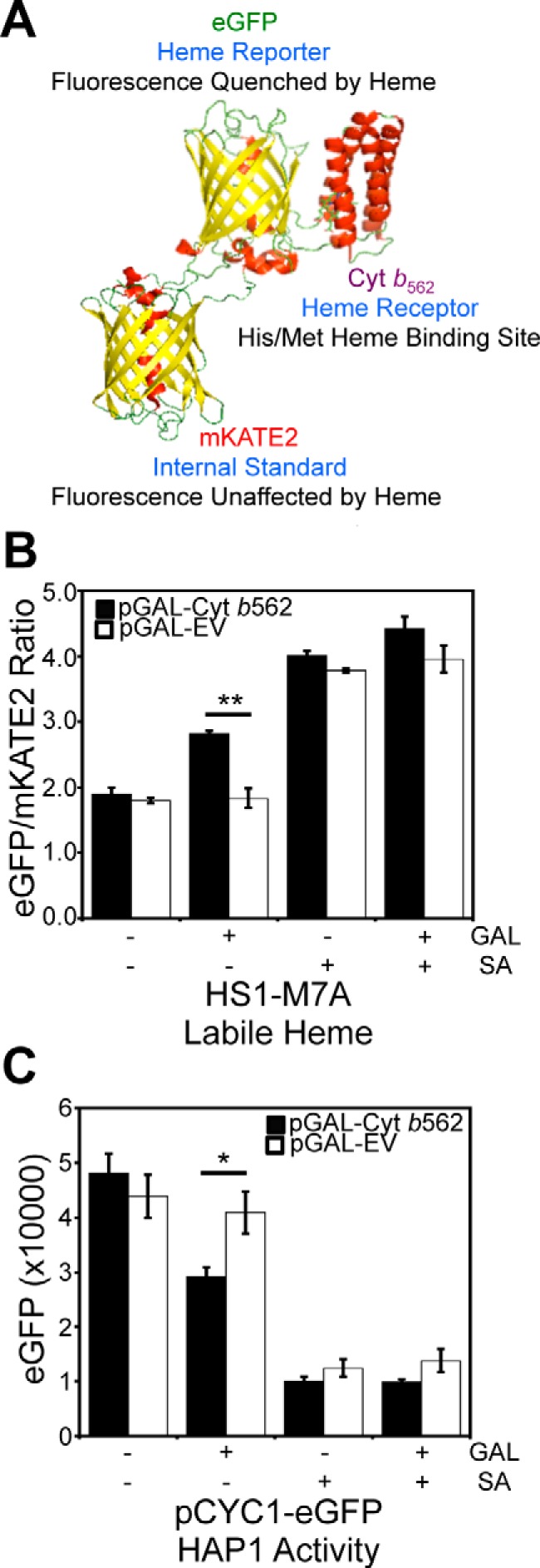
Overexpression of a high affinity hemoprotein, Cyt b562, attenuates labile heme and the activity of heme-regulated transcription factor, Hap1. A, molecular model and design principles of the heme sensor, HS1. Model derived from the X-ray structures of mKATE (Protein Data Bank code 3BXB) and CG6 (Protein Data Bank code 3U8P). B, cells expressing the heme sensor, HS1-M7A, and an allele of Cyt b562 on a galactose (GAL)-inducible promoter (pGAL-Cyt b562) or empty vector (pGAL-EV) were cultured in 2% RAF with or without 1.0% GAL or 500 μm SA for 16 h in SCE medium. After growth, HS1-M7A sensor eGFP (excitation 488 nm, emission 510 nm) and mKATE2 (excitation 588 nm, emission 620 nm) fluorescence emission ratios were recorded. C, Hap1 activity was measured in cells expressing pGAL-Cyt b562 or pGAL-EV and cultured in 2% RAF with or without 1.0% GAL or 500 μm SA for 16 h in SC medium using a transcriptional reporter consisting of an allele of eGFP driven by the CYC1 promoter (pCYC1-eGFP), a Hap1 target gene. All data represent the mean ± S.D. (error bars) of triplicate cultures, and the statistical significance was assessed using a two-sample t test. *, p < 0.01; **, p < 0.001.
WT cells expressing heme sensor HS1-M7A and GAL-inducible Cyt b562 (pGAL-Cyt b562) were cultured in 2% raffinose (RAF) with or without 1.0% GAL for 16 h. As demonstrated in Fig. 1B, induction with GAL results in an increase in the eGFP/mKATE2 fluorescence ratio, from 1.90 ± 0.09 to 2.82 ± 0.04, consistent with less heme binding to the sensor and diminished LH. By comparison, cells that express an empty pGAL vector (EV) or those that are heme-depleted with the heme biosynthetic inhibitor, succinylacetone (SA) (7, 21, 22), are unaffected by induction of Cyt b562. Using the aforementioned sensor calibration protocols, which are described in detail under “Experimental procedures,” the induction of Cyt b562 results in a decrease in the fractional saturation of the heme sensor from ∼60 to ∼35% heme-bound, which corresponds to a change in LH from 35 to 14 nm.
Having established that LH can be sequestered by overexpression of Cyt b562, we next sought to determine whether this would impact the activity of the heme-regulated transcription factor Hap1 (8–11). Heme binding to Hap1 alters its ability to promote or repress transcription of a number of target genes, including CYC1, which Hap1 positively regulates (7–11). To probe Hap1 activity, we used a transcriptional reporter that employs the promoter of a Hap1 target gene, pCYC1, driving the expression of eGFP (7). As demonstrated in Fig. 1C, not only does heme depletion with SA decrease eGFP fluorescence, as expected, but Cyt b562 induction with GAL also results in a decrease in eGFP fluorescence, both of which are consistent with reduced heme binding to Hap1 and diminished transcriptional activation of CYC1. Altogether, our results strongly suggest that overexpression of Cyt b562 can sequester LH, and this results in diminished heme signaling and Hap1 activity.
LH is preferentially consumed relative to total heme during heme depletion
We next sought to determine whether LH is consumed preferentially relative to total heme during conditions of heme deficiency to ascertain whether LH is mobilized for heme-dependent functions when cells are confronted with defects in heme synthesis. Toward this end, we titrated the heme biosynthetic inhibitor SA in WT cells expressing the high-affinity heme sensor, HS1, or the medium-affinity sensor, HS1-M7A, and measured total and labile heme (Fig. 2). Titration of SA over a broad concentration range, up to 500 μm, results in the depletion of both total (Fig. 2C) and labile heme (Fig. 2, A and B) as expected. However, most interestingly, LH is more sensitive than total heme to SA-mediated heme depletion (Fig. 2). When total heme is only modestly depleted by 25% using a relatively low 25 μm dose of SA (Fig. 2C), there is a much larger diminution of LH, as measured by HS1-M7A and HS1 (Fig. 2, A and B); heme loading of the medium affinity sensor, HS1-M7A, shifts from ∼26% bound to ∼0% bound, and the high-affinity sensor, HS1, shifts in heme loading from ∼100% bound to ∼60% bound. In terms of LH concentration, based on the ferrous heme dissociation constants of HS1-M7A, KDII = 25 nm (7), and HS1, assumed to be KDII ∼ 1 pm based on the estimated KDII for Cyt b562, we would estimate LH decreases from ∼10 nm to <1 nm (likely ∼2 pm based on the predicted KDII for HS1) in response to an SA concentration that modestly depletes total heme by 25%. Taken together, these data strongly suggest that heme deficiency mobilizes labile heme to nonexchangeable, and likely higher-affinity and/or buried, heme-binding sites.
Figure 2.
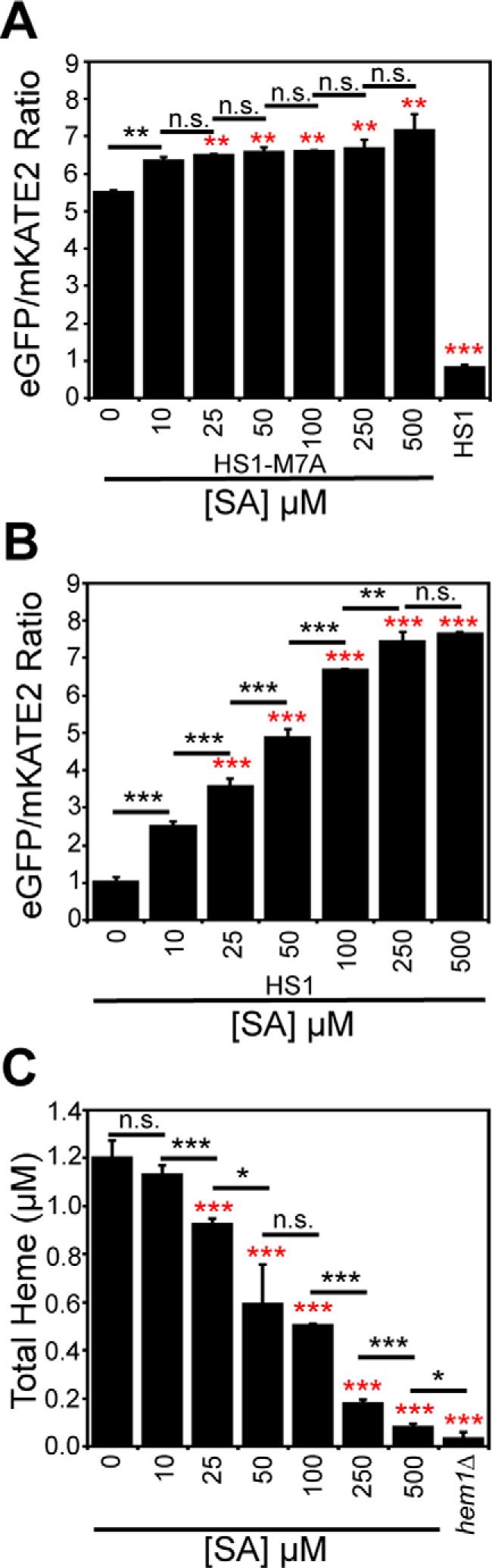
LH is more sensitive to heme depletion using the heme biosynthetic inhibitor, SA, than total heme. Labile heme was measured in HS1-M7A–expressing (A) or HS1-expressing (B) cells cultured in SCE medium for 16 h with the indicated concentration of SA before measurement of the eGFP/mKATE2 fluorescence ratios. C, total heme was measured in the cultures depicted in A or in heme-deficient hem1Δ cells. All data represent the mean ± S.D. (error bars) of triplicate cultures, and the statistical significance was assessed using a two-sample t test. Black asterisks, statistical significance between the indicated pairwise comparisons of conditions; red asterisks, statistical significance relative to 0 μm SA. *, p < 0.05; **, p < 0.005; ***, p < 0.001; n.s., not significant.
Pb2+ depletes total heme but increases labile heme and heme-mediated signaling
A number of xenobiotics and environmental toxins negatively impact heme homeostasis (23), including N-methylprotoporphyrins (24, 25), certain alkylating agents (e.g. 2-allyl-2-isopropylacetamide) (26), various polyhalogenated biphenyls (e.g. tetrachlorodibenzo-p-dioxin) (27, 28)), and heavy metals (29–34). Pb2+, in particular, is a major public health concern, given the ubiquity of Pb2+-based paints, piping, and munitions (34). Among other targets, Pb2+ is well-known to inhibit heme biosynthetic enzymes aminolevulinic acid (ALA) dehydratase (ALAD) and ferrochelatase, resulting in defects in heme synthesis, which in turn leads to anemia, cognitive decline, and other health problems (34). Further, Pb2+ and other heavy metals are known to induce heme oxygenase (HO) (35, 36), which may also contribute to decreased heme levels. However, whereas much is known about the role of Pb2+ in impacting heme synthesis and degradation, virtually nothing is known about the effects of Pb2+ on bioavailable labile heme. Given our findings that LH is important for maintaining heme signaling to Hap1 and is preferentially consumed relative to total heme during heme deficiency, we sought to determine the effects of Pb2+ on LH and heme-based signaling. Toward this end, we established a yeast model of Pb2+ toxicity and probed the effects of Pb2+ on total heme, LH, and heme signaling.
Due to the insolubility of Pb(NO3)2 in standard yeast growth media, we employed a Pb2+ toxicity model similar to what was previously described in which yeast cells are subjected to an acute 3-h exposure to varying concentrations of Pb2+ in 10 mm MES, pH 6.0, buffer, a medium in which Pb(NO3)2 is soluble and Pb2+ is not complexed by the buffer (37). Following Pb2+ exposure, cells were washed and allowed to recover for 4 h in an appropriate synthetic complete (SC) drop-out medium before subsequent analyses. Cell viability was measured by diluting cells into SC medium after the recovery period and monitoring growth for 20 h. Cell viability as measured by outgrowth is virtually identical to viability measurements using FUN-1, a fluorescent dye that exhibits red punctate emission in the vacuoles of metabolically active live cells and diffuse green cytosolic emission in dead cells (Fig. S1, A–C) (37). As shown in Fig. 3A, yeast viability decreases with increasing [Pb2+]. The concentration of Pb2+ that inhibits growth by 50%, lethal dose 50 (LD50), varied between 25 and 500 μm over the course of our studies with different batches of medium for reasons that are not completely understood. As a consequence, all of our analyses were done at the Pb2+ LD50 dose and not necessarily at a constant Pb2+ concentration.
Figure 3.
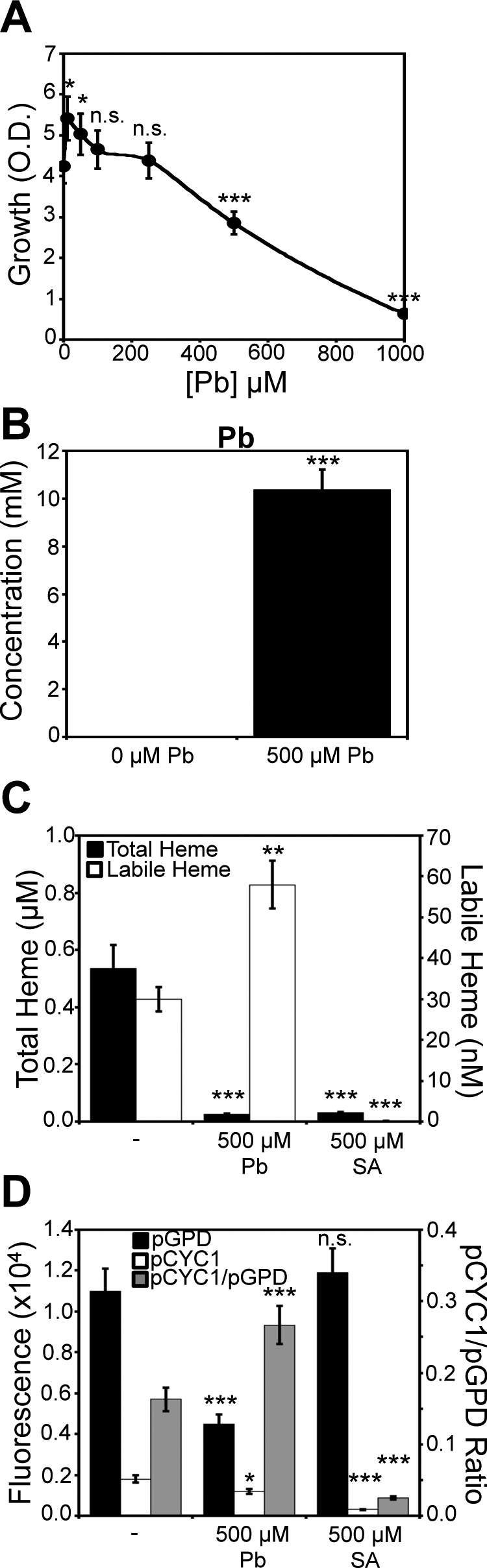
Labile heme and heme signaling is increased, but total heme is attenuated in response to Pb2+ toxicity. A, yeast cell viability, as measured by solution turbidity at an optical density (O.D.) of 600 nm, is diminished by exposure to Pb2+ in a dose-dependent manner. B, at the LD50 dose of Pb2+ (500 μm), cells hyperaccumulate up to 10 mm Pb2+, as measured by TXRF. C, a dose of 500 μm Pb2+ diminishes total heme to levels similar to 500 μm SA, but Pb2+ increases labile heme 2-fold, whereas SA does not. D, Hap1p activity in response to heme depletion by SA or Pb2+ is measured using the pCYC1-eGFP Hap1 reporter construct (pCYC1). To control for the Hap1-independent effects of Pb2+ on eGFP expression, we measured eGFP fluorescence in response to Pb2+ or SA using an allele of eGFP driven by the heme/Hap1-independent promoter, GPD (pGPD). The ratio of pCYC1 to pGPD eGFP expression (pCYC1/pGPD) is a measure of heme/Hap1-specific activation of CYC1. All data represent the mean ± S.D. (error bars) of triplicate cultures, and the statistical significance relative to untreated cells was assessed using a two-sample t test. *, p < 0.05; **, p < 0.005; ***, p < 0.001; n.s., not significant.
At the Pb2+ LD50 dose, elemental analysis using total reflection X-ray fluorescence (TXRF) spectroscopy confirms a significant enrichment of Pb2+ in yeast cells (Fig. 3B). In addition, a host of other bio-elements are impacted as a consequence of Pb2+ toxicity; phosphorus, sulfur, potassium, iron, and zinc are diminished, whereas calcium and copper are increased (Fig. S2). These effects are consistent with prior studies in a number of organisms and cell types demonstrating the impact of Pb2+ on various aspects of metal (37–42), phosphate (43, 44), and sulfur homeostasis (45–47).
Interestingly, whereas total heme is depleted due to Pb2+ exposure, as expected due to its known effects on inhibiting heme synthesis (34) and up-regulating heme degradation (35, 36), LH is completely maintained and, in fact, increases by 2-fold (Fig. 3C). In striking contrast, a dose of SA that depletes total heme to a similar degree as Pb2+ treatment attenuates both LH and total heme (Fig. 3C). Titration of Pb2+ over a broad concentration range indicates a dose-dependent increase in LH (Fig. 4A), decrease in total heme (Fig. 4B), and decrease in cell viability (Fig. 4C). The Pb2+-dependent depletion of total heme and increase in LH primarily occurs after the recovery period in SC medium; measurement of total heme and LH after the 3-h Pb2+ exposure in MES buffer has minimal effects on LH (Fig. 4D) and total heme (Fig. 4E) relative to cells allowed to recover in SC medium (Fig. 4, A and B). The changes in HS1-M7A sensor ratio are not due to artifacts associated with Pb2+-dependent changes in sensor expression, given that the emission of mKATE2, the heme-insensitive fluorophore, is constant over a wide range of Pb2+ doses (Fig. S3A). Furthermore, a variant of HS1 that cannot bind heme, HS1-M7A,H102A, which has the Met and His heme-coordinating residues mutated to Ala, does not exhibit Pb2+-dependent changes in fluorescence ratio (Fig. S3B). Altogether, these data indicate that the total and labile heme pools can be acted upon independently of each other in response to stress, and metabolically active cells are required for the observed changes in Pb2+-dependent LH and total heme.
Figure 4.

Pb2+ only perturbs total and labile heme after a recovery period following exposure to Pb2+. In cells expressing HS1-M7A, Pb2+ increases labile heme (A), decreases total heme (B), and diminishes cell viability (C) in a dose-dependent manner if cells are allowed to recover for 4 h in SCE medium following Pb2+ exposure in MES buffer. However, labile (D) and total heme (E) in cells expressing HS1-M7A immediately after the exposure to Pb2+ in MES buffer are not significantly affected. All data represent the mean ± S.D. (error bars) of triplicate cultures, and statistical significance was assessed using a two-sample t test. Black asterisks, statistical significance between the indicated pairwise comparisons of conditions; red asterisks, statistical significance relative to 0 μm Pb. *, p < 0.05; **, p < 0.005; ***, p < 0.001; n.s., not significant.
We next addressed whether the increase in LH in response to Pb2+ translates to changes in heme signaling. Toward this end, we measured Hap1 activity using the pCYC1-eGFP transcriptional reporter in cells conditioned with the LD50 dose of Pb2+. To account for the effects of Pb2+ on eGFP expression independently of Hap1 activation, we also expressed eGFP under control of the GPD promoter (pGPD), which is not a transcriptional target of Hap1. As shown in Fig. 3D, exposure to Pb2+ results in a ∼33% decrease in the eGFP fluorescence of the Hap1 reporter, whereas SA exposure results in a 6-fold decrease, despite a similar depletion in total heme.
Given that Pb2+ also affects eGFP fluorescence independently of Hap1, as evidenced by a ∼60% decrease in fluorescence of parallel cultures harboring the pGPD-eGFP construct, we normalized the fluorescence from pCYC1-driven expression of eGFP to the fluorescence from pGPD-driven expression of eGFP, giving a pCYC1/pGPD ratio. Exposure to Pb2+ results in a nearly 2-fold increase, from 0.16 to 0.26, in the pCYC1/pGPD ratio of eGFP fluorescence, an indicator of Hap1/pCYC1-specific activation. In striking contrast, cells conditioned with a dose of SA that results in a similar concentration of intracellular heme as the LD50 dose of Pb2+ exhibit a nearly ∼10-fold decrease in the pCYC1/pGPD ratio of eGFP fluorescence, with a nearly 10-fold decrease in fluorescence of the pCYC1-eGFP construct and minimal perturbation to fluorescence from the pGPD-eGFP construct.
Taken together, at minimum, these results suggest that, in response to Pb2+ toxicity, Hap1 activity is largely maintained, despite a >10-fold decrease in total heme. At maximum, these results suggest that “heme-specific” Hap1 activity, as recorded from the pCYC1/pGPD ratio, actually increases in response to Pb2+ stress. The maintenance of or increase in Hap1 activity, depending on the interpretation of the data, is presumably due to the Pb2+-induced increase in LH.
Pb2+-mediated heme depletion is largely due to a block in heme synthesis
We next sought to determine the mechanism by which intracellular heme is depleted in response to Pb2+ toxicity. The intracellular concentration of heme is governed by the relative rates of heme synthesis and degradation or export. To parse apart the effects of Pb2+ on these opposing processes, we tested the effects of Pb2+ on cells that had a defect in the ability to synthesize heme. First, we determined that Pb2+-dependent heme depletion occurs within the first 1.5 h of the 4-h recovery period in SC medium (Fig. 5A, 0 μm SA). Second, we found that in cells that had a defect in heme synthesis due to conditioning with 500 μm SA, Pb2+ had an attenuated effect on the loss of total heme. For instance, after 2.7 h, cells that could biosynthesize heme (Fig. 5A, 0 μm SA) exhibited a 2-fold and 6-fold decrease in total heme when treated with 125 and 250 μm Pb2+, respectively. On the other hand, in cells with ablated heme biosynthesis (Fig. 5A, 500 μm SA), both 125 and 250 μm Pb2+ resulted in a relatively modest ∼30% decrease in total heme. The diminished effect of Pb2+ on total heme in cells conditioned with the heme biosynthetic inhibitor SA suggested that Pb2+ depletes total heme by largely suppressing heme biosynthesis.
Figure 5.
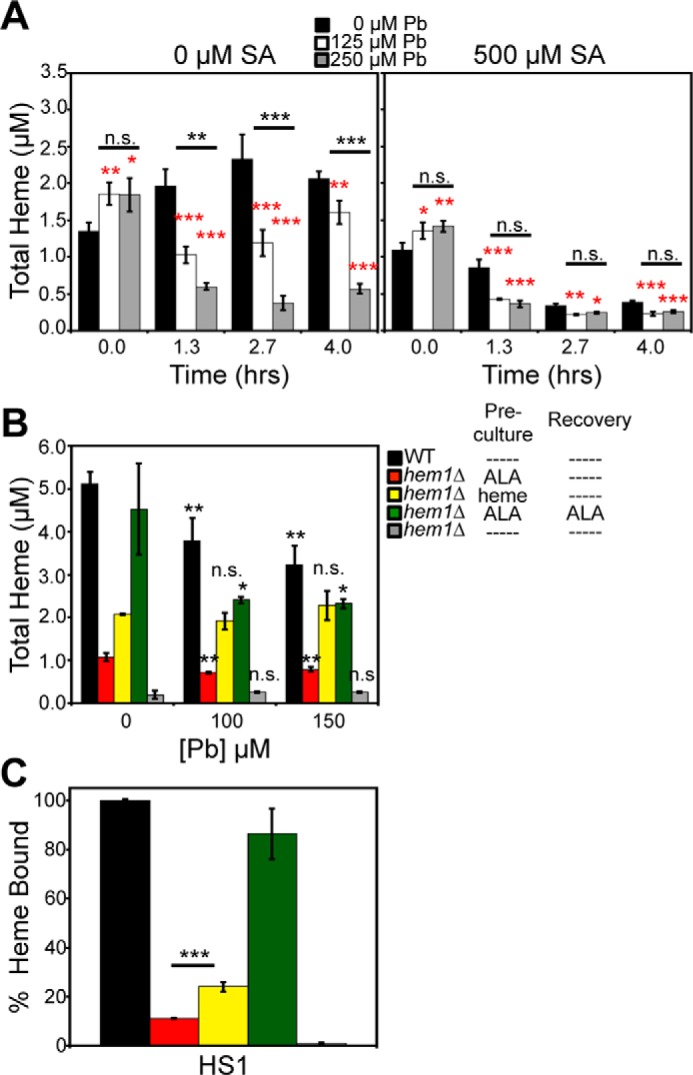
Pb2+-dependent attenuation of total heme is largely dependent on heme synthesis. A, WT cells untreated (left) or treated (right) with 500 μm SA and the indicated concentrations of Pb2+ demonstrate that Pb2+ has a larger effect on depleting total heme in cells that can properly synthesize heme. B and C, endogenous heme, but not exogenous heme, is degraded in a Pb2+-dependent manner. Total (B) and labile heme (C) were measured in HS1-expressing WT or hem1Δ cells conditioned with the indicated Pb2+ concentration and/or 200 μm ALA or 50 μm hemin chloride during the preculture and/or post-Pb2+ recovery period. All data represent the mean ± S.D. (error bars) of triplicate cultures, and statistical significance was assessed using a two-sample t test. In A, black asterisks represent the statistical significance between the indicated pairwise comparisons of conditions, and red asterisks represent the statistical significance relative to 0 μm Pb for each time point. In B, black asterisks represent the statistical significance relative to 0 μm Pb for each test condition. In C, the black asterisks represent the statistical significance between the indicated pairwise comparisons of strains. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
We next sought to further validate the observation that Pb2+ depletes heme by primarily affecting heme synthesis and not heme degradation or export. Toward this end, we utilized a hem1Δ strain, which lacks the first enzyme in the heme synthesis pathway, ALA synthase (ALAS), to test the effects of Pb2+ on the degradation of mitochondrially derived de novo synthesized heme or exogenously supplied heme. hem1Δ cells can only acquire heme by stimulating mitochondrial heme synthesis via supplementation with ALA, the product of ALAS, or by heme uptake via hemin supplementation. We supplemented hem1Δ cells with ALA or heme during a 16-h preculture in SCE medium and subjected them to Pb2+ exposure in MES buffer for 3 h, followed by a 4-h recovery in SCE medium and then analysis of total heme (Fig. 5B). WT and hem1Δ cells supplemented with ALA both in the preculture and during the recovery accumulated similar amounts of intracellular heme, underwent a Pb2+-dependent depletion of total heme (Fig. 5B, black and green columns), and exhibited >80% heme loading of the high-affinity heme sensor HS1 (Fig. 5C, black and green columns). hem1Δ cells treated with ALA only during the preculture, but not during the recovery, also exhibited a Pb2+-dependent attenuation in intracellular heme (Fig. 5B, red columns). However, the total amount of heme was ∼5-fold lower (Fig. 5B, red columns), and the heme-loading of HS1 was considerably reduced, ∼10% bound, compared with hem1Δ cells supplemented with ALA in both the preculture and the recovery medium (Fig. 5C, red columns), presumably because ALA was limiting because it is not supplied during the recovery phase. In striking contrast to ALA supplementation, hem1Δ cells supplemented with exogenous heme during the preculture, which contributed to a 2-fold increase in intracellular heme relative to ALA supplemented hem1Δ cells, did not exhibit the Pb2+-dependent depletion of heme (Fig. 5B, yellow columns). Exogenously supplied heme is still available to bind HS1, and in fact, its fractional saturation is similar to ALA-treated cells after normalizing for the change in total intracellular heme concentration (Fig. 5C, yellow columns). The observation that Pb2+ depletes endogenously synthesized heme and not exogenously supplied heme strongly suggests that Pb2+ primarily inhibits heme synthesis, with minor effects on heme degradation.
Given that heme synthesis is O2-dependent and our finding that Pb2+ largely inhibits heme synthesis, we next sought to test our prediction that Pb2+ should have minimal impact on total heme in the absence of O2. To test the O2 dependence of Pb2+-induced heme depletion, we subjected aerobically grown cells to Pb2+ exposure in MES buffer in an anoxic or normoxic environment, followed by recovery in anoxic or normoxic culture conditions and measurement of total heme, LH, and Pb2+ toxicity (Fig. 6). Quite strikingly, despite accumulating similar levels of Pb2+ between normoxic and anoxic Pb2+ exposures (Fig. S4), in the absence of O2, cells do not exhibit Pb2+-dependent heme depletion (Fig. 6A). On the other hand, the increase in LH in response to Pb2+ is similar between both normoxic and anoxic Pb2+ exposures (Fig. 6B). Notably, Pb2+-mediated cell toxicity is entirely O2-dependent, given that viability is not affected in anoxic cultures (Fig. 6C). Taken together, our data suggest that Pb2+-induced depletion of total heme only occurs in cells that are synthesizing heme de novo, further supporting the notion that Pb2+ largely inhibits heme synthesis.
Figure 6.
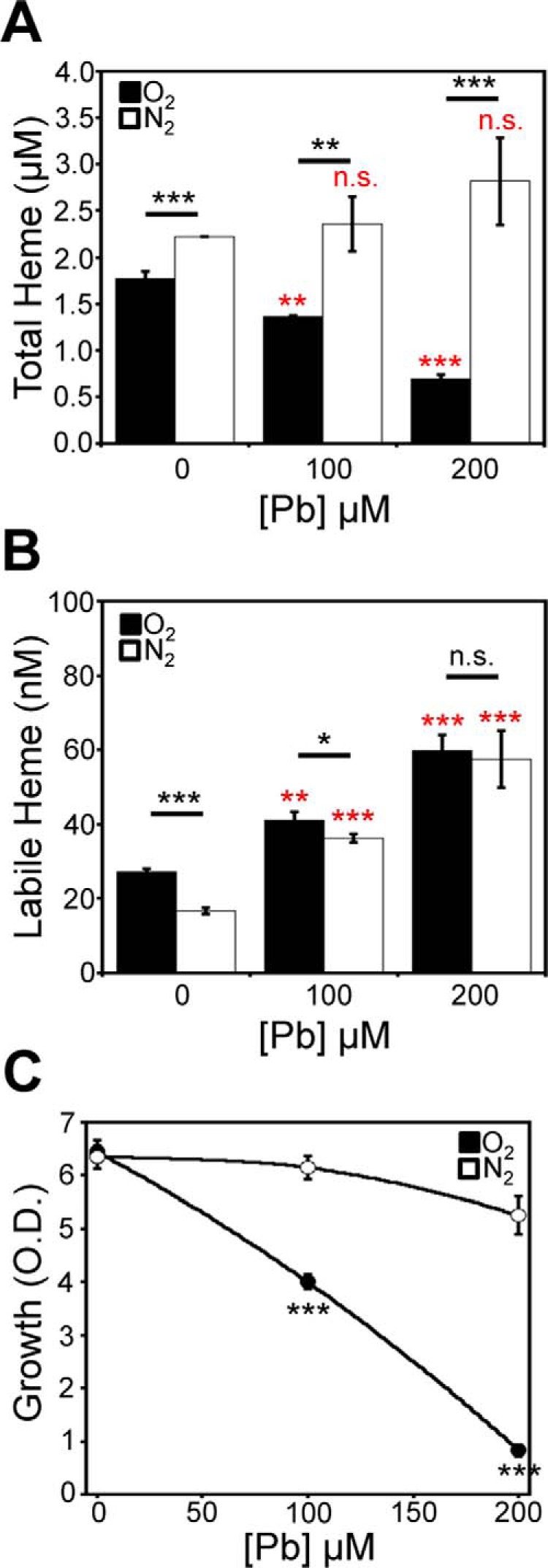
Pb2+-induced depletion of heme is O2-dependent. Cells expressing HS1-M7A were conditioned with the indicated concentration of Pb2+ and allowed to recover in an anoxic, nitrogen-rich (N2) atmosphere or in air (O2), and total heme (A), labile heme (B), and viability (C) were measured. All data represent the mean ± S.D. (error bars) of triplicate cultures, and statistical significance was assessed using a two-sample t test. Black asterisks, statistical significance between the indicated pairwise comparisons of conditions; red asterisks, statistical significance relative to 0 μm Pb. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
We next sought to determine whether a block in heme degradation or export could preserve total heme in the face of Pb2+ toxicity and affect labile heme. One established mechanism of heme degradation is via HO, an enzyme that oxidatively degrades heme into bilirubin, carbon monoxide (CO), and ferrous iron (Fe2+) (36). To test the role of HO in Pb2+-dependent heme depletion, we compared WT and hmx1Δ cells, which lack heme oxygenase 1 (48, 49), for Pb2+-dependent heme depletion. As shown in Fig. 7A, in the absence of Pb2+, hmx1Δ cells exhibit a ∼25% increase in total heme, consistent with prior studies in yeast and the known role of Hmx1 in heme degradation (48). However, both WT and hmx1Δ cells exhibit the Pb2+-induced depletion of heme to similar degrees, indicating that HO does not play a role in the loss of heme during Pb2+ toxicity. Both WT and hmx1Δ cells exhibited a Pb2+-dependent increase in LH (Fig. 7B). For reasons that are not entirely clear at this time, hmx1Δ cells appeared to be more resistant to Pb2+ toxicity (Fig. 7C). In contrast, prior work has demonstrated that Hmx1p confers resistance to various oxidative insults, including H2O2, diamide, and menadione (49), presumably due to released CO, bilirubin, and biliverdin.
Figure 7.

Pb2+-induced depletion of heme does not involve HMX1 or PUG1. A–C, hmx1Δ cells exhibit a Pb2+-dependent decrease in total heme (A) and increase in labile heme (B). C, hmx1Δ cells are more resistant to Pb2+ toxicity. D–F, pug1Δ cells exhibit a Pb2+-dependent decrease in total heme (D), increase in labile heme (E), and WT sensitivity to Pb2+ toxicity (F). All data represent the mean ± S.D. (error bars) of triplicate cultures, and statistical significance was assessed using a two-sample t test. Black asterisks, statistical significance between the indicated pairwise comparisons of conditions; red asterisks, statistical significance relative to 0 μm Pb. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant.
An alternative mechanism for cellular heme depletion is heme export. Many metazoans express heme exporters (e.g. FLVCR1) to aid in heme detoxification. S. cerevisiae expresses a porphyrin-heme exchanger, PUG1, which uptakes protoporphyrin IX and expels heme (50). To test the role of heme export via Pug1 during Pb2+ toxicity, we determined the extent to which heme is depleted in WT and pug1Δ cells exposed to increasing doses of Pb2+. As with hmx1Δ cells, pug1Δ cells exhibit a similar degree of Pb2+-induced heme depletion as WT cells (Fig. 7D), indicating that Pug1 does not affect the loss of heme during Pb2+ toxicity. Both WT and pug1Δ cells exhibit an increase in LH in response to Pb2+ (Fig. 7E) and similar sensitivities to Pb2+ toxicity (Fig. 7F).
Altogether, our data indicate that 1) Pb2+ depletes total heme primarily through its ability to inhibit heme synthesis, and 2) heme degradation and export pathways do not affect Pb2+-dependent changes in total and labile heme.
Pb2+-dependent increases in labile heme correlate with protein degradation
Most heme is associated with nonexchangeable binding sites in high-affinity hemoproteins. As such, we predicted that the increase in labile heme in response to Pb2+ may be associated with the degradation of hemoproteins and the release of heme into the labile heme pool. To test this hypothesis, we conducted one-dimensional PAGE analysis of cells conditioned with and without Pb2+ at the LD50 dose immediately after the 3-h exposure to Pb2+ in MES buffer and after the 4-h recovery phase (Fig. 8A). We found that immediately following exposure to Pb2+, protein expression is unaffected, whereas after the 4-h recovery, a number of proteins are degraded (Fig. 8A). The diminished protein expression during the recovery phase correlates with when we observe increased LH, suggesting that protein turnover and an increase in LH are linked. Notably, using tandem MS, we found that GAPDH, a component of the LH buffer (7), is retained during Pb2+ exposure (Fig. 8, A and B, and Table S2). Another glycolytic protein, enolase, is also maintained during Pb2+ exposure. On the other hand, Pb2+ has the capacity to degrade high-affinity hemoproteins. Ctt1, which is a heme-containing catalase enzyme, is degraded in a Pb2+-dependent manner (Fig. 8B). Altogether, our data are consistent with a model in which Pb2+-induced degradation of hemoproteins releases heme that contributes to the LH pool.
Figure 8.

Pb2+-dependent changes in heme homeostasis correlate with the degradation of a large fraction of the proteome and are affected by the proteosome. A, SDS-PAGE and Coomassie staining of lysates prepared from cells conditioned with or without an LD50 dose of Pb2+, 100 μm, that did or did not undergo a post-Pb2+ exposure recovery period. Tandem MS reveals that the high-intensity chromatic bands that are retained under Pb2+ stress, indicated by the arrows, are GAPDH and enolase (Table S2). B, the expression and activity of the high-affinity hemoprotein Ctt1p, a heme-catalase enzyme, is down-regulated in response to Pb2+ conditioning, whereas GAPDH expression, a constituent of the labile heme buffer, is maintained. All gels are representative of at least three independent cultures. C and D, effects of atg1Δ (C), rpn10Δ (C), and pep4Δ (D) deletion on Pb2+-dependent changes in labile heme were assessed. E and F, Pb2+-dependent changes in total heme (E) and growth (F) were assessed in WT and rpn10Δ cells. All labile and total heme measurements represent the mean ± S.D. (error bars) of triplicate cultures, whereas the growth data represent the mean ± S.D. of duplicate cultures. Statistical significance was assessed using a two-sample t test. Black asterisks, statistical significance between the indicated pairwise comparisons of conditions; red asterisks, statistical significance relative to 0 μm Pb. *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant; n.d., not detectable.
To test the hypothesis that Pb2+-dependent protein turnover releases heme from hemoproteins and increases LH, we assayed Pb2+-dependent changes in LH in yeast mutants defective in various protein degradation pathways, including vacuolar, autophagic, and proteasomal degradation. We found that a proteasome mutant, rpn10Δ (51), exhibited Pb2+-dependent and -independent changes in LH (Fig. 8C), whereas mutants defective in vacuolar (pep4Δ) (52) (Fig. 8D) or autophagic (atg1Δ) (53) (Fig. 8C) pathways did not have altered LH. Under nonstressed conditions, WT cells exhibited an LH concentration of ∼5 nm, corresponding to an HS1-M7A fractional saturation of 17%. On the other hand, HS1-M7A was ∼0% bound to heme when expressed in rpn10Δ cells, which corresponds to an LH concentration of <1 nm. However, Pb2+ induced a greater increase in LH (Fig. 8C) and decrease in total heme (Fig. 8E) in rpn10Δ cells relative to WT. Moreover, the growth of rpn10Δ cells was more sensitive to Pb2+ toxicity than WT (Fig. 8F). In total, these results indicate that the proteasome positively regulates LH, and loss of proteasomal function sensitizes cells to Pb2+-dependent effects on heme homeostasis.
Discussion
Heme synthesis and degradation are tightly coordinated so as to minimize the accumulation of potentially cytotoxic “free” or labile heme (LH) (4–6). As a consequence, the concentration and physiological role of LH in biology has been controversial (1, 2). Whereas estimates for the concentration of LH across various cell types have spanned subpicomolar to micromolar quantities (1, 2, 5, 54, 55), the use of genetically encoded heme sensors in yeast and various nonerythroid human cells lines has established a consensus range for cytosolic LH that spans 10–100 nm, representing up to 10% of the total heme concentration (7, 56). However, few studies have directly probed the contribution of steady-state LH as a heme source for hemoproteins and heme signaling. Herein, we established a functional role for LH in regulating heme signaling and demonstrated that LH is preferentially consumed relative to total heme when cells become heme-depleted.
We find that modestly depleting total heme by 25% with succinylacetone, an inhibitor of the second enzyme in the heme biosynthetic pathway, ALAD, results in a much larger perturbation to LH, depleting it more than 10-fold, from 10 nm to <1 nm, possibly as low as ∼2 pm (Fig. 2). The sensitivity of the LH pool toward heme depletion relative to total heme is an indication that LH is mobilized to support heme-dependent processes when cells are confronted with diminished heme synthesis. In other words, LH re-equilibrates with other high-affinity and/or poorly exchangeable heme binding sites that may become vacant when heme is depleted. These results are consistent with prior pulse-chase experiments that utilized radiolabeled ALA and/or heme sources to demonstrate that P450 enzymes equilibrate with LH (23). Altogether, these observations may have significant implications for conceptualizing new heme-based therapies for porphyrias, a family of inherited disorders associated with defects in heme biosynthesis. For instance, treatment methods designed to increase LH via supplementation with appropriate heme complexes may form the basis for alleviating the symptoms of heme scarcity (57).
To address the role of LH in heme-dependent functions, we induced a site-specific heme chelator in the cytosol via the overexpression of Cyt b562 and assessed its effect on heme signaling through the yeast heme-regulated transcription factor Hap1 (Fig. 1). We found that sequestration of LH inhibited Hap1 activity, demonstrating that LH can control heme-signaling processes. Importantly, these results indicate that increasing heme synthesis, a metabolically demanding process, is not the only means to activate heme signaling, as was previously suggested for activation of Hap1 (10, 58) and metabolic cycling in yeast (59, 60), as well as heme regulation of the circadian clock through the nuclear receptors, Rev-erb-α and Rev-erb-β, in mammals (61).
To understand the relationship between total heme, LH, and heme signaling during adaptation to cellular stress, we subjected yeast cells to an environmentally relevant toxicant well known to affect heme homeostasis, Pb2+ (34). In yeast, Pb2+ toxicity suppresses metabolic activity and proliferation through a mechanism that requires new protein synthesis (37). In addition, mitochondria are a major target of Pb2+ toxicity and are the source of Pb2+-induced reactive oxygen species production (62). Heavy metal sequestration by vacuoles and binding to GSH and metallothioneins are important detoxification pathways for Pb2+ (63).
A number of previously established yeast models for Pb2+ toxicity employ exposures spanning 0.1–10 mm, which is high relative to the 240 nm [Pb2+] threshold in patient blood samples used to initiate public health actions (64), the 1–5 μm blood [Pb2+] associated with inhibition of heme biosynthesis in humans (65, 66), or the 0.1–250 μm [Pb2+] used in mammalian cell culture models of Pb2+ toxicity (67, 68). However, it is important to note that the biochemical features of Pb2+ toxicity at these high concentrations in yeast phenocopy many of the hallmarks of Pb2+ toxicity in mammalian cell lines at lower concentrations (37, 62, 63, 69–71), including inhibition of heme synthesis (current work). The higher Pb2+ exposure concentrations required for yeast may reflect differences in Pb2+ uptake, efflux, or intracellular bioavailability.
Whereas Pb2+ has the capacity to deplete cellular heme through its ability to inhibit two enzymes in the heme biosynthetic pathway (ALAD, also the target of SA, and ferrochelatase) and up-regulate the expression of the heme-degrading enzyme HO (34–36), we found that, in yeast, Pb2+ significantly attenuates total heme primarily due to the inhibition of heme synthesis (Figs. 5 and 6). However, rather surprisingly and paradoxically, we found that Pb2+ significantly increases LH (Fig. 3C). Given our previous findings that thiol-specific alkylating agents impact the NO-mediated mobilization of LH (7) and the well-documented interactions between Pb2+ and cysteine residues (72), we propose that Pb2+ may liberate heme in cells from certain thiol-containing heme-binding sites. Alternatively, because Pb2+ induces the degradation of a large fraction of the proteome, the increase in LH could simply be a result of heme that is released from hemoproteins that are being turned over. Consistent with this model, we found that a mutant that has a defect in proteasomal function, rpn10Δ, has less LH than WT cells (Fig. 8C). However, it is unclear why rpn10Δ cells exhibit a greater magnitude increase in LH (Fig. 8C) and decrease in total heme (Fig. 8E) in response to Pb2+ stress. One explanation that could account for the former is that Pb2+ is directly or indirectly inducing the release of heme from a hemoprotein target that is stabilized due to the attenuation in proteasome function in rpn10Δ cells. Given that previous studies have demonstrated that Pb2+ positively regulates the proteasome (73) and heme and ALAD act as negative regulators of the proteasome (74, 75), there is a complex mosaic of competing effects that might account for the Pb2+-dependent changes in heme homeostasis in WT and rpn10Δ cells. We are currently elucidating the molecular mechanisms underlying heme regulation of LH and the proteasome.
The Pb2+-induced increase in LH seems to impact heme signaling via Hap1 (Fig. 3D). Indeed, despite the attenuation in total heme concentration due to Pb2+ toxicity (Fig. 3C), we still observe a level of Hap1 activity that is much greater relative to SA-mediated heme depletion (Fig. 3D), which attenuates both LH and total heme (Fig. 3C).
The observation of an increased LH pool in response to Pb2+ may have implications for the pathology of Pb2+ toxicity. For instance, the increase in LH in response to Pb2+ may contribute to a cytotoxic heme pool (5, 6) that leads to the oxidative stress associated with Pb2+ toxicity (76, 77). Alternatively, given that there are a number of heme-regulated transcription factors, kinases, and ion channels (1, 2), the increase in LH in response to Pb2+ may be required to activate heme-based signaling pathways important for adaptation to heavy metal stress. The specific physiological consequences of increased LH in response to Pb2+ stress remain to be fleshed out.
Altogether, our studies demonstrate the functional importance of LH in heme utilization, especially during stress associated with heme depletion and Pb2+ toxicity. Our future work will involve probing the specific mechanisms by which LH can be coupled to heme utilization as well as the mechanisms underlying the Pb2+-mediated increase in LH and the physiological consequences of this action.
Experimental procedures
Yeast strains, transformations, and growth conditions
S. cerevisiae strains used in this study were derived from BY4741 (MATa, his3Δ1, leu2Δ0, met15Δ0, ura3Δ0). pug1::KANMX4, hmx1::KANMX4, cta1::KANMX4, and ctt1::KANMX4 strains were obtained from the yeast gene deletion collection (Thermo Fisher Scientific), and the hem1::HIS3 strain was described previously (7). Yeast transformations were performed by the lithium acetate procedure (78). Strains were maintained at 30 °C on either enriched yeast extract (1%)-peptone (2%)–based medium supplemented with 2% glucose (YPD), or SC medium supplemented with 2% glucose and the appropriate amino acids to maintain selection (7). Cells cultured on solid medium plates were done so with YPD or SC medium supplemented with 2% agar (7). Selection for yeast strains containing the KanMX4 marker was done with YPD agar plates supplemented with G418 (200 μg/ml) (7). WT cells treated with the heme synthesis inhibitor, SA, and hem1Δ cells were cultured in YPD or SC medium supplemented with 50 μg/ml ALA or 15 mg/ml ergosterol and 0.5% Tween 80 (YPDE or SCE, respectively) (7, 79).
Labile heme and total heme depletion
To sequester labile heme in the cytosol, we generated a WT yeast strain expressing an episomal plasmid (p316-GAL) containing an allele of the high-affinity hemoprotein Cyt b562 driven by the galactose-inducible promoter (pGAL); the plasmid is referred to as pGAL-Cyt b562. A control strain expressing the EV (pGAL-EV) was also generated. For labile heme measurements, the strains expressing pGAL-Cyt b562 or pGAL-EV also co-expressed the previously described heme sensor, HS1-M7A, using an episomal plasmid (pRS415) that drives sensor expression with the GPD promoter (7). For Hap1 activity measurements, the strains expressing pGAL-Cyt b562 or pGAL-EV also co-expressed the previously described pCYC1-eGFP Hap1 reporter, which is an episomal plasmid (pRS415) that drives eGFP expression using the CYC1 promoter, a transcriptional target of Hap1 (7). To induce Cyt b562 expression, cells were cultured in SC-URA-LEU medium to maintain selection of both Cyt b562 and the heme sensor, HS1-M7A, or pCYC1-eGFP. Instead of using 2% glucose, which will repress the expression of the GAL-inducible promoter, we cultured cells in 2% raffinose and either 1.0% galactose (inducing conditions) or vehicle (sterile water, noninducing conditions). Parallel control cultures were treated with 500 μm SA to deplete intracellular heme. All cultures were seeded at an initial optical density of A600 nm = 0.005 and cultured until cells reached a final density of A600 nm ∼ 1.0, which typically took 14–16 h. Following growth, cells were harvested, washed, and resuspended in PBS, and sensor or eGFP fluorescence was measured as described under “Labile heme quantification” and “Hap1 activity.”
To deplete total heme, cells were cultured with the indicated concentrations of the heme biosynthetic inhibitor, SA, in an appropriate SCE drop-out medium. All cultures were seeded at an initial optical density of A600 nm = 0.005 and cultured until cells reached a final density of A600 nm ∼ 1.0, which typically took 14–16 h. Following growth, cells were harvested, washed, and processed for determination of labile or total heme as described under “Labile heme quantification” and “Total heme quantification.”
Yeast model of Pb2+ toxicity
Due to the insolubility of Pb(NO3)2 in yeast medium, we subjected exponential phase yeast cells to varying doses of Pb2+ in MES buffer for 3 h, a medium in which Pb(NO3)2 is soluble. Cells were precultured in an appropriate SC drop-out medium to a final density of A600 nm ∼ 1.0. Following washing with sterile water, cells were resuspended in 10 mm MES buffer containing varying concentrations of Pb(NO3)2 at a density of 1 OD/ml and mixed every 15 min at 25 °C for 3 h. Following exposure to Pb2+ in MES buffer, cells were thoroughly washed with sterile water, resuspended in an appropriate SC drop-out mixture to a final density of A600 nm ∼ 1.0, and allowed to recover for 4 h, while shaking at 220 rpm at 30 °C. Cell viability was measured by diluting the cells to an initial density of A600 nm = 0.01 in an appropriate SC drop-out medium after the recovery phase, and solution turbidity was recorded by measuring A600 nm after 20 h of growth shaking at 220 rpm and 30 °C.
To assess the effects of Pb2+ toxicity in an anaerobic environment, the Pb2+ toxicity model described above was modified as follows: the Pb2+ exposure in MES buffer was accomplished in degassed MES buffer with varying concentrations of Pb2+ in a COY anaerobic chamber maintained with an inert atmosphere of 95% N2 and 5% H2. Following Pb2+ exposure, cells were washed with sterile degassed water and allowed to recover in degassed SC medium, all anaerobically in the COY anaerobic chamber. Cell viability was measured by diluting the anaerobic cultures into in an appropriate SC drop-out medium after the recovery phase, and solution turbidity was recorded by measuring A600 nm after 16 h of growth shaking at 220 rpm and 30 °C in air.
All analytical analyses, including for labile or total heme, were conducted immediately after the 3-h Pb2+ exposure in MES buffer (pre-recovery) or after the 4-h recovery in SC medium (post-recovery).
Viability measurements using FUN-1
Cells were grown as indicated under “Yeast model of Pb2+ toxicity.” After conditioning cells in MES buffer with or without Pb2+, 1 OD of cells were pelleted and washed once with 1 ml of sterile MilliQ water and once with 500 μl of 10 mm HEPES with 2% glucose (w/v) (HG buffer). Cells were pelleted again, resuspended in 300 μl of HG buffer, and then treated with 24.5 μl of 200 μm FUN-1 (Thermo Fisher Scientific) to a final concentration of 15 μm. The 200 μm FUN-1 working stock solution was prepared by diluting a 10 mm DMSO FUN-1 stock solution into HG buffer. Cells were allowed to incubate in the dark at 30 °C for 30 min and then washed three times in HG buffer. Cells were imaged on glass slides with coverslips using the Cytation 3 imaging plate reader (Biotek) with eGFP and Texas Red Filter Cubes. In stained cells, the observation of red puncta was used to score viable cells, and the observation of diffuse green fluorescence was used to score dead cells. On average, ∼100 cells were analyzed per sample.
Labile heme quantification
Measurements of labile heme were accomplished as described previously (7). For all heme sensor fluorescence measurements, following cell growth, cells were washed in water and resuspended in PBS at a density between 3 and 5 A600 nm/ml, or 6 × 107 to 1 × 108 cells/ml. Fluorescence was recorded on a Synergy Mx multimodal plate reader using black Greiner Bio-one flat-bottom fluorescence plates. eGFP and mKATE2 fluorescence was recorded using excitation and emission wavelength pairs of 488 and 510 nm and 588 and 620 nm, respectively. Background fluorescence of cells not expressing the heme sensors was recorded and subtracted from the eGFP and mKATE2 fluorescence values. The sensor eGFP/mKATE2 fluorescence ratio (Rexpt) is a qualitative indicator of labile or bioavailable heme, with a low ratio indicating a high concentration of labile heme and a high ratio indicating a low concentration of labile heme.
For quantitative labile heme monitoring, we can convert Rexpt values to the fractional heme saturation of the sensor (% heme bound) (Equation 1) or, if the sensor heme dissociation constant is known (KD), the concentration of labile heme (Equation 2). Both metrics require that the eGFP/mKATE2 sensor fluorescence ratio is known when the sensor is 100% bound (Rmax) or 0% bound (Rmin) (7). Based on a 1:1 heme-binding model, the fractional saturation, % Bound, of the sensor can be calculated according to Equation 1 (7).
| (Eq. 1) |
The labile heme concentration can be calculated according to Equation 2 (7, 80).
| (Eq. 2) |
Rexpt is the eGFP/mKATE2 fluorescence ratio under any given experimental conditions, Rmin is the eGFP/mKATE2 fluorescence ratio when 0% of the sensor is bound to heme, Rmax is the eGFP/mKATE2 fluorescence ratio when 100% of the sensor is bound to heme, FminmKATE2 is the mKATE2 emission intensity when 0% of the sensor is bound to heme, and FmaxmKATE2 is the mKATE2 emission intensity when 100% of the sensor is bound to heme. The FminmKATE2/FmaxmKATE2 ratio is typically taken to be 1, given that mKATE2 fluorescence emission is not significantly perturbed upon heme binding to the sensor (7). Determination of Rmax and FmaxmKATE2 involves recording eGFP and mKATE2 fluorescence after digitonin permeabilization of cells and incubation with 50 μm heme. Briefly, 3–5 A600 nm/ml of cells are resuspended in PBS with 100 μg/ml of digitonin, 1 mm ascorbate, and 50 μm hemin chloride. After a 30-min incubation at 30 °C, cells are harvested, washed, and resuspended in PBS buffer before recording of fluorescence. Given that the high affinity heme sensor, HS1, is quantitatively saturated with heme and its fluorescence properties are virtually identical to HS1-M7A, we can also determine Rmax and FmaxmKATE2 from parallel WT cultures expressing HS1 (7). Determination of Rmin and FminmKATE2 involves recording eGFP and mKATE2 fluorescence after cells are treated with the heme biosynthesis inhibitor SA (21) or from hem1Δ cells cultured in parallel (7).
Total heme quantification
Measurements of total heme were accomplished using a fluorimetric assay designed to measure the fluorescence of protoporphyrin IX upon the release of iron from heme as described previously (81). For all total heme measurements, following cell growth, 2 × 108 cells were harvested, washed in sterile water, and resuspended in 500 μl of 20 mm oxalic acid and stored in a closed box at 4 °C overnight (16–18 h). Next, an equal volume (500 μl) of 2 m oxalic acid was added to the cell suspensions in 20 mm oxalic acid. The samples were split, with half the cell suspension transferred to a heat block set at 95 °C and heated for 30 min and the other half of the cell suspension kept at room temperature (∼25 °C) for 30 min. All suspensions were centrifuged for 2 min on a table-top microcentrifuge at 21,000 × g, and the porphyrin fluorescence (excitation 400 nm, emission 620 nm) of 200 μl of each sample was recorded on a Synergy Mx multimodal plate reader using black Greiner Bio-one flat-bottom fluorescence plates. Heme concentrations were calculated from a standard curve prepared by diluting 500–1500 μm hemin chloride stock solutions in 0.1 m NaOH into MilliQ water, which was then added back to extra cell samples as prepared above. To calculate heme concentrations, the fluorescence of the unboiled sample (taken to be the background level of protoporphyrin IX) is subtracted from the fluorescence of the boiled sample (taken to be the free base porphyrin generated upon the release of heme iron). The cellular concentration of heme is determined by dividing the moles of heme determined in this fluorescence assay and dividing by the number of cells analyzed, giving moles of heme per cell, and then converting to a cellular concentration by dividing by the volume of a yeast cell, taken to be 50 fl (7). This fluorescence assay gives similar qualitative trends between samples as an HPLC assay for heme we employed previously (7), but the absolute concentrations tend to be consistently 3–5-fold higher (data not shown).
Hap1 activity
After growth, cells expressing p415-CYC1-eGFP, or eGFP driven by the Hap1-regulated CYC1 promoter, were washed in sterile water and resuspended in PBS to a concentration of 1 × 108 cells/ml, and 100 μl was used to measure eGFP fluorescence (excitation 488 nm, emission 510 nm). Background autofluorescence of cells not expressing eGFP was recorded and subtracted from the p415-CYC1-eGFP-expressing strains. To account for heme/Hap1-independent changes in eGFP expression/fluorescence, we also cultured cells expressing p415-GPD-eGFP, a plasmid expressing eGFP under control of the heme/Hap1-independent GPD promoter.
Immunoblotting
After culturing, cells were harvested, washed in ice-cold MilliQ water, and lysed in two pellet volumes of phosphate buffer supplemented with protease inhibitors as described previously (7, 82). Lysis was achieved at 4 °C using one pellet volume of zirconium oxide beads and a bead beater (Bullet Blender, Next Advance) on a setting of 8 for 3 min (7). Lysate protein concentrations were determined by the Bradford method (Bio-Rad), and 14% Tris/glycine gels (Invitrogen) were employed for SDS-PAGE (7). α-GAPDH rabbit polyclonal antibodies (Genetex, GTX100118) and a goat α-rabbit secondary antibody conjugated to a 680-nm emitting fluorophore (Biotium) were used to probe for GAPDH. Yeast cytosolic catalase, Cttp1, was probed using a custom antibody generated by Genscript's custom antibody service (Poly Express Premium Service, SC1676). An α-Ctt1p antibody was raised in rabbits against an amino acid 1–320 fragment of Ctt1p. The α-Ctt1p antibody was validated in yeast by comparing immunoreactivity between WT, ctt1Δ, and cta1Δ cells, the latter being a deletion mutant of a peroxisomal/mitochondrial catalase, Cta1p, unrelated to Ctt1p (Fig. S5). All gels were imaged on a LI-COR Odyssey IR imager (7, 82).
Catalase activity
After culturing, cells were harvested and lysed in phosphate buffer, and 10 μg of protein lysate were subjected to native PAGE on a 10% Tris/glycine gel (Invitrogen). After electrophoresis, an in-gel activity stain was utilized to measure catalase activity (7, 83). Briefly, a catalase-staining solution containing 1 part dopamine (20 mg/ml) in pH 8 0.2 m KPi buffer, 1 part para-phenylenediamine (3.5 mg/ml) in pH 8 0.2 m KPi, 1 part 15% H2O2, and 2 parts DMSO were mixed in the order listed. The staining solution was added directly to the gel and allowed to stain for 2 min, followed by rinsing in MilliQ water and imaging.
Plasmids
All yeast expression plasmids used in this study are listed in Table S1 and were described previously, except for pDH039, the plasmid expressing cytochrome b562 (Cyt b562) driven by a GAL-inducible promoter, pGAL-Cyt b562. This plasmid was constructed by amplifying the coding sequence of Cyt b562 from the HS1 sensor (7). The following primers were utilized to amplify a BamHI/XbaI fragment of Cyt b562: prDH025, 5′-CCTTTGGTGGCTCTGGATCCATGGCAGATCTGGAAGACAACATGG-3′; prDH026, 5′-GGTCAGTTTGCCACCTCTAGATCATCTGTATTTCTGATGATATGC-3′.
Following amplification and digestion with BamHI and XbaI, the Cyt b562 coding sequence was ligated into BamHI- and XbaI-digested p316-GAL1 (17). The amino acid sequence of Cyt b562 is as follows, with the heme-coordinating residues in boldface type: ADLEDNMETLNDNLKVIEKADNAAQVKDALTKMRAAALDAQKATPPKLEDKSPDSPEMKDFRHGFDILVGQIDDALKLANEGKVKEAQAAAEQLKTTRNAYHQKYR.
TXRF spectroscopy
Elemental analysis of cells, and in particular metal analysis of lead, first row transition elements, phosphorus, and sulfur, was accomplished by TXRF on a Bruker S2 Picofox TXRF as described previously (7). Briefly, following growth, cells were washed sequentially in ice-cold Tris-EDTA, pH 8.0, buffer and MilliQ water and then finally resuspended in MilliQ water to a density of 2 × 109 cells/ml. 2 μl of a cell suspension, spiked with 1 ppm of a gallium internal standard, were spotted onto a quartz sample disc, and atomic fluorescence emission spectra were collected according to the manufacturer's recommendations (7). The cellular metal concentrations were determined by assuming a yeast cell volume of 50 fl and that a solution turbidity of A600 nm = 1.0 is equivalent to 2 × 107 cells/ml (7).
In-gel protein digestion
In-gel protein digestion was conducted as described previously (84), with modifications. Briefly, selected protein bands were excised from the Coomassie-stained gel, diced into small pieces, and then destained with HPLC-grade water (Avantor) and 1:1 acetonitrile (ACN)/ammonium bicarbonate (ABC) (Sigma-Aldrich). The destained gel pieces were then dehydrated with multiple ACN washes until rock hard, followed by air drying for ∼15 min. The gel pieces were rehydrated for 30 min with 10 mm DTT (Sigma-Aldrich) to reduce disulfide bonds, followed by replacement with 55 mm iodoacetic acid (Sigma-Aldrich) and 45 min shaking at 750 rpm in the dark to alkylate the reduced thiols. After reduction/alkylation, the gel pieces were once again washed and dehydrated as before and then chilled on ice for 15 min. To the chilled gel pieces, 30 μl of 20 μg/ml sequencing grade modified trypsin (Promega) was added for 30 min on ice. Excess trypsin solution was then removed and replaced with 100 μl of 50 mm ABC, and the pieces were incubated overnight at 37 °C with shaking at 750 rpm. Resultant proteolytic peptides were extracted by two rounds of dehydration using 100 μl of ACN and collection of the resulting extract into low-retention microcentrifuge tubes, which were frozen solid at −80 °C and then sublimated by centri-vapping. The dried peptides were reconstituted by sonication in 5% ACN, 0.1% formic acid and stored at −80 °C before analysis.
Mass spectrometry
LC-MS analysis of peptides produced by in-gel digestion was carried out with an UltiMateTM 3000 RSLCnano UPLC system (Dionex) with Acclaim PepMap RSLC column (75 μm × 25-cm nanoViper C18 2 μm, 100 Å) coupled to a Q-Exactive Plus Orbitrap mass spectrometer (Thermo Scientific) run in data-dependent acquisition mode (top 15). Resultant RAW files were analyzed using Proteome Discoverer version 2.1 with embedded SEQUEST search algorithm operating with an allowable 1% false discovery rate, wherein the S. cerevisiae protein sequence database was used as the target for spectral matching. Only high-confidence peptide spectral matches were used for protein identification, and they are reported in Table S2.
Author contributions
D. A. H., R. H., H. K., O. M.-G., M. P. T., and A. R. R. designed research. D. A. H., R. H., H. K., O. M.-G., and A. R. R. performed research. All authors analyzed data. D. A. H. and A. R. R. wrote the paper with input from all co-authors.
Supplementary Material
Acknowledgment
We thank Dr. Rebecca Donegan for assistance in developing the porphyrin fluorescence assay for total heme.
This work was supported by the National Institutes of Health Grants ES025661 (to A. R. R.) and GM118744 (to A. R. R. with a sub-award to M. P. T.), National Science Foundation Grant MCB-1552791 (to A. R. R.), the Blanchard Professorship (to A. R. R.), and start-up funding from the Georgia Institute of Technology (to A. R. R. and M. P. T.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Tables S1 and S2 and Figs. S1–S5.
- LH
- labile heme
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- Cyt b562
- cytochrome b562
- GAL
- galactose
- eGFP
- enhanced green fluorescent protein
- EV
- empty vector
- RAF
- raffinose
- SA
- succinylacetone
- ALA
- aminolevulinic acid
- ALAD
- ALA dehydratase
- ALAS
- ALA synthase
- SC
- synthetic complete
- LD50
- lethal dose 50
- TXRF
- total reflection X-ray fluorescence
- HO
- heme oxygenase
- ACN
- acetonitrile
- ABC
- ammonium bicarbonate
- NHE
- normal hydrogen electrode.
References
- 1. Reddi A. R., and Hamza I. (2016) Heme mobilization in animals: a metallolipid's journey. Acc. Chem. Res. 49, 1104–1110 10.1021/acs.accounts.5b00553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hanna D. A., Martinez-Guzman O., and Reddi A. R. (2017) Heme gazing: illuminating eukaryotic heme trafficking, dynamics, and signaling with fluorescent heme sensors. Biochemistry 56, 1815–1823 10.1021/acs.biochem.7b00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Severance S., and Hamza I. (2009) Trafficking of heme and porphyrins in metazoa. Chem. Rev. 109, 4596–4616 10.1021/cr9001116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hamza I., and Dailey H. A. (2012) One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochim. Biophys. Acta 1823, 1617–1632 10.1016/j.bbamcr.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sassa S. (2004) Why heme needs to be degraded to iron, biliverdin IXalpha, and carbon monoxide? Antioxid. Redox Signal. 6, 819–824 10.1089/ars.2004.6.819 [DOI] [PubMed] [Google Scholar]
- 6. Kumar S., and Bandyopadhyay U. (2005) Free heme toxicity and its detoxification systems in human. Toxicol. Lett. 157, 175–188 10.1016/j.toxlet.2005.03.004 [DOI] [PubMed] [Google Scholar]
- 7. Hanna D. A., Harvey R. M., Martinez-Guzman O., Yuan X., Chandrasekharan B., Raju G., Outten F. W., Hamza I., and Reddi A. R. (2016) Heme dynamics and trafficking factors revealed by genetically encoded fluorescent heme sensors. Proc. Natl. Acad. Sci. U.S.A. 113, 7539–7544 10.1073/pnas.1523802113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pfeifer K., Kim K. S., Kogan S., and Guarente L. (1989) Functional dissection and sequence of yeast HAP1 activator. Cell 56, 291–301 10.1016/0092-8674(89)90903-3 [DOI] [PubMed] [Google Scholar]
- 9. Zhang L., Bermingham-McDonogh O., Turcotte B., and Guarente L. (1993) Antibody-promoted dimerization bypasses the regulation of DNA binding by the heme domain of the yeast transcriptional activator HAP1. Proc. Natl. Acad. Sci. U.S.A. 90, 2851–2855 10.1073/pnas.90.7.2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang L., and Hach A. (1999) Molecular mechanism of heme signaling in yeast: the transcriptional activator Hap1 serves as the key mediator. Cell. Mol. Life Sci. 56, 415–426 10.1007/s000180050442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang L., Hach A., and Wang C. (1998) Molecular mechanism governing heme signaling in yeast: a higher-order complex mediates heme regulation of the transcriptional activator HAP1. Mol. Cell. Biol. 18, 3819–3828 10.1128/MCB.18.7.3819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Feng Y., Sligar S. G., and Wand A. J. (1994) Solution structure of apocytochrome b562. Nat. Struct. Biol. 1, 30–35 10.1038/nsb0194-30 [DOI] [PubMed] [Google Scholar]
- 13. Feng Y. Q., and Sligar S. G. (1991) Effect of heme binding on the structure and stability of Escherichia coli apocytochrome b562. Biochemistry 30, 10150–10155 10.1021/bi00106a011 [DOI] [PubMed] [Google Scholar]
- 14. Itagaki E., Palmer G., and Hager L. P. (1967) Studies on cytochrome b562 of Escherichia coli. II. Reconstitution of cytochrome b562 from apoprotein and hemin. J. Biol. Chem. 242, 2272–2277 [PubMed] [Google Scholar]
- 15. Nikkila H., Gennis R. B., and Sligar S. G. (1991) Cloning and expression of the gene encoding the soluble cytochrome b562 of Escherichia coli. Eur. J. Biochem. 202, 309–313 10.1111/j.1432-1033.1991.tb16377.x [DOI] [PubMed] [Google Scholar]
- 16. Robinson C. R., Liu Y., Thomson J. A., Sturtevant J. M., and Sligar S. G. (1997) Energetics of heme binding to native and denatured states of cytochrome b562. Biochemistry 36, 16141–16146 10.1021/bi971470h [DOI] [PubMed] [Google Scholar]
- 17. Kiktev D. A., Patterson J. C., Müller S., Bariar B., Pan T., and Chernoff Y. O. (2012) Regulation of chaperone effects on a yeast prion by cochaperone Sgt2. Mol. Cell. Biol. 32, 4960–4970 10.1128/MCB.00875-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barker P. D., Nerou E. P., Cheesman M. R., Thomson A. J., de Oliveira P., and Hill H. A. (1996) Bis-methionine ligation to heme iron in mutants of cytochrome b562. 1. Spectroscopic and electrochemical characterization of the electronic properties. Biochemistry 35, 13618–13626 10.1021/bi961127x [DOI] [PubMed] [Google Scholar]
- 19. Reddi A. R., Reedy C. J., Mui S., and Gibney B. R. (2007) Thermodynamic investigation into the mechanisms of proton-coupled electron transfer events in heme protein maquettes. Biochemistry 46, 291–305 10.1021/bi061607g [DOI] [PubMed] [Google Scholar]
- 20. Conant J. B., and Tongberg C. O. (1930) The oxidation-reduction potentials of hemin and related substances: I. The potentials of various hemins and hematins in the abscence and presence of pyridine. J. Biol. Chem. 86, 733–741 [Google Scholar]
- 21. Ebert P. S., Hess R. A., Frykholm B. C., and Tschudy D. P. (1979) Succinylacetone, a potent inhibitor of heme biosynthesis: effect on cell growth, heme content and δ-aminolevulinic acid dehydratase activity of malignant murine erythroleukemia cells. Biochem. Biophys. Res. Commun. 88, 1382–1390 10.1016/0006-291X(79)91133-1 [DOI] [PubMed] [Google Scholar]
- 22. Sassa S., and Kappas A. (1982) Succinylacetone inhibits δ-aminolevulinate dehydratase and potentiates the drug and steroid induction of δ-aminolevulinate synthase in liver. Trans. Assoc. Am. Physicians 95, 42–52 [PubMed] [Google Scholar]
- 23. Correia M. A., Sinclair P. R., and De Matteis F. (2011) Cytochrome P450 regulation: the interplay between its heme and apoprotein moieties in synthesis, assembly, repair, and disposal. Drug Metab. Rev. 43, 1–26 10.3109/03602532.2010.515222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. De Matteis F., Gibbs A. H., and Smith A. G. (1980) Inhibition of protohaem ferro-lyase by N-substituted porphyrins. Structural requirements for the inhibitory effect. Biochem. J. 189, 645–648 10.1042/bj1890645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Matteis F., and Marks G. S. (1983) The effect of N-methylprotoporphyrin and succinyl-acetone on the regulation of heme biosynthesis in chicken hepatocytes in culture. FEBS Lett. 159, 127–131 10.1016/0014-5793(83)80430-X [DOI] [PubMed] [Google Scholar]
- 26. Ortiz de Montellano P. R., Mico B. A., and Yost G. S. (1978) Suicidal inactivation of cytochrome P-450: formation of a heme-substrate covalent adduct. Biochem. Biophys. Res. Commun. 83, 132–137 10.1016/0006-291X(78)90407-2 [DOI] [PubMed] [Google Scholar]
- 27. Smith A. G., Clothier B., Carthew P., Childs N. L., Sinclair P. R., Nebert D. W., and Dalton T. P. (2001) Protection of the Cyp1a2(−/−) null mouse against uroporphyria and hepatic injury following exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol. Appl. Pharmacol. 173, 89–98 10.1006/taap.2001.9167 [DOI] [PubMed] [Google Scholar]
- 28. Urquhart A. J., Elder G. H., Roberts A. G., Lambrecht R. W., Sinclair P. R., Bement W. J., Gorman N., and Sinclair J. A. (1988) Uroporphyria produced in mice by 20-methylcholanthrene and 5-aminolaevulinic acid. Biochem. J. 253, 357–362 10.1042/bj2530357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Schauder A., Avital A., and Malik Z. (2010) Regulation and gene expression of heme synthesis under heavy metal exposure: review. J. Environ. Pathol. Toxicol. Oncol. 29, 137–158 10.1615/JEnvironPatholToxicolOncol.v29.i2.70 [DOI] [PubMed] [Google Scholar]
- 30. Sharma B., Singh S., and Siddiqi N. J. (2014) Biomedical implications of heavy metals induced imbalances in redox systems. Biomed. Res. Int. 2014, 640754 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 31. Lubran M. M. (1980) Lead toxicity and heme biosynthesis. Ann. Clin. Lab. Sci. 10, 402–413 [PubMed] [Google Scholar]
- 32. Cohen A. R., Trotzky M. S., and Pincus D. (1981) Reassessment of the microcytic anemia of lead poisoning. Pediatrics 67, 904–906 [PubMed] [Google Scholar]
- 33. Wani A. L., Ara A., and Usmani J. A. (2015) Lead toxicity: a review. Interdiscip. Toxicol. 8, 55–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Flora G., Gupta D., and Tiwari A. (2012) Toxicity of lead: a review with recent updates. Interdiscip. Toxicol. 5, 47–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Alam J., Cai J., and Smith A. (1994) Isolation and characterization of the mouse heme oxygenase-1 gene: distal 5′ sequences are required for induction by heme or heavy metals. J. Biol. Chem. 269, 1001–1009 [PubMed] [Google Scholar]
- 36. Gozzelino R., Jeney V., and Soares M. P. (2010) Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 50, 323–354 10.1146/annurev.pharmtox.010909.105600 [DOI] [PubMed] [Google Scholar]
- 37. Van der Heggen M., Martins S., Flores G., and Soares E. V. (2010) Lead toxicity in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 88, 1355–1361 10.1007/s00253-010-2799-5 [DOI] [PubMed] [Google Scholar]
- 38. Qian Z. M., and Morgan E. H. (1990) Effect of lead on the transport of transferrin-free and transferrin-bound iron into rabbit reticulocytes. Biochem. Pharmacol. 40, 1049–1054 10.1016/0006-2952(90)90492-4 [DOI] [PubMed] [Google Scholar]
- 39. Fu X., Zeng A., Zheng W., and Du Y. (2014) Upregulation of zinc transporter 2 in the blood-CSF barrier following lead exposure. Exp. Biol. Med. (Maywood) 239, 202–212 10.1177/1535370213509213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pounds J. G. (1984) Effect of lead intoxication on calcium homeostasis and calcium-mediated cell function: a review. Neurotoxicology 5, 295–331 [PubMed] [Google Scholar]
- 41. Quintanar-Escorza M. A., González-Martínez M. T., del Pilar I. O., and Calderón-Salinas J. V. (2010) Oxidative damage increases intracellular free calcium [Ca2+]i concentration in human erythrocytes incubated with lead. Toxicol In Vitro 24, 1338–1346 10.1016/j.tiv.2010.05.002 [DOI] [PubMed] [Google Scholar]
- 42. Zheng G., Zhang J., Xu Y., Shen X., Song H., Jing J., Luo W., Zheng W., and Chen J. (2014) Involvement of CTR1 and ATP7A in lead (Pb)-induced copper (Cu) accumulation in choroidal epithelial cells. Toxicol. Lett. 225, 110–118 10.1016/j.toxlet.2013.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dongre N. N., Suryakar A. N., Patil A. J., Hundekari I. A., and Devarnavadagi B. B. (2013) Biochemical effects of lead exposure on battery manufacture workers with reference to blood pressure, calcium metabolism and bone mineral density. Indian J. Clin. Biochem. 28, 65–70 10.1007/s12291-012-0241-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Schulze H., and Brand J. J. (1978) Lead toxicity and phosphate deficiency in chlamydomonas. Plant Physiol. 62, 727–730 10.1104/pp.62.5.727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Jaishankar M., Tseten T., Anbalagan N., Mathew B. B., and Beeregowda K. N. (2014) Toxicity, mechanism and health effects of some heavy metals. Interdiscip. Toxicol. 7, 60–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mendoza-Cózatl D., Loza-Tavera H., Hernández-Navarro A., and Moreno-Sánchez R. (2005) Sulfur assimilation and glutathione metabolism under cadmium stress in yeast, protists and plants. FEMS Microbiol. Rev. 29, 653–671 10.1016/j.femsre.2004.09.004 [DOI] [PubMed] [Google Scholar]
- 47. Ernst W. H., Krauss G. J., Verkleij J. A., and Wesenberg D. (2008) Interaction of heavy metals with the sulphur metabolism in angiosperms from an ecological point of view. Plant Cell Environ. 31, 123–143 [DOI] [PubMed] [Google Scholar]
- 48. Protchenko O., and Philpott C. C. (2003) Regulation of intracellular heme levels by HMX1, a homologue of heme oxygenase, in Saccharomyces cerevisiae. J. Biol. Chem. 278, 36582–36587 10.1074/jbc.M306584200 [DOI] [PubMed] [Google Scholar]
- 49. Collinson E. J., Wimmer-Kleikamp S., Gerega S. K., Yang Y. H., Parish C. R., Dawes I. W., and Stocker R. (2011) The yeast homolog of heme oxygenase-1 affords cellular antioxidant protection via the transcriptional regulation of known antioxidant genes. J. Biol. Chem. 286, 2205–2214 10.1074/jbc.M110.187062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Protchenko O., Shakoury-Elizeh M., Keane P., Storey J., Androphy R., and Philpott C. C. (2008) Role of PUG1 in inducible porphyrin and heme transport in Saccharomyces cerevisiae. Eukaryot. Cell 7, 859–871 10.1128/EC.00414-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Elsasser S., Chandler-Militello D., Müller B., Hanna J., and Finley D. (2004) Rad23 and Rpn10 serve as alternative ubiquitin receptors for the proteasome. J. Biol. Chem. 279, 26817–26822 10.1074/jbc.M404020200 [DOI] [PubMed] [Google Scholar]
- 52. Marques M., Mojzita D., Amorim M. A., Almeida T., Hohmann S., Moradas-Ferreira P., and Costa V. (2006) The Pep4p vacuolar proteinase contributes to the turnover of oxidized proteins but PEP4 overexpression is not sufficient to increase chronological lifespan in Saccharomyces cerevisiae. Microbiology 152, 3595–3605 10.1099/mic.0.29040-0 [DOI] [PubMed] [Google Scholar]
- 53. Papinski D., and Kraft C. (2016) Regulation of autophagy by signaling through the Atg1/ULK1 complex. J. Mol. Biol. 428, 1725–1741 10.1016/j.jmb.2016.03.030 [DOI] [PubMed] [Google Scholar]
- 54. Liu S. C., Zhai S., and Palek J. (1988) Detection of hemin release during hemoglobin S denaturation. Blood 71, 1755–1758 [PubMed] [Google Scholar]
- 55. Aich A., Freundlich M., and Vekilov P. G. (2015) The free heme concentration in healthy human erythrocytes. Blood Cells Mol. Dis. 55, 402–409 10.1016/j.bcmd.2015.09.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Song Y., Yang M., Wegner S. V., Zhao J., Zhu R., Wu Y., He C., and Chen P. R. (2015) A genetically encoded FRET sensor for intracellular heme. ACS Chem. Biol. 10, 1610–1615 10.1021/cb5009734 [DOI] [PubMed] [Google Scholar]
- 57. Bonkovsky H. L., Healey J. F., Lourie A. N., and Gerron G. G. (1991) Intravenous heme-albumin in acute intermittent porphyria: evidence for repletion of hepatic hemoproteins and regulatory heme pools. Am. J. Gastroenterol. 86, 1050–1056 [PubMed] [Google Scholar]
- 58. Zhang T., Bu P., Zeng J., and Vancura A. (2017) Increased heme synthesis in yeast induces a metabolic switch from fermentation to respiration even under conditions of glucose repression. J. Biol. Chem. 292, 16942–16954 10.1074/jbc.M117.790923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tu B. P., Mohler R. E., Liu J. C., Dombek K. M., Young E. T., Synovec R. E., and McKnight S. L. (2007) Cyclic changes in metabolic state during the life of a yeast cell. Proc. Natl. Acad. Sci. U.S.A. 104, 16886–16891 10.1073/pnas.0708365104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tu B. P., and McKnight S. L. (2009) Evidence of carbon monoxide-mediated phase advancement of the yeast metabolic cycle. Proc. Natl. Acad. Sci. U.S.A. 106, 14293–14296 10.1073/pnas.0907786106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Raghuram S., Stayrook K. R., Huang P., Rogers P. M., Nosie A. K., McClure D. B., Burris L. L., Khorasanizadeh S., Burris T. P., and Rastinejad F. (2007) Identification of heme as the ligand for the orphan nuclear receptors REV-ERBα and REV-ERBβ. Nat. Struct. Mol. Biol. 14, 1207–1213 10.1038/nsmb1344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sousa C. A., and Soares E. V. (2014) Mitochondria are the main source and one of the targets of Pb (lead)-induced oxidative stress in the yeast Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 98, 5153–5160 10.1007/s00253-014-5631-9 [DOI] [PubMed] [Google Scholar]
- 63. Perez R. R., Sousa C. A., Vankeersbilck T., Machado M. D., and Soares E. V. (2013) Evaluation of the role of glutathione in the lead-induced toxicity in Saccharomyces cerevisiae. Curr. Microbiol. 67, 300–305 10.1007/s00284-013-0364-z [DOI] [PubMed] [Google Scholar]
- 64. Lanphear B. P., Burgoon D. A., Rust S. W., Eberly S., and Galke W. (1998) Environmental exposures to lead and urban children's blood lead levels. Environ. Res. 76, 120–130 10.1006/enrs.1997.3801 [DOI] [PubMed] [Google Scholar]
- 65. Piomelli S., Seaman C., Zullow D., Curran A., and Davidow B. (1982) Threshold for lead damage to heme synthesis in urban children. Proc. Natl. Acad. Sci. U.S.A. 79, 3335–3339 10.1073/pnas.79.10.3335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Gillis B. S., Arbieva Z., and Gavin I. M. (2012) Analysis of lead toxicity in human cells. BMC Genomics 13, 344 10.1186/1471-2164-13-344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lidsky T. I., and Schneider J. S. (2003) Lead neurotoxicity in children: basic mechanisms and clinical correlates. Brain 126, 5–19 10.1093/brain/awg014 [DOI] [PubMed] [Google Scholar]
- 68. Yedjou C. G., Milner J. N., Howard C. B., and Tchounwou P. B. (2010) Basic apoptotic mechanisms of lead toxicity in human leukemia (HL-60) cells. Int. J. Environ. Res. Public Health 7, 2008–2017 10.3390/ijerph7052008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Chen C., and Wang J. (2007) Response of Saccharomyces cerevisiae to lead ion stress. Appl. Microbiol. Biotechnol. 74, 683–687 10.1007/s00253-006-0678-x [DOI] [PubMed] [Google Scholar]
- 70. Bussche J. V., and Soares E. V. (2011) Lead induces oxidative stress and phenotypic markers of apoptosis in Saccharomyces cerevisiae. Appl. Microbiol. Biotechnol. 90, 679–687 10.1007/s00253-010-3056-7 [DOI] [PubMed] [Google Scholar]
- 71. Wysocki R., and Tamás M. J. (2010) How Saccharomyces cerevisiae copes with toxic metals and metalloids. FEMS Microbiol. Rev. 34, 925–951 10.1111/j.1574-6976.2010.00217.x [DOI] [PubMed] [Google Scholar]
- 72. Magyar J. S., Weng T. C., Stern C. M., Dye D. F., Rous B. W., Payne J. C., Bridgewater B. M., Mijovilovich A., Parkin G., Zaleski J. M., Penner-Hahn J. E., and Godwin H. A. (2005) Reexamination of lead(II) coordination preferences in sulfur-rich sites: implications for a critical mechanism of lead poisoning. J. Am. Chem. Soc. 127, 9495–9505 10.1021/ja0424530 [DOI] [PubMed] [Google Scholar]
- 73. Grunberg-Etkovitz N., Lev N., Ickowicz D., Avital A., Offen D., and Malik Z. (2009) Accelerated proteasomal activity induced by Pb2+, Ga3+, or Cu2+ exposure does not induce degradation of α-synuclein. J. Environ. Pathol. Toxicol. Oncol. 28, 5–24 10.1615/JEnvironPatholToxicolOncol.v28.i1.20 [DOI] [PubMed] [Google Scholar]
- 74. Guo G. G., Gu M., and Etlinger J. D. (1994) 240-kDa proteasome inhibitor (CF-2) is identical to δ-aminolevulinic acid dehydratase. J. Biol. Chem. 269, 12399–12402 [PubMed] [Google Scholar]
- 75. Vallelian F., Deuel J. W., Opitz L., Schaer C. A., Puglia M., Lönn M., Engelsberger W., Schauer S., Karnaukhova E., Spahn D. R., Stocker R., Buehler P. W., and Schaer D. J. (2015) Proteasome inhibition and oxidative reactions disrupt cellular homeostasis during heme stress. Cell Death Differ. 22, 597–611 10.1038/cdd.2014.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Lopes A. C., Peixe T. S., Mesas A. E., and Paoliello M. M. (2016) Lead exposure and oxidative stress: a systematic review. Rev. Environ. Contam. Toxicol. 236, 193–238 [DOI] [PubMed] [Google Scholar]
- 77. Lee K. H., Lee S. K., Kim H. S., Cho E. J., Joo H. K., Lee E. J., Lee J. Y., Park M. S., Chang S. J., Cho C. H., Park J. B., and Jeon B. H. (2009) Overexpression of Ref-1 inhibits lead-induced endothelial cell death via the upregulation of catalase. Korean J. Physiol. Pharmacol. 13, 431–436 10.4196/kjpp.2009.13.6.431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Gietz R. D., and Schiestl R. H. (1991) Applications of high efficiency lithium acetate trasformation of intact yeast cells using single-stranded nucleic acids as carrier. Yeast 7, 253–263 10.1002/yea.320070307 [DOI] [PubMed] [Google Scholar]
- 79. Ness F., Achstetter T., Duport C., Karst F., Spagnoli R., and Degryse E. (1998) Sterol uptake in Saccharomyces cerevisiae heme auxotrophic mutants is affected by ergosterol and oleate but not by palmitoleate or by sterol esterification. J. Bacteriol. 180, 1913–1919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Grynkiewicz G., Poenie M., and Tsien R. Y. (1985) A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 260, 3440–3450 [PubMed] [Google Scholar]
- 81. Michener J. K., Nielsen J., and Smolke C. D. (2012) Identification and treatment of heme depletion attributed to overexpression of a lineage of evolved P450 monooxygenases. Proc. Natl. Acad. Sci. U.S.A. 109, 19504–19509 10.1073/pnas.1212287109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Reddi A. R., and Culotta V. C. (2013) SOD1 integrates signals from oxygen and glucose to repress respiration. Cell 152, 224–235 10.1016/j.cell.2012.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Baureder M., and Hederstedt L. (2012) Genes important for catalase activity in Enterococcus faecalis. PLoS One 7, e36725 10.1371/journal.pone.0036725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Shevchenko A., Tomas H., Havlis J., Olsen J. V., and Mann M. (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.