

Abstract
Partial loss-of-function variants in the TREM2 immune receptor are associated with increased risk for Alzheimer's disease (AD) and other forms of neurodegenerative disease, but the molecular bases for these connections are unknown. Three new structures of WT and R47H mutant TREM2 immunoglobulin-like (Ig-like) domain now reveal that R47 functions to correctly position elements of the ligand-binding surface. Intriguingly, the authors also demonstrate a disruption of receptor oligomerization by the R47H mutation, suggesting a role for ligand-induced clustering in receptor signaling and resultant plaque clearance.
Introduction
Alzheimer's disease (AD)2 is a devastating neurodegenerative disease with high economic cost and societal burden associated with the care of patients. Early genetic studies into AD uncovered variants in several genes involved in the production of β-amyloid (Aβ) peptide, leading to the formulation of the amyloid hypothesis: Extracellular deposits of Aβ are neurotoxic and are the underlying cause of the disease. However, more recent genome-wide association studies and whole genome sequencing approaches have uncovered several additional genes that modulate risk for AD, including a number functioning within the immune system, such as TREM2, CR1, and CD33 (1). In particular, the R47H variant of TREM2 is associated with a 3–5-fold increased risk for late-onset AD (2, 3). These genetic insights, together with functional experiments, have led to the hypothesis that AD is partially a disease of dysregulation of the immune system. However, the mechanisms linking immune system dysfunction to disease progression remain poorly characterized.
TREM2 is an immune receptor expressed on myeloid lineage cells, including the brain-resident microglia that constantly scavenge the brain, looking for and removing plaques and damaged cells. TREM2 forms a signaling complex with the adaptor protein DAP12 and is activated by ligand binding, which initiates phagocytosis. The extracellular region of the receptor contains a single V-set Ig-like domain, similar to an antibody-variable domain, on whose surface most AD-associated variants map (4). Data from the past 5 years indicate that TREM2 functions as a rather promiscuous pattern recognition receptor, binding to a range of targets, including membrane phospholipids (5), apoE, and Aβ itself (6), and that R47H TREM2 binds with lower affinity than does WT. Initial studies in TREM2 KO mice gave conflicting results, suggesting either beneficial (5) or detrimental effects of TREM2 expression in AD models, likely due to the idiosyncrasies of the models and ages of the mice studied. Over the past year, more convincing studies have used humanized or murine TREM2 WT and R47H mice to demonstrate a protective role for TREM2 (7–9). WT TREM2 appears critical for microglial modulation into a more phagocytic state, able to ameliorate AD via clearance of amyloid plaques and neuronal debris. One study also demonstrated that WT, but not R47H, TREM2 Ig-like domain shed from microglia in a soluble form is able to decorate the surfaces of amyloid plaques and neurons (8), although the function of these interactions is unclear. That TREM2 performs a critical role in regulating microglial activation is clear. However, the precise mechanisms leading to loss-of-function in R47H TREM2 and the functional consequences at the molecular level remain poorly defined.
In their study, Sudom et al. (10) have sought to answer some of these questions via a structural and biophysical analysis of the Ig-like domain. A previous publication (4) determined a structure for WT TREM2 at low resolution, defining the overall fold. Here, Sudom et al. (10) have determined structures for WT and R47H TREM2 and a ligand-bound complex, revealing substantial further insights into the impact of the R47H mutation on both structure and function. The authors screened several expression systems and constructs with differing termini and glycosylation site mutations, indicating the challenges of crystallizing this protein. WT TREM2 was expressed in mammalian HEK293-S cells, whereas the R47H protein was refolded following expression in an insoluble form in Escherichia coli. Overall, the WT and R47H structures are highly similar, conforming to the expected fold for a V-set Ig-like domain. By analogy with antibody-variable domains, the TREM2 domain possesses three complementarity-determining region (CDR) loops, and it is in the CDR2 loop that a striking difference is observed: The loop remodels from an irregular conformation in the WT structure to a short helix in the R47H structure (Fig. 1). This alteration can be directly explained by the set of interactions that the residue at position 47 in the neighboring CDR1 loop makes with residues in the CDR2 loop. Arg47 extends out from CDR1 toward CDR2 and makes several hydrogen bonds to residues in CDR2, acting to stabilize the conformation of this loop. In contrast, the side-chain of His47 is unable to make the same set of contacts, and, as a result, the R47H CDR2 loop swings out into a different conformation. The R47H CDR2 loop and surrounding regions of the structure display increased B-factors, suggesting increased flexibility, and the authors go on to show that this flexibility correlates with a reduced thermal stability in vitro and furthermore with a reduction in soluble TREM2 levels in a R47H TREM2 CRISPR/Cas9 gene-edited mouse.
Figure 1.
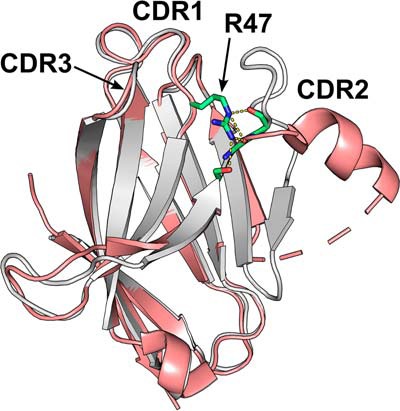
TREM2 R47H mutation disrupts ligand-binding CDR2 loop. Shown is an image of superimposed WT (gray) and R47H (pink) TREM2 Ig-like V-set domains. The stabilizing interactions (yellow dashes) made between Arg47 in CDR1 and residues in CDR2 (indicated in stick format) are shown. Mutation of Arg47 to histidine removes these interactions, causing CDR2 to alter to a partially helical conformation.
The previous publication describing a lower resolution WT TREM2 structure introduced the concept of a positive ligand-interacting surface (PLIS) on one face of TREM2 (4), including contributions from the CDR1 and CDR2 loops, capable of interaction with a range of anionic ligands. Sudom et al. (10) show that the R47H-driven disruption of CDR2 leads to a reduction of the surface area and net charge of the PLIS. This results in R47H TREM2 exhibiting reduced binding to phosphatidylserine (PS) and reduced signaling upon stimulation by PS in a reporter cell line. The authors also provide significant new insight into the nature of ligand binding through the observation of a hexameric arrangement of WT (but not R47H) TREM2 in the crystal structure. Although the interaction surface areas are not extensive, the authors were able to demonstrate that addition of PS induces a ligand-driven oligomerization of the WT protein in solution as observed by light-scattering measurements. To further investigate the interaction of PS with WT TREM2, the structure of the complex was determined. Three PS molecules were located per hexamer, with each binding primarily to a single protomer, but with additional contacts to a neighboring protomer, explaining the ligand-induced oligomerization. The negatively charged head group of PS interacts with several positively charged side chains, particularly in the CDR2 loop.
The structural basis for the effects of the R47H mutation on TREM2 function reported by Sudom et al. (10) dovetails nicely with recent functional data indicating that overexpression of WT TREM2 in transgenic mouse models can boost amyloid plaque clearance (7) and shows that the field may be approaching a consensus on the function of TREM2 in the context of AD. That is, activation of TREM2 drives a phagocytic microglial response enabling clearance of amyloid plaques and associated debris. Several other mutations associated with AD, such as R62H, also map to the PLIS described in the paper and likely lead to a similar disruption of this surface and consequent effects on stability and ligand binding. TREM2 binds a range of ligands, including the recently reported Aβ (6), and the methods reported by Sudom et al. (10) may provide the basis for structural studies of complexes with these ligands. The possibility of ligand-induced clustering of TREM2 is intriguing, and future studies should look to confirm this finding and explore its functional consequences. The big question facing the field now is if therapeutic activation of endogenous TREM2 or overexpression in vivo of WT TREM2 can form a viable basis for treatment or prevention of AD.
R. D. is a full-time employee of MedImmune, a company generating therapeutic biologics for the treatment of a variety of diseases.
- AD
- Alzheimer's disease
- PS
- phosphatidylserine
- PLIS
- positive ligand–interacting surface
- Aβ
- amyloid-β
- CDR
- complementarity-determining region.
References
- 1. Heneka M. T., Golenbock D. T., and Latz E. (2015) Innate immunity in Alzheimer's disease. Nat. Immunol. 16, 229–236 10.1038/ni.3102 [DOI] [PubMed] [Google Scholar]
- 2. Guerreiro R., Wojtas A., Bras J., Carrasquillo M., Rogaeva E., Majounie E., Cruchaga C., Sassi C., Kauwe J. S. K., Younkin S., Hazrati L., Collinge J., Pocock J., Lashley T., Williams J., Lambert J.-C., Amouyel P., Goate A., Rademakers R., Morgan K., Powell J., St George-Hyslop P., Singleton A., Hardy J., and Alzheimer Genetic Analysis Group (2013) TREM2 variants in Alzheimer's disease. N. Engl. J. Med. 368, 117–127 10.1056/NEJMoa1211851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P. V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A. I., Lah J. J., Rujescu D., Hampel H., Giegling I., Andreassen O. A., Engedal K., Ulstein I., Djurovic S., Ibrahim-Verbaas C., Hofman A., Ikram M. A., van Duijn C. M., Thorsteinsdottir U., Kong A., and Stefansson K. (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N. Engl. J. Med. 368, 107–116 10.1056/NEJMoa1211103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kober D. L., Alexander-Brett J. M., Karch C. M., Cruchaga C., Colonna M., Holtzman M. J., and Brett T. J. (2016) Neurodegenerative disease mutations in TREM2 reveal a functional surface and distinct loss-of-function mechanisms. Elife 5, e20391 10.7554/eLife.20391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wang Y., Cella M., Mallinson K., Ulrich J. D., Young K. L., Robinette M. L., Gilfillan S., Krishnan G. M., Sudhakar S., Zinselmeyer B. H., Holtzman D. M., Cirrito J. R., and Colonna M. (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071 10.1016/j.cell.2015.01.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao Y., Wu X., Li X., Jiang L.-L., Gui X., Liu Y., Sun Y., Zhu B., Piña-Crespo J. C., Zhang M., Zhang N., Chen X., Bu G., An Z., Huang T. Y., and Xu H. (2018) TREM2 is a receptor for β-amyloid that mediates microglial function. Neuron 97, 1023–1031.e7 10.1016/j.neuron.2018.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lee C. Y. D., Daggett A., Gu X., Jiang L.-L., Langfelder P., Li X., Wang N., Zhao Y., Park C. S., Cooper Y., Ferando I., Mody I., Coppola G., Xu H., and Yang X. W. (2018) Elevated TREM2 gene dosage reprograms microglia responsivity and ameliorates pathological phenotypes in Alzheimer's disease models. Neuron 97, 1032–1048.e5 10.1016/j.neuron.2018.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Song W. M., Joshita S., Zhou Y., Ulland T. K., Gilfillan S., and Colonna M. (2018) Humanized TREM2 mice reveal microglia-intrinsic and -extrinsic effects of R47H polymorphism. J. Exp. Med. 215, 745–760 10.1084/jem.20171529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cheng-Hathaway P. J., Reed-Geaghan E. G., Jay T. R., Casali B. T., Bemiller S. M., Puntambekar S. S., von Saucken V. E., Williams R. Y., Karlo J. C., Moutinho M., Xu G., Ransohoff R. M., Lamb B. T., and Landreth G. E. (2018) The Trem2 R47H variant confers loss-of-function-like phenotypes in Alzheimer's disease. Mol. Neurodegener. 13, 29 10.1186/s13024-018-0262-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sudom A., Talreja S., Danao J., Bragg E., Kegel R., Min X., Sharkov N., Marcora E., Thibault S., Bradley J., Wood S., Lim A.-C., Chen H., Wang S., Foltz I. N., Sambashivan S., and Wang Z. (2018) Molecular basis for the loss-of-function effects of the Alzheimer's disease–associated R47H variant of the immune receptor TREM2. J. Biol. Chem. 293, 12634–12646 10.1074/jbc.RA118.002352 [DOI] [PMC free article] [PubMed] [Google Scholar]