

Abstract
Hydrogen sulfide (H2S) is a signaling molecule with many beneficial effects. However, its cellular concentration is strictly regulated to avoid toxicity. Persulfide dioxygenase (PDO or ETHE1) is a mononuclear non-heme iron–containing protein in the sulfide oxidation pathway catalyzing the conversion of GSH persulfide (GSSH) to sulfite and GSH. PDO mutations result in the autosomal-recessive disorder ethylmalonic encephalopathy (EE). Here, we developed γ-glutamyl-homocysteinyl-glycine (GHcySH), in which the cysteinyl moiety in GSH is substituted with homocysteine, as a mechanism-based PDO inhibitor. Human PDO used GHcySH as an alternative substrate and converted it to GHcy-SO2H, mimicking GS-SO2H, the putative oxygenated intermediate formed with the natural substrate. Because GHcy-SO2H contains a C–S bond rather than an S–S bond in GS-SO2H, it failed to undergo the final hydrolysis step in the catalytic cycle, leading to PDO inhibition. We also characterized the biochemical penalties incurred by the L55P, T136A, C161Y, and R163W mutations reported in EE patients. The variants displayed lower iron content (1.4–11-fold) and lower thermal stability (1.2–1.7-fold) than WT PDO. They also exhibited varying degrees of catalytic impairment; the kcat/Km values for R163W, L55P, and C161Y PDOs were 18-, 42-, and 65-fold lower, respectively, and the T136A variant was most affected, with a 200-fold lower kcat/Km. Like WT enzyme, these variants were inhibited by GHcySH. This study provides the first characterization of an intermediate in the PDO-catalyzed reaction and reports on deficits associated with EE-linked mutations that are distal from the active site.
Keywords: hydrogen sulfide, iron, dioxygenase, enzyme kinetics, enzyme mechanism, ethylmalonic acid, ethylmalonic encephalopathy, persulfide dioxygenase, sulfide oxidation
Introduction
Ethylmalonic encephalopathy (EE)3 is an autosomal-recessive disorder that is associated with pathological effects in the brain, gastrointestinal tract, and peripheral vessels (1–3). It results in acrocyanosis, petechiae, hemorrhagic diarrhea, developmental delay, and progressive neurological failure, leading to necrotic lesions in the deep gray matter of the brain. Patients with EE usually succumb to the disease within the first decade of life (4). The clinical profile of EE includes elevated ethylmalonic acid in urine, C4 and C5 acrylcarnitines in blood, and a deficiency of cytochrome c oxidase in brain and muscle (5). EE is caused by mutations in the ethe1 gene that encodes persulfide dioxygenase (PDO or ETHE1). Over 20 mutations have been described in the ethe1 gene, of which a subset represents missense mutations (3, 4, 6).
PDO is a mitochondrial matrix protein that participates in the mitochondrial sulfide oxidation pathway, which converts H2S to the end products thiosulfate and sulfate (7, 8). In addition to PDO, the other enzymes involved in the mitochondrial sulfur oxidation pathway are sulfide quinone oxidoreductase (9), rhodanese (10), and sulfite oxidase (11). PDO catalyzes the second step in the pathway (i.e. the oxidation of GSH persulfide (GSSH) to sulfite (Reaction 1) (12).
Sulfite is either oxidized by sulfite oxidase, forming sulfate, or undergoes sulfur transfer by rhodanese in the presence of the sulfur donor GSSH to produce thiosulfate.
PDO is a member of the metallo-β-lactamase family (13) and contains a mononuclear non-heme iron. It exists as a monomer or a dimer in solution (12, 14, 15). Human PDO displays a typical αββα metallo-β-lactamase-type fold consisting of two central β-sheet clusters enclosed by three helices on each side (Fig. 1A) (14). A 2-His-1-carboxylate facial triad comprising residues His-79, His-135, and Asp-154 coordinates the mononuclear iron. Three water molecules occupy the remaining coordination sites. The active site iron is located inside a large groove that is framed by positively charged residues on one side and a polar tyrosine on the other (14). From the crystal structures of PDO homologs with GSH bound (16, 17) and of human PDO with GSSH docked (14), the substrate binding residues have been identified as Arg-163, Tyr-197, and, potentially, Arg-214, which is predicted to be within hydrogen-bonding distance to GSSH.
Figure 1.
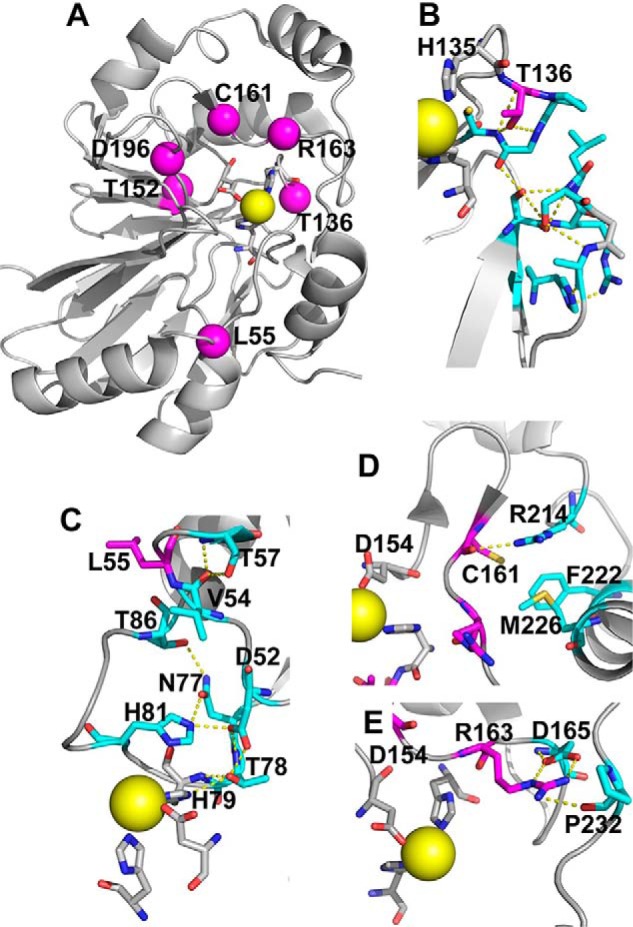
Structural analysis of PDO mutations. The structure of human PDO (Protein Data Bank entry 4CHL) was used to map the locations of PDO mutations characterized in this study. A, iron (yellow sphere) ligands (His-79, His-135, and Asp-154) that form the 2-His-1-Asp facial triad are shown in stick representation. Locations of the six residues mutated in patients that were characterized previously and/or herein, Leu-55, Thr-136, Thr-152, Cys-161, Arg-163, and Asp-196, are shown by magenta spheres. B, close-up showing the side-chain interactions of Thr-136, which would be lost in the T136A mutation. C, close-up of Leu-55 displays interactions that would potentially be lost due to the L55P mutation. D, close-up of the interaction involving Cys-161, which would be perturbed in the C161Y mutant. E, close-up of Arg-163 highlights the interactions that might be lost in the R163W mutant.
Whereas the mechanisms of several members of the mononuclear non-heme iron dioxygenases have been studied in detail (18, 19), the mechanism of PDO has not been investigated. Based on the general mechanism of non-heme iron dioxygenases and, more specifically, cysteine dioxygenase (20), a mechanism was proposed for PDO (Fig. 2A) in which binding of GSSH to ferrous PDO (1) results in displacement of one of the three coordinated water molecules (2). Binding of O2 displaces a second water ligand and results in the formation of a ferric-superoxo complex (3). The latter, via resonance, gives a radical cation character to the coordinated sulfane sulfur on the substrate (4). Recombination of the superoxo and sulfur radical species yields a cyclic peroxo intermediate (5), followed by homolytic cleavage of the O–O bond, resulting in a sulfoxy-cation intermediate and an Fe(II)-bound activated oxygen (6). Recombination of the activated oxygen and the sulfoxy-cation yields the GSSO2− intermediate (7), which is hydrolyzed to give the products sulfite and GSH (8). The postulated intermediates form transiently and are reactive. None has been characterized directly.
Figure 2.

Proposed reaction mechanism for PDO. A, in the resting state, Fe(II) is coordinated by a 2-His-1-Asp facial triad, and the remaining three coordination sites are occupied by waters (1). Binding of GSSH displaces a water molecule as the sulfane sulfur coordinates to the iron (2). Oxygen binding displaces a second water molecule and gives rise to the Fe(III)-superoxo complex (3), which exists in resonance with a structure (4) in which the coordinated sulfur has a radical cation character. Recombination of the iron-coordinated sulfur leads to formation of a cyclic-peroxo intermediate (5). Homolytic cleavage of the O–O bond results in a sulfoxy-cation and an Fe(II)-activated oxygen (6). Recombination (7) followed by hydrolysis yields the products, sulfite and GSH (8), and restores the enzyme to its resting state (1). B, structures of GSSH, GHcySH, and GSH.
Mutations in PDO lead to an increase in the levels of thiosulfate and sulfide in EE patients and in ethe1−/− mice (4, 5). The majority of the patient mutations clusters around the iron-coordinating residues and the active site pocket, suggesting that they disrupt metal and substrate binding and lead to enzyme destabilization. To date, two patient mutations (T152I and D196N) that are proximal to the iron ligands have been characterized and were shown to adversely impact the thermal stability, iron content, and kinetic properties of PDO (12). Two other pathological mutations, R163Q and R163W, were also shown to have lower thermal stabilities and lower activity (15). The redox potential of the iron center was decreased in R163Q (−310 mV) and R163W (−370 mV) PDO compared with the WT enzyme (−272 mV). However, the kinetic properties of the Arg-163 mutations were not characterized in detail (15).
Herein, we report the impact of four patient mutations, L55P, T136A, C161Y, and R163W (Fig. 1, B–E), and characterize γ-Glu-homocysteine-Gly (GHcySH), a substrate analog designed to initiate but not complete the catalytic cycle. The persulfide moiety (-SSH) in GSSH is substituted by a methylene thiol (-CH2SH) in GHcySH (Fig. 2B). The resulting derivative is of approximately the same length as GSSH but with a terminal C–S versus an S–S bond. Hence, it cannot undergo hydrolysis in the final step of the reaction mechanism. We show that GHcySH undergoes dioxygenation, forming GHcy-SO2H, which remains bound and inhibits PDO. GHcySH also inhibits the PDO mutants. This study reports the first characterization of a reaction intermediate in the PDO catalytic cycle.
Results
Properties of PDO mutants
The mutant proteins were purified using a one-step protocol and were >95% pure, which is comparable with the purity obtained for WT PDO (not shown). Preparations of L55P, T136A, C161Y, and R163W PDO yielded an average of ∼60, 20, 40, and 50 mg of protein, respectively, per 6 liters of culture. The Y38C and L185R mutants were only expressed in insoluble form and were not characterized further. The thermal denaturation profiles for the mutants were altered compared with WT PDO. The Tm value for WT PDO was 64 ± 5 °C, as described previously (12). The mutants were less stable and exhibited Tm values of 47 ± 6 °C (L55P), 44 ± 2 °C (T136A), 38 ± 3 °C (C161Y), and 53 ± 2 °C (R163W) (Table 1).
Table 1.
Comparison of the kinetic parameters for WT and mutant PDOs
The kinetic parameters were determined by monitoring O2 consumption in the presence of GSSH at 22 °C in 100 mm sodium phosphate, pH 7.4, with 0.5–170 μg of enzyme as described under “Experimental procedures.”
| Enzyme | Tm | Iron content | Specific activity | Km (GSSH) | kcat | kcat/Km |
|---|---|---|---|---|---|---|
| °C | mol Fe/mol enzyme | μmol min−1 mg−1 | mm | s−1 | mm−1 s−1 | |
| WTa | 64 ± 5 | 0.82 ± 0.5 | 113 ± 4 | 0.34 ± 0.03 | 47 | 140 |
| L55P | 47 ± 6 | 0.078 ± 0.005 | 6.3 ± 0.8 | 0.86 ± 0.07 | 2.8 | 3.3 |
| T136A | 44 ± 2 | 0.58 ± 0.2 | 1.7 ± 0.2 | 1.1 ± 0.3 | 0.8 | 0.7 |
| C161Y | 38 ± 3 | 0.073 ± 0.001 | 5.8 ± 0.5 | 1.2 ± 0.2 | 2.6 | 2.2 |
| R163W | 53 ± 2 | 0.27 ± 0.04 | 12.9 ± 0.8 | 0.73 ± 0.09 | 5.6 | 7.6 |
a The values for WT PDO have been reported previously (12).
The iron content of PDO was determined using the 2,4,6-tripyridyl-S-triazine–based colorimetric method (21). The mutants exhibited between 9 and 71% of the ferrous iron content of the WT enzyme (Table 1). The L55P and C161Y mutants, in particular, were significantly depleted in iron. The rate of oxygen consumption during enzyme-catalyzed oxygenation of GSSH (Reaction 1) was monitored as a measure of PDO activity. The mutants displayed varying degrees of catalytic impairment with specific activities ranging from 1.5 to 11% of WT PDO (Table 1), which was independent of their metal ion content. The mutants exhibited modest increases (∼2–3-fold) in the Km value for GSSH (Fig. 3), yielding kcat/Km values that were 18–200-fold lower than WT PDO (Table 1).
Figure 3.
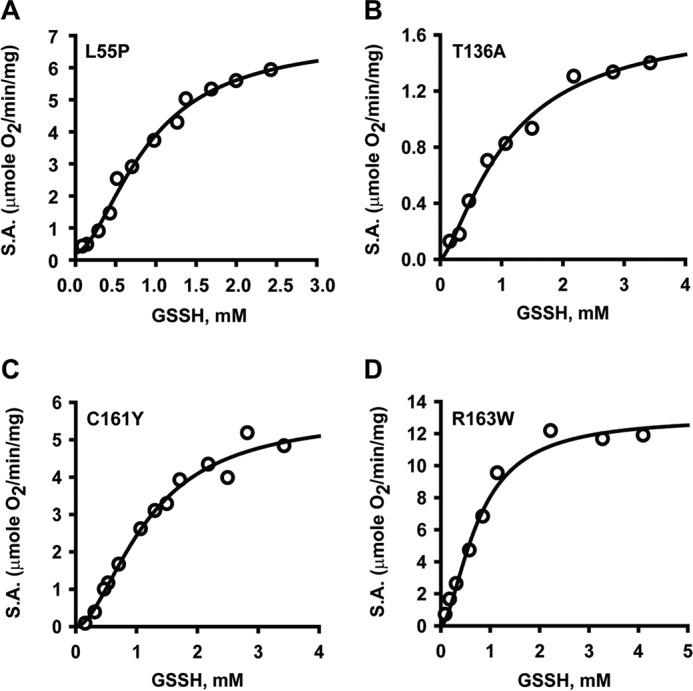
Kinetic analysis of PDO mutants. A–D, the dependence of PDO activity on GSSH concentration for L55P (A), T136A (B), C161Y (C), and R163W (D) PDO. Oxygen consumption by PDO in 100 mm sodium phosphate buffer, pH 7.4, was monitored at 22 °C in the presence of varying concentrations of GSSH. The data are representative of three independent experiments. Data were fitted with the Hill equation as described under “Experimental procedures.” S.A., specific activity.
Binding of GHcySH and GSH to ferric PDO
GHcySH was synthesized as a potential substrate mimic to probe the reaction mechanism of PDO. In GHcySH, the inner sulfur of GSSH is replaced by a methylene group, which results in a derivative having approximately the same side-chain length as GSSH but presents a thiol rather than a persulfide ligand to the iron. With a C–S bond replacing the S–S bond in GSSH, GHcySH was expected a priori to undergo oxygenation but to be unable to proceed through the last step (i.e. hydrolysis), thus leading to entrapment of a reaction intermediate in the active site of PDO.
As a first step toward its characterization, binding of GHcySH to PDO was assessed. For this, the ferric form of the enzyme was used because formation of a charge transfer species is observed in the presence of thiols but oxygen-dependent chemistry does not occur. The addition of GHcySH or GSH, the reaction product, resulted in the formation of a charge-transfer species with an absorbance maximum at 600 nm (Fig. 4, A and B). From the dependence of the absorbance change at 600 nm on the concentration of GHcySH (0.004–0.84 mm) or GSH (0.007–4 mm), Kd values of 17 ± 3 μm (for GHcySH) and 550 ± 20 μm (for GSH) were estimated for ferric PDO (Fig. 4, C and D).
Figure 4.
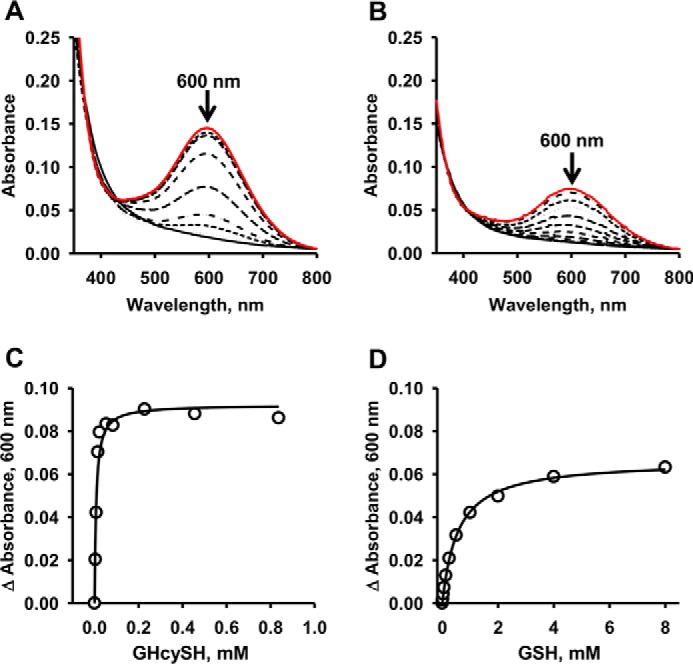
Spectral analysis of GSH and GHcySH binding to ferric-PDO. UV-visible spectra of ferric PDO solutions (150 μm) in 100 mm HEPES, pH 8, were recorded in the presence of increasing concentrations of GSH (A) and GHcySH (B) at 22 °C under aerobic conditions. The initial and final spectra are shown in solid lines in black and red, respectively. The change in absorbance at 600 nm was used to estimate the Kd for GSH (C) and GHcySH (D). Data are representative of three or four independent experiments and were fit to the Michaelis–Menten equation as described under “Experimental procedures.”
GHcySH oxygenation by PDO
In the presence of GHcySH, PDO appeared to catalyze a single turnover based on O2 consumption, which was stoichiometric with the active sites present. WT PDO exhibits a Km for O2 of 25 ± 3 μm. The mixture from the single-turnover reaction in the presence of GHcySH was characterized by HPLC. After incubation for 90 min, the reaction was derivatized with iodoacetic acid (IAA) to block thiols and dinitrofluorobenzene (DNFB) to fluorescently label amino groups. Oxidation of GHcySH to GHcy-SO2H precludes IAA derivatization. Because GHcy-SO2H was not available as a standard, we reasoned that DNB-labeled GHcySH could be used as a first approximation to bracket the retention time of the sulfinic acid derivative. HPLC analysis of the reaction mixture revealed the appearance of a peak with retention time of 7.5 min, which is similar to that of DNB-GHcySH (Fig. 5). In contrast, IAA-blocked DNB-GHcySH (i.e. DNB-GHcyS-CAA) exhibited a retention time of 21 min. The peaks from the reaction mixture with retention times of 7.5 and 21 min were collected for MS analysis.
Figure 5.
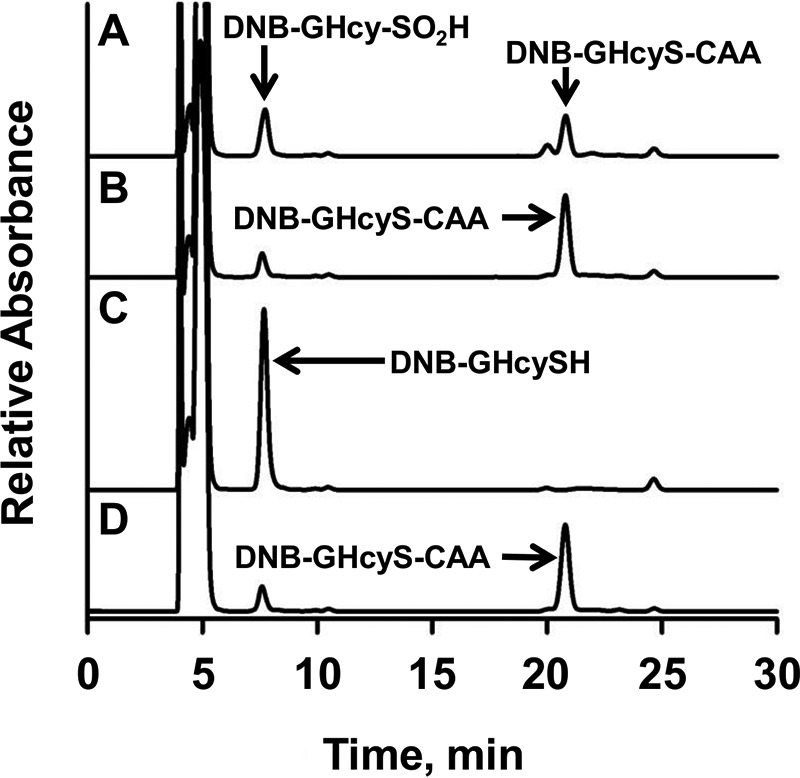
HPLC analysis of the PDO reaction mixture with GHcyS. A, HPLC analysis of a reaction mixture containing PDO (500 μm) and GHcySH (1 mm). The peaks with retention times of 7.5 min and 21 min represent DNB-GHcy-SO2H and DNB-GHcyS-CAA, respectively. B, a control sample was prepared as in A, but lacking PDO, and it showed DNB-GHcyS-CAA. In C and D, standards for DNB-GHcySH (C) and DNB-GHcyS-CAA (D) are shown. The peak at 4–5 min is associated with DNFB, which was present in excess.
The peak at 7.5 min in the reaction mixture showed the presence of DNB-GHcy-SO2H (m/z = 520) and DNB-GHcySH (m/z = 488) by LC-MS analysis (Fig. 6A). Neither GHcy-SO2H nor the DNB-labeled derivatives were observed in the control, lacking enzyme (Fig. 6B). The fragmentation pattern of the reaction mixture confirmed assignment of DNB-GHcy-SO2H. A peak corresponding to CH2CH2(CO)NH-(CH2)3SO2H (m/z = 177), representing the loss of DNB, glycine, and the CH(NH2)COOH moiety of glutamate, was observed (Fig. 6C). Other fragments observed in the MS/MS spectrum correspond to loss of the γ-glutamyl and ethyl sulfinic moieties (m/z = 133), loss of the amino-DNB and glycyl moieties (m/z = 265), and loss of CO2 from the m/z = 265 ion (m/z = 221). Assignment of DNB-GHcyS-CAA (m/z = 546) in the control sample was also confirmed by the presence of a peak representing the loss of glycine (m/z = 471 in Fig. 6D). Other fragments observed in this sample correspond to the loss of the DNB-γ-glutamyl chain (m/z = 251), loss of glycine and CO from the m/z = 251 ion (m/z = 148.5), and finally, loss of glycine, CO, and 2-thia-acetic acid (m/z = 351.5).
Figure 6.

MS analysis of the reaction of PDO with GHcySH. LC/MS spectra were extracted for the reaction mixture at 4.8 min and the control (minus PDO) sample at 20.3 min. A, for the reaction sample, peaks corresponding to DNB-labeled GHcySH (m/z = 488) and DNB-labeled GHcy-SO2H (m/z = 520) were observed. B, for the control sample, a peak corresponding to DNB-GHcyS-CAA (m/z = 546) was seen. C, MS/MS spectra were extracted at 4.8 min for DNB-GHcy-SO2H. Fragmentation produced peaks corresponding to the loss of DNB, glycine, and CH(NH2)COOH moiety of glutamate (m/z = 177). The CO2HCH2CH2NH2CHCO2H fragment of glutamate was also observed (m/z = 133). D, the MS/MS spectra were extracted at 21 min for DNB-GHcyS-CAA. Fragmentation produced peaks corresponding to the loss of glycine (m/z = 471). The fragment corresponding to protonated glutamate was also observed (m/z = 148).
GHcySH inhibits PDO
In the presence of GHcySH (0.5–2.0 mm), PDO was maximally inhibited to ∼30% of the initial activity (Fig. 7A, inset). Because the oxygenation of GHcySH forming GHcy-SO2H is slow, the effect of pre-incubating PDO for varying times with 1 mm GHcySH was examined. Under these conditions, ∼90% inhibition was observed within 1 h (Fig. 7A).
Figure 7.
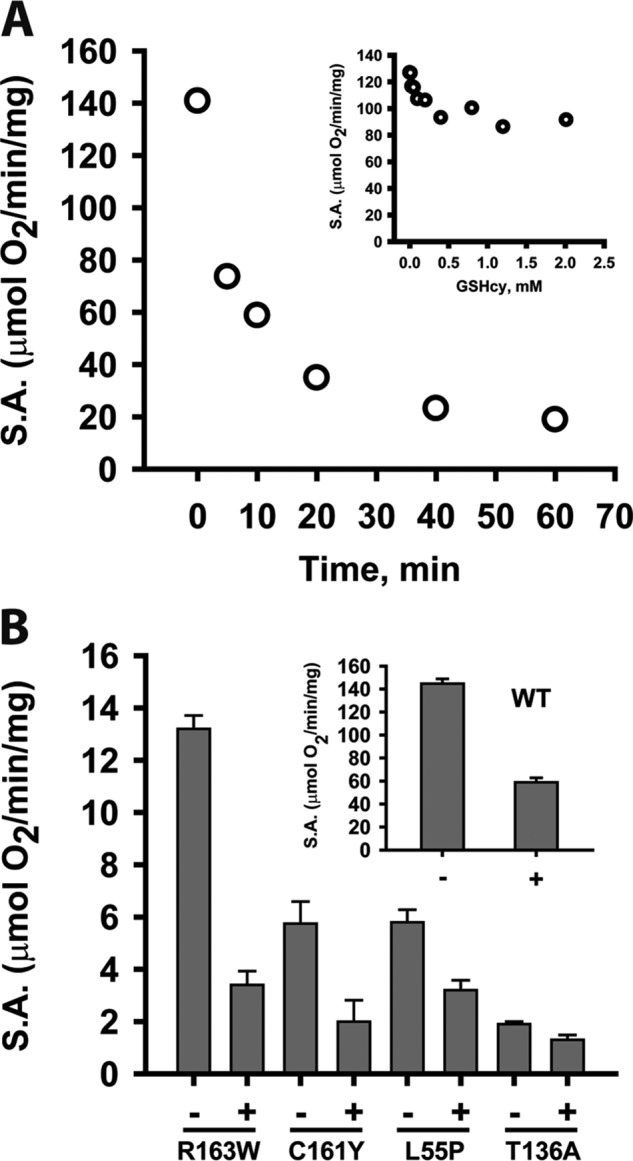
Inhibition of WT and mutant PDOs by GHcySH. A, time-dependent inhibition of PDO by GHcySH. PDO activity was measured in the presence of GHcySH using the standard oxygen consumption assay as described under “Experimental procedures.” Solutions of 2 μg of PDO in 100 mm sodium phosphate buffer, pH 7.4, were pre-incubated aerobically with 1 mm GHcySH for various times (0–60 min) before sealing the reaction chamber and initiating the reaction by injecting GSSH (3 mm final concentration). The data are representative of three independent experiments. Inset, dependence of PDO inhibition on GHcySH concentration. PDO activity was measured in the presence of GHcySH as described under “Experimental procedures.” B, inhibition of WT and mutant PDOs with GHcySH. Reactions were performed under Vmax conditions for WT (inset) and mutant PDOs as described under “Experimental procedures” with 0.5–170 μg of enzyme pre-incubated with 1 mm GHcySH for 10 min before initiating the reaction with 3 mm GSSH. Reactions were performed at 22 °C. The data are representative of three independent experiments. S.A., specific activity.
The PDO mutants were also sensitive to inhibition by GHcySH (Fig. 7B). Like WT PDO, which lost ∼60% of its activity when pre-incubated with 1 mm GHcySH for 10 min (Fig. 7B, inset), the R163W and C161Y mutants displayed 75 and 65% loss, respectively. The L55P and T136A mutants showed slightly lower sensitivity and were inhibited 47 and 30%, respectively, under the same conditions.
Oxidation of GSH by PDO
Because coordination of the thiol group of GSH was observed to ferric iron in PDO (Fig. 4A), we asked whether GSH could also be oxygenated by PDO. We found that GSH stimulated low oxygen consumption activity (3.0 ± 0.4 nmol of O2 min−1 mg−1 protein), which is 30,000-fold lower than the corresponding activity observed in the presence of GSSH (113 μmol of O2 min−1 mg−1; Table 1). Under these conditions, GSH disulfide (GSSG) was the only oxidation product that was detected (data not shown). We also tested homocysteine and cysteine as alternative substrates. Whereas both bind to ferric PDO, forming charge transfer species similar to GSH (data not shown), neither stimulated oxygen consumption in the standard in vitro assay nor inhibited oxygenation in the presence of the substrate, GSSH. Cysteine sulfinic acid also did not inhibit PDO activity (not shown).
Discussion
The reaction catalyzed by human PDO bears striking similarity to isopencillin N-synthase, an unusual member of the superfamily of iron- and α-ketoglutarate-dependent oxygenases that does not, however, use a 2-oxo acid co-substrate. Like PDO, the reaction mechanism of isopencillin N-synthase involves Fe–S–peptide interactions as the linear cysteinyl-tripeptide substrate is transformed into isopenicillin N via desaturative ring closure and concomitant reduction of O2 to water (22). Fe–S–Cys interactions are also important in the cysteine dioxygenase–catalyzed dioxygenation of cysteine to cysteinesulfinic acid (23). Both isopencillin N-synthase and cysteine dioxygenase involve Fe–SCH2R complexes versus an Fe–S–SCH2R complex in PDO. In the latter, the coordinating sulfur is connected via a scissile bond to an adjoining sulfur atom in GSSH. We reasoned that the GHcySH derivative, acting as a substrate mimic, would form the Fe-SCH2R species and initiate, but not complete, the reaction cycle (Fig. 8).
Figure 8.
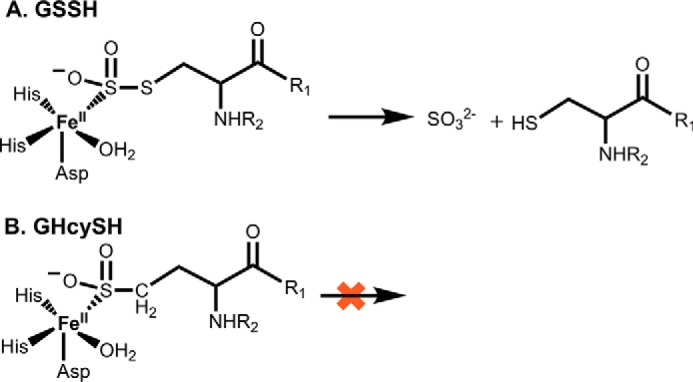
Dioxygenation of GSSH versus GHcySH. A, with GSSH, the sulfinic acid intermediate undergoes hydrolysis, presumably by the iron-bound water, to give sulfite and GSH. B, with GHcySH, the corresponding sulfinic acid species represents a dead end product.
GHcySH, in fact, turned out to be a mechanism-based inhibitor as designed, allowing, for the first time, isolation of an intermediate in the PDO catalytic cycle. Pre-incubation of the WT or mutant PDOs with chemically synthesized GHcySH led to its time-dependent inhibition (Fig. 7A). Formation of GHcy-SO2H from GHcySH demonstrated that replacement of the inner sulfur in GSSH did not preclude dioxygen insertion at the terminal sulfur. Oxygen consumption was stoichiometric with the concentration of active sites and suggested that the sulfinic acid product GHcy-SO2H remained tightly bound, inhibiting PDO. GHcy-SO2− is analogous to the GS-SO2− intermediate that is predicted to form along the reaction coordinate (Fig. 2 (7)). The presence of GHcy-SO2H as the only detectable product is consistent with the inability of PDO to process GHcySH through the ultimate hydrolysis step.
Crystal structures of PDO homologs indicate that only the sulfur atom of the substrate coordinates to the iron, displacing a single water molecule (16, 17). This is similar to isopencillin N-synthase (22) but contrasts with cysteine dioxygenase, in which the substrate serves as a bidentate ligand donating both the sulfur and the α-amino group (20). We speculate that upon diooxygen binding, the water molecule that remains coordinated to iron is used to hydrolyze the GSSO2H intermediate (Fig. 2). Chemically, the PDO reaction is, in a sense, a hybrid between the reactions catalyzed by the structurally unrelated non-heme iron–dependent oxygenases (18) and the structurally related metallo-β-lactamases, which are hydrolases (13).
GSH, which also provides a thiol ligand to the iron center, as indicated by formation of the charge transfer complex with the ferric enzyme (Fig. 4A), stimulates oxygen consumption ∼30,000-fold more slowly than GSSH and forms GSSG. GSH binds weakly to ferric PDO and is unlikely to be an effective inhibitor of the active, ferrous enzyme. In fact, in a previous study, we reported that GSH enhances PDO activity 2.2-fold by a mechanism that was not understood (12). The crystal structure of human PDO revealed the oxidative susceptibility of a surface-exposed residue, Cys-247, which was present as cysteine sulfinic acid. We speculate that GSH could react with the sulfenic acid form of Cys-247 (i.e. before it is further oxidized to the sulfinic acid) and give rise to glutathionylated PDO. Reaction of the latter with a second GSH would lead to GSSG formation and elimination of the thiol form of Cys-247 on PDO. This model, which warrants testing, suggests the potential for redox regulation of PDO via a reactive cysteine residue and indicates that the enzyme in which the reactive cysteine is in the reduced state is more active.
Alternatively, PDO might utilize GSH as a substrate but aborts the reaction after monooxygenation due to poor stereoelectronic reaction control with the shorter analog. The expected product in this case would be GSOH. However, we were unable to detect formation of a sulfenic acid derivative using dimedone (not shown). This could be due to either its lack of formation or its further reaction with GSH, generating GSSG. At present, we are unable to distinguish between the two mechanisms.
Reduced PDO activity is observed in EE, albeit the cause of this inhibition is genetic. In EE, H2S and thiosulfate levels are increased (5). Inhibition of cytochrome c oxidase due to elevated H2S (24) impairs the sulfide oxidation pathway overall, as seen in EE patients and in ethe−/− mice (25). The observed increase in thiosulfate could result from the increased utilization of GSSH by rhodanese due to reduced competition from PDO. Alternatively or additionally, increased H2S could stimulate its oxidation by heme proteins. We have shown that globins like hemoglobin, myoglobin, and neuroglobin catalyze oxidation of H2S to thiosulfate (26–29).
The PDO mutants investigated in this study exhibited pleiotropic effects from decreased thermal stability to lower iron content and significantly lower catalytic efficiency compared with the WT enzyme. The crystal structure of human PDO (14) provides a framework for interpreting the functional effects of each of the missense mutations characterized in this study. Thr-136 is located immediately downstream of the iron ligand His-135 (Fig. 1B). It participates in hydrogen bonds with the backbone amide groups of Gly-138 and Cys-139, which probably stabilize the loop and position His-135 for coordination. Thr-136 also participates in a hydrogen bond network that stabilizes an adjacent loop in the β-sheet core. The T136A mutation would disrupt this network, probably leading to displacement of His-135, loss of iron, and decreased activity (Table 1).
Leu-55 is located on a loop that is >16 Å away from the active site (Fig. 1C). Several residues both up- and downstream from Leu-55 are involved in a hydrogen bond network that probably helps position the iron ligand, His-79. The L55P mutation could result in repositioning of the loop and disruption of the network, leading to loss of iron. Cys-161 is located on the same loop as the iron-coordinating ligand Asp-154 and is involved in a hydrogen bond with Arg-214 (Fig. 1D), which would be lost in the C161Y mutant. Additionally, the tyrosine residue in the C161Y mutant could clash sterically with Phe-222 and Met-226 on the adjacent α-helix (Fig. 1D), perturbing the iron ligand Asp-153 as well as Arg-163, which interacts with substrate. These changes could, in turn, explain the observed loss of iron and the 4-fold increase in Km for GSSH.
Studies on the Pseudomonas putida PDO and the Burkholderia phytofirmans PRF, a fusion protein containing a PDO domain, have shown that the substrate is within hydrogen-bonding distance to the residue corresponding to Arg-163 in human PDO (14, 16, 17). Arg-163 is involved in hydrogen-bonding interactions with Asp-165 and Ser-230 (Fig. 1E). Mutation of Arg-163, which resides on a loop, to a bulky tryptophan residue, could perturb the upstream iron-binding ligand Asp-154, explaining the 70% lower iron content and the 2-fold increase in the Km for GSSH. Whereas an earlier characterization of the R163W mutant reported a similar reduction in specific activity as determined in our study, a detailed kinetic characterization was not reported (15). The discrepancy between the substoichiometric iron content of the R163W mutant in our study and the full complement of iron reported previously (15) is not understood.
In summary, we have demonstrated that GHcySH functions as a mechanism-based inhibitor of PDO. The resulting GHcy-SO2H product cannot undergo the final hydrolysis step and inhibits the enzyme by remaining bound. Additionally, we have shown that four PDO missense mutations identified in EE patients adversely impact multiple features of PDO that could be transmitted via secondary perturbations to residues involved in iron coordination and/or substrate binding. Like WT enzyme, the four PDO mutants are also susceptible to inhibition by GHcySH. Membrane-permeable derivatives of GHcySH that target PDO but spare other GSH-dependent reactions could potentially be developed as inhibitors of the sulfide oxidation pathway.
Experimental procedures
Materials
The sodium salts of sulfide (Na2S), GSH, GSSG, iodoacetic acid, meta-phosphoric acid, and 2,4,6-tripyridyl-S-triazine were purchased from Sigma. DNFB was purchased from Sigma.
Expression and purification of WT and mutant human PDO
The PDO mutations were generated by site-directed mutagenesis using the WT PDO expression construct as a template (12). Mutagenesis was performed using the QuikChange kit (Stratagene). The resulting vectors were transformed into BL21 Escherichia coli cells. Recombinant PDO was expressed in BL21 E. coli. A 200-ml culture in lysogeny broth medium was grown overnight at 37 °C and used to inoculate 3 × 1 liter of the same medium. Cultures were grown at 28 °C, and expression was induced with 100 μm isopropyl-β-d-thiogalactopyranoside when the optical density at 600 nm reached 0.5. At the time of induction, cultures were supplemented with ferrous ammonium sulfate to a final concentration of 250 μm, and growth was continued for an additional 14 h at 28 °C. Cells were harvested by centrifugation at 2,683 × g for 20 min at 4 °C.
PDO were purified as follows: the cell pellet from a 3-liter culture was suspended in 500 ml of 50 mm Tris buffer, pH 8, containing 0.5 m NaCl (Buffer A), 0.1% Tween 20, one tablet of protease inhibitor mixture (Roche Applied Science), and 100 mg of lysozyme (Sigma). DNase (50 mg) and MgCl2 (10 mm final concentration) were added to the cell suspension and stirred at 4 °C for 60 min, followed by sonication on ice with the following pulse sequence: 30-s burst, 1-min rest for a total burst time of 5 min at a power output setting of 6. The sonicate was centrifuged at 8,217 × g for 15 min at 4 °C. The resulting supernatant was diluted to a final volume of 1 liter with Buffer A and loaded onto a 20-ml nickel-nitrilotriacetic acid column equilibrated with the same buffer. The column was washed with 500 ml of Buffer A containing 20 mm imidazole. PDO was eluted from the column with a linear gradient ranging from 20 to 500 mm imidazole. PDO-containing fractions were pooled, concentrated, and dialyzed overnight against 50 mm Tris, pH 8.0, 0.25 m NaCl (4 liters) and stored at −80 °C. Protein concentration was determined using the Bradford reagent with BSA standards.
Metal analysis
Iron content was measured using a colorimetric assay described previously (21). Briefly, protein (10–50 μm) was denatured with 250 μl of 0.5 n HCl and 5% (w/v) TCA, mixed for 30 s, and subsequently centrifuged for 10 min at 16,000 × g in a microcentrifuge. To determine the total iron content, the supernatant (700 μl) was mixed with 300 μl of a 1:2:1 mixture of 4 mm 2,4,6-tripyridyl-S-triazine (TPTZ), 50% ammonium acetate, and 10% hydroxylamine chloride and incubated at room temperature for 5 min. To determine the residual ferrous iron content, if any, in H2O2-oxidized PDO, the supernatant (700 μl) was mixed with 300 μl of a 1:2:1 mixture of 4 mm TPTZ, 50% ammonium acetate, and water and incubated at room temperature for 5 min. The absorbance of the resulting Fe(II)–TPTZ complex was measured at 596 nm. The concentration of iron was calculated using an extinction coefficient of 22,600 m−1 cm−1 for the Fe(II)–TPTZ complex (21). The ferric iron content was calculated by subtracting the ferrous iron concentration from the total iron concentration.
Thermal stability assay
WT and mutant PDO stabilities were determined as described previously (12) by monitoring the increase in absorbance at 600 nm with temperature. For this, enzyme (100 μg) in Buffer A (final volume 200 μl) was placed in a cuvette housed in a Cary 100 Bio UV-visible spectrophotometer equipped with a heating block connected to a water bath. The temperature was increased from 25 to 70 °C in 5 °C increments.
GHcySH synthesis
Solid-supported chemical synthesis reactions were performed in a 10-ml plastic syringe with a porous polypropylene disc as a filter. Chemicals were obtained from commercial suppliers and used without further purification. Solvents were dried according to standard procedures and stored over molecular sieves (3 Å).
Resin loading
Chlorotrityl resin (0.1 g, 1.4 mmol/g loading) was treated with SOCl2/CH2Cl2 (1:9, Vtot = 2 ml, 5 × 1 min). Excess chemicals were removed by filtration, and the resin was washed with CH2Cl2 (5 × 2 ml for 2 min). Subsequently, a solution of Fmoc-Gly (2 eq) and N,N-diisopropylethylamine (DIPEA; 2.5 equivalents) in CH2Cl2 (2 ml) was added to the resin, and the mixture was gently agitated at room temperature for 30 min. Excess chemicals were removed by filtration, and the resin was washed with dimethylformamide (DMF) (4 × 2 ml for 2 min) and CH2Cl2 (3 × 2 ml for 2 min). Unreacted resin sites were capped with a mixture of CH2Cl2/MeOH/DIPEA (80:15:5, 2 ml, 2 times for 10 min), and the resin was washed with CH2Cl2 (4 × 2 ml for 2 min).
Peptide chain assembly
For Fmoc deprotection, the resin was treated with 20% piperidine in N-methyl-2-pyrrolidone (2 × 2 ml for 10 min) followed by subsequent washing with DMF (4 × 2 ml for 2 min). Coupling of amino acid residues was achieved by treating the resin with a solution of Fmoc-blocked amino acid (3 eq), 1-hydroxybenzotriazole (3 eq), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate (2.9 eq), and DIPEA (6 eq) in DMF (V = 2 ml) for 3 h. Excess chemicals were removed by filtration, and the resin was washed with DMF (4 × 2 ml for 2 min) and CH2Cl2 (3 × 2 ml for 2 min). After final Fmoc deprotection, the resin was washed with DMF (4 × 2 ml for 2 min), CH2Cl2 (3 × 2 ml for 2 min), and diethylether (3 × 2 ml for 2 min) and was dried in vacuo. Dry peptidyl resin was treated with a mixture of TFA/phenol/triisopropylsilane/1,2-ethanedithiol (90:5:2.5:2.5, Vtot = 0.5 ml) for 2 h. The solution was separated, and the resin was washed with TFA (2 × 0.3 ml for 1 min). Combined TFA fractions were added to an ice-cold mixture of ether/hexane (1:1, 30 ml), and the formed precipitate was isolated by centrifugation and washed with ice-cold ether (2 × 20 ml). The obtained white solid (10.1 mg, 22%) was lyophilized from H2O (20 ml): 1H NMR (D2O, 250 MHz): 4.64–4.53 (m, 1H, HαHCys), 4.14–4.00 (m, 3H, HαGlu, HαGly), 2.79–2.50 (m, 4H, HγGlu, HγHCys), 2.33–2.02 (m, 4H, HβGlu, HβHCys). MS (ESI+): m/z = 321.8 (calculated 322.1 for [M + H]+). GHcySH stock solutions were prepared anaerobically in 100 mm HEPES, pH 7.4, and sealed to prevent air oxidation of the thiol.
Preparation of GSH persulfide
GSSH was prepared as described previously (12) by reacting GSSG with sodium sulfide (Na2S) in a Coy anaerobic chamber (atmosphere of 95:5 N2/H2) (Reaction 2).
Briefly, solid Na2S was added in 4-fold excess to an anaerobic solution of 50 mm GSSG in 350 mm sodium phosphate, pH 7.4 (final volume 5 ml). The reaction vial was immediately sealed to prevent loss of Na2S and incubated at 37 °C for 25–30 min. The concentration of GSSH was measured using the cold cyanolysis method as described previously (30).
Oxygen consumption assays
PDO activity was measured as described previously (12). The reaction mixture contained 100 mm sodium phosphate, pH 7.4, and 0.5–170 μg of PDO (final volume 1.5 ml) mixed in a Gilson-type chamber containing a Clark oxygen electrode and a magnetic stir bar. The reaction was initiated by the addition of GSSH. GSSH concentration was varied from 0.02 to 4 mm to determine the dependence of enzyme activity on substrate concentration. The reactions were performed at room temperature (22 °C), and the data were fit using SigmaPlot (Systat Software, San Jose, CA).
The Km for oxygen was determined at a saturating GSSH concentration (3 mm) in 100 mm phosphate, pH 7.4. Buffer containing dissolved oxygen ranging in concentration from 2 to 250 μm was injected into the Gilson-type chamber placed in an inflatable Glove Bag (Cole-Parmer), under a nitrogen (99%) atmosphere. The known solubility of oxygen and its partial pressure were used to calculate the concentration of oxygen in air-saturated buffer. The electrode was calibrated to generate a standard curve using a serial dilution of oxygenated buffer (and anaerobic buffer as a diluent). The oxygen concentration at the start of each reaction was estimated using the standard curve.
The activity of PDO with GHcySH was examined under multiple- and single-turnover conditions. Multiple turnover of PDO in the presence of GHcySH was performed with 2.5 μg of enzyme (final volume 1.5 ml) in 100 mm sodium phosphate, pH 7.4. Reactions were initiated with GHcySH (2 mm final concentration). For the single-turnover experiments, PDO (250 μm final concentration) in 100 mm sodium phosphate, pH 7.4 (final volume 1.5 ml) was mixed with GHcySH (250 μm) in the Gilson-type chamber equipped with an oxygen electrode. All reactions were performed at room temperature (22 °C).
Oxygen consumption in the presence of GSH was examined by mixing 1.1–2.2 mg (25–50 μm) of PDO in a final volume of 1.5 ml in 100 mm sodium phosphate, pH 7.4. Reactions were initiated by the addition of 0.25–25 mm GSH, and oxygen consumption was monitored as described above.
Inhibition of PDO by GHcySH
PDO inhibition by GHcySH was monitored using an oxygen electrode as described above with the following modifications. The reaction mixture consisted of 100 mm sodium phosphate, pH 7.4, and 2 μg of WT PDO enzyme (final volume 1.6 ml) and GHcySH. Immediately after the addition of GHcySH (0.01–2 mm), the reaction was initiated by the addition of GSSH (3 mm final concentration).
To examine the dependence of pre-incubation time on PDO inhibition, 1 mm GHcySH was pre-incubated with PDO (2 μg in a final volume of 1.5 ml) in 100 mm sodium phosphate, pH 7.4, for 0–60 min. Then 3 mm GSSH (final concentration) was added, and oxygen consumption was monitored using an oxygen electrode as described above.
To compare the inhibition of WT and mutant PDOs, 0.5–170 μg of protein was pre-incubated with 1 mm GHcySH (final volume 1.5 ml) in 100 mm sodium phosphate, pH 7.4, for 10 min. The reaction was then initiated with 3 mm GSSH as above and monitored at room temperature (22 °C).
Preparation of ferric PDO
H2O2 was added to a final concentration of 4 mm to PDO (270 μm, 5 ml) in 100 mm HEPES, pH 7.4, and incubated overnight at 22 °C. The buffer was then exchanged with fresh 100 mm HEPES, pH 7.4, until the H2O2 concentration was estimated to be in the picomolar range.
Binding of GHcySH and GSH to ferric PDO
The dissociation constant for GHcySH and GSH binding to ferric PDO was determined spectrophotometrically at 600 nm. For this, increasing concentrations of GHcySH (0.004–0.8 mm) or GSH (0.007–4 mm) were added to ferric-PDO (150 μm) in 100 mm sodium phosphate, pH 7.4. The spectra were monitored after a 5-min incubation using a Cary spectrophotometer at room temperature (22 °C). Data were fitted to the Michaelis–Menten equation using SigmaPlot (Systat Software).
HPLC analysis of GHcySH reaction product
The reaction mixture contained either 250 μm PDO for the reaction with GSH or 500 μm PDO for the reaction with GHcySH in 100 mm sodium phosphate, pH 7.4 (50-μl final volume). The reaction was initiated by the addition of either 1 mm GHcySH or 250 μm GSH. After either 60 min (for GSH) or 90 min (for GHcySH) of incubation, the reactions were quenched by the addition of 50 μl of a meta-phosphoric acid solution containing 68.3 mm EDTA, 154 mm NaCl, and 210 mm meta-phosphoric acid in water. Next, the pH was neutralized with 2 μl of saturated K2CO3. The samples were then alkylated with 10 μl of 75 mm IAA (final concentration, 7.5 mm) and incubated at 22 °C, protected from light for 1 h. The IAA-labeled samples were then derivatized by the addition of 100 μl of DNFB (60 mm) and incubated for 6 h. For this, a 120 mm stock solution of DNFB was prepared in ethanol and protected from light during handling. GHcySH standards were prepared as described above and were labeled with both IAA and DNFB or with DNFB only. Each standard (250 μm) was prepared in 100 mm sodium phosphate, pH 7.4, at 22 °C and incubated either with IAA for 1 h followed by DNFB for 6 h or with only DNFB for 6 h.
The derivatized samples were centrifuged at 10,000 × g at 4 °C for 3 min. The resulting supernatants were separated on a 3.9 × 300-mm μBondpak-NH2 HPLC column (10 μm packing; Waters, Milford MA) using an Agilent 110 series HPLC system equipped with a multisignal UV-visible detector. The column was equilibrated with the following solution: 80% solvent A (800 ml of water and 200 ml of methanol) and 20% solution B (1 liter of solution A and 500 ml of ammonium acetate buffer consisting of 154 g of ammonium acetate dissolved in glacial acetic acid). Samples (50 μl) were injected onto the column and eluted using the following buffer B gradient: 20% isocratic from 0 to 10 min, 20–65% from 10 to 30 min, 65–100% from 30 to 32 min, 100% isocratic from 32 to 35 min, 100 to 20% from 35 to 40 min, and 20% isocratic from 40 to 45 min. The flow rate was 0.75 ml/min. The absorbance of the DNFB adducts was detected at 355 nm.
MS analysis of GHcySH PDO reaction product
HPLC samples of the PDO reaction with GHcySH were prepared and run as described above. Fractions corresponding to the peak of interest were collected and analyzed by LC-MS and LC-MS/MS as described previously (27). The samples were separated on an amide X-Bridge column (Waters; 150 × 2.1 mm, 3.5 μm) at a flow rate of 0.25 ml/min with mobile phase A comprising 20 mm ammonium acetate and 20 mm ammonia (22% (w/w) ammonium hydroxide in water) and mobile phase B, 100% acetonitrile. Other ion source parameters for the LC-MS experiments (4000 QTrap, Sciex, Framingham, MA) were as follows: T = 650 °C, gas source 1 = gas source 2 = 60 liters/min, declustering potential = 70 V, and collision energy = 30 V).
Author contributions
N. M. and O. K. designed, performed, and analyzed the experiments. N. M. and O. K. wrote the manuscript. N. M.-N. helped conceive the synthesis, and M. S. performed the synthesis of GHcySH. J. S. performed the MS analysis. R. B. helped conceive the experiments, analyzed the data, and co-wrote the manuscript. All authors edited the manuscript and approved the final version of the manuscript.
This work was supported in part by National Institutes of Health Grant GM112455 (to R. B.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
- EE
- ethylmalonic encephalopathy
- DIPEA
- N,N-diisopropylethylamine
- DMF
- dimethylformamide
- DNFB
- dintrofluorobenzene
- Fmoc
- fluorenylmethyloxycarbonyl
- IAA
- iodoacetic acid
- PDO
- persulfide dioxygenase
- GHcySH
- γ-Glu-homocysteine-Gly
- TPTZ
- 2,4,6-tripyridyl-S-triazine
- DNB
- 1,3 dinitrobenzene
- GHcy-SO2H
- γ-Glu-homocysteine sulfinyl-Gly.
References
- 1. Burlina A., Zacchello F., Dionisi-Vici C., Bertini E., Sabetta G., Bennet M. J., Hale D. E., Schmidt-Sommerfeld E., and Rinaldo P. (1991) New clinical phenotype of branched-chain acyl-CoA oxidation defect. Lancet 338, 1522–1523 10.1016/0140-6736(91)92338-3 [DOI] [PubMed] [Google Scholar]
- 2. García-Silva M. T., Campos Y., Ribes A., Briones P., Cabello A., Santos Borbujo J., Arenas J., and Garavaglia B. (1994) Encephalopathy, petechiae, and acrocyanosis with ethylmalonic aciduria associated with muscle cytochrome c oxidase deficiency. J. Pediatr. 125, 843–844 10.1016/S0022-3476(06)80197-6 [DOI] [PubMed] [Google Scholar]
- 3. Tiranti V., Briem E., Lamantea E., Mineri R., Papaleo E., De Gioia L., Forlani F., Rinaldo P., Dickson P., Abu-Libdeh B., Cindro-Heberle L., Owaidha M., Jack R. M., Christensen E., Burlina A., and Zeviani M. (2006) ETHE1 mutations are specific to ethylmalonic encephalopathy. J. Med. Genet. 43, 340–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tiranti V., D'Adamo P., Briem E., Ferrari G., Mineri R., Lamantea E., Mandel H., Balestri P., Garcia-Silva M. T., Vollmer B., Rinaldo P., Hahn S. H., Leonard J., Rahman S., Dionisi-Vici C., et al. (2004) Ethylmalonic encephalopathy is caused by mutations in ETHE1, a gene encoding a mitochondrial matrix protein. Am. J. Hum. Genet. 74, 239–252 10.1086/381653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tiranti V., Viscomi C., Hildebrandt T., Di Meo I., Mineri R., Tiveron C., Levitt M. D., Prelle A., Fagiolari G., Rimoldi M., and Zeviani M. (2009) Loss of ETHE1, a mitochondrial dioxygenase, causes fatal sulfide toxicity in ethylmalonic encephalopathy. Nat. Med. 15, 200–205 10.1038/nm.1907 [DOI] [PubMed] [Google Scholar]
- 6. Valente L., Piga D., Lamantea E., Carrara F., Uziel G., Cudia P., Zani A., Farina L., Morandi L., Mora M., Spinazzola A., Zeviani M., and Tiranti V. (2009) Identification of novel mutations in five patients with mitochondrial encephalomyopathy. Biochim. Biophys. Acta 1787, 491–501 10.1016/j.bbabio.2008.10.001 [DOI] [PubMed] [Google Scholar]
- 7. Hildebrandt T. M., and Grieshaber M. K. (2008) Three enzymatic activities catalyze the oxidation of sulfide to thiosulfate in mammalian and invertebrate mitochondria. FEBS J. 275, 3352–3361 10.1111/j.1742-4658.2008.06482.x [DOI] [PubMed] [Google Scholar]
- 8. Kabil O., and Banerjee R. (2010) The redox biochemistry of hydrogen sulfide. J. Biol. Chem. 285, 21903–21907 10.1074/jbc.R110.128363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Libiad M., Yadav P. K., Vitvitsky V., Martinov M., and Banerjee R. (2014) Organization of the human mitochondrial H2S oxidation pathway. J. Biol. Chem. 289, 30901–30910 10.1074/jbc.M114.602664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Libiad M., Sriraman A., and Banerjee R. (2015) Polymorphic variants of human rhodanese exhibit differences in thermal stability and sulfur transfer kinetics. J. Biol. Chem. 290, 23579–23588 10.1074/jbc.M115.675694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garrett R. M., Johnson J. L., Graf T. N., Feigenbaum A., and Rajagopalan K. V. (1998) Human sulfite oxidase R160Q: identification of the mutation in a sulfite oxidase-deficient patient and expression and characterization of the mutant enzyme. Proc. Natl. Acad. Sci. U.S.A. 95, 6394–6398 10.1073/pnas.95.11.6394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kabil O., and Banerjee R. (2012) Characterization of patient mutations in human persulfide dioxygenase (ETHE1) involved in H2S catabolism. J. Biol. Chem. 287, 44561–44567 10.1074/jbc.M112.407411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pettinati I., Brem J., Lee S. Y., McHugh P. J., and Schofield C. J. (2016) The chemical biology of human metallo-β-lactamase fold proteins. Trends Biochem. Sci. 41, 338–355 10.1016/j.tibs.2015.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pettinati I., Brem J., McDonough M. A., and Schofield C. J. (2015) Crystal structure of human persulfide dioxygenase: structural basis of ethylmalonic encephalopathy. Hum. Mol. Genet. 24, 2458–2469 10.1093/hmg/ddv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Henriques B. J., Lucas T. G., Rodrigues J. V., Frederiksen J. H., Teixeira M. S., Tiranti V., Bross P., and Gomes C. M. (2014) Ethylmalonic encephalopathy ETHE1 R163W/R163Q mutations alter protein stability and redox properties of the iron centre. PLoS One 9, e107157 10.1371/journal.pone.0107157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sattler S. A., Wang X., Lewis K. M., DeHan P. J., Park C. M., Xin Y., Liu H., Xian M., Xun L., and Kang C. (2015) Characterizations of two bacterial persulfide dioxygenases of the metallo-β-lactamase superfamily. J. Biol. Chem. 290, 18914–18923 10.1074/jbc.M115.652537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Motl N., Skiba M. A., Kabil O., Smith J. L., and Banerjee R. (2017) Structural and biochemical analyses indicate that a bacterial persulfide dioxygenase-rhodanese fusion protein functions in sulfur assimilation. J. Biol. Chem. 292, 14026–14038 10.1074/jbc.M117.790170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Costas M., Mehn M. P., Jensen M. P., and Que L. Jr. (2004) Dioxygen activation at mononuclear nonheme iron active sites: enzymes, models, and intermediates. Chem. Rev. 104, 939–986 10.1021/cr020628n [DOI] [PubMed] [Google Scholar]
- 19. Bruijnincx P. C., van Koten G., and Klein Gebbink R. J. (2008) Mononuclear non-heme iron enzymes with the 2-His-1-carboxylate facial triad: recent developments in enzymology and modeling studies. Chem. Soc. Rev. 37, 2716–2744 10.1039/b707179p [DOI] [PubMed] [Google Scholar]
- 20. McCoy J. G., Bailey L. J., Bitto E., Bingman C. A., Aceti D. J., Fox B. G., and Phillips G. N. Jr. (2006) Structure and mechanism of mouse cysteine dioxygenase. Proc. Natl. Acad. Sci. U.S.A. 103, 3084–3089 10.1073/pnas.0509262103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fischer D. S., and Price D. C. (1964) A simple serum iron method using the new sensitive chromogen tripyridyl-S-triazine. Clin. Chem. 10, 21–31 10.1016/0009-8981(64)90210-4 [DOI] [PubMed] [Google Scholar]
- 22. Roach P. L., Clifton I. J., Fülöp V., Harlos K., Barton G. J., Hajdu J., Andersson I., Schofield C. J., and Baldwin J. E. (1995) Crystal structure of isopenicillin N synthase is the first from a new structural family of enzymes. Nature 375, 700–704 10.1038/375700a0 [DOI] [PubMed] [Google Scholar]
- 23. Blaesi E. J., Gardner J. D., Fox B. G., and Brunold T. C. (2013) Spectroscopic and computational characterization of the NO adduct of substrate-bound Fe(II) cysteine dioxygenase: insights into the mechanism of O activation. Biochemistry 52, 6040–6051 10.1021/bi400825c [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nicholls P., and Kim J. K. (1982) Sulphide as an inhibitor and electron donor for the cytochrome c oxidase system. Can. J. Biochem. 60, 613–623 10.1139/o82-076 [DOI] [PubMed] [Google Scholar]
- 25. Di Meo I., Fagiolari G., Prelle A., Viscomi C., Zeviani M., and Tiranti V. (2011) Chronic exposure to sulfide causes accelerated degradation of cytochrome c oxidase in ethylmalonic encephalopathy. Antioxid. Redox Signal. 15, 353–362 10.1089/ars.2010.3520 [DOI] [PubMed] [Google Scholar]
- 26. Vitvitsky V., Yadav P. K., An S., Seravalli J., Cho U. S., and Banerjee R. (2017) Structural and mechanistic insights into hemoglobin-catalyzed hydrogen sulfide oxidation and the fate of polysulfide products. J. Biol. Chem. 292, 5584–5592 10.1074/jbc.M117.774943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vitvitsky V., Yadav P. K., Kurthen A., and Banerjee R. (2015) Sulfide oxidation by a noncanonical pathway in red blood cells generates thiosulfate and polysulfides. J. Biol. Chem. 290, 8310–8320 10.1074/jbc.M115.639831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bostelaar T., Vitvitsky V., Kumutima J., Lewis B. E., Yadav P. K., Brunold T. C., Filipovic M., Lehnert N., Stemmler T. L., and Banerjee R. (2016) Hydrogen sulfide oxidation by myoglobin. J. Am. Chem. Soc. 138, 8476–8488 10.1021/jacs.6b03456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ruetz M., Kumutima J., Lewis B. E., Filipovic M. R., Lehnert N., Stemmler T. L., and Banerjee R. (2017) A Distal ligand mutes the interaction of hydrogen sulfide with human neuroglobin. J. Biol. Chem. 292, 6512–6528 10.1074/jbc.M116.770370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wood J. L. (1987) Sulfane sulfur. Methods Enzymol. 143, 25–29 10.1016/0076-6879(87)43009-7 [DOI] [PubMed] [Google Scholar]