

Abstract
Normally folded prion protein (PrPC) and its functions in healthy brains remain underappreciated compared with the intense study of its misfolded forms (“prions,” PrPSc) during the pathobiology of prion diseases. This impedes the development of therapeutic strategies in Alzheimer's and prion diseases. Disrupting the zebrafish homologs of PrPC has provided novel insights; however, mutagenesis of the zebrafish paralog prp2 did not recapitulate previous dramatic developmental phenotypes, suggesting redundancy with the prp1 paralog. Here, we generated zebrafish prp1 loss–of–function mutant alleles and dual prp1−/−;prp2−/− mutants. Zebrafish prp1−/− and dual prp1−/−;prp2−/− mutants resemble mammalian Prnp knockouts insofar as they lack overt phenotypes, which surprisingly contrasts with reports of severe developmental phenotypes when either prp1 or prp2 is knocked down acutely. Previous studies suggest that PrPC participates in neural cell development/adhesion, including in zebrafish where loss of prp2 affects adhesion and deposition patterns of lateral line neuromasts. In contrast with the expectation that prp1's functions would be redundant to prp2, they appear to have opposing functions in lateral line neurodevelopment. Similarly, loss of prp1 blunted the seizure susceptibility phenotypes observed in prp2 mutants, contrasting the expected exacerbation of phenotypes if these prion gene paralogs were serving redundant roles. In summary, prion mutant fish lack the overt phenotypes previously predicted, and instead they have subtle phenotypes similar to mammals. No evidence was found for functional redundancy in the zebrafish prion gene paralogs, and the phenotypes observed when each gene is disrupted individually are consistent with ancient functions of prion proteins in neurodevelopment and modulation of neural activity.
Keywords: neurodevelopment, zebrafish, prion disease, transcription activator-like effector nuclease, TALEN, PrPC, excitotoxicity, neurodegeneration, gene editing, neural hyperactivity, targeted mutagenesis
Introduction
Reduced normal cellular prion protein (PrPC)3 function(s) are a likely contributor to prion disease progression (despite its loss not being sufficient for disease (1, 2)); thus, it is important to understand the normal physiological functions of PrPC to devise effective disease therapies. Furthermore, removal of PrPC is considered a promising therapeutic avenue for prion and Alzheimer's diseases (3, 4), and knowing its function is critical to predicting safety and mitigating detrimental effects of treatment. Although the normal functions of PrPC are not well characterized, it is known to interact with numerous extracellular matrix and cell-surface proteins including laminin (5) and NCAM (6, 7). Further investigation of these interactions revealed participation of PrPC in processes such as neuritogenesis and neurite outgrowth (8–10). Recently, it was also found that PrPC is involved in polysialylation of NCAM during epithelial–to–mesenchymal transitions (11). Understanding the putative PrPC functions that are relevant in vivo has been hindered by the lack of overt phenotypes in Prnp knockout mice (12, 13), highlighting the need for alternative in vivo systems and methods. Zebrafish have emerged as genetically tractable disease models and can be used to complement studies performed in other model organisms such as rats and mice (14); here, we used zebrafish as a model system to further uncover functions of PrPC.
Zebrafish possess two copies of the prion gene (15), and this type of duplication relative to mammalian homologs is common due to a whole-genome duplication in the fish lineage. Both paralogs of zebrafish prion genes, prp1 and prp2, are similar in possessing all the recognizable linear domains of mammalian prion proteins (Fig. 1) (15–18); specific gene identifiers are listed under “Experimental procedures” and see “Discussion” for dismissal of a putative third paralog). The mammalian and fish homologs do not share high percent identity, as the latter are longer, but both are predicted to have the repeat domains, hydrophobic domains, glycine zippers, and post-translational modifications (GPI-anchor, disulfide bridge, and N-linked glycosylations) that are present in the canonical mammalian prion protein (15, 16, 19–21). The functional conservation among these genes is strongly supported by experiments where mammalian prion proteins rescue phenotypes observed following disruption of fish prion genes (18, 22, 23). The reciprocal approach expressing fish PrPC in mammalian cells demonstrates that fish PrPC has post-translational modifications (glycosylation) similar to its mammalian homologs (20, 24). Predicted protein products of prp1 and prp2 are present in zebrafish brain, as detected by proteomics, including at least prp2 being in synaptosomal preparations (25, 26).
Figure 1.

Two stable mutant lines of prp1−/− zebrafish were engineered and both lack overt developmental phenotypes. A, targeted mutagenesis of the prion protein 1 (prp1) gene via TALENs resulted in deletions within the first 50 bp of the translation start site. The coding region of the prp1 gene (gray box) is within a single exon, homologous to the mammalian PRNP gene structure; ∼4.25 kb of chromosome 10 is schematized. TALENs were designed to cut the zebrafish genome specifically at the start of the prp1-coding region within exon 2. Two frameshift alleles were isolated: zebrafish with the prp1ua5003 allele have an 8-bp deletion (Δ8bp), and zebrafish with the prp1ua5004 have a 19-bp deletion (Δ19bp). The predicted protein products of these mutants are truncated because both of these frameshifts lead to a stop codon early in each allele, and the remaining peptides have no similarity to prion proteins. B, heterozygous prp1+/ua5003 and zygotic prp1ua5003/ua5003 larvae are similar in appearance to WT larvae at 50 h post-fertilization. C, predicted wildtype (WT) Prp1 protein shares most predicted domains with mammalian prion protein (PrPC), although they differ in length. All recognizable PrPC domains are lost in the mature predicted protein products of these mutant fish (bottom). These include signal peptides (S), repetitive region, a central hydrophobic domain (H), a GPI anchor, glycine zipper (GXXXG), disulfide bridge (S—S), and N-linked glycosylation sites. Putative proteins from the ua5003 allele and ua5004 allele have a signal peptide, followed by nonsense sequence (36 and 2 amino acids (a.a.), respectively) and early stop codons; the signal peptide is cleaved off of the mature PrPC protein.
We recently engineered mutations of prp2 and were surprised to observe that the fish developed normally, with phenotypes that included deeply conserved roles in NMDA receptor dysregulation, learning/memory, and seizure susceptibility (16, 27). This contrasted several works (including our own) using prion knockdown to produce dramatic early defects and suggested that PrPC is required for embryonic development (16, 22, 28). To reconcile this discrepancy, we hypothesized that the two PrPC paralogs in zebrafish, prp1 and prp2, might have partially redundant roles such that prp1 was masking phenotypes in our prp2 mutants. This work addresses this hypothesis, using targeted mutagenesis to disrupt prp1, and seeks to resolve whether PrPC mutation in zebrafish can fulfill its promise of revealing key roles in neurophysiology and neurodevelopment that have been lacking in similar experiments using PRNP knockout mammals.
Indeed, previous studies in zebrafish have helped to uncover conserved in vivo functions of PrPC, including roles in early development and neuroprotective functions. Transient knockdown of prp1 with a high dose of prp1 morpholino (MO) was found to arrest development during gastrulation (18), and at a lower dose caused developmental delay, CNS malformations, and apoptosis (22). These phenotypes could be reversed through ectopic delivery of zebrafish and mammalian Prnp mRNA (18, 22). Similarly, prp2 morphants had differential expression of genes linked to apoptosis, neurogenesis, and embryonic development (29) and exhibited developmental deficits (18, 22, 29, 30). However, ectopic delivery of prp2 mRNA was not readily able to rescue these developmental phenotypes (18, 22, 30); thus, specificity of prp2 morpholinos remains unverified. Zebrafish studies have also revealed that PrPC participates in cell adhesion in vivo. Prp1 knockdown has revealed a role for prp1 in mediating cell adhesion through E-cadherin and Src kinases (18, 23, 28). Furthermore, we found that prp1 and appa loss have synergistic roles in neuroprotection and cell adhesion and that PrPC and APP physically interact (22), thus arguing for a specific niche role for PrP1 in its interaction with APP that can be extended to mammals and expands the research space for considering a role for PrPC in Alzheimer disease. Therefore, morpholino knockdown of prion paralogs in zebrafish, when accompanied by stringent controls of mRNA rescue, has generated enticing support for novel hypotheses about the role of PrPC loss–of–function in neurodegenerative diseases (2).
Our first aim was to deploy targeted mutagenesis to engineer prp1 loss–of–function alleles. We generated two lines of fish with frameshift mutations in prp1 that are predicted to be null alleles. Both lines of prp1−/− mutants as well as compound prp1−/−;prp2−/− mutants resembled mouse Prnp knockouts in that they appeared overtly normal, except that prp1−/− mutants were slightly smaller at larval stages. Interestingly, concerted loss of both prp1 and prp2 mutants reversed this reduction in larval size. Similarly, a combined loss of prp1 and prp2 did not exacerbate defects in neural hyperexcitability and instead led to a blunting of seizure susceptibility phenotypes. We also characterized the lateral line, as this is an accessible site to assess role(s) for PrPC in cell cohesion during neurodevelopment (30). Rather than prp1 and prp2 having the hypothesized redundant roles in neuromast patterning, the lateral line phenotypes in mutants were contrasting and nonadditive. Overall, these data suggest that prp1 and prp2 might have opposing/competing roles in the signaling pathway(s) underlying neurodevelopment and neural excitability in at least some tissues.
Results
prp1 TALENs induced somatic and germline mutations in prp1
We sought to disrupt the function of prp1 via targeted mutagenesis by targeting the 5′ end of the first (and only) coding exon with Tal Effector Nucleases (TALENs; custom restriction enzymes designed to engineer dsDNA breaks), such that small insertions or deletions producing frameshift mutations would lead to loss of all predicted protein domains (Fig. 1). To ensure that TALENs were capable of producing mutations in vivo, we first analyzed somatic cutting in embryos injected with the prp1 TALEN reagents. High-resolution melt analysis (HRM) on genomic DNA from three pools of 20 injected embryos revealed different melt profiles compared with the uninjected controls. Somatic cutting of the target genomic DNA was confirmed, in that cloning and HRM analysis on 12 of these clones identified two clones (17%) where sequencing revealed 1- or 6-bp deletions (Fig. S1A).
Germline transmission of similar mutations was observed, allowing us to generate frameshift (predicted null) mutations in the prp1 gene of adult fish. F0 generation fish showed interesting melt profiles compared with controls (sample melt profiles shown in Fig. S1B). These interesting clones originated from three pairs of fish, and sibling fish from these crosses were raised to adulthood and genotyped.
Identification of fish heterozygous for the prp1 ua5003 and ua5004 frameshift alleles
To identify F1 generation fish carrying mutations in prp1, 48 of their adult progeny in the F2 generation were fin-clipped; these were progeny of fish injected with prp1 TALENS as above. Two males and three females had different HRM profiles than the controls. Upon sequencing clones from these individual fish, it was found that one male had an 8-bp deletion and an I10T missense mutation (designated as allele ua5003, Fig. 1A), whereas three females and one male had a 19-bp deletion (designated as allele ua5004, Fig. 1A). “ua” in our allele names is the identifier assigned by ZFin.org, the Zebrafish Model Organism Database, to denote “University of Alberta.” These frameshift alleles are predicted to produce truncated nonsense proteins (Fig. 1C) that lack all recognizable domains of the mature prion protein.
Both prp1 mutant alleles appear to be phenotypically normal, except being slightly smaller than prp1+/+ at larval stages
To test whether prp1 is required for zebrafish development, we bred fish with the prp1ua5003/ua5003 or prp1ua5004/ua5004 alleles to homozygosity. We observed no overt phenotypes at larval stages (Fig. 1B). Post-larval growth and maintenance also appeared normal, such that adult zygotic mutants were indistinguishable from WT (adult fish with the ua5003 or ua5004 alleles are shown in Fig. S2, B and C). To test the hypothesis that maternally provided prp1 mRNA is sufficient to mask early developmental phenotypes, we raised homozygous maternal zygotic mutants for the prp1 ua5003 and ua5004 alleles. Again, we observed no overt phenotypes (Fig. 2), except that prp1−/− maternal zygotic mutants with the ua5003 and ua5004 alleles were ∼3 and 2.5%, respectively, shorter than prp1+/+ fish at 50 h post-fertilization (hpf) (Fig. 2B; p < 0.05). Thus, reduced body size is a consistent phenotype across two independent disruptions of prp1, but its biological significance (if any) remains to be determined.
Figure 2.

Maternal zygotic prp1ua5003/ua5003 and prp1ua5004/ua5004 zebrafish are predicted to be nulls and have no overt phenotypes except being slightly smaller than WT as larvae. A, maternal zygotic prp1ua5003/ua5003 and prp1ua5004/ua5004 zebrafish larvae have no overt phenotypes at 50 hpf. B, mean lengths of maternal zygotic prp1ua5003/ua5003 larvae and maternal zygotic prp1ua5004/ua5004 larvae are reduced by 3 and 2.5%, respectively, compared with prp1+/+ larvae at 50 hpf. *, p < 0.05 with the Kruskal Wallis test. n refers to the number of zebrafish. C and D, maternal zygotic prp1ua5003/ua5003 (C) and maternal zygotic prp1ua5004/ua5004 zebrafish (D) survive to adulthood and have no overt phenotypes compared with WT fish. prp1 transcript abundance is reduced in both mutant alleles. E, prp1 transcript abundance was reduced by ∼10-fold in 3 dpf prp1ua5003/ua5003 larvae compared with 3 dpf WT larvae. Data are normalized to the WT fish. **, p = 0.0027 with the unpaired t test. n refers to the number of biological replicates (15–20 larvae/biological replicate. See also supporting Fig. S2A and E). F, prp2 transcript abundance was not altered in prp1ua5003/ua5003 larvae. G and H, prp1 and prp2 are both substantially decreased in compound maternal zygotic prp1ua5003/ua5003;prp2ua5001/ua5001 mutants. ***, p < 0.001 with the unpaired t test. This dramatically reduced gene product abundance is also apparent when quantified by RNA-Seq (supporting Fig. S11) and is complemented by lack of evidence for cryptic splicing of mutant transcripts (supporting Figs. S10 and S11), all of which should be considered within the context that the predicted mutant PrP1 and PrP2 proteins lack all recognizable PrPC domains (Fig. 1).
Maternal zygotic prp1−/− fish have reduced prp1 transcript abundance
Because we did not observe overt phenotypes in our prp1 mutants as has been observed in prp1 morphants (18, 22), we tested the alternative hypothesis that the prp1 alleles were not null alleles. We found that prp1 transcript abundance was reduced 10-fold in 3 dpf maternal zygotic prp1ua5003/ua5003 larvae compared with WT (Fig. 2E, p = 0.0027). In contrast, prp2 transcript abundance was not altered in these prp1ua5003/ua5003 larvae (Fig. 2F; although note that prp2 transcript abundance is reduced in prp2 mutants (see Fig. 2H and Fig. S2E) (16). Similarly, compound mutant maternal zygotic prp1−/−;prp2−/− larvae had dramatically reduced transcript abundance for both prp1 and prp2 compared with WT (Fig. 2, G and H; n = 21; p < 0.001). Considering the other allele, prp1 transcript abundance was reduced in 2 dpf maternal zygotic prp1ua5004/ua5004 larvae by about 50% compared with WT larvae (Fig. S2A, p = 0.0012). Overall, it appears that prp1 transcript levels are reduced in fish with the prp1 ua5003 and ua5004 alleles, perhaps through nonsense-mediated decay (31).
These transcripts were characterized further, with special attention to the possibility that the deletions we engineered in mutant prp1 or prp2 transcripts might produce unexpected mRNA splicing in ways that might allow functional protein to be produced. Two approaches were used. First, transcripts were characterized with 5′-rapid amplification of cDNA ends (RACE) using gene-specific primers positioned in the middle of the only coding exon of each gene and positioned 3′ of the mutations. Compound prp1ua5003/ua5003;prp2ua5001/ua5001 mutant larvae at 2 dpf had prp1 and prp2 transcripts with 5′ ends that were indistinguishable from WT in length (Fig. S10), and this was confirmed via sequencing.
Second, transcripts were characterized by examining the alignment of RNA-sequencing reads to the chromosomal locations containing prp1 and prp2 genes (on chromosomes 10 and 25, respectively); we predicted that any unexpected transcript configuration in the mutants would be revealed by reads aligning outside of the exons, and this would occur differentially in mutant but not in WT RNA-Seq reads. RNA-sequencing to a large depth (>41 million reads) revealed no out-of-place RNA-Seq reads from prp1−/−;prp2−/− mutants relative to the exons of prp1 or prp2 genes (Fig. S11). Incidentally, the abundance of read alignments (Fig. S11) independently confirmed that the absolute abundances of both prp1 and prp2 transcripts are reduced in prp1−/−;prp2−/− mutants (to 21 and 10% of WT abundances, respectively, at 3 dpf). Overall, following careful characterization, we could find no evidence that prp1 or prp2 transcripts in our engineered mutants have any opportunity to make a functional protein.
Several attempts to make custom monoclonal and polyclonal antibodies in mice and rabbits, using in-house and commercial services, were not successful in producing reagents that gave specific bands on Western blottings; therefore, we have been unable to query whether our mutants have reduced abundance of PrP1 protein. The reduced prp1 transcript abundance, combined with the N-terminal frameshift deletion that predicts absence of all recognizable prion protein domains (Fig. 1C), is strong evidence that mature PrP1 protein is either nearly or completely absent in our maternal zygotic mutants. The same arguments apply for prp2 likely being a null allele, as we reported previously (16), and data herein (including 5′-RACE and RNAseq) strengthen this conclusion.
Maternal zygotic prp1−/−;prp2−/− fish have no overt phenotypes
As neither prp1−/− mutants nor prp2−/− mutants exhibited overt phenotypes in larval or adult stages, we hypothesized that prp1 and prp2 have partially redundant functions in zebrafish development. We therefore created maternal zygotic compound prp1−/−;prp2−/− mutants. These fish did not have overt phenotypes, compared with WT fish at larval (50 hpf, Fig. 3A) or adult (Fig. 3C) stages. Considering the second prp1 allele, maternal zygotic compound prp1ua5004/ua5004;prp2ua5001/ua5001 mutants also survive into adulthood without displaying overt phenotypes (Fig. S2D). Loss of prp2 also appeared to rescue the smaller size of prp1−/− larvae, because compound maternal zygotic prp1−/−;prp2−/− mutants were instead ∼2% longer than WT fish at 50 hpf (Fig. 3B; p = 0.0123).
Figure 3.
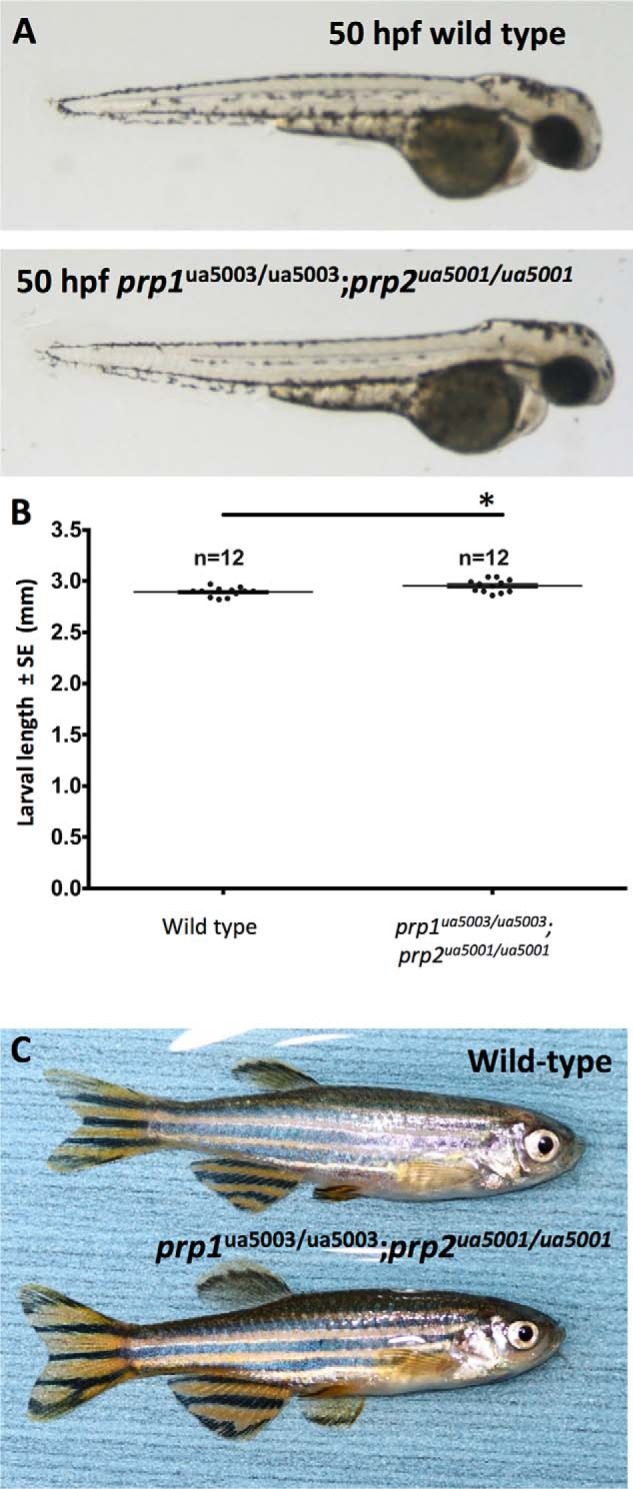
Putative redundancy between zebrafish prion gene paralogs does not account for lack of overt phenotypes. A, maternal zygotic compound prp1ua5003/ua5003;prp2ua5001/ua5001 mutants (bottom) have no overt phenotypes compared with WT fish (top) at 50 hpf. B, prp1ua5003/ua5003;prp2ua5001/ua5001 mutants have a mean length that is increased by ∼2% compared with WT fish. *, p = 0.0123 with the unpaired t test. Sample size (n =) refers to the number of fish. C, maternal zygotic compound prp1ua5003/ua5003;prp2ua5001/ua5001 mutants (bottom) survive to adulthood and have no overt phenotypes compared with WT fish (top).
Loss of prp1 reduces the number of neuromasts in the developing zebrafish posterior lateral line
PrPC has previously been shown to contribute to neurite outgrowth (8, 9, 32), and transient disruption of the zebrafish prp2 paralog with either morpholinos or mutagenesis was found to affect lateral posterior lateral line (PLL) primordium migration and neuromast number (30) using an established alkaline phosphatase staining protocol (33, 34). WT larvae typically have five primary trunk neuromasts and two to three terminal neuromasts at the tip of the tail arising from the primI primordium (Fig. 4A) (35). In some cases, the sixth primary neuromast is deposited before the primordium reaches the tip of the tail (36). 2 dpf maternal zygotic prp1ua5004/ua5004 mutants had fewer trunk neuromasts than age-matched WT AB strain zebrafish (Fig. S12, A and B, p < 0.05). To verify this phenotype, we crossed our prp1 ua5004 allele into the Tg(cldnb:gfp) line. In this line, GFP labels the cell membranes of all PLL cells, including those of the neuromasts, the interneuromast cells, and the migrating primordium (37). The trunk neuromast number was reduced in 50 hpf cldnb:gfp-labeled zygotic prp1ua5004/ua5004 fish and prp1+/ua5004 fish compared with age-matched cldnb:gfp-labeled prp1+/+ fish (Fig. 4, C and D, and Fig. S12C, p < 0.05). The difference in neuromast number between WT cldnb:gfp fish compared with nontransgenic WT fish stained with alkaline phosphatase is likely attributable in large part to the labeling method. Both mature and immature neuromasts express gfp mRNA under the cldnb promoter, whereas mature neuromasts produce more alkaline phosphatase, accounting for different absolute counts (33), although note that the trends in our phenotypes were consistent regardless of method. To ensure that the observed phenotype was not due to developmental delay, the neuromast number was also examined in 3 dpf larvae. Again, trunk neuromast number was reduced in prp1ua5004/ua5004 larvae compared with age-matched WT AB strain larvae (Fig. 4E, p < 0.05), as assessed through alkaline phosphatase labeling. Upon counter-staining with phalloidin, to more readily visualize the somites and thus document neuromast position, it was found that the L1 neuromast was deposited more posteriorly (near somites 7–8) in 20% of the 3 dpf prp1ua5004/ua5004 fish compared with the WT fish examined, wherein the L1 neuromast was deposited near somites 5 and 6 (although 10% of WT fish examined on a different day had L1 positioned near somites 7 and 8 as well; data not shown). Unexpectedly, when we compared the number of neuromasts in 3 dpf maternal zygotic prp1ua5003/ua5003 to age-matched WT larvae, we found approximately the same number of neuromasts in both genotypes (Fig. 4E). For this reason, it is possible that developmental delay contributes to the lateral line phenotype observed in 2 dpf prp1−/− larvae. Altogether, these data support that prp1 is involved in normal development of the zebrafish PLL.
Figure 4.

Neurodevelopment, as revealed by neuromast number, is altered in zebrafish prion mutants, and the prp1 and prp2 paralogs have opposing phenotypes. A, schematic of the development of the zebrafish PLL at 48 hpf, when Prim I (solid red line) has completed its migration toward the tail. It originated as a placode posterior to the otic vesicle (o.v.). During its migration, it deposited five lateral proneuromasts (L1–L5) and a stream of interneuromast cells on each side of the body. At about 40 hpf, PrimI reached the tip of the tail to produce 2–3 terminal neuromasts (Ter.), and a second primordium (dashed blue line) formed near the otic vesicle. This primordium deposited the first two dorsal neuromasts (D1 and D2) and the first two secondary neuromasts (LII.1 and LII.2) (35). B, neuromasts at 72 hpf. C and D, 2 dpf cldnb:gfp prp1+/+ WT larvae (left) have six trunk neuromasts, and a 2-dpf maternal zygotic cldnb:gfp prp1ua5004/ua5004 mutant (right) has four trunk neuromasts (quantified in supporting Fig. S12). E, loss of prp2 increased neuromasts, whereas loss of prp1 decreased neuromasts. Loss of prp2 rescued the defects caused by loss of prp1. **, p < 0.05; ***, p < 0.01; ****, p < 0.0001 with the Kruskal-Wallis test. Alternatively, these symbols represent **, p < 0.01; ***, p < 0.001; ****, p < 0.0001 calculated with one-way ANOVA. ns = not significant. † refers to one instance where nonparametric statistics disagree with parametric statistics, as Kruskal-Wallis reports p = 0.065 on this small sample size; however, a biologically and statistically significant reduction in this abundance is apparent with larger sample size (as per supporting Fig. S12) and by ANOVA and in the ua5003 allele. Sample sizes (n =) refer to the number of fish. Raw data plotted in E is provided in supporting Table S4, and endogenous alkaline phosphatase labeling was used to visualize these neuromasts.
Loss of prp2 increases the number of neuromasts in the developing zebrafish posterior lateral line
We hypothesized that prp1 and prp2 have redundant roles in PLL development, and we expected that loss of prp2 would also disrupt PLL neuromast deposition. As neuromast patterning was previously found to be disrupted in prp2 morphants and mutants (30), we examined neuromast position and number in maternal zygotic prp2ua5001/ua5001 mutants. We previously reported that the L1 neuromast was prematurely deposited in maternal zygotic prp2ua5001/ua5001 mutants (30). Here, we confirmed and extended this conclusion, finding that the L1 neuromast was deposited prematurely (near somites 1–3) in 92% of maternal zygotic prp2ua5001/ua5001 mutants. In WT fish, the L1 neuromast was typically found near somites 5 and 6, with a range between the 3rd and 8th somites. Additionally, the maternal zygotic prp2ua5001/ua5001 mutants had extra primI trunk neuromasts at 3 dpf compared with age-matched WT AB strain (prp2+/+) fish (Fig. 4E and Fig. S12D, p < 0.0001). Comparing these data with that of the previous section, an unexpected contrast emerges: prp1 promotes neuromast formation and/or deposition, whereas prp2 restricts the number of neuromasts in the PLL. Our hypothesis that prp1 and prp2 principally have redundant roles in PLL development was therefore rejected.
Combined loss of prp1 and prp2 restores the number of posterior lateral line neuromasts to WT levels
Considering that prp1 and prp2 have opposite effects on neuromast abundance when they are lost, as reported above, we next examined whether a genetic interaction between prp1 and prp2 might exist in neuromast number and patterning. Heterozygous loss of both prp1 and prp2 in 3 dpf compound prp1+/ua5004;prp2+/ua5001 fish did not disrupt the neuromast number compared with age-matched WT AB strain (prp1+/+; prp2+/+ fish) (Fig. 4E). Additionally, the L1 neuromasts in compound heterozygous prp1+/ua5004;prp2+/ua5001 mutants were positioned near somites 4–6, matching the pattern seen in WT AB strain zebrafish. To further test whether an interaction between prp1 and prp2 might exist in neuromast number and patterning, we bred to generate compound homozygous fish. The neuromast number in 3 dpf compound prp1ua5003/ua5003;prp2ua5001/ua5001 larvae was also reduced (although not significantly) relative to the number of neuromasts in 3 dpf prp2ua5001/ua5001 fish (Fig. 4E). Overall, concerted loss of prp1 and prp2 in prp1−/−;prp2−/− double mutants tended to revert the apparently opposing phenotypes that were observed in single prp1−/− and prp2−/− mutants; these data further reject our hypothesis that the prion gene paralogs are principally redundant during PLL neurodevelopment.
Combined loss of prp1 and prp2 does not exacerbate seizure susceptibility phenotypes
A role for PrPC in seizures has been debated, due to disparate seizure susceptibility phenotypes in various Prnp knockout mice (1, 38, 39), which may (or may not) be related to seizures observed in Alzheimer's and prion disease patients (40–43). Zebrafish, as a disparate system, contributed to resolving this controversy when it was reported that prp2ua5001/ua5001 mutant zebrafish larvae have increased susceptibility to convulsant, perhaps via modulating NMDA receptors, among other mechanisms (16). We sought to further clarify this issue, and we asked whether control of neural excitability is a shared function of ancient prion gene products, by investigating whether our prp1 mutants exhibit seizure susceptibility phenotypes. Measuring larval locomotion in response to a low-dose range of the convulsant pentylenetetrazole (PTZ) induces stage II seizures resembling a whirling motion as larvae swim erratically around the well of a 96-well plate (Fig. 5A) (44). We predicted that loss of prp1 would increase responses to convulsant, as this appears to be a common effect of PrPC disruption in various systems (see debate above). We also expected that loss of prp1 would synergize with loss of prp2 in this regard, a prediction emanating from our hypothesis that these paralogous genes serve redundant roles.
Figure 5.

Product of the prion gene prp1 protects from seizures akin to its paralog prp2 but through separate pathways. Seizures in zebrafish larvae responding to the convulsant PTZ were quantified as increased locomotor activity. A, loss of prp1 increases response to convulsant. Top, zebrafish larvae (4 days post-fertilization) were monitored in 96-well plates over the course of hours (representative movement during 2 min of PTZ exposure reported as red traces). Graph, typical activity traces of two individual larvae are shown, one of each genotype, before and during the presence of PTZ. Typical prp1−/− larvae (blue trace) have approximately double the activity of typical WT (black trace), and in these particular individuals the ratio of PTZ-induced activity relative to basal activity was 13.29 and 32.61 for WT and prp1−/−, respectively. B, disruption of either prion gene paralog increases seizure susceptibility, represented here as the mean of many activity tracings from larvae before and during exposure of 10 mm PTZ. Lines represent mean activity as % change in pixels within each minute, and shaded ribbons display ±S.E. C, applying PTZ in a dose-response format reveals loss of prp1 significantly increases seizure susceptibility. Loss of prp2 is more impactful on seizure susceptibility compared with loss of prp1. Concerted loss of prp1 and prp2 does not exacerbate seizure susceptibility and instead significantly reduces it compared with loss if prp2 alone. D, loss of prp2 reduced baseline activity of zebrafish larvae, whereas loss of prp1 did not measurably impact basal activity. Average baseline activity for each individual was recorded over 1 h (lines represent mean and error bars represent ± S.E.). Data in B and C were assessed by two-way ANOVA and in D by one-way ANOVA. Sample sizes (n =) represent the number of larvae tested. * indicates statistically significant compared with WT. # indicates statistically significant compared with prp1−/−;prp2−/−. One symbol (* or #) indicates p < 0.05; two symbols indicate p < 0.01; three symbols indicate p < 0.001; and four symbols indicate p < 0.0001. Allele ua5003 of prp1 used throughout; see supporting Fig. S8 for other alleles. The same behavioral traces were used to calculate velocity as an alternative metric of seizure intensity, which broadly shows the same relationships (supporting Fig. S7).
Larvae lacking prp1 were found to be more susceptible to the convulsant PTZ compared with WT (representative example in Fig. 5A). This was especially apparent during responses to low doses of PTZ, where maternal zygotic prp1 mutants had approximately double the activity compared with WT (Fig. 5C; p < 0.05 at 5 and 10 mm PTZ). Larvae lacking prp1 did not present with significant changes in basal activity levels (Fig. 5D) compared with WT. This increased susceptibility to convulsant in prp1 mutants was robust, being apparent when seizure susceptibility was assessed with various methods, including swim velocity (Fig. S7), or when considering a second allele of prp1 in various metrics of seizure intensity and using altered equipment parameters (Fig. S8 and see “Experimental procedures”), or when assessing sensitivity to convulsants via measuring abundance of the immediate early gene c-fos (Fig. S9). This seizure susceptibility in prp1 mutants was reminiscent of phenotypes following loss of prp2 (see new data in Fig. 5 and Figs. S7 and S9) (16) although lesser in its impact upon the phenotype. Indeed, at all doses of the convulsant we investigated, loss of prp2 significantly increased seizure susceptibility above WT (Fig. 5C, p < 0.0001) and was severalfold more impactful on seizures than loss of prp1 (Fig. 5C).
The similarity of these seizure susceptibility phenotypes is consistent with our hypothesis that prp1 and prp2 play redundant roles in regulating neuron excitability. To test this further, we assessed concerted loss of prp1 and prp2 and predicted an exacerbation of the seizure phenotype. Contrary to this expectation, compound prp1ua5003/ua5003;prp2ua5001/ua5001 larvae were found to have blunted seizure susceptibility compared with the prp2ua5001/ua5001 larvae at most doses (Fig. 5C and Figs. S7 and S9). Indeed the seizure activity of prp2−/− mutants was significantly higher than that of compound prp1−/−;prp2−/− mutants (Fig. 5C, p < 0.0001) at three of the four doses tested. Loss of prp1 did not significantly impact the baseline stochastic activity levels of larvae, nor did it significantly alter the impact of prp2 mutation on baseline activity (Fig. 5D and Fig. S7).
In sum, loss of prp1 increases seizure susceptibility in zebrafish larvae akin to observations on prp2 mutants made previously (16) and confirmed herein. However the impact of prp1 and prp2 on seizures does not appear to be redundant, and instead it appears that loss of prp1 blunts the impact of prp2 loss, suggesting opposing actions of these gene products in regulating neural or synaptic activity.
Discussion
Engineering prion mutant zebrafish refines the cadre of conserved functions for prion protein
The functions of PrPC in healthy brains has remained a matter of interest and debate, in part due to the complexity of its numerous putative functions and in part due to the potential impact of PrPC functions being lost during disease or during treatment thereof. Zebrafish, and their purported phenotypes when PrPC homologs are disrupted, offer very good potential to continue contributing to this debate. Here, we successfully used TALENs to engineer two lines of zebrafish that have frameshift mutations in prp1, a homolog of the mammalian gene encoding PrPC. We also generated compound maternal zygotic prp1−/−;prp2−/− zebrafish, such that all recognizable homologs of PrPC are predicted to be absent. The lack of overt developmental phenotypes in these zebrafish prion mutants is in line with what has been observed in Prnp knockout mice, goats, and cattle (12, 13, 45, 46) and in goats with a naturally occurring Prnp null allele (47). The phenotypes that were observed in these mutants include defects in neurodevelopment and neurophysiology at the level of seizure susceptibility. The details of these phenotypes suggest conserved, ancient, and nonredundant actions of prion genes in these spheres of broad interest for revealing prion protein function and evolution.
Importantly, the mutant phenotypes characterized herein differ substantially in severity from the developmental defects and CNS cell death found by several groups, including our own, in prp1 morphants (1, 2, 18, 19, 21–23, 28, 48, 49). These morphant phenotypes have included defects in cell adhesion and molecular interactions that inform hypotheses about the importance of PrPC in neurodevelopment and disease. Loss of prp1, as induced by high doses of morpholino, is also reported to halt embryo gastrulation, which has been one of several key observations that inspires framework hypotheses about the early evolution of PrPC homologs from zinc transporter proteins (50–53) and their role in cell adhesion during epithelial–to–mesenchymal transition (11, 54–57). The differences between the morphant and mutant phenotypes could be for technical reasons. For example, 1) either we have not created null alleles; 2) phenotypes in the mutants are masked by maternal WT transcripts deposited in the egg; or 3) the morpholinos have off-target effects. A separate, biological explanation for the disparity is also important to consider; the acute loss of Prnp homologs could produce different phenotypes compared with chronic loss in Prnp−/− homolog mutants (e.g. through genetic compensation). Below, we consider these alternatives in turn.
First, although we have not definitively demonstrated that we have created null alleles due to lack of suitable antibodies (see “Results”), the two frameshift mutations we engineered are both predicted to produce truncated proteins lacking all recognizable domains of prion proteins (Fig. 1). Furthermore, we report here that the prp1 transcript levels are substantially reduced in both of our engineered lines of prp1−/− mutants (but prp2 transcript abundance is normal in prp1 mutants; the reciprocal is also true, only prp2 is reduced in prp2 mutants). Because the total abundance of prp1 in WT zebrafish larvae is not large (e.g. compared with prp2), measuring decreases in abundance begins to be similar to reporting baseline noise when the transcript is absent: a 90% decrease in prp1 abundance is comparable with decreasing the RT-qPCR signal into the region of the baseline noise. Notably, there is no formal reason to expect the small deletions we engineered (e.g. 8 bp deleted, producing a frameshift mutation, see Fig. 1) to result in any decreased transcript abundance as would be expected following gene knockout. In this context, the reduction in prp1 transcripts, presumably by nonsense-mediated decay, is impressively thorough and perhaps indicates a substantial deficit. Similarly, we previously reported a marked reduction in prp2 transcript levels in prp2ua5001/ua5001 fish (16) and that remains apparent in our prp1−/−;prp2−/− fish (independently assessed by RT-qPCR and RNA-Seq). Furthermore, our 5′-RACE and RNA-Seq analyses imply that the transcripts made are the predicted loss–of–function alleles, i.e. no unexpected transcript splicing is observed. Thus, prp1−/−;prp2−/− fish are expected to have, at minimum, a substantial loss of PrPC function via both truncated predicted proteins and decreased abundance: if they are not null mutants, then they are reasonably assumed herein to be strong hypomorphs. Arguably, the loss of gene function in the strong hypomorphs ought be comparable with or greater than that produced by morpholino-based gene knockdown. Two independent mutant alleles of prp1 were generated, each producing a different frameshift in the sole coding exon; therefore, each is predicted to truncate the protein into a different null allele lacking all recognizable prion protein domains (Fig. 1). That many of the phenotypes we predicted are indeed observable in our larvae, although not as dramatic as perhaps expected, also argues in favor of gene disruption having been successful. Indeed, although the various phenotypes in the prp1 mutants (neurodevelopment, larval size, and seizure susceptibility) seem subtle, they were consistent with predictions and are also robust and specific, insomuch that they can be reversed or blunted in a coordinate manner following disruption of a related gene paralog (prp2).
Second, another possible reason for the discrepancy between mutant and morphant phenotypes is that the morpholinos deployed against prp1 and prp2 are expected to impact maternally deposited mRNA in the embryo, whereas mutants generated from an incross of heterozygous parents might have WT transcripts present in their embryos. Indeed, maternal zygotic deposition of transcript has been documented for both prp1 (18) and prp2 (16). To address this, we generated maternal zygotic embryos by breeding fish that were homozygous-mutant for the prion gene paralogs such that the embryos are expected to completely lack WT transcript. We completed this for multiple prp1 alleles by breeding these with fish carrying the prp2 ua5001 allele. We observed that maternal zygotic embryos were normal in the double prp1ua5003/ua5003;prp2ua5001/ua5001 mutants, and also with an alternative prp1 allele generating maternal zygotic prp1ua5004/ua5004;prp2ua5001/ua5001 embryos. Indeed, these maternal zygotic embryos and larvae were viable and raised to adulthood without displaying overt phenotypes. Therefore, it is unlikely that maternal contribution of WT transcript is masking phenotypes in our mutants, removing this potential difference compared with morphants.
Third, although we and others have presented data that argue strongly in favor of the specificity of the prp1 MO, some off-target toxicity may yet exist, and this would likely be exacerbated upon delivery of higher morpholino doses. Rescue of a morphant phenotype with cognate mRNA is an important control to test for morpholino specificity (58, 59). We previously showed that prp1 morphant phenotypes, using low doses of MO, can be partially rescued by injection of cognate prp1 mRNA or homologous mammalian Prnp mRNA, but not by similar mRNAs encoding prp2 or shadoo (22). Therefore, mRNA rescue experiments in past efforts were thorough (thousands of embryos were phenotyped in dozens of replicated mRNA + MO rescue combinations; control mRNA injections with engineered STOP codons were used, and mRNA rescue using paralogs was successful but only in certain contexts, etc.) (22) and are strongly supportive of MO specificity at these low MO doses.
An additional test of MO specificity is to assess the prediction that MO-induced phenotypes will be reduced when the MO is injected into embryos that are loss–of–function mutants for the MO target gene. This strategy was attempted here; we sought to define prp1 MO specificity by delivering it to prp1 mutants, although this required higher doses of MO than we used previously (22) so that a MO-induced phenotype could be observed (in our hands, disruptions to gastrulation are very rare with these MO reagents, but other morphological defects are apparent, see Fig. S13). High-dose prp1 MO did not have any less impact on prp1 mutants compared with WT embryos (Fig. S13). Thus, this test does not support prp1 MO specificity at high MO doses. Testing MO specificity in this fashion at low prp1 MO dosage is precluded by lack of phenotypes produced; therefore, the thorough tests reviewed above remain as the best available evidence and strongly support prp1 MO specificity at the low doses previously used (22).
In this instance, one reasonable interpretation of the data is that both the mutagenesis and morpholino approaches toward disrupting prp1 have good efficacy and specificity, at least at some doses, despite producing disparate results (see below). On the other hand, both approaches present with sufficient complexity that it is not possible to draw final conclusions. Our interpretations of these phenotypes, with respect to the role of PrPC in development and in healthy brains, thus intentionally avoids contrasting mutant versus morphant data sets and centers instead on the phenotypes that were found to be in common during disparate disruptions of PrPC homologs in zebrafish. Going forward, and in consideration of this complexity, we do not expect to use morpholino reagents to address questions regarding PrPC loss–of–function.
Moving beyond the putative technical intricacies discussed above, there may also be more fundamental (more biologically interesting) reasons why prion loss–of–function phenotypes would differ between techniques. For context, it is worth considering how varied the phenotypes have been among the various Prnp knockout mice (60), so some variation in outcomes when we are instead applying distinct methods of gene disruption might not be surprising. Beyond such (un)expected variations, it is notable that morpholino knockdown acutely exposes the larvae to loss of prion protein function; this contrasts the mutagenesis or prion knockout approaches, wherein gene compensation is likely. Mutagenesis and breeding toward homozygosity have an unavoidable consequence of selecting for individuals that can survive (and thrive to breeding age) with disrupted prion protein (e.g. consider the multiple generations of heterozygous animals that preceded the homozygous animals characterized herein; consider that homozygous animals failing to compensate may never successfully develop). Thus, gene compensation is reasonably likely, and individuals with prion genes mutated or knocked out likely have compensatory alterations in other gene products in the prion network. Indeed an interesting parallel was observed following disruption of Shadoo (“shadow of prion protein,” Sprn, a closely related family member of PrPC) in mice. Acute loss of Shadoo by lentiviral delivery of shRNA led to lethality, and this was dependent upon a Prnp KO background, whereas Shadoo knockout mice reveal no overt phenotypes even when combined with Prnp KO (61–63). Thus, in both mice and zebrafish loss of prion gene paralogs by acute methods leads to embryonic lethality, whereas stable mutation or knockout produces only subtle phenotypes. One approach to reconcile the disparate impacts following acute loss versus long-term loss of PrPC would be via conditional ablation such as in the Cre-Lox system. In this strategy, fish or mice with floxed Prnp alleles would not be expected to experience any Prnp loss–of–function (or undergo selection pressure due to loss of PrPC function) prior to being bred into animals with a Cre-driver line. This would allow acute disruption (although maybe not complete loss) of these proteins while avoiding the eccentricities of gene knockdown and the selection pressure leading to compensation inherent in gene knockout. Conditional knockout of mouse Prnp has indeed produced intriguing results (64); however, we are not aware of this experiment being done using a driver line with the broad Cre expression as our experimental design would demand (2).
In the meantime, we have generated zebrafish loss–of–function Prnp mutants that can now be used to interrogate the normal molecular and physiological functions of PrPC. Several of these intriguing functions may be partially lost or subverted during prion diseases and Alzheimer's disease.
The zebrafish genome contains a third gene that was annotated as a prion protein gene due to some recognizable motifs in a similar gene in Fugu (15, 65), although this was prior to a reliable genome assembly and the identification of prp1 or prp2; however, this gene (“prnpa” or “PrP3,” ZFIN ID: ZDB-GENE-041217-6) is not an obvious paralog of prp1 or prp2 and is not apparently present in the genomes of animals beyond some fishes. The predicted gene product lacks the critical domains and organization that would assign it even as a member of related Doppel or Shadoo protein families or in the broader ZIP (Zrt-Irt-like protein) family from which PrPC evolved. Thus, as noted during its original description, prnpa is not recognizable to a prion biologist due to structural inconsistencies (65), and although it may indeed emerge to be of interest elsewhere, it is not expected that it will contribute to understanding prion biology and therefore is not considered further here.
The phenotypes observed in our prion mutants include marginally reduced larval size, disruptions to neurodevelopment revealed in the lateral line, and susceptibility to seizures. We discuss the latter two topics below, and we broadly note that in all three of these phenotypes it was observed that the coordinate loss of both prp1 and prp2 either reduced or blunted the phenotypes compared with losing either gene alone; this demonstrates that the prion gene paralogs are not redundant in (several of) their function(s) as we had predicted. The apparent oppositional actions of the paralogs inspires some speculations about their roles, which remain untested but are introduced below.
Prion proteins contribute to cell cohesion during neurodevelopment, as revealed by neuromast patterning within the zebrafish PLL. Upon finding that prp1−/− mutants and compound prp1−/−;prp2−/− fish have no overt phenotypes, we proceeded to use these fish to study the role of PrPC in neural development. We hypothesized that the zebrafish prp1 and prp2 would have redundant functions in PLL development. We found that both prp1 and prp2 contributed to neuromast patterning, but they appeared to have opposing roles: loss of prp1 (in prp1−/− mutants) led to a reduced number of neuromasts, consistent with the phenotype previously observed in prp2 morphants (30), whereas loss of Prp2 in prp2−/− mutants led to an increase in neuromast number and premature deposition of the L1 neuromast. These results countered our expectation that prp1 and prp2 would be redundant in development of the PLL such that their concerted disruption would exacerbate the observed phenotypes.
Hypothetical mechanisms underlying abnormal neuromast patterning in zebrafish PrP mutants
We speculate that zebrafish prp1 or prp2 loss–of–function could interfere with neuromast formation/deposition at multiple levels. First, loss of Prp1 function could reduce neuromast number at the level of Wnt/β-catenin signaling. Inhibition of β-catenin has been found to reduce the number of proneuromasts in the developing lateral line primordium (66). GSK-3β phosphorylates cytosolic β-catenin, inducing its destruction by the proteasome (67). As PrPC has been shown to inactivate GSK-3β through caveolin/Lyn (68), reduction in neuromast number in prp1−/− mutants could be due to increased degradation of β-catenin and subsequent reduction in β-catenin signaling. Furthermore, loss of Prp2 function could increase neuromast number and cause premature deposition of the L1 neuromast by disrupting Notch signaling. Notch signaling normally restricts hair cell progenitor formation (69). In part, this is because atoh1a, expressed by hair cells, restricts E-cadherin expression (69). Therefore, it is possible that prp2 loss–of–function causes an increase in neuromast number by impacting the Notch pathway. Finally, differential localization and/or levels of cell adhesion molecules in prp1−/− and prp2−/− mutants may influence proneuromast rosette cohesion (and hence neuromast number and patterning). E-cadherin is one candidate cell adhesion protein that may be mislocalized in prp1−/− and/or prp2−/− mutants. PrPC down-regulation has been shown to contribute to abnormal adherens junctions in human A431 epidermoid carcinoma cells (70). The authors proposed that PrPC participates in activating Src kinases that in turn reduce macropinocytosis of E-cadherin from adherens junctions by promoting epidermal growth factor receptor endocytosis (70). Testing these hypotheses within the zebrafish PLL will provide further insight into the functions of PrPC.
Prion protein has an ancient and conserved role in regulating neural activity
PrPC is linked to several neuroprotective functions, although the molecular mechanisms typically remain to be revealed. One such case is a role for PrPC in regulating neural excitability, perhaps on a continuum toward excitotoxicity, such that a lack of PrPC leads to disruptions of various channel physiologies and seizure susceptibility. Intriguingly, seizures occur in a subset of patients with prion and Alzheimer's disease, which might coordinate with a loss of PrPC function (40–43). Observations in Prnp knockout mice have revealed a seizure susceptibility phenotype, and controversy surrounding this topic appears to be partially reconciled by considering differences in how the knockout mice were engineered (38, 39). Furthermore, data from zebrafish prp2 mutants supported a role for PrPC in regulating seizures in an independent system (16). Intriguingly, this also implies that the PrPC in the last common ancestor of fish and mammals had a role in regulating neural excitability. Indeed that notion was partially supported by examining the impact of PrPC loss on NMDA receptors, wherein the kinetics of channel closing were pushed toward hyperexcitability in both fish and mice (16). Further roles for PrPC at the synapse appear to be shared between mice and zebrafish, because its loss in either species leads to deficits in learning and memory (27, 71).
It is therefore of substantial interest to appreciate whether other divergent PrPC variants may impact seizure susceptibility. Broadly, our characterization of increased seizure susceptibility in prp1−/− mutant zebrafish confirms this in a second fish paralog and adds good support to the notion that an ancient PrPC had some role in regulating neurophysiology.
Intriguingly, loss of prp1 did not exacerbate the similar seizure susceptibility phenotype we documented for prp2 mutants. Indeed, we had predicted the impacts would be additive or synergistic, such that compound mutants would present with more dramatic responses to convulsants. Our data clearly reject this, and the loss of prp1 significantly blunted the seizure susceptibility of prp2 mutant fish. This suggests that the impacts of prp1 and prp2, while producing similar outcomes, are occurring through disparate mechanisms. Future work should focus on comparing and contrasting these mechanisms, as they have the potential to reveal the nuances of how PrPC impacts upon neurophysiology, including excitotoxicity, learning, and memory. It may be that future work documenting differences in the PrPC paralog structures would lead to novel hypotheses. Some speculation about such mechanisms are detailed below, in hope they suggest some new experimental designs.
Hypothetical mechanisms to explain the apparent rescue of developmental phenotypes and hyperactivity in compound prp1−/−;prp2−/− compound mutants
One alternative hypothesis to explain the apparent rescue of neuromast patterning in compound prp1−/−;prp2−/− mutants is that Prp1 and Prp2 act at different stages of development (e.g. have sub-functionalized roles in neuromast patterning), and a disruption in one phase of development is countered by a second disruption later in development. An example was discussed above, and it may be that loss of Prp1 may cause a partial reduction in Wnt/β-catenin signaling leading to an initial reduction in the number of proneuromast rosettes in the primordium. Later, however, loss of Prp2 function may reduce Delta/Notch lateral inhibition producing an extra hair cell progenitor (and eventually an extra proneuromast rosette).
An alternative explanation that could account for the apparent rescue of phenotypes observed in single mutants (neuromast patterning defects, body size, and hyperactivity) in compound prp1−/−;prp2−/− mutants is that the gene paralogs have evolved antagonistic roles (e.g. see schema in Fig. S6). Antagonistic roles have been observed for other gene paralog pairs. For example, in zebrafish the ribosomal protein L22 (Rpl22) represses SmadI expression and hematopoietic stem cell emergence, whereas its paralog ribosomal protein L22 like1 (Rpl22l1) activates SmadI expression and hematopoietic stem cell emergence (72). Furthermore, in mammals it has been found that the regulator of nonsense transcripts 3B (UPF3B) activates nonsense-mediated decay through its interaction with the exon–junction complex (EJC). In contrast, EPF3A acts as a repressor of nonsense-mediated decay due to its weaker interaction with EJC. Hence, gene paralogs may develop antagonistic roles of the parent gene due to disruption of an important functional domain (73). Therefore, it is possible that loss of prp1 dampens sensitivity to convulsant in prp2−/− larvae because Prp1 and Prp2 have an antagonistic effect on a common pathway. Furthermore, it is possible that prp2 cannot replace prp1 in regulating larval size because Prp2 cannot effectively interact with a protein with which Prp1 normally interacts. In single mutants, the remaining prp paralog outcompetes with a third protein for a place in the relevant protein complex. However, in double prp mutants, this third protein would be expected to be able to compensate for loss of the active prp paralog. It remains to be determined what protein(s) might be at the center of this putative antagonism, although candidates may include molecules such as metabotropic glutamate receptor 5 and APP that are thought to transduce extracellular signals bound to PrPC through to the intracellular compartment (2, 8, 22, 74, 75). Identifying commonalities and differences among the functions of zebrafish prp1, prp2, and mammalian Prnp is expected to inspire new hypotheses about the key functions of PrPC and their relevance to the etiology of neurodegeneration.
Conclusions
In summary, we have generated new genetic resources to study the function of PrPC in healthy organisms (zebrafish prp1−/− and compound prp1−/−;prp2−/− mutants), through early development and in adulthood, and demonstrated that zebrafish prion proteins participate in neuromast patterning and regulating neurophysiology. As such, loss of functional PrPC during prion and Alzheimer's disease may impair adult neurogenesis, neuron survival, or contribute to producing disease symptoms such as memory loss. Further study of the mechanisms through which prion proteins contribute to neuromast patterning and neurophysiology in the zebrafish will provide new insights into the role of PrPC in neural development and maintenance. Considering the disruption/subversion of the function of PrPC in both prion disease and Alzheimer's disease (1, 2), these studies will likely uncover putative therapeutic targets for prion diseases and Alzheimer's disease.
Experimental procedures
Animal ethics and zebrafish husbandry
Zebrafish were raised and maintained using protocols approved by the Animal Care and Use Committee: Biosciences at the University of Alberta, operating under the guidelines of the Canadian Council of Animal Care. The fish were raised and maintained within the University of Alberta fish facility at 28 °C under a 14:10 light/dark cycle as described previously (76).
Fish lines/strains
Zebrafish of the AB strain were used as the WT fish in this study and were the background strain for the targeted mutagenesis. Our previously published prp2ua5001 allele (ZFin ID: ZDB-ALT-130724-2), which has a 4-bp deletion and is predicted to produce a truncated protein lacking all recognizable protein domains (16), was maintained on an AB background. Tg(cldnb:gfp) larvae (Tg (−8.0cldnb:Ly-EGFP, ZFin ID: ZDB-ALT-060919-2); referred to herein as cldnb:gfp (37)) were kindly provided by Pierre Drapeau and were bred into fish with the newly generated prp1 ua5004 allele upon reaching adulthood.
Targeted mutagenesis
Targeted mutagenesis was performed on the zebrafish prp1 gene (Ensemble ENSDARG00000044048, ZFIN ZDB-GENE-041221-2) using TALENs. The target sequence within the prp1 genes is shown in Fig. S3A, and a flowchart summarizing the steps involved in the targeted mutagenesis process is shown in Fig. S3B. The location of the target sequence within the prp1 gene is schematized in Fig. 1.
Production of TALEN plasmids
Custom TAL blocksTM and heterodimeric backbone plasmids were ordered from Transposagen (Lexington, KY). The backbone contains the first half-site of the DNA-binding domain, the sequence that recognizes the final base of the target site, and the FokI cleavage domain. The TAL blocks, which contain the remainder of the DNA-binding domain, were assembled via Transposagen's FLASH build process. After digestion with BsmBI (New England Biolabs catalog no. R0580S, Ipswich, MA), the vectors were purified using an Agencourt AMPure XP-PCR purification kit (Beckman Coulter catalog no. A63880, Indianapolis, IN). The custom TAL blocksTM of the forward TALEN were then ligated into the appropriate vectors (JDS 82 KKR heterodimer with NN to recognize the final cytosine in the prp1 target sequence) with T4 ligase (Invitrogen/ThermoFisher Scientific catalog no. 15224-017, Waltham, MA) and transformed into Stbl3 cells (Invitrogen/ThermoFisher Scientific catalog no. C7373-03). Because of changes in Transposagen's manufacturing process, the prp1 reverse TAL blockTM was provided by Transposagen as pre-ligated plasmids. Colony PCR was performed to screen for colonies with the correct number of repeats (Fig. S4), and those yielding PCR products of the appropriate length were sequenced to ensure that they contained the correct TALEN sequence (see Table S1 for primer sequences; sequencing performed by the University of Alberta's Molecular Biology Service Unit). Clones with the full TALEN sequence were then prepared using a Plasmid Maxiprep kit (Qiagen catalog no. 12163, Toronto, Ontario, Canada).
Production of TALEN mRNA and its delivery to zebrafish embryos
mRNA was synthesized from maxi-prepped plasmid DNA that had been linearized with Fast Digest MssI (ThermoFisher Scientific catalog no. FD1344) and purified by ethanol precipitation. Briefly, 1 μl of 0.5 m EDTA, pH 8, 2 μl of 3 m sodium acetate, and 40 μl of 100% ethanol were added to the linearized plasmid and frozen overnight at −80 °C. Linearized plasmid was then centrifuged for 15 min at 13,000 rpm at 4 °C, and the supernatant was removed. The pellet was then suspended in 6 μl of nuclease-free water. mRNA was synthesized using the mMESSAGE mMachine T7 ultra kit (Invitrogen/ThermoFisher Scientific catalog no. AM1345, Waltham, MA) including the poly(A) tailing reaction as per manufacturer's protocol. 100 pg each of prp1 forward and reverse TALENs were injected into AB strain WT zebrafish embryos. 25 pg of egfp mRNA was co-injected with the TALEN mRNA so that fish that were successfully injected could be identified and raised to adulthood. egfp mRNA was produced from pCS2+egfp by first linearizing the plasmid with Fast Digest NotI (ThermoFisher Scientific catalog no. FD0593) and then transcribing mRNA using the mMESSAGE MMachine SP6 kit (Invitrogen/ThermoFisher Scientific catalog no. AM1340).
Detection of larvae with somatic and germline mutations in prp1
The first step in our analyses of TALEN effectiveness was to determine whether TALENs induced somatic mutations in injected embryos (F0 generation). Siblings of successfully mutated F0 fish were grown to adulthood. F0 fish were then bred, and pools of F1 generation embryos were screened for successful germline transmission of TALEN-induced mutations. For detection of both somatic and germline-transmitted mutations in embryos, genomic DNA was isolated from pools of test fish (injected F0 embryos for detection of somatic mutations or offspring of F0 embryos for detection of germline-transmitted mutations) or un-injected WT AB strain fish at either 24 hpf or 3 dpf using a protocol modified from Ref. 77. Briefly, samples (pools of up to 20 embryos) were boiled for 15 min in 50 mm NaOH (5 μl/embryo), cooled on ice for 5 min, and neutralized with 1 m Tris-HCl, pH 8 (0.5 μl/embryo). Genomic DNA was then diluted 10-fold in sterile Milli-Q water prior to HRM analysis. Diluted genomic DNA was amplified using HRM primers (Table S2) and MeltDoctorTM HRM Master Mix (ThermoFisher Scientific catalog no. 4415440). HRM data were generated using the Applied Biosystems 7500 Fast Real-Time PCR system with MeltDoctorTM HRM Master Mix. Data were then analyzed using the High ReSolution Melt (Version 2.0, High ReSolution Melt, ThermoFisher Scientific).
Upon identification of pools of genomic DNA with distinct melt profiles compared with un-injected WT AB strain samples, those pools (and WT control pools) were PCR-amplified with amplicon primers (Table S2) and cloned into the pCR2.1 Topo vector as per the instructions in the TOPO TA cloning kit (Invitrogen/ThermoFisher Scientific catalog no. K4500-01). Clones were dissolved in 25 μl of sterile Milli-Q water for HRM analysis, and a portion of each clone was streaked on agar plates for subsequent analysis. Clones with a different melt profile compared with control clones were mini-prepped with a Qiaprep Spin Miniprep Kit (Qiagen catalog no. 27106, Toronto, Ontario, Canada) and submitted to the Molecular Biology Service Unit, University of Alberta, for sequencing.
Identification of adult F1 generation fish that are heterozygous for TALEN-induced mutations in prp1
Siblings of F1 embryos that had germline-transmitted TALEN mutations were grown to adulthood. These adult F1 fish were then screened to identify carriers of TALEN mutations. Fish were anesthetized with 4.1% tricaine, and a small piece of caudal fin was harvested. Genomic DNA extraction was performed as above but with 15 μl of 50 mm NaOH and 1.5 μl Tris-HCl per sample. DNA was diluted either 20- or 30-fold in sterile Milli-Q water prior to HRM analysis. A PCR product (primers shown in Table S2) containing the target site was amplified from genomic DNA and was cloned into the pCR2.1 Topo vector using a TOPO TA cloning kit (Invitrogen/ThermoFisher Scientific catalog no. K4500-01). The construct was then mini-prepped with a Qiaprep Spin Miniprep kit (Qiagen catalog no. 27106, Toronto, Ontario, Canada) and sequenced using a T7 primer sequence within the vector (5′-TAA TAC GAC TCA CTA TAG GG-3′).
Genotyping
HRM to identify fish with mutant prp1 alleles
Once stably inherited alleles had been identified (see under “Experimental procedures” and “Results”), methods were developed for genotyping individual fish. Genomic DNA was amplified using HRM as described above. Sample melt curves are shown in Fig. S5. One round of HRM (primers listed in Table S2) was used to distinguish the genotype of fish for the prp1 ua5004 allele. The accuracy of this method was confirmed by sequencing the area around the target site. Briefly, a 453-bp region around the target site was amplified (primers in Table S2). The amplicon was either cloned into pCR2.1, followed by sequencing of mini-prepped plasmid with a T7 vector-specific primer (5′-TAA TAC GAC TCA CTA TAG GG-3′), or the PCR product was treated with Illustra ExoProstar (Sigma catalog no. US78210, St. Louis, MO), as specified in the manufacturer's instructions, and directly sequenced using the same primers used to generate the amplicon. HRM to screen for prp1+/ua5003 and prp1ua5003/ua5003 fish was performed in a two-step process because genomic DNA from WT and prp1ua5003/ua5003 fish had overlapping melt profiles (see Fig. S5A). Thus, heterozygous individuals could be discriminated from homozygous individuals, but homozygous fish were not clearly mutant or WT. In the second round of HRM, 0.5 μl of sample genomic DNA was added to 0.5 μl of known DNA (WT or homozygous mutant) and diluted 20-fold in sterile Milli-Q water. The accuracy of this method was confirmed by sequencing as described above for the ua5004 allele.
RFLP to detect fish heterozygous and homozygous for the ua5001 allele
For ease of genotyping fish with the prp2ua5001 allele previously isolated (16), a restriction fragment length polymorphism (RFLP) assay was developed, as reported recently (27). Genomic DNA was amplified using prp2 RFLP primers (Table S2) and then digested with Fast Digest MvaI (ThermoFisher Scientific, catalog no. FD0554). As the ua5001 mutation disrupted the Mva I cut site, PCR products from mutant and WT DNA produced different banding patterns on an ethidium bromide-agarose gel (WT allele yields three bands with sizes of 21, 36, and 54 bp; ua5001 allele yields two bands with sizes of 36 and 71 bp; Fig. S5C). The accuracy of this genotyping assay was confirmed by sequencing. Briefly, a 1039-bp region around the target site was amplified (primers in Table S2) and sequenced using the same primers used to generate the amplicon after treatment with Illustra ExoProstar (Sigma catalog no. US78210) to remove unincorporated nucleotides.
RT-qPCR to quantify prp1, prp2, or c-fos transcripts abundance
Experiments were performed in compliance with the MIQE guidelines (Minimum Information for Publication of Quantitative Real-Time PCR Experiments) (78). RNA samples for comparing prp1 and/or prp2 transcript abundance were obtained from pools of larvae at the ages noted (each biological replicate represents 15–20 larvae) that had been stored in RNAlater (Ambion/ThermoFisher Scientific, catalog no. AM7021). RNA samples for comparing prp1 transcript abundance in prp1ua5004/ua5004 mutants versus prp1 transcript abundance in WT fish were obtained from pools of 2 dpf larvae (each biological replicate represents five larvae) and stored in RNAlater. Total RNA was extracted from pools of embryos using the RNeasy kit (Qiagen catalog no. 74104, Toronto, Ontario, Canada) as outlined in the manufacturer's protocols. The samples were homogenized in the appropriate lysis buffer (Buffer RLT for larvae) with a rotor stator homogenizer (VWR catalog no. 47747-370, Radnor, PA), and on-column DNA digestion was performed using Qiagen DNase I (Qiagen catalog no. 79254, Toronto, Ontario, Canada). RNA quantity was determined using a Nanodrop 2000 spectrophotometer (ThermoFisher Scientific). All of the samples had rRNA profiles with strong 28S and 18S bands, and RNA integrity numbers of at least 7/10 as determined using an Agilent RNA 6000 NanoChip and Agilent 2100 Bioanalyzer. cDNA was then generated using a qScript Supermix kit (Quanta BioSciences catalog no. 95048-100, Beverly, MA).
qPCR was performed using a 7500 Real-Time PCR system (ABI Applied Biosystems). Primers were designed using Primer Express (Version 3.0, Primer Express, ThermoFisher Scientific), and the prp1, prp2, and β-actin primers were previously verified with standard curves and melt curves (16). These primers were further verified here by confirming that no product was apparent when reverse transcriptase was left out of the cDNA synthesis. The c-fos qPCR primers (forward, 5′-GCAAAGACCTCCAACAAGAGA-3′; reverse, 5′-TTTCGCAGCAGCCATCTT-3′) span introns 2–3 of the c-fos gene (PMID Gene ID:394198; ZFIN ID: ZDB-GENE-031222-4) and produce a 102-bp product from cDNA. qPCRs were performed with three technical replicates of each biological replicate. Each reaction contained 5 μl of Dynamite Master Mix (prepared and supplied by Molecular Biology Service Unit, University of Alberta). The mix included SYBR Green and platinum Taq hot start enzyme), 2.5 μl of pre-mixed primer working stocks (final concentrations of the β-actin, c-fos, and prp1 primers were 800, 800, and 200 nm, respectively), and 2.5 μl cDNA for a total volume of 10 μl. After an initial denaturation step (2 min at 95 °C), cycling consisted of 40 cycles of 95 °C for 15 s followed by 60 °C for 1 min. One cycle for melting dissociation curve analysis followed these 40 cycles and consisted of 95 °C for 15 s, 60 °C for 1 min, 95 °C for 15 s, and finally 60 °C for 1 min. Data analysis was performed using 7500 Software for 7500 and 7500 Fast Real Time PCR Systems version 2.0.1 (AB Applied Biosystems) with auto Ct calling. Transcript abundance was normalized to β-actin levels. Relative fold change in transcript abundance was statistically analyzed on the resulting relative quantification values.
Characterizing mutant transcripts
To assess whether prp1 and prp2 transcripts had the expected length and content, RACE was performed as per the kit instructions (Clontech catalog no. 634860) using mRNA isolated as per RT-qPCR methods above. 5′-RACE used gene-specific primers reported in supporting Table S3. Transcript lengths were compared between mutant and WT by gel electrophoresis and validated by sequencing. Transcripts were also characterized by examining the alignment of RNA-Seq reads around both the prp1 and prp2 genes by comparing 3 dpf WT and prp1−/−;prp2−/− larvae. Pools of 50 larvae from each genotype were homogenized in TRIzol (Invitrogen/ThermoFisher Scientific catalog no. 15596026) and shipped to Otogenetics (Atlanta GA) for Illumina RNA-Seq, and DNAnexus Platform standard RNAseq analysis at a depth of >41 million reads each.
Measuring the length of larval zebrafish
2 dpf larvae were fixed overnight in 4% paraformaldehyde (PFA) in phosphate buffer, pH 7.4, with 5% sucrose at 4 °C. Larvae were then rinsed several times with 1× PBS and imaged and photographed with a Leica M165 FC dissecting microscope and a Leica DFC 400 camera. The scale bar feature in the Leica software was then used to measure the length of each fish from the forebrain to the tip of the caudal fin.
Alkaline phosphatase staining
Neuromasts are rich in endogenous alkaline phosphatase and staining for this allows for their observation. Zebrafish larvae were fixed in 4% PFA in phosphate buffer, pH 7.4, with 5% sucrose for 3–3.5 h at room temperature. They were subsequently washed four times in phosphate-buffered saline (PBS; pH 7.4) with 0.1% Tween (PBST) and then for 15 min in fresh alkaline phosphatase buffer (pH 9.5; 100 mm Tris-HCl, 100 mm NaCl, 50 mm MgCl2) with 0.1% Tween. They were developed in alkaline phosphatase buffer containing 0.225% NBT and 0.175% BCIP (Roche Applied Science catalog nos. 11383213001 and 11383221001, Basel, Switzerland) for ∼10 min. Fish were then washed for 30 min in alkaline phosphatase wash buffer (pH 7.5; 154 mm NaCl, 11 mm Tris-HCl, 1 mm EDTA) with 0.1% Tween, fixed in 4% PFA with 5% sucrose, and washed three times in PBST. Some fish were counterstained with phalloidin 488 or 555 (catalog nos. A12379 and A34055, Invitrogen Molecular Probes, Eugene, OR) prior to imaging. For this, fish were incubated in PBS with 2% Triton X-100 for 2 h. Phalloidin 488 was diluted 20-fold in PBS with 2% Triton X-100 and left overnight at 4 °C. Fish were then washed three times for 20 min in PBS with 2% Triton X-100 and then washed into PBS with 0.1% Tween. Whole mounts were transferred to a PBST/glycerol mixture and imaged with a Leica M165 FC dissecting microscope and Leica DFC 400 camera.
Analysis of neuromast number and position
Trunk neuromasts of the PLL were visualized by alkaline phosphatase NBT/BCIP staining as described above or by detection of GFP fluorescence in Tg(cldnb:gfp) using a Leica M165 FC dissecting microscope. An observer, who was blinded to the genotype of the fish, counted the number of neuromasts. In some analyses, we examined neuromast number with respect to somite number. In some 3 dpf larvae, we noticed lighter stained neuromasts between the L1 and L2 neuromasts. These were likely neuromasts of the secondary posterior lateral line system (see Fig. 4). We considered these to be part of the secondary posterior lateral line system, and excluded them from our counts if they were ≤5 somite lengths posterior to the L1 neuromasts.
Locomotor activity measurements of PTZ-induced seizures
Assays for measuring stage I and II seizures were performed according to established methods (44, 79), and behavioral tracking software was used to quantify the activity and velocity of zebrafish larvae arrayed in 96-well plates. Larvae at age 4 dpf were carefully pipetted into 96-well plate (7701-1651; Whatman) containing 650 μl of their typical embryonic growth media (E3 media) and were acclimatized for 3 h prior to baseline recording for 1 h. This was followed by treatment with various doses of PTZ (2.5–15.0 mm) for 2 h. Plates were placed on top of an IR backlight source and below a Basler GenICaM (Basler acA 1300-60) scanning camera, both provided by Noldus (Wageningen, Netherlands), which were used to track locomotor activity of an individual larvae within each well. Etho Vision® XT-11.5 software (Noldus) was used to quantify locomotor activity, which was defined as % pixel change within a corresponding well between samples (motion was captured by taking 25 samples (frames)/s), similar to described previously (80). Thus, the absolute numbers recorded can appear to be small, as only a small percentage of pixels in the region of interest (a well in a 96-well plate) might be changing at any one time, and bursts of activity may occur such that total movement is small within any given minute. Thus, the absolute values are not especially meaningful in these assays, which operationally are both sensitive and robust insomuch that the data are reproducible between trials and give rational outcomes in dose-response formats. The results are consistent with those attained when the same behavioral traces were assessed for their mean velocity (Fig. S7). For baseline activity (Fig. 5D) and PTZ time-course experiments (Fig. 5B), data were calculated directly, whereas for PTZ dose-response (Fig. 5C) the mean activity following 30 min of PTZ treatment was normalized to 1 h baseline activity prior PTZ addition. All data reported from mutant larvae herein are from maternal zygotic larvae.
Further analysis of seizure behavior (Fig. S9) used different optics on the camera (a 75-mm f2.8 C-mount lens, Noldus) that improved fidelity of larval tracking, allowed assessment of further parameters, and permitted use of 96-well plates with round wells (Costar catalog no. 3599). Larvae were kept in 100 μl of E3 media and tracked with daniovision software. To exclude background noise, active movement was defined as >0.2 mm at >2 mm/s velocity. Outliers were removed objectively using ROUT Q = 0.1 for velocity, number of movements, and movement duration. For activity, Q was set to 10, but only three outliers were detected in WT.
Detecting c-fos by in situ hybridization
For probe production, RNA was extracted from WT zebrafish larvae using an RNeasy mini kit (Qiagen catalog no. 74104, Toronto, Ontario, Canada). A 602-bp cDNA product was then produced from WT zebrafish larvae using a Qiagen LongRange Two-step RT-PCR kit (Qiagen catalog no. 205920, Toronto, Ontario, Canada). For the initial PCR, primers from a previous publication were used: forward, 5′-TCTCCTCTGTGGCGCCCTCC-3′, and reverse, 5′-GTCTGGAACCGAGCGAGCCG-3′ (81). The cDNA product was then cloned into the pCr2.1 Topo vector (Invitrogen) and sequenced to verify the insert orientation in the plasmids. The resulting plasmid was linearized with FastDigest KpnI (ThermoFisher Scientific catalog no. FD0524) and used to template production of digoxigenin-labeled riboprobe with T7 RNA polymerase (Roche Applied Science/Sigma catalog no. 10881775001). After PTZ or vehicle treatment, fish were rinsed with E3 medium and fixed overnight in 4% PFA in phosphate buffer, pH 7.4, with 5% sucrose. Fish were then washed in 50% methanol/diethyl pyrocarbonate-treated water for 5 min, rinsed in 100% MeOH, and stored at least overnight in 100% MeOH at −20 °C. In situ hybridization was performed as described previously (16), except that the hybridization temperature was 60 °C.
Statistical analysis
All statistics were performed using GraphPad Prism Software (Version 6, GraphPad, San Diego). Prior to performing pairwise comparisons between groups, the F-test was used to assess variance. If variance within groups was not statistically significant, pairwise comparisons were performed using paired t-tests. If variance was statistically significantly different, Mann-Whitney U tests were performed instead. For multiple group comparisons, groups were assessed for variance and normal distribution using the Brown Forsythe and Bartlett's tests. If variance was not significant, data were analyzed using one-way ANOVA, and if variance was significant, data were analyzed using a Kruskal Wallis test. RT-qPCR data were analyzed by performing statistics on the RQ values. For graphical presentation, data were normalized to the WT samples and plotted as a percentage.
Statistical analysis of basal and seizure activity following exposure to convulsant used one-way ANOVA to compare basal activities across genotype and Dunnett's multiple comparison test. Two-way ANOVAs with Holm-Šídák multiple comparison test were used to compare the seizures across phenotypes and time, or across phenotypes and dose. Outlier data points were objectively removed, where indicated, using the ROUT function in the Prism software with Q = 0.01.
Author contributions
P. L. L., R. K., and W. T. A. formal analysis; P. L. L., R. K., G. J. N., N. M. P., and W. T. A. investigation; P. L. L., R. K., and W. T. A. visualization; P. L. L. and R. K. methodology; P. L. L. and W. T. A. writing-original draft; P. L. L., R. K., N. M. P., and W. T. A. writing-review and editing; W. T. A. conceptualization; W. T. A. supervision; W. T. A. funding acquisition; W. T. A. project administration.
Supplementary Material
Acknowledgments
We thank Laszlo Locksai, Jordyn Ko, Natasha Lifeso, and Kirklin Maclise for technical assistance.
This work was supported in part by operating grants from the Alberta Prion Research Institute–Alberta Innovates BioSolutions and the Alzheimer Society of Alberta and the Northwest Territories (to W. T. A.). The authors declare that they have no conflicts of interest with the contents of this article.
 This article contains Figs. S1–S13 and Tables S1–S7.
This article contains Figs. S1–S13 and Tables S1–S7.
- PrPC
- normal cellular prion protein
- APP
- amyloid-β precursor protein
- dpf
- days post-fertilization
- HRM
- high-resolution melt analysis
- MO
- morpholino
- NCAM
- neural cell adhesion molecule
- PLL
- posterior lateral line
- PTZ
- convulsant pentylenetetrazole
- RT-qPCR
- reverse transcriptase quantitative PCR
- TALEN
- Tal Effector Nuclease
- hpf
- h post-fertilization
- ANOVA
- analysis of variance
- GPI
- glycosylphosphatidylinositol
- RNA-Seq
- RNA-sequencing
- RACE
- rapid amplification of cDNA ends
- PFA
- paraformaldehyde
- EJC
- exon–junction complex
- RFLP
- restriction fragment length polymorphism
- NMDA
- N-methyl-d-aspartic acid
- CNS
- central nervous system
- NBT
- nitro blue tetrazolium
- BCIP
- 5-bromo-4-chloro-3-indolyl phosphate.
References
- 1. Leighton P. L., and Allison W. T. (2016) Protein misfolding in prion and prion-like diseases: reconsidering a required role for protein loss–of–function. J. Alzheimers Dis. 54, 3–29 10.3233/JAD-160361 [DOI] [PubMed] [Google Scholar]
- 2. Allison W. T., DuVal M. G., Nguyen-Phuoc K., and Leighton P. L. A. (2017) Reduced abundance and subverted functions of proteins in prion-like diseases: gained functions fascinate but lost functions affect aetiology. Int. J. Mol. Sci. 18, E2223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reimann R. R., Sonati T., Hornemann S., Herrmann U. S., Arand M., Hawke S., and Aguzzi A. (2016) Differential toxicity of antibodies to the prion protein. PLoS Pathog. 12, e1005401 10.1371/journal.ppat.1005401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Laurén J. (2014) Cellular prion protein as a therapeutic target in Alzheimer's disease. J. Alzheimers Dis. 38, 227–244 [DOI] [PubMed] [Google Scholar]
- 5. Graner E., Mercadante A. F., Zanata S. M., Forlenza O. V., Cabral A. L., Veiga S. S., Juliano M. A., Roesler R., Walz R., Minetti A., Izquierdo I., Martins V. R., and Brentani R. R. (2000) Cellular prion protein binds laminin and mediates neuritogenesis. Mol. Brain Res. 76, 85–92 10.1016/S0169-328X(99)00334-4 [DOI] [PubMed] [Google Scholar]
- 6. Schmitt-Ulms G., Legname G., Baldwin M. A., Ball H. L., Bradon N., Bosque P. J., Crossin K. L., Edelman G. M., DeArmond S. J., Cohen F. E., and Prusiner S. B. (2001) Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol. 314, 1209–1225 10.1006/jmbi.2000.5183 [DOI] [PubMed] [Google Scholar]
- 7. Watts J. C., Huo H., Bai Y., Ehsani S., Jeon A. H., Won A. H., Shi T., Daude N., Lau A., Young R., Xu L., Carlson G. A., Williams D., Westaway D., and Schmitt-Ulms G. (2009) Interactome analyses identify ties of PrPC and its mammalian paralogs to oligomannosidic N-glycans and endoplasmic reticulum-derived chaperones. PLoS Pathog. 5, e1000608 10.1371/journal.ppat.1000608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beraldo F. H., Arantes C. P., Santos T. G., Machado C. F., Roffe M., Hajj G. N., Lee K. S., Magalhães A. C., Caetano F. A., Mancini G. L., Lopes M. H., Américo T. A., Magdesian M. H., Ferguson S. S., Linden R., et al. (2011) Metabotropic glutamate receptors transduce signals for neurite outgrowth after binding of the prion protein to laminin γ1 chain. FASEB J. 25, 265–279 10.1096/fj.10-161653 [DOI] [PubMed] [Google Scholar]
- 9. Santuccione A., Sytnyk V., Leshchyns'ka I., and Schachner M. (2005) Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol. 169, 341–354 10.1083/jcb.200409127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Parrie L. E., Crowell J. A. E., Telling G. C., and Bessen R. A. (2018) The cellular prion protein promotes olfactory sensory neuron survival and axon targeting during adult neurogenesis. Dev. Biol. 438, 23–32 10.1016/j.ydbio.2018.03.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mehrabian M., Brethour D., Wang H., Xi Z., Rogaeva E., and Schmitt-Ulms G. (2015) The prion protein controls polysialylation of neural cell adhesion molecule 1 during cellular morphogenesis. PLoS ONE 10, e0133741 10.1371/journal.pone.0133741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Manson J. C., Clarke A. R., Hooper M. L., Aitchison L., McConnell I., and Hope J. (1994) 129/Ola mice carrying a null mutation in PrP that abolishes mRNA production are developmentally normal. Mol. Neurobiol. 8, 121–127 10.1007/BF02780662 [DOI] [PubMed] [Google Scholar]
- 13. Büeler H., Fischer M., Lang Y., Bluethmann H., Lipp H. P., DeArmond S. J., Prusiner S. B., Aguet M., and Weissmann C. (1992) Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577–582 10.1038/356577a0 [DOI] [PubMed] [Google Scholar]
- 14. Lieschke G. J., and Currie P. D. (2007) Animal models of human disease: zebrafish swim into view. Nat. Rev. Genet. 8, 353–367 10.1038/nrg2091 [DOI] [PubMed] [Google Scholar]
- 15. Cotto E., André M., Forgue J., Fleury H. J., and Babin P. J. (2005) Molecular characterization, phylogenetic relationships, and developmental expression patterns of prion genes in zebrafish (Danio rerio). FEBS J. 272, 500–513 10.1111/j.1742-4658.2004.04492.x [DOI] [PubMed] [Google Scholar]
- 16. Fleisch V. C., Leighton P. L., Wang H., Pillay L. M., Ritzel R. G., Bhinder G., Roy B., Tierney K. B., Ali D. W., Waskiewicz A. J., and Allison W. T. (2013) Targeted mutation of the gene encoding prion protein in zebrafish reveals a conserved role in neuron excitability. Neurobiol. Dis. 55, 11–25 10.1016/j.nbd.2013.03.007 [DOI] [PubMed] [Google Scholar]
- 17. Rivera-Milla E., Oidtmann B., Panagiotidis C. H., Baier M., Sklaviadis T., Hoffmann R., Zhou Y., Solis G. P., Stuermer C. A., and Málaga-Trillo E. (2006) Disparate evolution of prion protein domains and the distinct origin of Doppel- and prion-related loci revealed by fish-to-mammal comparisons. FASEB J. 20, 317–319 10.1096/fj.05-4279fje [DOI] [PubMed] [Google Scholar]
- 18. Málaga-Trillo E., Solis G. P., Schrock Y., Geiss C., Luncz L., Thomanetz V., and Stuermer C. A. (2009) Regulation of embryonic cell adhesion by the prion protein. PLoS Biol. 7, e55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Málaga-Trillo E., and Sempou E. (2009) PrPs: proteins with a purpose: lessons from the zebrafish. Prion 3, 129–133 10.4161/pri.3.3.9651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Salta E., Kanata E., Ouzounis C. A., Gilch S., Schätzl H., and Sklaviadis T. (2014) Assessing proteinase K resistance of fish prion proteins in a scrapie-infected mouse neuroblastoma cell line. Viruses 6, 4398–4421 10.3390/v6114398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Málaga-Trillo E., Salta E., Figueras A., Panagiotidis C., and Sklaviadis T. (2011) Fish models in prion biology: underwater issues. Biochim. Biophys. Acta 1812, 402–414 10.1016/j.bbadis.2010.09.013 [DOI] [PubMed] [Google Scholar]
- 22. Kaiser D. M., Acharya M., Leighton P. L., Wang H., Daude N., Wohlgemuth S., Shi B., and Allison W. T. (2012) Amyloid β precursor protein and prion protein have a conserved interaction affecting cell adhesion and CNS development. PLoS ONE 7, e51305 10.1371/journal.pone.0051305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sempou E., Biasini E., Pinzón-Olejua A., Harris D. A., and Málaga-Trillo E. (2016) Activation of zebrafish Src family kinases by the prion protein is an amyloid-β-sensitive signal that prevents the endocytosis and degradation of E-cadherin/β-catenin complexes in vivo. Mol. Neurodegener. 11, 18 10.1186/s13024-016-0076-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miesbauer M., Bamme T., Riemer C., Oidtmann B., Winklhofer K. F., Baier M., and Tatzelt J. (2006) Prion protein-related proteins from zebrafish are complex glycosylated and contain a glycosylphosphatidylinositol anchor. Biochem. Biophys. Res. Commun. 341, 218–224 10.1016/j.bbrc.2005.12.168 [DOI] [PubMed] [Google Scholar]
- 25. Bayés À., Collins M. O., Reig-Viader R., Gou G., Goulding D., Izquierdo A., Choudhary J. S., Emes R. D., and Grant S. G. (2017) Evolution of complexity in the zebrafish synapse proteome. Nat. Commun. 8, 14613 10.1038/ncomms14613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Singh S., Ramamoorthy K., Saradhi A. P., and Idris M. (2010) Proteome profile of zebrafish brain based on Gel LC-ESI MS/MS analysis. J. Proteomics Bioinformatics 3, 135–142 10.4172/jpb.1000132 [DOI] [Google Scholar]
- 27. Leighton P. L. A., Nadolski N. J., Morrill A., Hamilton T. J., and Allison W. T.. An ancient conserved role for prion protein in learning and memory. Biol. Open 7, bio025734 10.1242/bio.025734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Solis G. P., Radon Y., Sempou E., Jechow K., Stuermer C. A., and Málaga-Trillo E. (2013) Conserved roles of the prion protein domains on subcellular localization and cell-cell adhesion. PLoS ONE 8, e70327 10.1371/journal.pone.0070327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nourizadeh-Lillabadi R., Seilø Torgersen J., Vestrheim O., König M., Aleström P., and Syed M. (2010) Early embryonic gene expression profiling of zebrafish prion protein (Prp2) morphants. PLoS ONE 5, e13573 10.1371/journal.pone.0013573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Huc-Brandt S., Hieu N., Imberdis T., Cubedo N., Silhol M., Leighton P. L., Domaschke T., Allison W. T., Perrier V., and Rossel M. (2014) Zebrafish prion protein PrP2 controls collective migration process during lateral line sensory system development. PLoS ONE 9, e113331 10.1371/journal.pone.0113331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Culbertson M. R. (1999) RNA surveillance. Unforeseen consequences for gene expression, inherited genetic disorders and cancer. Trends Genet. 15, 74–80 10.1016/S0168-9525(98)01658-8 [DOI] [PubMed] [Google Scholar]
- 32. Pantera B., Bini C., Cirri P., Paoli P., Camici G., Manao G., and Caselli A. (2009) PrPc activation induces neurite outgrowth and differentiation in PC12 cells: role for caveolin-1 in the signal transduction pathway. J. Neurochem. 110, 194–207 10.1111/j.1471-4159.2009.06123.x [DOI] [PubMed] [Google Scholar]
- 33. Dambly-Chaudière C., Cubedo N., and Ghysen A. (2007) Control of cell migration in the development of the posterior lateral line: antagonistic interactions between the chemokine receptors CXCR4 and CXCR7/RDC1. BMC Dev. Biol. 7, 23 10.1186/1471-213X-7-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Villablanca E. J., Renucci A., Sapède D., Lec V., Soubiran F., Sandoval P. C., Dambly-Chaudière C., Ghysen A., and Allende M. L. (2006) Control of cell migration in the zebrafish lateral line: implication of the gene “tumour-associated calcium signal transducer,” tacstd. Dev. Dyn. 235, 1578–1588 10.1002/dvdy.20743 [DOI] [PubMed] [Google Scholar]
- 35. Ghysen A., and Dambly-Chaudière C. (2007) The lateral line microcosmos. Genes Dev. 21, 2118–2130 10.1101/gad.1568407 [DOI] [PubMed] [Google Scholar]
- 36. Gompel N., Cubedo N., Thisse C., Thisse B., Dambly-Chaudière C., and Ghysen A. (2001) Pattern formation in the lateral line of zebrafish. Mech. Dev. 105, 69–77 10.1016/S0925-4773(01)00382-3 [DOI] [PubMed] [Google Scholar]
- 37. Haas P., and Gilmour D. (2006) Chemokine signaling mediates self-organizing tissue migration in the zebrafish lateral line. Dev. Cell 10, 673–680 10.1016/j.devcel.2006.02.019 [DOI] [PubMed] [Google Scholar]
- 38. Carulla P., Llorens F., Matamoros-Angles A., Aguilar-Calvo P., Espinosa J. C., Gavín R., Ferrer I., Legname G., Torres J. M., and del Río J. A. (2015) Involvement of PrP(C) in kainate-induced excitotoxicity in several mouse strains. Sci. Rep. 5, 11971 10.1038/srep11971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Striebel J. F., Race B., and Chesebro B. (2013) Prion protein and susceptibility to kainate-induced seizures: genetic pitfalls in the use of PrP knockout mice. Prion 7, 280–285 10.4161/pri.25738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Vossel K. A., Tartaglia M. C., Nygaard H. B., Zeman A. Z., and Miller B. L. (2017) Epileptic activity in Alzheimer's disease: causes and clinical relevance. Lancet Neurol. 16, 311–322 10.1016/S1474-4422(17)30044-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ryan N. S., Nicholas J. M., Weston P. S. J., Liang Y., Lashley T., Guerreiro R., Adamson G., Kenny J., Beck J., Chavez-Gutierrez L., de Strooper B., Revesz T., Holton J., Mead S., Rossor M. N., and Fox N. C. (2016) Clinical phenotype and genetic associations in autosomal dominant familial Alzheimer's disease: a case series. Lancet Neurol. 15, 1326–1335 10.1016/S1474-4422(16)30193-4 [DOI] [PubMed] [Google Scholar]
- 42. Wieser H. G., Schindler K., and Zumsteg D. (2006) EEG in Creutzfeldt-Jakob disease. Clin. Neurophysiol. 117, 935–951 10.1016/j.clinph.2005.12.007 [DOI] [PubMed] [Google Scholar]
- 43. Bertani I., Iori V., Trusel M., Maroso M., Foray C., Mantovani S., Tonini R., Vezzani A., and Chiesa R. (2017) Inhibition of IL-1β signaling normalizes NMDA-dependent neurotransmission and reduces seizure susceptibility in a mouse model of Creutzfeldt-Jakob disease. J. Neurosci. 37, 10278–10289 10.1523/JNEUROSCI.1301-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baraban S. C., Taylor M. R., Castro P. A., and Baier H. (2005) Pentylenetetrazole induced changes in zebrafish behavior, neural activity and c-fos expression. Neuroscience 131, 759–768 10.1016/j.neuroscience.2004.11.031 [DOI] [PubMed] [Google Scholar]
- 45. Yu G., Chen J., Xu Y., Zhu C., Yu H., Liu S., Sha H., Chen J., Xu X., Wu Y., Zhang A., Ma J., and Cheng G. (2009) Generation of goats lacking prion protein. Mol. Reprod. Dev. 76, 3 10.1002/mrd.20960 [DOI] [PubMed] [Google Scholar]
- 46. Richt J. A., Kasinathan P., Hamir A. N., Castilla J., Sathiyaseelan T., Vargas F., Sathiyaseelan J., Wu H., Matsushita H., Koster J., Kato S., Ishida I., Soto C., Robl J. M., and Kuroiwa Y. (2007) Production of cattle lacking prion protein. Nat. Biotechnol. 25, 132–138 10.1038/nbt1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Benestad S. L., Austbø L., Tranulis M. A., Espenes A., and Olsaker I. (2012) Healthy goats naturally devoid of prion protein. Vet. Res. 43, 87 10.1186/1297-9716-43-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Málaga-Trillo E., and Ochs K. (2016) Uncontrolled SFK-mediated protein trafficking in prion and Alzheimer's disease. Prion 10, 352–361 10.1080/19336896.2016.1221873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ochs K., and Málaga-Trillo E. (2014) Common themes in PrP signaling: the Src remains the same. Front. Cell Dev. Biol. 2, 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Schmitt-Ulms G., Ehsani S., Watts J. C., Westaway D., and Wille H. (2009) Evolutionary descent of prion genes from the ZIP family of metal ion transporters. PLoS ONE 4, e7208 10.1371/journal.pone.0007208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ehsani S., Huo H., Salehzadeh A., Pocanschi C. L., Watts J. C., Wille H., Westaway D., Rogaeva E., St George-Hyslop P. H., and Schmitt-Ulms G. (2011) Family reunion–the ZIP/prion gene family. Prog. Neurobiol. 93, 405–420 10.1016/j.pneurobio.2010.12.001 [DOI] [PubMed] [Google Scholar]
- 52. Ehsani S., Tao R., Pocanschi C. L., Ren H., Harrison P. M., and Schmitt-Ulms G. (2011) Evidence for retrogene origins of the prion gene family. PLoS ONE 6, e26800 10.1371/journal.pone.0026800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ehsani S., Mehrabian M., Pocanschi C. L., and Schmitt-Ulms G. (2012) The ZIP-prion connection. Prion 6, 317–321 10.4161/pri.20196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mehrabian M., Hildebrandt H., and Schmitt-Ulms G. (2016) NCAM1 polysialylation: the prion protein's elusive reason for being? ASN Neuro 8, 1759091416679074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mehrabian M., Ehsani S., and Schmitt-Ulms G. (2014) An emerging role of the cellular prion protein as a modulator of a morphogenetic program underlying epithelial–to–mesenchymal transition. Front. Cell Dev. Biol. 2, 53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yamashita S., Miyagi C., Fukada T., Kagara N., Che Y. S., and Hirano T. (2004) Zinc transporter LIVI controls epithelial-mesenchymal transition in zebrafish gastrula organizer. Nature 429, 298–302 10.1038/nature02545 [DOI] [PubMed] [Google Scholar]
- 57. Brethour D., Mehrabian M., Williams D., Wang X., Ghodrati F., Ehsani S., Rubie E. A., Woodgett J. R., Sevalle J., Xi Z., Rogaeva E., and Schmitt-Ulms G. (2017) A ZIP6-ZIP10 heteromer controls NCAM1 phosphorylation and integration into focal adhesion complexes during epithelial–to–mesenchymal transition. Sci. Rep. 7, 40313 10.1038/srep40313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Bedell V. M., Westcot S. E., and Ekker S. C. (2011) Lessons from morpholino-based screening in zebrafish. Brief. Funct. Genomics 10, 181–188 10.1093/bfgp/elr021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Stainier D. Y. R., Raz E., Lawson N. D., Ekker S. C., Burdine R. D., Eisen J. S., Ingham P. W., Schulte-Merker S., Yelon D., Weinstein B. M., Mullins M. C., Wilson S. W., Ramakrishnan L., Amacher S. L., Neuhauss S. C. F., et al. (2017) Guidelines for morpholino use in zebrafish. PLoS Genet. 13, e1007000 10.1371/journal.pgen.1007000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wulf M. A., Senatore A., and Aguzzi A. (2017) The biological function of the cellular prion protein: an update. BMC Biol. 15, 34 10.1186/s12915-017-0375-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Daude N., Wohlgemuth S., Brown R., Pitstick R., Gapeshina H., Yang J., Carlson G. A., and Westaway D. (2012) Knockout of the prion protein (PrP)-like Sprn gene does not produce embryonic lethality in combination with PrP(C)-deficiency. Proc. Natl. Acad. Sci. U.S.A. 109, 9035–9040 10.1073/pnas.1202130109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Young R., Passet B., Vilotte M., Cribiu E. P., Béringue V., Le Provost F., Laude H., and Vilotte J. L. (2009) The prion or the related Shadoo protein is required for early mouse embryogenesis. FEBS Lett. 583, 3296–3300 10.1016/j.febslet.2009.09.027 [DOI] [PubMed] [Google Scholar]
- 63. Daude N., and Westaway D. (2012) Shadoo/PrP (Sprn(0/0) /Prnp(0/0)) double knockout mice: more than zeroes. Prion 6, 420–424 10.4161/pri.21867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Mallucci G., Dickinson A., Linehan J., Klöhn P. C., Brandner S., and Collinge J. (2003) Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302, 871–874 10.1126/science.1090187 [DOI] [PubMed] [Google Scholar]
- 65. Suzuki T., Kurokawa T., Hashimoto H., and Sugiyama M. (2002) cDNA sequence and tissue expression of Fugu rubripes prion protein-like: a candidate for the teleost orthologue of tetrapod PrPs. Biochem. Biophys. Res. Commun. 294, 912–917 10.1016/S0006-291X(02)00546-6 [DOI] [PubMed] [Google Scholar]
- 66. Aman A., and Piotrowski T. (2008) Wnt/beta-catenin and Fgf signaling control collective cell migration by restricting chemokine receptor expression. Dev. Cell 15, 749–761 10.1016/j.devcel.2008.10.002 [DOI] [PubMed] [Google Scholar]
- 67. Komiya Y., and Habas R. (2008) Wnt signal transduction pathways. Organogenesis 4, 68–75 10.4161/org.4.2.5851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Hernandez-Rapp J., Martin-Lannerée S., Hirsch T. Z., Pradines E., Alleaume-Butaux A., Schneider B., Baudry A., Launay J. M., and Mouillet-Richard S. (2014) A PrP(C)-caveolin-Lyn complex negatively controls neuronal GSK3β and serotonin 1B receptor. Sci. Rep. 4, 4881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Matsuda M., and Chitnis A. B. (2010) Atoh1a expression must be restricted by Notch signaling for effective morphogenesis of the posterior lateral line primordium in zebrafish. Development 137, 3477–3487 10.1242/dev.052761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Solis G. P., Schrock Y., Hülsbusch N., Wiechers M., Plattner H., and Stuermer C. A. (2012) Reggies/flotillins regulate E-cadherin–mediated cell contact formation by affecting EGFR trafficking. Mol. Biol. Cell 23, 1812–1825 10.1091/mbc.e11-12-1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Schmitz M., Greis C., Ottis P., Silva C. J., Schulz-Schaeffer W. J., Wrede A., Koppe K., Onisko B., Requena J. R., Govindarajan N., Korth C., Fischer A., and Zerr I. (2014) Loss of prion protein leads to age-dependent behavioral abnormalities and changes in cytoskeletal protein expression. Mol. Neurobiol. 50, 923–936 10.1007/s12035-014-8655-3 [DOI] [PubMed] [Google Scholar]
- 72. Zhang Y., Duc A. C., Rao S., Sun X. L., Bilbee A. N., Rhodes M., Li Q., Kappes D. J., Rhodes J., and Wiest D. L. (2013) Control of hematopoietic stem cell emergence by antagonistic functions of ribosomal protein paralogs. Dev. Cell 24, 411–425 10.1016/j.devcel.2013.01.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Shum E. Y., Jones S. H., Shao A., Dumdie J., Krause M. D., Chan W. K., Lou C. H., Espinoza J. L., Song H. W., Phan M. H., Ramaiah M., Huang L., McCarrey J. R., Peterson K. J., De Rooij D. G., et al. (2016) The antagonistic gene paralogs Upf3a and Upf3b govern nonsense-mediated RNA decay. Cell 165, 382–395 10.1016/j.cell.2016.02.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Salazar S. V., and Strittmatter S. M. (2017) Cellular prion protein as a receptor for amyloid-β oligomers in Alzheimer's disease. Biochem. Biophys. Res. Commun. 483, 1143–1147 10.1016/j.bbrc.2016.09.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Um J. W., Kaufman A. C., Kostylev M., Heiss J. K., Stagi M., Takahashi H., Kerrisk M. E., Vortmeyer A., Wisniewski T., Koleske A. J., Gunther E. C., Nygaard H. B., and Strittmatter S. M. (2013) Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer Aβ oligomer bound to cellular prion protein. Neuron 79, 887–902 10.1016/j.neuron.2013.06.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Westerfield M. (2000) The Zebrafish Book. A Guide for the Laboratory Use of Zebrafish (Danio rerio). University of Oregon Press, Eugene, OR [Google Scholar]
- 77. Meeker N. D., Hutchinson S. A., Ho L., and Trede N. S. (2007) Method for isolation of PCR-ready genomic DNA from zebrafish tissues. BioTechniques 43, 610 10.2144/000112619 [DOI] [PubMed] [Google Scholar]
- 78. Bustin S. A., Benes V., Garson J. A., Hellemans J., Huggett J., Kubista M., Mueller R., Nolan T., Pfaffl M. W., Shipley G. L., Vandesompele J., and Wittwer C. T. (2009) The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55, 611–622 10.1373/clinchem.2008.112797 [DOI] [PubMed] [Google Scholar]
- 79. Baraban S. C., Dinday M. T., Castro P. A., Chege S., Guyenet S., and Taylor M. R. (2007) A large-scale mutagenesis screen to identify seizure-resistant zebrafish. Epilepsia 48, 1151–1157 10.1111/j.1528-1167.2007.01075.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bhinder G., and Tierney K. B. (2012) in Neuromethods (Kalueff A. V., and Stewart A. V., eds) pp. 71–84, Humana Press, Totowa, NJ [Google Scholar]
- 81. deCarvalho T. N., Akitake C. M., Thisse C., Thisse B., and Halpern M. E. (2013) Aversive cues fail to activate fos expression in the asymmetric olfactory-habenula pathway of zebrafish. Front. Neural Circuits 7, 98 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.