

Abstract
ADP-ribosyl-acceptor hydrolase 3 (ARH3) plays important roles in regulation of poly(ADP-ribosyl)ation, a reversible post-translational modification, and in maintenance of genomic integrity. ARH3 degrades poly(ADP-ribose) to protect cells from poly(ADP-ribose)–dependent cell death, reverses serine mono(ADP-ribosyl)ation, and hydrolyzes O-acetyl-ADP-ribose, a product of Sirtuin-catalyzed histone deacetylation. ARH3 preferentially hydrolyzes O-linkages attached to the anomeric C1″ of ADP-ribose; however, how ARH3 specifically recognizes and cleaves structurally diverse substrates remains unknown. Here, structures of full-length human ARH3 bound to ADP-ribose and Mg2+, coupled with computational modeling, reveal a dramatic conformational switch from closed to open states that enables specific substrate recognition. The glutamate flap, which blocks substrate entrance to Mg2+ in the unliganded closed state, is ejected from the active site when substrate is bound. This closed-to-open transition significantly widens the substrate-binding channel and precisely positions the scissile 1″-O-linkage for cleavage while securing tightly 2″- and 3″-hydroxyls of ADP-ribose. Our collective data uncover an unprecedented structural plasticity of ARH3 that supports its specificity for the 1″-O-linkage in substrates and Mg2+-dependent catalysis.
Keywords: ADP-ribosylation, conformational change, structural biology, substrate specificity, hydrolase, ARH3, PARP1
Introduction
Regulation of poly(ADP-ribosyl)ation (PARylation),4 a reversible post-translational modification (PTM) of proteins, is required for maintaining genomic integrity and cellular responses to DNA damage (1, 2). The addition of poly(ADP-ribose) (PAR) to proteins by PAR polymerase 1 (PARP1; also known as ARTD1) plays a pivotal role in the repair of DNA single- and double-strand breaks (3–6). However, excessive PARylation by PARP1 often interferes with protein function and may activate parthanatos, a PAR-dependent cell death pathway, which involves the translocation of PAR to the cytoplasm, eventually triggering the PAR-dependent release of apoptosis-inducing factor (AIF) from mitochondria (7, 8). AIF released from mitochondria then goes to the nucleus, leading to DNA cleavage and cell death. The cellular level of PARylation and PAR is therefore tightly controlled by PAR synthesis and turnover.
In mammals, two enzymes, ADP-ribosyl-acceptor hydrolase 3 (ARH3; also known as ADPRHL2) and PAR glycohydrolase (PARG), function in tandem to reverse PARylation (9). These hydrolytic enzymes commonly cleave the α(1″-2′) O-glycosidic linkages in PAR chains (Fig. 1a) (10, 11). PARG has both exo- and endoglycohydrolase activities, acting at terminal and internal sites of PAR chains, respectively (9, 12–14). Thus, in addition to free ADP-ribose (ADPR), PARG generates short chains of PAR that serve as a potent cell death signal (9). In contrast, ARH3 appears to catalyze primarily exocytic cleavage of PAR, generating free ADPR (9). Consistent with this difference in biochemical activity, ARH3 protects cells from oxidative stress–induced parthanatos by lowering the cytoplasmic PAR level (9). ARH3 knockout cells are healthy under unstressed conditions. However, following H2O2-induced DNA damage, these cells show accumulation of cellular PAR, in particular in the cytoplasm where it interacts with mitochondria, leading to AIF cleavage and release, enhanced nuclear accumulation of AIF, and overall increased activity of the parthanatos pathway (9). Taken together, these findings indicate that ARH3 is a key enzyme that not only controls PAR content but also determines cell fate during the DNA damage response.
Figure 1.

Structure of full-length human ARH3 bound to ADP-ribose and Mg2+. a, structures of ARH3 substrates: a linear unbranched poly(ADP-ribose) (left), O-acetyl-ADP-ribose (middle), and α-ADP-ribosylserine (right). ARH3 cleaves the 1″-O-linkage in substrates. The exoglycohydrolase activity of ARH3 cleaves the α(1″-2′) O-glycosidic bond between n and n − 1 ADP-ribose, releasing ADP-ribose as a product. b, left, Mg2+ enhances the ADP-ribosyl-acceptor hydrolase activity of ARH3. The ARH3-mediated hydrolysis of PAR on PARylated PARP1C was monitored in the presence and absence of Mg2+ using a gel-based assay. PARG has stronger PAR turnover activity than ARH3 and was used as a positive control. Right, metal preference of ARH3. ARH3 prefers Mg2+ for catalysis followed by Mn2+ and Ca2+. c, difference electron density maps (Fo − Fc) for ADPR and Mg2+ ions contoured at 3.0 σ (blue, ADPR; orange, Mg2+). d, structure of the ARH3–ADPR–Mg2+ complex revealing two unique and flexible structural elements, adenine cap (green) and glutamate flap (Glu41-flap) (red), that undergo conformational changes and strongly contribute to specific substrate recognition. The 1″-OH of ADPR (blue circled), corresponding to the scissile 1″-O-linkage in substrates, is exposed to solvent, consistent with ARH3 specificity for 1″-O-linkage for cleavage. A putative binding site for the leaving group is highlighted with a black ellipse. The N and C termini of ARH3 are indicated by N and C, respectively.
PARG is unable to reverse a protein-bound mono(ADP-ribosyl)ation (MARylation), the last step in completely erasing poly(ADP-ribosyl)ation (15). This MARylation mark, particularly on acidic residues, is removed by macrodomain-containing proteins with MAR hydrolytic activity such as terminal ADP-ribose protein glycohydrolase (TARG1), MacroD1, and MacroD2 (16–18). It has been shown that serine-specific ADP-ribosylation, a PTM specifically generated by PARP1/HPF1 and PARP2/HPF1 complexes, is the major cellular PTM following DNA damage (19–21). Importantly, in addition to PAR degradation, ARH3 was identified as the cellular eraser of serine MARylation marks (20, 22). Furthermore, ARH3 can specifically digest O-acetyl-ADPR, a product of Sirtuin-catalyzed NAD+-dependent histone deacetylase reactions, which regulate diverse biological processes, including chromatin remodeling (11, 23, 24). Therefore, ARH3 is the only known enzyme that can specifically hydrolyze PAR, MAR PTMs, and O-acetyl-ADPR. For all these substrates, ARH3 preferentially hydrolyzes the scissile α-O-linkage attached to the anomeric C1″ position of ADPR (24) (Fig. 1a). However, the structural basis for the specificity of ARH3 for the 1″-O-linkage is unknown. Collectively, ARH3 is a distinctive, multitasking enzyme that controls two biologically important NAD+-dependent cellular signaling pathways.
The structure of the unliganded human ARH3, lacking the N-terminal 16 residues (ARH3ΔN16), shows a compact all-α-helical fold with a binuclear Mg2+ center, which constitutes an archetype of the ARH superfamily (25). This di-Mg2+–containing catalytic center (MgA and MgB) is not found in PARG and is consistent with the Mg2+-dependent ADP-ribosyl-acceptor hydrolase activity of ARH3 (11, 23). Consistently, mutations of Mg2+-coordinating residues of ARH3 led to a drastic decrease in PAR- and MAR-acceptor–hydrolyzing activities (22, 25). However, we lack a fundamental mechanistic understanding of how ARH3 specifically recognizes and cleaves structurally diverse substrates in a Mg2+-dependent manner.
To better understand the molecular mechanism of ARH3 activities, we determined high-resolution crystal structures of full-length human ARH3 bound to Mg2+ and ADPR, a product and an effective inhibitor of ARH3 activities (23, 24). Coupled with computational analysis of ARH3–PAR substrate interactions, these structures reveal that substrate binding drives a large-scale conformational transition from an unliganded closed state to an open incision-competent enzyme state. Glu41 from the “glutamate flap,” which blocks the entry of substrate in the closed state, is completely ejected from the binuclear Mg2+ center. The concomitant restructuring of the active site uncovers the catalytically important MgB, widens the leaving group–binding channel, and precisely positions the scissile 1″-O-linkage for hydrolysis while tightly securing the 2″- and 3″-OH groups of ADPR. Unique structural features and conformational flexibility of ARH3 strongly support ARH3's specificity for the 1″-O-linkage in structurally diverse substrates and Mg2+-dependent catalysis.
Results
Structure of human ARH3FL–ADP-ribose–Mg2+ complex
The crystal structure of apo-ARH3ΔN16 explained the lack of ADPR binding (25) (Fig. S1). We reasoned that the full-length ARH3 (ARH3FL) might provide opportunities to capture ARH3 in an ADPR-bound active form. The purified ARH3FL is likely metal-free given its comparable basal activity without the addition of Mg2+ and in the presence of EDTA (Fig. 1b). The addition of Mg2+ markedly enhances ARH3FL-mediated PAR hydrolysis (Fig. 1b), which is consistent with a previous report (11). ARH3FL shows maximal activity with Mg2+ followed by Mn2+ and Ca2+ (Fig. 1b). Unlike DraG, the closest functional homolog that has a strong preference for Mn2+ over Mg2+ (25–27), ARH3 exhibits rather comparable activity with either Mg2+ or Mn2+. We crystallized ARH3FL in complex with ADPR and Mg2+ and determined its crystal structure at 1.7-Å resolution (Fig. 1d). Thus, the numbering of amino acid residues in this report deviates by 16 from that used for the apo-ARH3ΔN16 structure (25), but it is consistent with those used by Oka et al. (11) and Abplanalp et al. (22). ARH3FL binds to one ADPR as evidenced by a clear and strong difference electron density at the active site (Fig. 1c).
ARH3FL adopts a compact all-α-helical fold with a central deep ADPR-binding cleft, a signature of the ARH3 superfamily (Figs. 1 and 2). The ARH3–ADPR–Mg2+ complex structure identifies two unique structural elements of ARH3, the “adenine cap” and glutamate flap, which undergo a structural rearrangement upon ADPR binding and strongly contribute to the specific substrate recognition of ARH3 (Figs. 1d and 2). The binuclear Mg2+ catalytic center lies at the heart of a long, J-shaped substrate-binding channel. This structural feature enables ARH3 to bind and align both ADPR and the leaving group, e.g. two ADPR units (n ADPR and the n − 1 ADPR leaving group) in PAR substrates, for specific cleavage (Fig. 1, a and d).
Figure 2.

Structure-based alignment of ARH3 and DraG with structural elements and residues that are important for function. The Glu41 residue of the Glu41-flap of ARH3 that undergoes a large conformational change upon ADPR binding is indicated by a red box. A part of L2 of DraG (L2 wall) completely blocks the conformational change of α1 and restricts its activity to cleavage of mono(ADP-ribosyl)ated substrates. The end of α1 in ARH3, which exists as 310-helix in the unliganded form and undergoes 310-to-α transition upon ADPR binding (Fig. 4a), is indicated by a blue bar. Two aromatic residues in DraG that stabilize the L2 wall are indicated by a red triangle. Asp97 in DraG that is essential for the cleavage of MARylated arginine is indicated by a blue triangle.
The N-terminal 13 amino acids in the N-terminal extension are disordered, which is consistent with their dispensable role in ARH3 function (25). However, unexpectedly, Arg18 in the N-terminal extension, previously replaced by alanine in the apo-ARH3ΔN16 structure (25), contributes to ARH3 folding. Arg18 makes helix-capping interactions with the main-chain carbonyls of Ser161 and Leu162 in α7 to stabilize α7 (Fig. S2). Arg18 further stabilizes ARH3 folding by forming a hydrogen bond with the side-chain carbonyl of Gln361 of α19 and a van der Waals contact with the side chain of Phe23 of α1. However, given the fully functional PAR hydrolysis activity of ARH3ΔN16 (25) and a remarkably long distance between Arg18 and Glu41 (∼ 32 Å), it is unlikely that Arg18 contributes to the observed conformational changes.
Structural comparison of the unliganded and ADPR-bound forms of ARH3 reveals dramatic conformational changes in the Glu41-containing flap motif, which we named the glutamate flap (Glu41-flap) (r.m.s.d. of 2.5 Å, comparing 52 Cα atoms in the Glu41-flap and its flanking residues of the unliganded ARH3ΔN16 and complex D of the ARH3–ADPR–Mg2+ complex) (Fig. 1d). The Glu41-flap is composed of the end of α1 that exists as a 310-helix in the unliganded ARH3, α2, and a flexible loop connecting α1 and α2 (L1) (Figs. 1d and 2). This substrate-induced conformational transition fully exposes the bimetallic catalytic center for substrate engagement and to allow efficient hydrolysis to occur as described below. Together, these findings imply that ARH3 can exist in at least two states: a substrate-bound “open” state and an unliganded “closed” state.
Four ARH3–ADPR–Mg2+ complexes were found in the asymmetric unit (Fig. S3). The r.m.s.d. difference between the structures of four ARH3–ADPR–Mg2+ complexes (complexes A–D) and the unliganded ARH3 is summarized in Fig. S3b. Complexes B and D have nearly identical structures with a large-scale conformational switch in the Glu41-flap (Fig. S3a). Complex D shows the largest degree of conformational transition, which defines a fully open state, and therefore was primarily used for structural analyses. By contrast, complexes A and C show an intermediate conformational step between the unliganded ARH3 and complex D. A part of L1 in the Glu41-flap is disordered in complexes A and C, further supporting its structural flexibility (Fig. S3a).
ARH3 specifically exposes 1″-OH of ADP-ribose
ADPR is located at the deep ADPR-binding cleft of ARH3. All three parts of ADPR (adenosine, diphosphate, and the distal ribose″) make extensive contacts with ARH3 (Figs. 1d and 3). ADPR has a surface area of 694 Å2 of which ∼80% (555 Å2 on average in complexes A–D) is buried by direct contacts with ARH3. Overall, this matrix of ARH3–ADPR–Mg2+ interactions specifically exposes 1″-OH, corresponding to the scissile O-linkage in substrates (Figs. 1a and 3c), toward the catalytic MgB, strongly supporting its specificity for the 1″-O-linkage for substrate cleavage.
Figure 3.

ARH3 specifically exposes the scissile 1″-O-linkage in substrates for cleavage. a, structural superposition of ADPR-bound forms of ARH3 (wheat) and DraG (gray) reveals a distinctive ADPR-binding mode in ARH3. The adenine cap of ARH3 (green) grasps the adenine ring and is essential for ARH3 activities. Hydrogen bonds contributed by the main-chain atoms of the adenine cap to N6 and N7 of the adenine ring impart specificity. b, diagram showing interactions between ADPR and ARH3. c, close-up of the binuclear Mg2+ catalytic center and ADPR binding in ARH3. A matrix of Mg2+-mediated coordination and hydrogen-bonding interactions secure ADPR in the active site, which is consistent with Mg2+-dependent catalysis by ARH3. The 1″-OH of ADPR (blue circled), corresponding to the scissile 1″-O-linkage, is specifically exposed to solvent, strongly supporting ARH3 specificity for the 1″-O-linkage as a site for cleavage. d, close-up of the ARH3–di-ADPR–Mg2+ complex model. The energy-minimized computational model for the ARH3–di-ADPR–Mg2+ complex was generated using the ARH3–ADPR–Mg2+ complex as a starting model (see “Experimental procedures”). In this model, 3′-OH of the n − 1 ADPR leaving group is directly coordinated by MgB, replacing the W1 water ligand, and MgB tightly secures both n and n − 1 ADPR units for efficient cleavage to occur.
The distal ribose″ of ADPR lies adjacent to the binuclear Mg2+ center where a group of acidic and polar residues extensively contact Mg2+ ions and ADPR. The interactions between the ribose″ and two Mg2+ ions are asymmetrical with more extensive contacts on MgB. 3″-OH of the ribose″ is directly coordinated by MgB, and it is additionally hydrogen-bonded with the side chain of Asn151 (Fig. 3, b and c). A water molecule (μ-aqua ligand) that bridges MgA and MgB simultaneously engages 2″-OH of the ribose″ with an unusually short distance (2.2 Å). These Mg2+-mediated ARH3 interactions with 2″- and 3″-OH appear to secure tightly the ribose″ to facilitate efficient cleavage of substrates at the 1″-O-linkage. In support of this hypothesis, in contrast to 2″- and 3″-OH groups, 1″-OH of ADPR that corresponds to the 1″-O-linkage in substrates is solvent-accessible and exposed to the leaving group–binding site (Figs. 1d and 3c), consistent with cleavage at the C1″ position (11, 20, 24). Another water molecule (W1) is axially liganded to MgB and makes a hydrogen bond with 1″-OH (Fig. 3c). This W1 ligand is located proximal to the anomeric C1″ and appears well aligned for nucleophilic attack of C1″ during catalysis. At the other side of the binuclear metal center, the carboxyl group of Asp77 is coordinated by MgA and further secures 2″-OH (Fig. 3c).
Despite the structural similarity between ARH3 superfamily members, the binding orientation of ADPR in ARH3 is remarkably different from that in DraG (28) (r.m.s.d. of 2.6 Å, comparing all Cα positions of complex D and the DraG–ADPR complex (Protein Data Bank code 2WOE)) (Figs. 3a and S4a). This distinct mode of ADPR binding can be accounted for by a unique adenine-binding module of ARH3, the adenine cap, which consists of α6, α12, and a loop flanking the C terminus of α6 (L6). Notably, α6 and α12 are missing in DraG (Figs. 2 and 3a). Two aromatic amino acids from the adenine cap, Phe143 and Tyr149, sandwich the adenine base through extensive π–π stacking interactions (Fig. 3a). In line with this finding, the ARH3-Y149A mutant showed a drastic decrease in PAR- and MAR-acceptor–hydrolyzing activities (22, 25). Leu235 from the flexible α12 in the adenine cap moved ∼2.7 Å toward the adenine ring relative to the unliganded ARH3, forming a van der Waals contact with N6 of the adenine base (Fig. 3, a and b). The chemical selectivity of the adenine base is further enhanced by hydrogen bonds between the backbone carbonyl of Gly147 and N6 and between the backbone nitrogen of Tyr149 and N7 (Fig. 3, a and b). The diphosphate moiety interacts with the side chains of Ser148 and His182 and the main-chain nitrogen of Gly119. Consistently, substitution of Ser148 and His182 with alanine led to loss of ADP-ribosyl-acceptor hydrolase activities (22, 25).
A conformational switch of ARH3 enables specific substrate recognition
The most striking feature in the ARH3–ADPR–Mg2+ complex is the dramatic conformational change of the Glu41-flap (Fig. 4). The end of α1, which is at the beginning of the Glu41-flap, exists as a kinked 310-helix structure in the unliganded ARH3 (Fig. 4a). ADPR binding drives a 310-to-α transition, leading to ∼27° of rotation from the helical axis of the 310-helix. This rotation induces a concomitant ∼8.5-Å displacement of L1 away from the active site, which accompanies ∼24° of rotation in α2 (Fig. 4a). Consequently, ADPR binding results in an ∼4.5-Å movement of the carboxylate of Glu41 of the Glu41-flap away from the catalytic MgB. The straightened conformation of α1 is stabilized by new intrahelical stacking interactions within α1 among residues Phe39, Tyr40, and His43 (Fig. 4a).
Figure 4.

A conformational change of ARH3 enables specific substrate recognition and cleavage. a, structural comparison of the unliganded (gray/black) and ADPR-bound forms (orange/red) of ARH3 reveals a conformational switch in the Glu41-flap. The end of α1 undergoes a 310-to-α transition upon ADPR binding, which results in displacement of Glu41 away from MgB and significantly widens the leaving group–binding site of ARH3. b and c, ARH3 switches conformations to enable specific poly(ADP-ribose) recognition. The structure of the ARH3–di-ADPR–Mg2+ complex model is superimposed onto those of the ARH3–ADPR–Mg2+ complex and the apo-ARH3ΔN16, and di-ADPR is shown on the surfaces of ARH3. Comparison of solvent-accessible surfaces reveals an open platform that can accommodate n − 1 ADPR in the ARH3–ADPR–Mg2+ complex (b), whereas n − 1 ADPR binding is completely obstructed by the Glu41-flap in the unliganded ARH3 (c).
Mechanistically, the observed conformational switch of the Glu41-flap is required for specific substrate recognition. In the unliganded state, Glu41 of the Glu41-flap completely masks MgB from access to substrate (Fig. 4c). This closed state is therefore enzymatically inactive, and Glu41 must be ejected away from MgB to allow substrate to enter the bimetallic catalytic center. In support of this hypothesis, the ARH3–ADPR–Mg2+ complex shows a conformational switch in the Glu41-flap from a closed state to an open state. This substantial active-site restructuring unmasks MgB and significantly widens the leaving group–binding site (Figs. 1d and 4, b and c), which now allows substrate entrance to the dimetallic catalytic center. This open conformation of ARH3 provides an optimal alignment between the scissile 1″-O-linkage and catalytic groups (Mg2+ and catalytic residues) and therefore constitutes an “incision-competent” enzyme state (Figs. 3d and 4b). Collectively, the Glu41-flap controls substrate access to the binuclear Mg2+ center, and its closed-to-open conformational switch enables specific binding and cleavage of substrates.
To gain further mechanistic insights into substrate hydrolysis, we modeled di-ADPR, a substrate with the largest leaving group (Fig. 1a), to the active site of ARH3 in catalytic position (Fig. 4, b and c). In this ARH3–di-ADPR–Mg2+ model, the n − 1 ADPR leaving group snugly fits into the curved J-shaped substrate binding channel (Fig. 4b), whereas n ADPR occupies the identical site as that seen in the ARH3–ADPR–Mg2+ complex. Notably, 3′-OH of the adenosine ribose′ of n − 1 ADPR is directly coordinated to MgB, replacing the axially coordinated water ligand (W1), and it is positioned within hydrogen-bonding distance to Asp314 and Glu41 (Figs. 3d and 4b). These findings suggest that MgB is important for catalysis by securing ADPR and the leaving group to promote the subsequent cleavage at the C1″ position.
Structural superposition of ADPR-bound forms of ARH3 and DraG reveals that the observed 310-to-α transition of α1 and the conformational flexibility of the Glu41-flap are unique in ARH3. Analysis of secondary structures using Dictionary of Secondary Structure of Proteins (DSSP) (29) indicates that DraG lacks the flexible 310-helix structure at the end of α1 (Fig. 2). Furthermore, a part of L2 of DraG (named the “L2 wall”) tightly caps α1 through an edge-stacking π–π interaction between the side chains of Trp50 from L2 and Phe29 from α1 (Figs. 3a and S4). This structural arrangement of DraG effectively restricts the conformations of α1 and L1 (corresponding to the Glu41-flap in ARH3) to the closed state in which substrate access to the leaving group–binding site is prevented (Fig. 4a). The DraG mechanism instead involves a ring opening of the ribose″, which positions the scissile 1″-N-linkage in close proximity to Asp97 of DraG for cleavage (28). This structural feature would enforce DraG substrate specificity to MARylated arginine (28). In ARH3, Phe29 and Trp50 of DraG are replaced with alanine and serine, respectively, relieving the constraining interaction. Consequently, the L2 of ARH3, equivalent to the L2 wall of DraG, is disordered. Furthermore, Asp97 of DraG is replaced with glycine in ARH3 (Fig. 2). Taken together, the conformational flexibility of the Glu41-flap, the lack of the L2 wall, metal preference for Mg2+, and the lack of the catalytic residue equivalent to DraG Asp97 in ARH3 (25) (Fig. 2), all suggest a distinct catalytic mechanism for ARH3 and explain different substrate specificity in ARH3.
Binuclear metal center of the ARH3–ADPR–Mg2+ complex
In the ARH3–ADPR–Mg2+ complex, two Mg2+ ions (MgA and MgB) have an octahedral coordination geometry and are fully occupied with only 3.3-Å intermetal distance (3.8 Å in the unliganded ARH3 structure). They are bridged by the bidentate carboxyl group of Asp316 as well as by the μ-aqua ligand (Fig. 3c). The bridging μ-aqua ligand is nearly symmetrically coordinated by two Mg2+ ions (1.9 and 1.8 Å to MgA and MgB, respectively) and fixes 2″-OH of the ribose″ of ADPR (Fig. 3c).
In contrast to MgA, whose coordination ligands remain identical (including Thr76, Asp77, Asp78, and Asp316), the MgB coordination setup is dynamically changed. In the unliganded ARH3, Glu41 of the Glu41-flap is coordinated by MgB. Upon PAR substrate binding, 3′-OH of the adenosine ribose′ of the n − 1 ADPR leaving group replaces Glu41 and directly coordinates MgB. Thus, in the substrate-bound state, MgB engages both ADPR and the leaving group to precisely expose the scissile 1″-O-linkage for cleavage (Figs. 3d and 4b). Asp314 and Glu41 further aid in the correct positioning of the n − 1 ADPR leaving group by interacting with 3′-OH of the adenosine ribose′. Finally, in the ARH3–ADPR (product) complex, the leaving group departs, and a new water ligand (W1) is axially coordinated by MgB (Figs. 3c and 4b). This dynamic switching of the metal-site makeup in each catalytic step of ARH3 is consistent with Mg2+-dependent ARH3 activity.
Asp314 is essential for the formation of binuclear metal center
The structure of the ARH3–ADPR–Mg2+ complex shows that the dimetallic catalytic center of ARH3 is tightly packed against ADPR and appears ideally optimized for cleavage of the scissile 1″-O-linkage in substrates. Therefore, a subtle change in the active-site arrangement may result in a detrimental effect on ARH3 functions. Consistently, a conservative substitution of Asp314 with glutamate led to the loss of enzymatic activity (22, 25) (Fig. S5a). To gain further insights into this structure–function relationship, we determined the crystal structure of ARH3D314E bound to ADPR and Mg2+ at 1.6-Å resolution (Figs. 5 and S5b). The overall structure and ADPR-binding mode are nearly identical to those for the ARH3WT–ADPR–Mg2+ complex (r.m.s.d. of 0.2 Å, comparing all Cα positions of complexes D of ARH3WT and ARH3D314E). Strikingly, however, MgB is completely missing in the ARH3D314E–ADPR–Mg2+ complex, and the side chain of Glu314 is flipped out from the binuclear metal center, presumably due to a steric clash caused by the one-carbon–longer side chain in Glu314 (Fig. 5). The distance between Glu314 and the corresponding MgB in the ARH3WT–ADPR–Mg2+ complex is beyond the range of direct coordination (3.7 Å), explaining the loss of MgB and the loss of enzymatic activity. Consistent with the important role of MgB in securing ADPR (Fig. 3c), the overall B factors for atoms in the ribose″ of ADPR in the ARH3D314E–ADPR–Mg2+ complex are significantly higher than those in the ARH3WT–ADPR–Mg2+ complex. Collectively, these findings indicate that Asp314 is required for the formation of the binuclear Mg2+ center and suggest a critical role of MgB for catalysis.
Figure 5.
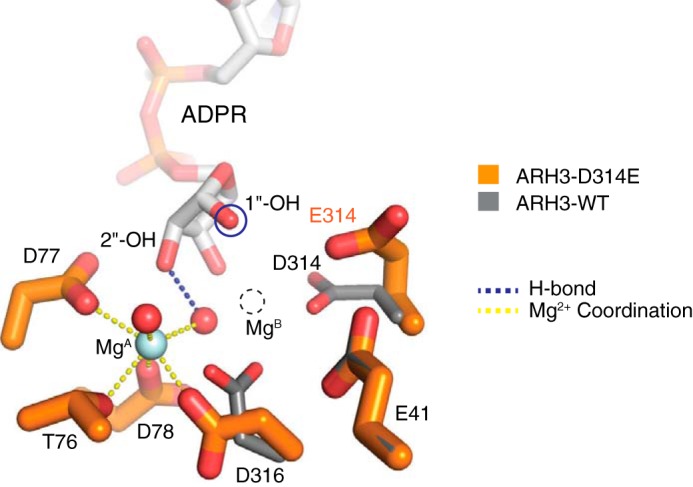
Asp314 is essential for the formation of the binuclear metal center. Structural comparison of the WT and catalytically inactive ARH3-D314E mutant reveals a side-chain flipping of Glu314, which leads to the loss of MgB. This suggests that Asp314 is essential for the formation of the binuclear metal center in ARH3 and that MgB is required for catalysis.
Discussion
Our structures of the ARH3–ADPR–Mg2+ complex and a computational model of the ARH3–di-ADPR–Mg2+ complex reveal the previously unknown conformational plasticity of the Glu41-flap, which strongly supports ARH3 specificity for the 1″-O-linkage for cleavage. Our studies also explain the published mutational analysis data for ARH3. These structural findings are consistent with Mg2+-dependent hydrolysis and provide important clues to the catalytic mechanism of ARH3. First, the observed conformational switch in ARH3 is required to specifically recognize substrates. In the closed unliganded state, Glu41 coordinates MgB and prevents substrates from entering into the dimetallic catalytic center (Fig. 6). To engage substrates, the Glu41-flap is completely moved away from MgB, inducing substantial active-site rearrangement from a closed to an open state. This conformational transition now allows substrates to enter the active site of ARH3. Second, our structures strongly support the observation that ARH3 favors the 1″-O-linkage in substrates for cleavage (24). In the ARH3–ADPR–Mg2+ complex, 2″- and 3″-OH of ADPR are secured by Mg2+ ions, the bridging μ-aqua ligand, and active-site residues, whereas 1″-OH corresponding to the scissile 1″-O-linkage in substrates is specifically exposed to the open leaving group–binding site (Fig. 3c). Substrates containing 2″- or 3″-O-linkage therefore would result in a serious steric clash with the tightly packed binuclear metal center of ARH3, leading to inefficient substrate cleavage. Consistently, IC50 values for ARH3 inhibition by 2″- or 3″-N-acetyl-ADPR, analogs of O-acetyl-ADPR, were significantly higher than that for ADPR (24). In our ARH3–di-ADPR–Mg2+ model, the di-ADPR is slightly bent due to the simultaneous engagement of 3″-OH of n ADPR and 3′-OH of n − 1 ADPR by MgB (Figs. 3d and 6). This structural feature would likely further reinforce the correct positioning of the scissile 1″-O-linkage for efficient cleavage (Fig. 6). Third, the substrate-induced widening of the leaving group–binding site enables ARH3 to specifically bind and cleave substrates with a structurally diverse leaving group (Figs. 1a and 4), supporting ARH3's broad substrate specificity.
Figure 6.

ARH3 switches between closed and open conformations to specifically recognize and cleave substrates. ARH3 switches between a self-inhibited and an incision-competent conformation for specific substrate recognition and cleavage. In the absence of PAR substrates, the Glu41-flap caps MgB, constituting a closed, self-inhibited enzyme state. Substrate binding induces a conformational switch in the Glu41-flap, allowing substrate entrance to the active site. This active-site rearrangement in ARH3 specifically exposes the scissile 1″-O-linkage to the catalytic Asp314 for cleavage, explaining ARH3 specificity for the 1″-O-linkage in structurally diverse substrates.
The flexible Glu41-flap dynamically switches between closed and open conformations and plays important roles in substrate recognition (Fig. 6). In the unliganded ARH3, Glu41 of the Glu41-flap serves as the key residue that constitutes the closed enzyme state by masking MgB. In the PAR substrate–bound state, Glu41 is released from MgB and instead interacts both with ADPR and the leaving group (Fig. 6), which presumably contributes to the precise alignment of the scissile O-linkage for subsequent cleavage and constitutes the open enzyme state. Therefore, we propose the Glu41-flap as the gate that controls substrate entrance to the active site. Given the structural flexibility and solvent accessibility of the Glu41-flap, it is also possible that the Glu41-flap functions as a protein–protein interaction module. Proteins that specifically interact with the closed conformation of the Glu41-flap of ARH3 might conformationally lock ARH3 in an inactive state and thereby regulate ARH3 function.
A notable ARH3 inhibition by ADP-HPD (Fig. S6a) (30), an analog of the oxocarbenium ion intermediate, raises the possibility that the ARH3 mechanism might involve an oxocarbenium intermediate in the distal ribose″ of ADPR in a similar way to PARG (15, 31), which is followed by water-mediated nucleophilic attack at the anomeric C1″ position. The observed 18O incorporation to C1″ during hydrolysis of O-acetyl-ADPR (24) is also consistent with this model. In glycosidase superfamily members, a catalytic acidic residue is typically found near the scissile bond, e.g. Glu756 in rat PARG (Glu752 in human PARG) (31, 32), to activate a water molecule for nucleophilic attack on the anomeric carbon (33). In the ARH3–ADPR–Mg2+ complex, Asp314 is located proximal to both 1″-OH (corresponding to the scissile 1″-O-linkage) and the axial water ligand (W1) of MgB (Fig. 3c). It is plausible that Asp314 might function as a general acid or base to protonate the leaving group and then activate W1 for backside attack of the anomeric C1″ in a manner similar to Glu756 in rat PARG (Fig. S6b) (31). In support of this catalytic role of Asp314, the ARH3-D314N mutant shows a dramatic reduction in ARH3 activities (22, 25). Although more work is needed to determine the catalytic mechanism of ARH3, it is unlikely that ARH3 has a redox chemistry step for catalysis given its preference for Mg2+ over Mn2+ (Fig. 1b).
ARH3 is a unique multitasking enzyme that regulates cellular concentrations of both PAR, either free or attached to proteins, and mono(ADP-ribosyl)ated substrates. Elevated PARylation levels are often found in many human cancers, including BRCA-deficient breast cancers (34, 35) and triple-negative breast cancers (36, 37). ARH3−/− cells undergo enhanced PAR-dependent cell death upon genotoxic stresses but remain healthy under unstressed conditions. We suggest that pharmacological intervention in ARH3-dependent pathways could be a safe and efficient therapeutic strategy for cancers with up-regulated PARylation by increasing the cytoplasmic PAR level and triggering parthanatos, either alone or in combination with current chemotherapeutic agents. Our structures reveal extensive adenine interactions with the adenine cap and a large conformational switch of the Glu41-flap that are crucial for specific engagement of structurally diverse substrates. These unique structural elements of ARH3 can be exploited for the development of specific ARH3 inhibitors, which have potential therapeutic applications, as well as the ability to advance our understanding of the role of ARH3 in regulation of PARylation. A concern is that ARH3 also catalyzes hydrolysis of O-acetyl-ADPR, which may be involved in other cellular pathways such as chromatin remodeling. This activity also relies on the ability of ARH3 to act at the C1″ position of ADPR. ARH3's contribution to the O-acetyl-ADPR–dependent pathways, in contrast to other proteins in the ARH-macrodomain superfamily, is not known. Thus, some ARH3 inhibitors may affect multiple signaling pathways due to its multiple enzymatic activities and substrates.
Experimental procedures
Plasmids and protein purification
Human ARH3FL was cloned into a modified pET21b vector with an N-terminal His6 tag and a following cleavage site for PreScission protease (pET21b-His6-pps). The pET21b-His6-pps-ARH3FL plasmid was introduced into Escherichia coli Rosetta 2 (DE3) cells, and ARH3FL was induced by adding 1 mm isopropyl β-d-thiogalactoside overnight at 16 °C. ARH3FL was purified by affinity capture on a nickel-nitrilotriacetic acid (GE Healthcare) column. After elution with imidazole (250 mm), the protein was loaded onto a heparin column (GE Healthcare) and eluted with a NaCl gradient (0.1–1 m). Fractions with ARH3FL were pooled, and PreScission protease was added to cleave the N-terminal histidine tag. Finally, ARH3FL was loaded to a Sephacryl 200 (GE Healthcare) size-exclusion column using a buffer containing 25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1 mm DTT, and 5% glycerol. The purified ARH3FL was concentrated to ∼30 mg/ml and then stored at −80 °C. A gene encoding the ARH3-D314E mutant was synthesized (GeneUniversal, Inc.) and cloned into pET-21b-pps vector. The ARH3-D314E mutant was purified using the same protocol as the WT protein. The human PARG catalytic domain (residues 448–976) was cloned in pET21b-His6-pps, expressed in E. coli Rosetta (DE3) cells, and purified by nickel-nitrilotriacetic acid–affinity chromatography, heparin chromatography, and Sephacryl 200 size-exclusion chromatography as described previously (32). For preparation of PARylated PARP1, the DNA-binding domain (residues 1–374) of human PARP1 and the PARP1C catalytic domain (residues 375–1014) were purified as described previously (6, 31).
Crystallization and data collection
The WT and the D314E mutant of ARH3 (10 mg/ml) were cocrystallized with 5 mm ADPR by hanging-drop vapor diffusion. ADPR binding is required for crystallization as the unliganded ARH3FL did not yield any crystals. The protein solution was mixed with an equal volume of well solution (22% PEG 4000, 0.1 m sodium acetate, pH 4.5, and 0.1 m MgSO4) and incubated at 22 °C. Crystals were briefly equilibrated in a harvesting solution (26% PEG 4000, 0.1 m sodium acetate, pH 4.5, 0.1 m MgSO4, and 5 mm ADPR), transferred to a cryoprotectant solution (26% PEG 4000, 0.1 m sodium acetate, pH 4.5, 0.1 m MgSO4, 5 mm ADPR, and 15% glycerol), and then flash-cooled in liquid nitrogen for data collection.
X-ray diffraction data were collected at the NE-CAT 24ID-E beamline at the Advanced Photon Source. The ARH3WT–ADPR–Mg2+ complex crystals (P1 symmetry; four ARH3–ADPR–Mg2+ complexes per asymmetric unit) diffracted to 1.7-Å resolution, and ARH3D314E–ADPR–Mg2+ complex crystals (P1 symmetry; four ARH3–ADPR–Mg2+ complexes per asymmetric unit) diffracted to 1.6-Å resolution. X-ray data sets were collected with an Eiger 16M detector and processed using HKL2000 (38) and SCALEPACK (38, 39). Data collection statistics are shown in Table S1.
Structure determination
The full-length human ARH3WT–ADPR complex structure was determined by molecular replacement using MolRep (40) in the CCP4 suite with the apo-ARH3ΔN16 structure (25) as a search model. The asymmetric unit contains four ARH3 molecules, and they all show a strong difference electron density for the bound ADPR at the active site, which was supported by polder map calculations. The restraints for ADPR were generated using Monomer Library Sketcher in the CCP4i suite (41). The model was manually rebuilt using Coot (42) and refined with PHENIX (43) to an Rfactor of 18.4% and an Rfree of 21.8%. The ARH3D314E–ADPR complex structure was determined by molecular replacement using MolRep (40) with the ARH3WT–ADPR–Mg2+ complex structure as a search model. The ARH3D314E–ADPR–Mg2+ complex model was rebuilt using Coot (42) and refined with PHENIX (43) to an Rfactor of 16.7% and an Rfree of 19.9%. Crystallographic data statistics are shown in Table S1. The Ramachandran plot shows that >98% of the residues in both ARH3WT–ADPR–Mg2+ and ARH3D314E–ADPR–Mg2+ complexes are in the favored regions, and all the others are in the allowed regions. No outlier residue was observed.
ARH3 activity assay
ARH3 activity was measured against PARylated PARP1 using a method similar to that for PARG activity measurement as described previously (31). The C-terminal catalytic domain of PARP1 (PARP1C; residues 375–1014) containing the automodification domain was PARylated in the presence of the N-terminal DNA-binding domain of PARP1, a nicked DNA, and NAD+ as described previously (6, 31). Briefly, PARylation of PARP1 was performed at 37 °C in a reaction buffer containing 50 mm Tris-HCl, pH 7.5, 100 mm NaCl, and 2 mm DTT. To PARylate PARP1, PARP1C (2 μm), DNA-binding domain (residues 1–374) (2 μm), and a nicked DNA (2 μm) were preincubated for 10 min on ice. 200 μm NAD+ was then added to the reaction, and it was then incubated at 37 °C for 30 min. After desalting with a buffer containing 50 mm Tris-HCl, pH 7.5, 100 mm NaCl, and 5% glycerol, purified human ARH3 proteins (WT and D314E) or human PARG was treated with PARylated PARP1 substrates in the presence or absence of 5 mm EDTA or divalent metals (Mg2+, Mn2+, and Ca2+) and incubated for 60 min at 37 °C. The level of modification of PARP1 was visualized by Coomassie Blue staining of SDS-polyacrylamide gels.
Computational modeling of the ARH3–di-ADPR–Mg2+ complex
The di-ADP-ribose model was generated by covalently linking 2′-OH of the adenosine ribose′ of n − 1 ADPR to the anomeric C1″ of the distal ribose″ of n ADPR in the ARH3–ADPR–Mg2+ complex using YASARA (44). The n ADPR molecule, two Mg2+ ions, and the bridging μ-aqua ligand remained anchored to the experimentally identified site in the ARH3–ADPR–Mg2+ complex. Then n − 1 ADPR was docked to the putative n − 1 ADPR-binding site (leaving group–binding site) of the J-shaped substrate-binding channel of ARH3. The ARH3–di-ADPR–Mg2+ model was then subjected to energy minimization using the AMBER ff14SB protein force field in the Chimera package (45). All atoms in the protein and di-ADPR ligand were allowed to move during energy minimization. All structural figures were prepared using PyMOL.
Author contributions
Y. P. validation; Y. P., J. V., I. K., and A. C. investigation; Y. P., J. V., and J. M. writing-original draft; J. V., A. C., and J. M. formal analysis; I. K., J. M., and I.-K. K. writing-review and editing; I.-K. K. supervision.
Supplementary Material
Acknowledgments
This work used NE-CAT beamlines (National Institutes of Health Grant P41 GM103403) and an Eiger 16M detector on 24-ID-E beam line (National Institutes of Health Grant S10OD021527) at the APS (United States Department of Energy Grant DE-AC02-06CH11357).
The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6 and Table S1.
The atomic coordinates and structure factors (codes 6D36 and 6D3A) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- PARylation
- poly(ADP-ribosyl)ation
- ARH
- ADP-ribosyl-acceptor hydrolase
- PAR
- poly(ADP-ribose)
- PARP
- PAR polymerase
- PARG
- PAR glycohydrolase
- PTM
- post-translational modification
- MAR
- mono(ADP-ribose)
- ADPR
- ADP-ribose
- AIF
- apoptosis-inducing factor
- MARylation
- mono(ADP-ribosyl)ation
- FL
- full-length
- r.m.s.d.
- root mean square deviation
- ADP-HPD
- adenosine diphosphate (hydroxymethyl)pyrrolidine-2′,3′-diol.
References
- 1. Hakmé A., Wong H. K., Dantzer F., and Schreiber V. (2008) The expanding field of poly(ADP-ribosyl)ation reactions. 'Protein modifications: beyond the usual suspects' review series. EMBO Rep. 9, 1094–1100 10.1038/embor.2008.191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Gibson B. A., and Kraus W. L. (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat. Rev. Mol. Cell Biol. 13, 411–424 10.1038/nrm3376 [DOI] [PubMed] [Google Scholar]
- 3. Mégnin-Chanet F., Bollet M. A., and Hall J. (2010) Targeting poly(ADP-ribose) polymerase activity for cancer therapy. Cell. Mol. Life Sci. 67, 3649–3662 10.1007/s00018-010-0490-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. El-Khamisy S. F., Masutani M., Suzuki H., and Caldecott K. W. (2003) A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 31, 5526–5533 10.1093/nar/gkg761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Beck C., Robert I., Reina-San-Martin B., Schreiber V., and Dantzer F. (2014) Poly(ADP-ribose) polymerases in double-strand break repair: focus on PARP1, PARP2 and PARP3. Exp. Cell Res. 329, 18–25 10.1016/j.yexcr.2014.07.003 [DOI] [PubMed] [Google Scholar]
- 6. Kim I. K., Stegeman R. A., Brosey C. A., and Ellenberger T. (2015) A quantitative assay reveals ligand specificity of the DNA scaffold repair protein XRCC1 and efficient disassembly of complexes of XRCC1 and the poly(ADP-ribose) polymerase 1 by poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 290, 3775–3783 10.1074/jbc.M114.624718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Andrabi S. A., Kim N. S., Yu S. W., Wang H., Koh D. W., Sasaki M., Klaus J. A., Otsuka T., Zhang Z., Koehler R. C., Hurn P. D., Poirier G. G., Dawson V. L., and Dawson T. M. (2006) Poly(ADP-ribose) (PAR) polymer is a death signal. Proc. Natl. Acad. Sci. U.S.A. 103, 18308–18313 10.1073/pnas.0606526103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yu S. W., Andrabi S. A., Wang H., Kim N. S., Poirier G. G., Dawson T. M., and Dawson V. L. (2006) Apoptosis-inducing factor mediates poly(ADP-ribose) (PAR) polymer-induced cell death. Proc. Natl. Acad. Sci. U.S.A. 103, 18314–18319 10.1073/pnas.0606528103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mashimo M., Kato J., and Moss J. (2013) ADP-ribosyl-acceptor hydrolase 3 regulates poly(ADP-ribose) degradation and cell death during oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 110, 18964–18969 10.1073/pnas.1312783110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Miwa M., Tanaka M., Matsushima T., and Sugimura T. (1974) Purification and properties of glycohydrolase from calf thymus splitting ribose-ribose linkages of poly(adenosine diphosphate ribose). J. Biol. Chem. 249, 3475–3482 [PubMed] [Google Scholar]
- 11. Oka S., Kato J., and Moss J. (2006) Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J. Biol. Chem. 281, 705–713 10.1074/jbc.M510290200 [DOI] [PubMed] [Google Scholar]
- 12. Davidovic L., Vodenicharov M., Affar E. B., and Poirier G. G. (2001) Importance of poly(ADP-ribose) glycohydrolase in the control of poly(ADP-ribose) metabolism. Exp. Cell Res. 268, 7–13 10.1006/excr.2001.5263 [DOI] [PubMed] [Google Scholar]
- 13. Braun S. A., Panzeter P. L., Collinge M. A., and Althaus F. R. (1994) Endoglycosidic cleavage of branched polymers by poly(ADP-ribose) glycohydrolase. Eur. J. Biochem. 220, 369–375 10.1111/j.1432-1033.1994.tb18633.x [DOI] [PubMed] [Google Scholar]
- 14. Hatakeyama K., Nemoto Y., Ueda K., and Hayaishi O. (1986) Purification and characterization of poly(ADP-ribose) glycohydrolase. Different modes of action on large and small poly(ADP-ribose). J. Biol. Chem. 261, 14902–14911 [PubMed] [Google Scholar]
- 15. Slade D., Dunstan M. S., Barkauskaite E., Weston R., Lafite P., Dixon N., Ahel M., Leys D., and Ahel I. (2011) The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 477, 616–620 10.1038/nature10404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jankevicius G., Hassler M., Golia B., Rybin V., Zacharias M., Timinszky G., and Ladurner A. G. (2013) A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struct. Mol. Biol. 20, 508–514 10.1038/nsmb.2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sharifi R., Morra R., Appel C. D., Tallis M., Chioza B., Jankevicius G., Simpson M. A., Matic I., Ozkan E., Golia B., Schellenberg M. J., Weston R., Williams J. G., Rossi M. N., Galehdari H., et al. (2013) Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J. 32, 1225–1237 10.1038/emboj.2013.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rosenthal F., Feijs K. L., Frugier E., Bonalli M., Forst A. H., Imhof R., Winkler H. C., Fischer D., Caflisch A., Hassa P. O., Lüscher B., and Hottiger M. O. (2013) Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 20, 502–507 10.1038/nsmb.2521 [DOI] [PubMed] [Google Scholar]
- 19. Bonfiglio J. J., Fontana P., Zhang Q., Colby T., Gibbs-Seymour I., Atanassov I., Bartlett E., Zaja R., Ahel I., and Matic I. (2017) Serine ADP-ribosylation depends on HPF1. Mol. Cell 65, 932–940.e6 10.1016/j.molcel.2017.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fontana P., Bonfiglio J. J., Palazzo L., Bartlett E., Matic I., and Ahel I. (2017) Serine ADP-ribosylation reversal by the hydrolase ARH3. Elife 6, e28533 10.7554/eLife.28533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Palazzo L., Leidecker O., Prokhorova E., Dauben H., Matic I., and Ahel I. (2018) Serine is the major residue for ADP-ribosylation upon DNA damage. Elife 7, e34334 10.7554/eLife.34334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Abplanalp J., Leutert M., Frugier E., Nowak K., Feurer R., Kato J., Kistemaker H. V. A., Filippov D. V., Moss J., Caflisch A., and Hottiger M. O. (2017) Proteomic analyses identify ARH3 as a serine mono-ADP-ribosylhydrolase. Nat. Commun. 8, 2055 10.1038/s41467-017-02253-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ono T., Kasamatsu A., Oka S., and Moss J. (2006) The 39-kDa poly(ADP-ribose) glycohydrolase ARH3 hydrolyzes O-acetyl-ADP-ribose, a product of the Sir2 family of acetyl-histone deacetylases. Proc. Natl. Acad. Sci. U.S.A. 103, 16687–16691 10.1073/pnas.0607911103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kasamatsu A., Nakao M., Smith B. C., Comstock L. R., Ono T., Kato J., Denu J. M., and Moss J. (2011) Hydrolysis of O-acetyl-ADP-ribose isomers by ADP-ribosylhydrolase 3. J. Biol. Chem. 286, 21110–21117 10.1074/jbc.M111.237636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mueller-Dieckmann C., Kernstock S., Lisurek M., von Kries J. P., Haag F., Weiss M. S., and Koch-Nolte F. (2006) The structure of human ADP-ribosylhydrolase 3 (ARH3) provides insights into the reversibility of protein ADP-ribosylation. Proc. Natl. Acad. Sci. U.S.A. 103, 15026–15031 10.1073/pnas.0606762103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nordlund S., and Noren A. (1984) Dependence on divalent cations of the activation of inactive Fe-protein of nitrogenase from Rhodospirillum rubrum. Biochim. Biophys. Acta 791, 21–27 10.1016/0167-4838(84)90276-0 [DOI] [Google Scholar]
- 27. Li X. D., Huergo L. F., Gasperina A., Pedrosa F. O., Merrick M., and Winkler F. K. (2009) Crystal structure of dinitrogenase reductase-activating glycohydrolase (DraG) reveals conservation in the ADP-ribosylhydrolase fold and specific features in the ADP-ribose-binding pocket. J. Mol. Biol. 390, 737–746 10.1016/j.jmb.2009.05.031 [DOI] [PubMed] [Google Scholar]
- 28. Berthold C. L., Wang H., Nordlund S., and Högbom M. (2009) Mechanism of ADP-ribosylation removal revealed by the structure and ligand complexes of the dimanganese mono-ADP-ribosylhydrolase DraG. Proc. Natl. Acad. Sci. U.S.A. 106, 14247–14252 10.1073/pnas.0905906106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kabsch W., and Sander C. (1983) Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 22, 2577–2637 10.1002/bip.360221211 [DOI] [PubMed] [Google Scholar]
- 30. Finch K. E., Knezevic C. E., Nottbohm A. C., Partlow K. C., and Hergenrother P. J. (2012) Selective small molecule inhibition of poly(ADP-ribose) glycohydrolase (PARG). ACS Chem. Biol. 7, 563–570 10.1021/cb200506t [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kim I. K., Kiefer J. R., Ho C. M., Stegeman R. A., Classen S., Tainer J. A., and Ellenberger T. (2012) Structure of mammalian poly(ADP-ribose) glycohydrolase reveals a flexible tyrosine clasp as a substrate-binding element. Nat. Struct. Mol. Biol. 19, 653–656 10.1038/nsmb.2305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tucker J. A., Bennett N., Brassington C., Durant S. T., Hassall G., Holdgate G., McAlister M., Nissink J. W., Truman C., and Watson M. (2012) Structures of the human poly(ADP-ribose) glycohydrolase catalytic domain confirm catalytic mechanism and explain inhibition by ADP-HPD derivatives. PLoS One 7, e50889 10.1371/journal.pone.0050889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vasella A., Davies G. J., and Böhm M. (2002) Glycosidase mechanisms. Curr. Opin. Chem. Biol. 6, 619–629 10.1016/S1367-5931(02)00380-0 [DOI] [PubMed] [Google Scholar]
- 34. Gottipati P., Vischioni B., Schultz N., Solomons J., Bryant H. E., Djureinovic T., Issaeva N., Sleeth K., Sharma R. A., and Helleday T. (2010) Poly(ADP-ribose) polymerase is hyperactivated in homologous recombination-defective cells. Cancer Res. 70, 5389–5398 10.1158/0008-5472.CAN-09-4716 [DOI] [PubMed] [Google Scholar]
- 35. Bieche I., Pennaneach V., Driouch K., Vacher S., Zaremba T., Susini A., Lidereau R., and Hall J. (2013) Variations in the mRNA expression of poly(ADP-ribose) polymerases, poly(ADP-ribose) glycohydrolase and ADP-ribosylhydrolase 3 in breast tumors and impact on clinical outcome. Int. J. Cancer 133, 2791–2800 10.1002/ijc.28304 [DOI] [PubMed] [Google Scholar]
- 36. Ossovskaya V., Koo I. C., Kaldjian E. P., Alvares C., and Sherman B. M. (2010) Upregulation of poly(ADP-ribose) polymerase-1 (PARP1) in triple-negative breast cancer and other primary human tumor types. Genes Cancer 1, 812–821 10.1177/1947601910383418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rojo F., García-Parra J., Zazo S., Tusquets I., Ferrer-Lozano J., Menendez S., Eroles P., Chamizo C., Servitja S., Ramírez-Merino N., Lobo F., Bellosillo B., Corominas J. M., Yelamos J., Serrano S., et al. (2012) Nuclear PARP-1 protein overexpression is associated with poor overall survival in early breast cancer. Ann. Oncol. 23, 1156–1164 10.1093/annonc/mdr361 [DOI] [PubMed] [Google Scholar]
- 38. Otwinowski Z., and Minor W. (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 39. Pflugrath J. W. (1999) The finer things in X-ray diffraction data collection. Acta Crystallogr. D Biol. Crystallogr. 55, 1718–1725 10.1107/S090744499900935X [DOI] [PubMed] [Google Scholar]
- 40. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 41. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 43. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krieger E., Koraimann G., and Vriend G. (2002) Increasing the precision of comparative models with YASARA NOVA—a self-parameterizing force field. Proteins 47, 393–402 10.1002/prot.10104 [DOI] [PubMed] [Google Scholar]
- 45. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.