

Abstract
Bacterial lactate racemase is a nickel-dependent enzyme that contains a cofactor, nickel pyridinium-3,5-bisthiocarboxylic acid mononucleotide, hereafter named nickel-pincer nucleotide (NPN). The LarC enzyme from the bacterium Lactobacillus plantarum participates in NPN biosynthesis by inserting nickel ion into pyridinium-3,5-bisthiocarboxylic acid mononucleotide. This reaction, known in organometallic chemistry as a cyclometalation, is characterized by the formation of new metal–carbon and metal–sulfur σ bonds. LarC is therefore the first cyclometallase identified in nature, but the molecular mechanism of LarC-catalyzed cyclometalation is unknown. Here, we show that LarC activity requires Mn2+-dependent CTP hydrolysis. The crystal structure of the C-terminal domain of LarC at 1.85 Å resolution revealed a hexameric ferredoxin-like fold and an unprecedented CTP-binding pocket. The loss-of-function of LarC variants with alanine variants of acidic residues leads us to propose a carboxylate-assisted mechanism for nickel insertion. This work also demonstrates the in vitro synthesis and purification of the NPN cofactor, opening new opportunities for the study of this intriguing cofactor and of NPN-utilizing enzymes.
Keywords: bacterial metabolism, nickel, lactic acid, natural product biosynthesis, protein structure, cyclometalation, lactate racemase, pincer complex
Introduction
Lactate racemase (LarA), the ninth discovered nickel-dependent enzyme (1), was shown to contain a newly identified cofactor, nickel pyridinium-3,5-bisthiocarboxylic acid mononucleotide, hereafter named nickel-pincer nucleotide (NPN),5 covalently attached to an active-site lysine residue (2). Synthesis of NPN occurs through a pathway involving three proteins encoded in the lar operon, i.e. LarB, LarC, and LarE (3). A bioinformatics study revealed that these genes are found in almost one-quarter of the analyzed prokaryotic genomes, showing the wide distribution of the NPN biosynthetic pathway (3). In 15% of the genomes, the genes encoding the NPN biosynthetic proteins were even found without any LarA homolog gene (3), indicating that other enzymes probably use the NPN cofactor as well.
NPN biosynthesis begins with LarB, a carboxylase/hydrolase that produces pyridinium-3,5-biscarboxylic acid mononucleotide (P2CMN) from nicotinic acid adenine dinucleotide (NaAD) (4). P2CMN is converted into pyridinium-3,5-bisthiocarboxylic acid (P2TMN) by LarE through two successive sacrificial sulfur transfer reactions (5). Finally, LarC inserts nickel and forms the final NPN cofactor with its five-membered nickellacycle structure (Fig. 1a) (4) in which the metal is σ-bonded to a carbon atom and a heteroatom (a metallacycle). We therefore call LarC a cyclometallase. In organometallic chemistry, cyclometalation refers to the transition metal-mediated and heteroatom-assisted activation of a C–R or C–H bond to form a metallacycle (6). Cyclometalation has become one of the most popular organometallic reactions, providing a straightforward method for activating strong C–R or C–H bonds and creating a metal–carbon bond. This reaction has been successfully applied in organic transformations, catalysis, and for the stabilization of reactive intermediates (6).
Figure 1.

Cell lysates and CTP have similar effects on NPN cofactor biosynthesis. a, NPN cofactor biosynthesis starts with NaAD that is carboxylated and hydrolyzed by LarB forming P2CMN. Two LarE proteins each catalyze a sacrificial sulfur transfer reaction to synthesize P2TMN from P2CMN. LarC inserts nickel into P2TMN, forming the NPN cofactor containing a metallacycle (bold blue lines) with Ni-C and Ni-S σ bonds. b, schematic representation of the larC gene. LarC1 and LarC are the two open reading frames that code for LarC. c, effect of 10% (v/v) Lc. lactis cell lysates on in vitro NPN cofactor biosynthesis by LarB, LarE, and LarC. Lar activity with cell lysates after 30 min was set to 100%. d, effect of 10% cell lysates, 10% cell lysates deproteinized with heat treatment (lysate HT), or 1 mm nucleotides on the in vitro NPN cofactor biosynthesis by LarB, LarE, and LarC after 30 min. Lar activity with cell lysates was set to 100%. b and c, NPN cofactor biosynthesis was assessed after addition of LarATt apoprotein and l-lactate by assaying for d-lactate production. The lines indicate the mean values, and the points indicate the individual data points.
In Lactobacillus plantarum, two open reading frames, larC1 and larC2, encode the full-length LarC protein through a programmed ribosomal frameshift (PRF) (Fig. 1b) (7). The reason for this PRF is unknown, but only 8% of larC genes show this feature, and the PRF can be bypassed by gene fusion without affecting the activity (3). So, this process is not an essential requirement for LarC activity. Consistent with a nickel-dependent function, a His-rich region is present in its N-terminal region, and nickel is found associated with the purified LarC (3). The LarC sequence shows no homology with any protein of known function, and its catalytic activity remains unexplored. Here, we show that LarC hydrolyzes CTP to CMP while inserting nickel into P2TMN, forming the metallacycle NPN that is stabilized in solution by the presence of thiols. The CTPase activity requires Mn2+ or Mg2+. We determined the crystal structure of LarC2, the C-terminal domain of LarC, revealing a hexameric ferredoxin-like fold and an unprecedented CTP-binding pocket involving three subunits, establishing LarC2 as a new member of the GlnB-like superfamily. Extensive mutagenesis studies suggested that LarC1, the N-terminal part of LarC, likely catalyzes the cyclometalation reaction somehow assisted by the CTPase activity of LarC2.
Results
CTP enhances NPN cofactor biosynthesis
We overproduced LarC in Lactococcus lactis cells grown in the presence of divalent nickel and purified the nickel-containing protein. Prior studies had shown LarC to be required in 2-fold excess over LarE to achieve maximum NPN cofactor biosynthesis in vitro (4), suggesting that LarC activity was not optimal in these conditions. We observed that inclusion of cell lysates strongly enhanced NPN cofactor biosynthesis by LarB, LarE, and LarC as shown by the >50-fold increase in Lar activity after 1 min of incubation preceding addition of LarATt, i.e. LarA apoprotein from Thermoanaerobacterium thermosaccharolyticum (Fig. 1c). Because removal of the proteins by heat treatment and centrifugation of the lysates did not abolish the stimulation (Fig. 1d), the involvement of a small molecule was suspected. Several small organic molecules were tested with CTP showing a similar effect to cell lysates, whereas other nucleotides had no effect (Fig. 1d), thus indicating that LarB, LarE, or LarC activity is CTP-dependent.
LarC is a CTP-dependent enzyme
To analyze the activity of LarC, we developed a method to purify its substrate, P2TMN, by reacting NaAD with LarB and LarE (Fig. 1a). After the combined LarB/LarE reaction, the proteins were heat-denatured and removed by centrifugation with the supernatant loaded onto an anion-exchange column. P2TMN was eluted using a NaCl gradient, and its identity was confirmed by mass spectrometry (MS) (Fig. 2a). LarC was reacted with P2TMN, MgCl2, and CTP, followed by addition of LarATt and l-lactate, with Lar activity observed (Fig. 2b). Lactate racemization was not detected when P2TMN or LarC was omitted, but a residual 2 or 15% activity was observed in the absence of CTP or MgCl2, respectively. These residual activities suggest that some CTP and Mg2+ co-purify with LarC. In agreement with this hypothesis, the mass of purified LarC includes an additional peak that is 505 Da larger than the LarC apoprotein, corresponding to the mass of CTP + Mg2+ − 2H+ (505.4 Da) (Fig. 2c). The absence of bound nickel in the observed spectrum (Fig. 2c) is probably due to its dissociation during the LC step preceding MS. These results demonstrate that LarC is a CTP-dependent enzyme that uses P2TMN as substrate.
Figure 2.
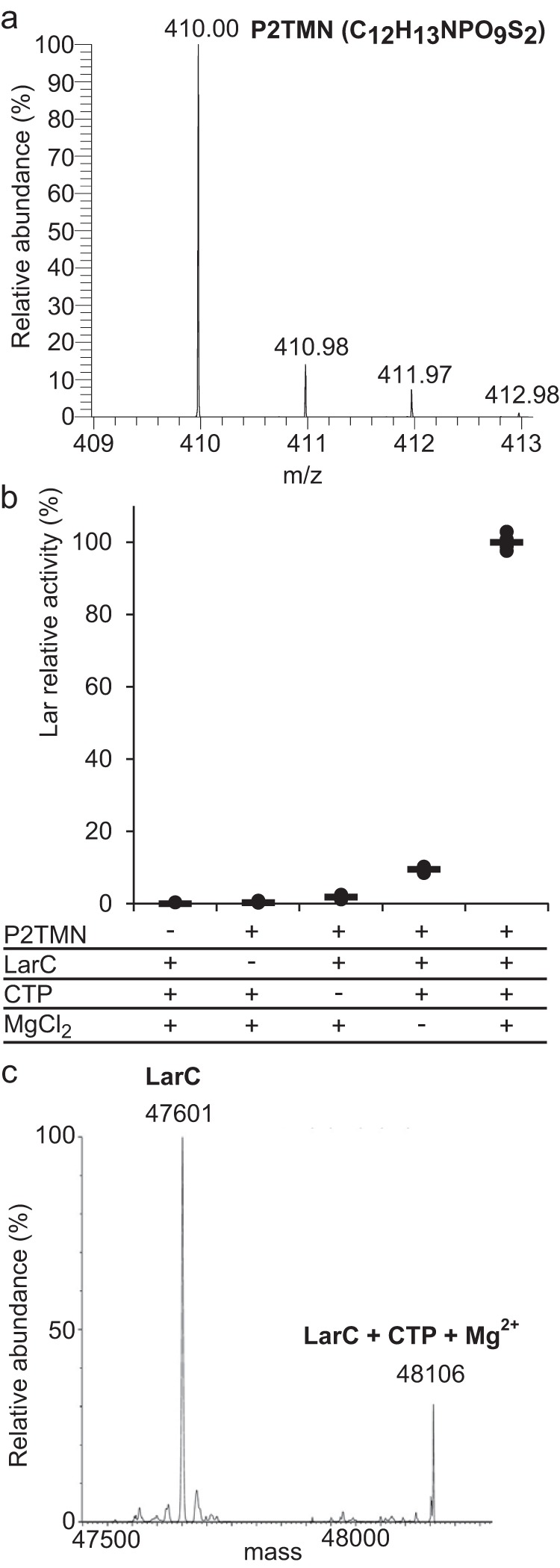
P2TMN is the substrate of LarC. a, P2TMN identification by MS in negative ionization mode with the molecular formula in parentheses. b, NPN cofactor biosynthesis using purified P2TMN, LarC, CTP, and MgCl2. Lar activity with all compounds was set to 100%. NPN cofactor biosynthesis was assessed after addition of LarATt apoprotein and l-lactate and by assaying for d-lactate production. c, mass of purified LarC. The expected mass of LarC is 47,601.5 Da, and the mass of LarC + CTP + Mg2+ − 2H+ is 48,107 Da.
Hydrolysis of CTP by LarC
The enzymatic properties of LarC were investigated in more detail. The nucleotide specificity for CTP was confirmed using purified P2TMN as a substrate (Fig. 3a). In addition, Mn2+ was shown to be 5-fold more efficient than Mg2+ to activate LarC and at 1/20th of the concentration (Fig. 3b), indicating that Mn2+ is the preferred cation for LarC in vivo. This result was surprising but not unexpected as Lb. plantarum accumulates approximately 30 mm Mn2+ in its cytoplasm (8). Lc. lactis, however, does not accumulate Mn2+ in its cytoplasm (8), which explains why Mg2+ was found instead of Mn2+ in LarC purified from Lc. lactis cells (Fig. 2c). To test whether and how LarC hydrolyzes the triphosphate, we performed thin-layer chromatography (TLC) of the LarC reaction products that form using α-32P-labeled CTP. When P2TMN and MgCl2 were added to the reaction, CMP accumulated over time (Fig. 3c) showing that LarC hydrolyzes CTP to CMP. In the absence of divalent cation, aberrant hydrolysis took place with CDP formed instead of CMP (Fig. 3c). Thus, LarC is a CTP-hydrolyzing nickel cyclometallase.
Figure 3.
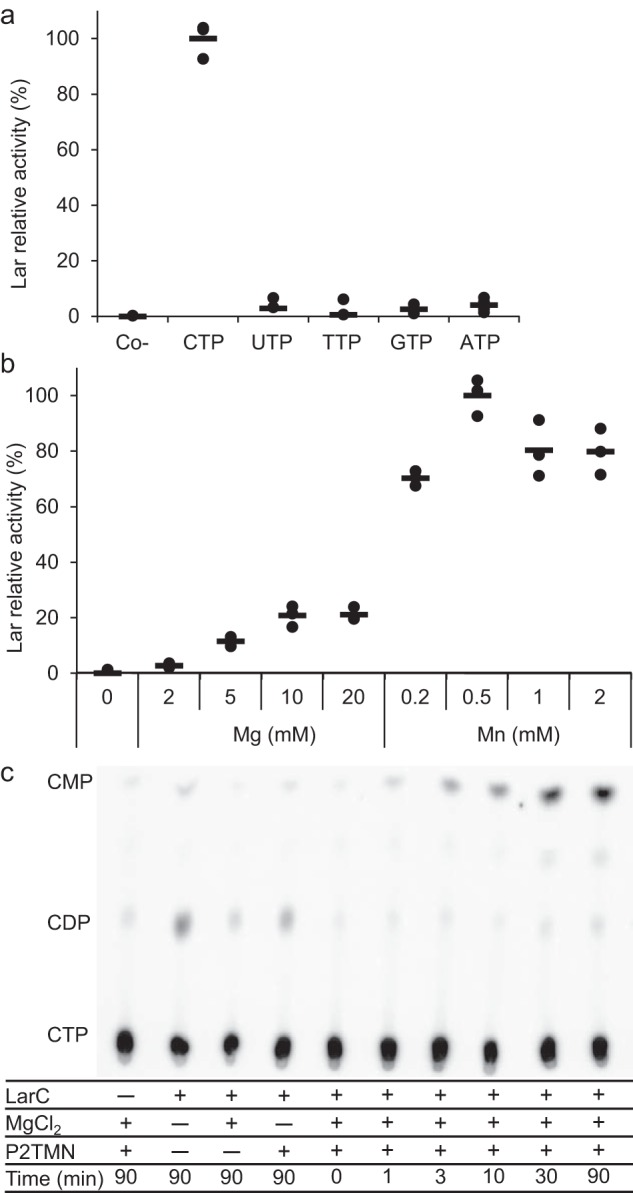
LarC is a CTP-hydrolyzing nickel cyclometallase. a, dependence of LarC reaction on nucleotide identity. Lar activity with CTP was set to 100%. b, dependence of LarC reaction on MgCl2 and MnCl2 concentration. Lar activity with 0.5 mm MnCl2 was set to 100%. Lar activity was assessed after addition of LarATt apoprotein in excess and l-lactate by assaying for d-lactate production. The lines indicate the mean values, and the points indicate the individual data points. c, radiogram from TLC of the LarC reaction using P2TMN, MgCl2, and [α-32P]CTP. The positions of CDP and CMP were confirmed by UV detection.
Soluble NPN produced by LarC directly activates LarA
The expected product of the LarC reaction, NPN, may be released as the free cofactor or it may require a protein that transfers and perhaps activates it for binding to LarALp, i.e. LarA from Lb. plantarum. LarALp forms a covalent thioamide bond with NPN, which is why its activation is time-dependent, in contrast to LarATt that interacts noncovalently with NPN (4). To address this question, we obtained NPN by reaction of NaAD with LarB, LarE, and LarC, the proteins were heat-denatured and removed by centrifugation, and the supernatant was tested for its ability to generate Lar activity in the presence of LarATt (Fig. 4a). Heat treatment of the LarB, LarE, and LarC reaction strongly decreased activation, but the activation could be restored, or even enhanced, when free thiols such as cysteine or β-mercaptoethanol (BME) were added before heat treatment (Fig. 4a). This observation suggests that free thiols stabilize NPN in solution, possibly by serving as a fourth ligand in the square–planar nickel coordination sphere.
Figure 4.

Characterization of the LarA activation by NPN. a, activation of LarATt apoprotein. b, time-dependent activation of LarATt and LarALp apoproteins by in vitro-synthesized NPN, with or without (Ctrl) heat denaturation of LarB, LarC, and LarE and with or without addition of cysteine (+Cys) and β-mercaptoethanol (+BME) (1 or 10 mm in a and 10 mm in b). LarA was incubated with in vitro-synthesized NPN for 1 min in a and for the indicated time in b. Lar activity of LarATt with 10 mm BME was set to 100%. Lar activity was assessed after addition of l-lactate by assaying for d-lactate production. The lines indicate the mean values, and the points indicate the individual data points. c, LarC reaction summary.
The activation of LarALp was then compared with LarATt activation. LarALp activation was lower and much slower, compared with LarATt (Fig. 4b), as expected by the requirement to form a covalent bond between LarALp and NPN. LarALp activation was still observed, and even enhanced three times, after heat denaturation of LarB, LarE, and LarC when BME was added (Fig. 4b), demonstrating that the thiol-stabilized free NPN activates LarA better without the synthetic proteins LarB, LarE, and LarC (Fig. 4c). The binding of the cofactor to a biosynthetic protein may slow down the activation of LarA.
The supernatant of the combined LarB, LarE, and LarC reactions was chromatographed on an anion-exchange column in the presence of 10 mm BME, and the identity of the purified NPN was confirmed by MS (Fig. 5a). The optical spectra of purified P2TMN and NPN show broad absorbance peaks around 260 and 450 nm, respectively (Fig. 5b). The NPN peak at 450 nm was observed previously in purified LarA holoprotein (2).
Figure 5.
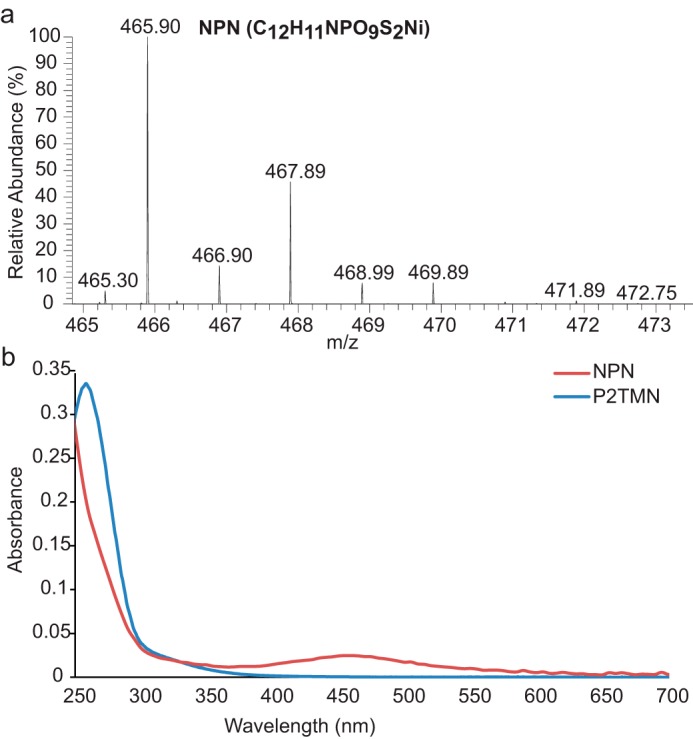
Spectra of NPN. a, NPN identification by MS in negative ionization mode with the molecular formula in parentheses. b, UV-visible absorbance spectra of purified P2TMN (blue) and NPN (red).
LarC behaves as a single-turnover enzyme hydrolyzing one CTP per nickel insertion
We next examined whether LarC is capable of catalyzing multiple turnovers. This protein does not contain nickel when purified from cells grown in the absence of this metal, and previous studies had shown that subsequent Ni2+ addition had only a slight positive effect on LarC activity (4). When LarC was limiting and the reaction time extended, added nickel could only slightly activate LarC and only at stoichiometric concentrations. Higher nickel concentration showed no effect (Fig. 6a). Moreover, nickel-devoid LarC cannot be activated by adding Ni2+ to the solution (Fig. 6a). In summary, added nickel enhances NPN biosynthesis when LarC is already nickel-loaded but cannot activate the LarC apoprotein. These results show that LarC cannot catalyze multiple nickel insertions into P2TMN using the conditions of the in vitro test.
Figure 6.
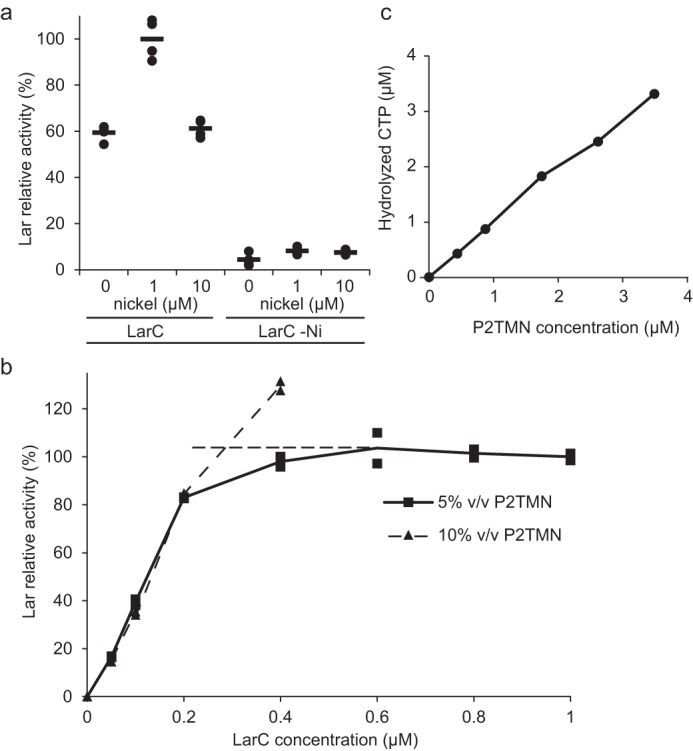
Characterization of LarC reaction. a, dependence of LarC reaction on nickel. Lar activity was measured using P2TMN, MgCl2, CTP, and either 1 μm LarC purified from cells cultivated in the presence (LarC) or absence (LarC −Ni) of 1 mm nickel, with or without further addition of nickel to the assay. NPN biosynthesis was performed for 10 min, in order to allow for several turnovers to take place. The Lar activity of LarC with 1 μm nickel was set to 100%. b, titration of P2TMN by LarC. Lar activity with 1 μm LarC and 5% (v/v) of purified P2TMN was set to 100%. Dashed lines represent the conditions of complete P2TMN consumption (horizontal) or of complete LarC consumption (vertical); the intersection is the point where all LarCs and all substrates are consumed with 5% (v/v) P2TMN. Lar activity was assessed after addition of LarATt apoprotein in excess and l-lactate by assaying for d-lactate production. The lines indicate the mean values, and the points indicate the individual data points. c, amount of CTP hydrolysis versus substrate addition, based on densitometry of the TLC autoradiograph.
Given that the LarC purified from nickel-replete cells was ∼90% loaded with nickel, the P2TMN concentration could be estimated based on the amount of LarC needed to completely react with all of the substrate (Fig. 6b). This P2TMN concentration could then be compared with the estimated CTP hydrolysis based on densitometry measurement of the TLC autoradiograph. The amount of CTP hydrolyzed equaled the amount of P2TMN added (Fig. 6c). This one-to-one ratio showed that CTP hydrolysis is stoichiometric with NPN biosynthesis.
Crystal structure of the CTP-binding domain of LarC
To gain better insight into the LarC catalytic mechanism, we attempted to crystallize the full-length (N-terminal StrepII-tagged) fused LarC protein (Table 1), first purified from Lc. lactis and later from Escherichia coli. In both cases, we obtained only LarC2 crystals (without the eight N-terminal residues and with the last five C-terminal residues being disordered). To investigate the cleavage of the full-length protein into LarC1 and LarC2 over time, we incubated the protein in purification buffer at ∼1 mg·ml−1 at room temperature for 2 weeks. The precipitate was separated from the supernatant, which was subjected to MS analysis. The supernatant only contained LarC2 with three MS peaks of 17,286, 17,074, and 16,804 Da (Fig. 7), corresponding to LarC2 with 4, 6, or 8 residues missing at the N terminus. The precipitate appeared to contain the full-length LarC and the degraded LarC1. This result shows that the full-length protein is cleaved at three different positions over time when purified from either Lc. lactis or E. coli, resulting in a very stable LarC2 that is crystallizable. In contrast, the remaining LarC1 and the full-length protein are less stable, precipitate with time, and do not form crystals. We overexpressed just LarC1 in Lc. lactis and in E. coli, but no soluble protein was produced or LarC1 purified as an aggregate according to gel-filtration analysis, respectively, so we focused on the LarC2 structure.
Table 1.
Strains, plasmids, and primers
| Strain, plasmid, or primer | Characteristic(s) or sequence | Source or Ref. | |
|---|---|---|---|
| Strain | |||
| Lc. lactis | |||
| NZ3900 | MG1363 derivative | 33 | |
| E. coli | |||
| DH10B | F− endA1 recA1 galE15 galK16 nupG rpsL ΔlacX74 Φ80lacZΔM15 araD139 Δ(ara,leu)7697 mcrA Δ(mrr-hsdRMS-mcrBC) λ− | Invitrogen | |
| DH5α | fhuA2 lacΔU169 phoA glnV44 Φ80lacZΔM15 gyrA96 recA1 relA1 endA1 thi-1 hsdR17 | Invitrogen | |
| ArticExpress | Contains Cpn60 and Cpn10 from Oleispira antarctica | Agilent | |
| Rosetta 2 | F− ompT hsdSB(rB− mB−) gal dcm (DE3) pRARE2 (CamR) | Novagen | |
| Plasmid | |||
| pNZ8048 | Cmr, PnisA | 34 | |
| pGIR012 | Cmr; pNZ8048 with a 1.31-kb insert after PnisA containing larALp translationally fused to DNA encoding the StrepII-tag | LarALp purification | 3 |
| pGIR082 | Cmr; pNZ8048 with a 1.31-kb insert after PnisA containing larATt translationally fused to DNA encoding the StrepII-tag | LarATt purification | 3 |
| pGIR026 | Emr Apr; pGIZ6603 with DNA encoding the StrepII-tag translationally fused at the 3′-end of the larB ORF | LarB purification | 4 |
| pGIR031 | Cmr; pNZ8048 with a 1.30-kb insert after PnisA containing DNA encoding the StrepII-tag translationally fused to larC | LarC purification | 3 |
| pGIR041 | Cmr; pNZ8048 with a 0.79-kb insert after PnisA containing DNA encoding the StrepII-tag translationally fused to larC1 | LarC1 purification | This study |
| pGIR051 | Cmr; pNZ8048 with a 1.30-kb insert containing the translational fusion between PnisA and Strep-tag--larC fusion with an 1 bp insertion between larC1 and larC2 | LarCfused purification | 3 |
| pGIR043 | Ampr; pET22b with a 0.79-kb insert containing DNA encoding the StrepII-tag translationally fused to larC1 | LarC1 purification | This study |
| pGIR053 | Ampr; pET22b with a 1.30-kb insert containing DNA encoding the Strep-tag--larC fusion with an 1 bp insertion between larC1 and larC2 | LarCfused purification | This study |
| pGIR076 | Ampr; pBADHisA with a 0.89-kb insert after PAra containing larE translationally fused to the StrepII-tag sequence | LarE purification | 5 |
| pGIR031 _MutCX | pGIR031 encoding the LarC variant X (see primers for detail) | LarC variants purification | This study |
| Primers for pGIR031 modification | |||
| MutC1_A | 3′-GATCATGAAGCGCGTCATTTAGCTG-5′ | ΔH75-H86 variant | This study |
| MutC1_B | 3′-TTCCACAAAACCATGGTCCTTACC-′ | This study | |
| MutC10_A | 3′-CAAGCGGCAGCAGCGGTTCATCACATGCCAC-5′ | E124A variant | This study |
| MutC10_B | 3′-GCTGCTGCCGCTTGCGCAATTTCCATAAAG-5′ | This study | |
| MutC11_A | 3′-TCCATGCAGTGGGCGCGTTGGATTCGATTGTTG-5′ | E139A variant | This study |
| MutC11_B | 3′-CCACTGCATGGAAATGAACTTCAGCTAGTGGC-5′ | This study | |
| MutC12_A | 3′-CGTTGGCTTCGATTGTTGATATTGTGGGCTGCTG-5′ | D144A variant | This study |
| MutC12_B | 3′-ATCGAAGCCAACGCGCCCACTTCATGGAAATG-5′ | This study | |
| MutC13_A | 3′-ATACTGCATTGATTACCCCCACTGGGATGGGC-5′ | E210A variant | This study |
| MutC13_B | 3′-ATCAATGCAGTATGCACATCCAACCGTTGTTGAATC-5′ | This study | |
| MutC14_A | 3′-CTAATCTGGCTGATCAGACGGGTGAAGGACTGG-5′ | D284A variant | This study |
| MutC14_B | 3′-CTGATCAGCCAGATTAGCTTCAATCATTAAGACAGCATC-5′ | This study | |
| MutC5_A | 3′-TCAGATGGCGAAAGATCGGCCGGCGACAAAG-5′ | K314A variant | This study |
| MutC5_B | 3′-CGATCTTTCGCCATCTGAATGGGCGTGAAAAACAC-5′ | This study | |
| MutC6_A | 3′-AGATGAAGGCAGATCGGCCGGCGACAAAG-5′ | K315A variant | This study |
| MutC6_B | 3′-GCCGATCTGCCTTCATCTGAATGGGCGTG-5′ | This study | |
| MutC7_A | 3′-AAGAAAGATGCGCCGGCGACAAAGTTAACGG-5′ | R317A variant | This study |
| MutC7_B | 3′-GTCGCCGGCGCATCTTTCTTCATCTGAATGG-5′ | This study | |
| MutC8_A | 3′-ATTGGTGTAGCTTACCAGACGTGGCAACGTAC-5′ | R348A variant | This study |
| MutC8_B | 3′-TCTGGTAAGCTACACCAATCGTCGTCGTTTCTTG-5′ | This study | |
| MutC24_A | 3′-TGCAAGCGCACTTCCTGACGGTCGCAAC-5′ | R359A variant | This study |
| MutC24_B | 3′-GTGCGCTTGCATAATAGTACGTTGCCACGTC-5′ | This study | |
| MutC15_A | 3′-CAAGTTGCAGTGGCGACATATCAAGATATTGAG-5′ | K374A variant | This study |
| MutC15_B | 3′-CCACTGCAACTTGAACGTCACCGTACG-5′ | This study | |
| MutC16_B | 3′-CCGGCCTATGCGGATTGTGCCCAG-5′ | E387A variant | This study |
| MutC16_B | 3′-GCATAGGCCGGCATCTTCTTCTCAATATC-5′ | This study | |
| UP_PNZ8048′ | 3′-ACAATGATTTCGTTCGAAGGAACTAC-5′ | Sequencing | 3 |
| DW_PNZ8048 | 3′-AAGCCTTGGTTTTCTAATTTTGG-5′ | Sequencing | This study |
Figure 7.
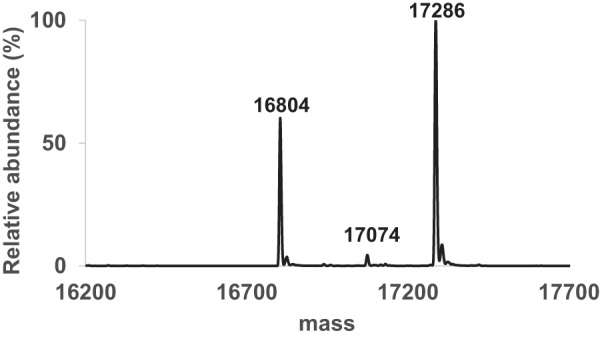
Masses of LarC2 after cleavage of the purified full-length LarC protein.
We solved the LarC2 structure under multiple crystallization conditions, which always revealed the same hexamer composed of a dimer of trimers (Fig. 8a). Each LarC2 unit comprises two distinct domains (Fig. 8b) as follows: the N-terminal domain 1 (residues 272–356) with a ferredoxin-like fold (β1α1β2β3α2β4); and the C-terminal domain 2 (residues 357–412) containing three successive β-strands (β5–7) followed by two α-helices (α3–4). Domain 1 forms a symmetric trimer with orthogonally packed β-sheets, reminiscent of the typical fold of the GlnB-like superfamily. Two domain 1 trimers stack vertically and share the 3-fold symmetry axis, forming the central part of the hexamer. Domain 2, which is extended from the C terminus of domain 1, forms a protrusion from the central hexamer and contributes to the deep CTP-binding pocket (see below). The substantial differences for β2 and β3 compared with known family members and the unique C-terminal extension (domain 2) establish LarC2 as a new member of GlnB-like superfamily (Fig. 8c). A three-dimensional protein structure similarity search using the Dali server (9) and a sequence search in the Protein Data Bank only resulted in one similar structure of a protein from Methanopyrus kandleri AV19 with unknown function (PDB code 3c19).
Figure 8.
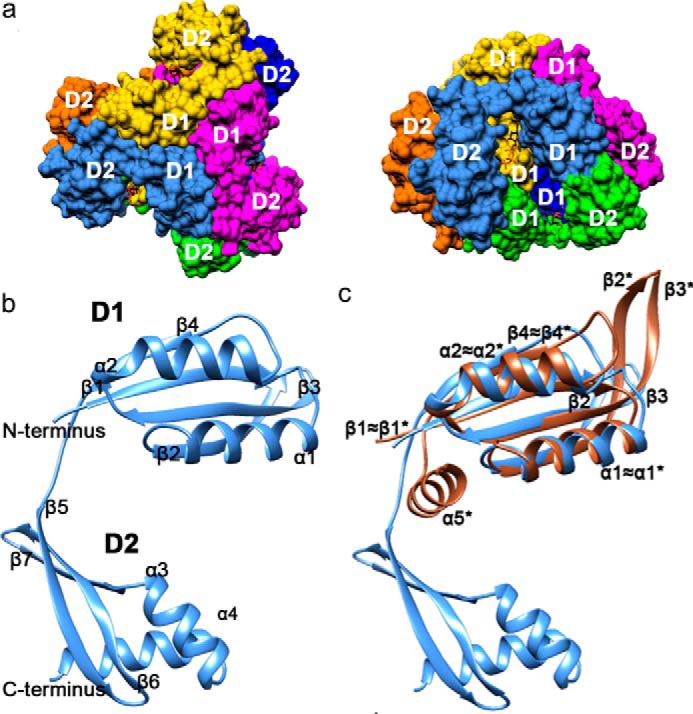
LarC2 structure. Individual chains and their symmetry-related copies are colored as follows: A, green; A′, orange; A″, dark blue; B, light blue; B′, yellow; B″, magenta. N-terminal domain 1 (D1, residues 272–356) forms the center of the trimer (top view), and C-terminal domain 2 (D2, residues 357–412) protrudes outward. The CTP molecules are shown in stick representation with carbon atoms in black, nitrogen in blue, oxygen in red, and phosphorus in orange. a, hexameric structure of LarC2. Surface representation of the top view (left) and side view (right). b, secondary structure of LarC2 (helices = α, strands = β). c, comparison of LarC2 chain B to the most similar match identified by Dali, a GlnB-like putative CutA protein (brown) from Thermotoga maritima (PDB code 1o5j). CutA helices and sheets are labeled with an asterisk.
Crystal soaking with the P2TMN analog nicotinic acid mononucleotide followed by CTP-Mg or soaking just with Ni2+ did not reveal meaningful binding, but CTP binding was identified in a magnesium- or manganese-dependent manner (Figs. 8a and 9a). The MnCl2 datasets were of higher quality and are discussed here; however, magnesium binds the triphosphate of CTP in exactly the same way. Three subunits contribute to the CTP-Mn2+–binding pocket, two from one trimer and, to a lesser extent, one from the other. CTP-Mn2+ is sandwiched by domain 1 and domain 2 of one subunit, with domain 1′ from a neighboring subunit providing additional key interacting residues with the triphosphate of CTP (Lys-314′, Lys-315′, and Arg-317′ on the loop between β2′ and β3′, where the symbol “′” refers to the components from the second unit) (Fig. 9, a and b). Mn1 is always hexa-coordinated with α- and γ-phosphates of CTP and four water molecules. Mn2 is also hexa-coordinated in chain A, chelated by the three phosphate groups, Glu-387 from domain 2, and two water molecules. In chain B, however, only two phosphate groups, Glu-387 from domain 2 and one water molecule chelate Mn2 (Fig. 9, b–d). Loss of the Mn2 interaction with the α-phosphate hypothetically represents a conformational state relevant for catalysis from CTP to CMP. A cluster of highly conserved acidic residues (Asp-284′, Asp-285′, and Glu-289″ (of the third unit)) forms a negatively-charged patch, indirectly facilitating Mn1 association by stabilizing Mn2+-coordinating water molecules within a well-ordered hydrogen-bond network. The triphosphate of CTP is further stabilized by Lys-374 from domain 2 electrostatically associating with the α-phosphate, making it the most ordered portion of CTP. In contrast, the ribose is the least ordered part with only Arg-359 (domain 2) being within hydrogen-bond distance. Arg-354 (on the linker between domain 1 and 2) forms hydrogen bonds with the cytidine, implying a role in determining the specificity toward CTP. Near the cytidine, an atom with about the same electron density peak height as found for sulfur or phosphate atoms in the structure is surrounded by nitrogen atoms from CTP, Arg-354, and Arg-348′ (domain 1′), and therefore we interpreted it as a chloride ion. Comparison between apoprotein (PDB code 6BWO) and CTP-Mn2+-bound (PDB code 6BWQ) structures did not reveal any major structural changes upon CTP binding, with an r.m.s.d. of ∼0.5 Å between corresponding chains.
Figure 9.

CTP-bound LarC2 structure. a, CTP-binding pocket in the chain B trimer. Ribbon and protein C atoms are colored by chain as described above. Mn2+ ions and chelation are shown in purple, water molecules in red, and chloride ion and chelation in light green. The 2mFo − DFc electron density map for ligands is shown as blue mesh at 1 σ. b, 2D representation of CTP binding in the LarC2–CTP complex for trimer A (above) and B (below). Atoms are shown with carbon in black, nitrogen in blue, oxygen in red, manganese in purple, and water in cyan; hydrogen bonds/metal chelation are shown as red dashes, protein bonds in brown, and CTP bonds in black. c, comparison of CTP binding in trimer A and trimer B where the ligands of trimer A are transparent. d, metal chelation differences between trimers A and B.
Site-directed mutagenesis in LarC2 confirms its CTP-binding site
By aligning seven divergent LarC sequences, we found that most of the above-cited residues are substantially invariant (Fig. 10). Arg-354 is not absolutely conserved, but an Arg residue is most prevalent at this position. To examine their importance, these conserved residues were substituted by Ala, and the in vitro activity of the LarC variants was evaluated. Most LarC variants, except R317A, showed reduced activity compared with the WT enzyme (WT) (Fig. 11a). Two LarC variants, D284A and K315A, showed around 10% activity compared with the WT, and the last five LarC variants, K314A, R348A, R359A, K374A, and E387A, showed only residual activity (Fig. 11a). It is interesting to note that for two variants with only residual LarC activity, K314A and K374A, a lysine residue pointing toward the α-phosphate of CTP was substituted (Fig. 9a), suggesting that these residues could be involved in catalysis. The LarC variant E387A has probably lost its ability to chelate metal ion (Mn2), which suggests that the metal ion coordinated by Glu-387 is necessary. The reduced, but not completely abolished, activity of D284A might indicate that the Asp-284–facilitated metal coordination (Mn1) is contributive to, but not essential for, catalysis. Arg-359 potentially stabilizes the ribose and is an important residue, given the effect of its substitution by Ala (Fig. 11a). To further confirm that LarC2 plays a key role in CTP binding, two LarC variants, D284A and K315A, were tested for their CTP-binding affinity (Fig. 11b). The initial rate of LarC reaction was measured as a function of CTP concentration, allowing the estimation of the CTP concentration required to attain 50% of the maximum LarC activity (K50). As shown in Fig. 11b, this K50 is around 0.05 mm for the WT and increased to around 0.5 mm in both variants. The large effect of the substitutions on the affinity for CTP shows that the CTP-binding site is affected by these substitutions and confirms that the crystal structure revealed the native CTP-binding site.
Figure 10.

Alignment of LarC homologs from Lb. plantarum NCIMB 8826, Staphylococcus epidermidis ATCC 12228, T. maritima MSB8, Thermosinus carboxydivorans NOR1, Desulfotomaculum carboxydivorans CO-1-SRB, Synechocystis sp. PCC 6803, and Methanosarcina acetivorans C2A. The numbering above the sequence corresponds to the residue numbering of Lb. plantarum LarC. The sequence of Lb. plantarum LarC was constructed assuming a PRF (7) at position 263, marking the end of LarC1. The residues conserved in all seven sequences are indicated with an asterisk, and the less conserved residues are indicated with one or two dots (as detailed in the ClustalX program (32)). The residues that were substituted by mutagenesis are highlighted in gray. The residues interacting with CTP in the LarC2 structure are boxed with black lines. The alignment was performed using ClustalX (32).
Figure 11.

LarC variants affect activity. a–c, analysis of selected substitutions in LarC2 (a and b) and LarC1 (c). Lar activity using P2TMN, CTP (0.1 mm in a and c), MgCl2 and WT LarC (WT), or the indicated LarC variants after addition of LarATt apoprotein plus l-lactate and assaying the d-lactate production was measured. Lar activity of the WT with 0.1 mm CTP was set to 100%. d, nickel content of LarC variants. The lines indicate the mean values, and the points indicate the individual data points. e, hypothetical model for the mechanism of cyclometalation by LarC. Dotted lines indicate broken and newly formed bonds, and straight arrows indicate coordination bonds.
Essential Asp/Glu residues in LarC1
With the knowledge that LarC2 is involved in CTP binding, we hypothesize that LarC1 functions in binding nickel and P2TMN. To test the involvement of the His-rich region and conserved negatively-charged residues of LarC1 in nickel binding or other functions of LarC, several variants were analyzed. The LarC variant with a deletion of the entire His-rich region showed decreased LarC activity and nickel content (Δ75–86, Fig. 11, c and d), indicating that the His-rich region binds nickel but is not strictly required for LarC activity, as also suggested by the low conservation of this sequence (Fig. 10). However, the three alanine variants of highly conserved acidic residues E124A, E139A, and D144A (Fig. 10) exhibited complete loss of LarC activity and variable nickel content (Fig. 11, c and d), potentially indicating a functional role. Thus, several carboxylic acid or carboxylate residues are critical for activity.
To rule out LarC2 binding of nickel, we soaked LarC2 crystals in a high concentration of this metal ion. The structure revealed four nickel-binding sites (Fig. 12), but none of the chelating residues are conserved (Fig. 10), and the sites were not observed in both trimers.
Figure 12.
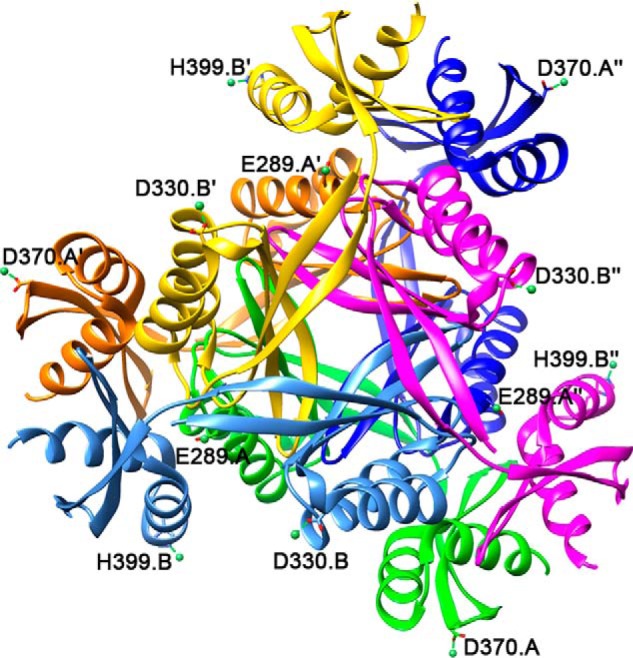
Hexameric view of the nickel-bound LarC2 structure (PDB code 6BWR). Nickel atoms and coordination are in green, and chelating side chains are labeled.
Discussion
We described here a previously uncharacterized CTP-hydrolyzing nickel cyclometallase, LarC. Perhaps the closest functionally-related enzyme is HcgD, a Nif3-like protein that is thought to act as iron chaperone in the biosynthesis of the iron-guanylylpyridinol (FeGP) cofactor of [Fe]-hydrogenase (10). In the case of the FeGP cofactor, the enzymatic system involved in the iron–acyl formation is unknown, but an Fe(0) or Fe(I)–carbonyl complex is thought to be the precursor of the acyl–iron bond formation and has been used for the chemical synthesis of FeGP cofactor model compounds (11). By contrast, LarC inserts nickel in the +2 oxidation state, without the need for reduction.
We showed that LarC activity is energy-dependent and requires hydrolysis of the rarely used nucleotide CTP (Fig. 1d). The function of CTP hydrolysis remains unclear. Perhaps the nickel is brought into close proximity to the P2TMN through a CTP-dependent conformational change. Alternatively, the P2TMN thioacid may be activated by cytidylylation, although our attempts to accumulate this covalent intermediate using LarC WT and variants were unsuccessful. A direct binding of CTP or CMP to nickel at some stage of the reaction also cannot be excluded. This hypothesis may relate to the basis of LarC's specificity for CTP; cytidine is the only pyrimidine not protonated at N3 at physiological pH and, hence, would be available for metal ion binding (12).
The observation that LarC behaves as a single-turnover enzyme in vitro may reflect the fact that LarC may not need to catalyze multiple turnovers in vivo. Indeed, LarC acts downstream of LarE, which is itself a single-turnover enzyme (4). Moreover, the limited amount of NPN synthesized will prevent the exhaustion of the NaAD precursor, which is required for NAD biosynthesis. Moreover, as NPN is a catalyst derived from a precursor of NAD, the physiological need in biosynthesis must be low and overproduction may be toxic.
We identified several residues involved in LarC function. First, we showed that the previously identified His-rich sequence (3) is a probable region for nickel binding (Fig. 11d), although not strictly required for LarC activity (Fig. 11c). This site may be used for nickel storage before its transfer to the active site and its insertion into P2TMN. Three conserved carboxylic residues, Glu-124, Glu-139, and Asp-144, are required for LarC activity (Fig. 11c). The essentiality of these residues leads us to postulate a mechanism for LarC activity involving carboxylate groups (Fig. 11e). In this hypothesis, two carboxylate residues, presumably Glu-124 and/or Glu-139 and/or Asp-D144, coordinate a nickel ion close to P2TMN. This binding might be unstable and might only take place temporarily during catalysis, which would explain why some of these variants show a higher nickel content than the WT (Fig. 11d). A pericyclic reaction involving the concerted movement of six electrons would then result in nickel replacing the hydrogen at the C4 position of the pyridinium ring. Such carboxylate-assisted C–H activation is a classical mechanism in organometallic cyclometalations (6). Asp and Glu residues have also been suggested to be involved in the transfer of iron into ferrochelatase (13) and in transmetalation of porphyrin (14).
The structure of the C-terminal domain of LarC (LarC2) was solved in the presence and absence of CTP, and the binding site of CTP was identified at a deep cleft at the interface of three LarC2 units (Figs. 8 and 9). The importance of CTP binding and catalysis was demonstrated by mutagenesis and activity assays (Fig. 11, a and b). Interestingly, the LarC sequence is devoid of known nucleotide-binding motifs according to the nucleotide-binding database (15). We note that the PII signaling transduction proteins of the GlnB-like superfamily also bind a nucleotide, but in that case it is ATP. ATP is not hydrolyzed, but its binding induces a conformational change that appears to be the structural basis of PII activation for its nitrogen-sensing role in bacteria (16, 17). The conserved ATP-binding pocket in the PII signaling transduction proteins only partially overlaps with the CTP-binding site of LarC2, and significant differences can be identified through side-by-side structural comparison (Fig. 13). Particularly, domain 2 of LarC2, which is absent in known members in the GlnB-like superfamily, provides key residues in CTP binding, supporting the notion that LarC2 has evolved to constitute a critical part of the unique nucleotide-binding pocket. Because CTP binding does not lead to any meaningful conformational change of LarC2, it is unlikely that CTP acts as a regulator of LarC as found for ATP in activation of the PII signaling transduction proteins. Instead, we have shown that CTP is a substrate of LarC, indicating that the function of LarC2 is to bind and present CTP to LarC1.
Figure 13.
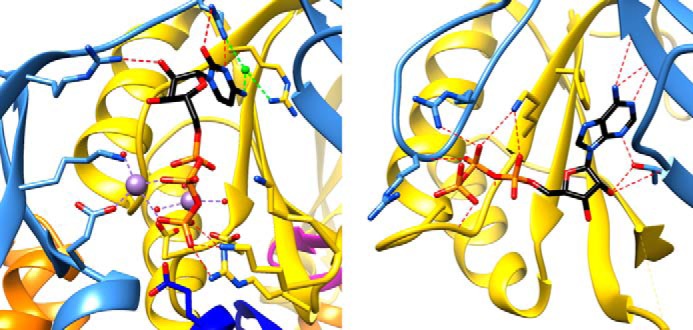
Comparison of CTP and ATP binding within two members of the GlnB-like family. Left, CTP binding to LarC2. Right, ATP binding to PII of E. coli (PDB code 5L9N (17)), with corresponding chains colored the same way as LarC2. Side chains at the ATP-binding pocket are shown with hydrogen bonds as red dashes, and the manganese ions and chelation are shown in purple, water molecules are in red, and chloride ion and chelation are in light green.
LarC is the first identified cyclometallase catalyzing the formation of a stable nickel–carbon bond. We have unveiled some characteristics of this intriguing enzyme, including that it is a CTP-hydrolyzing enzyme. We also performed the first in vitro synthesis and purification of the NPN cofactor, which will open the way to the direct investigation of this particular cofactor in vitro.
Experimental procedures
Materials and growth conditions
Bacterial strains and plasmids used in this study are listed in Table 1. Chemicals were purchased from Sigma. We grew Lc. lactis in M17 broth supplemented with 0.5% glucose at 28 °C. NiSO4 (1 mm) was added to cultures expressing pGIR031 (3), and chloramphenicol (10 mg·liter−1) was provided when expressing plasmids were derived from pNZ8048. For the induction of genes under control of the nisA expression signals, nisin A was added during the early exponential phase (A600 = 0.3–0.4) at a concentration of 1 μg·liter−1, and the cells were collected after 4 h. E. coli DH10B was grown with agitation at 37 °C in LB containing erythromycin (200 mg·liter−1). For LarE expression in E. coli ArticExpress, 10 ml of an overnight culture in LB with ampicillin (200 mg·liter−1) and gentamycin (40 mg·liter−1) was transferred to 1 liter of LB and grown for 3 h at 30 °C with agitation, and induction was started by addition of l-arabinose (0.2%) and grown at 13 °C for 24 h with agitation.
For determining the apoprotein structure, Lc. lactis cells containing PGIR051 were grown following the above-mentioned protocol, except that no NiSO4 was added. For the selenomethionine-containing crystal from Lc. lactis, we grew the cells in chemically defined medium containing selenomethionine instead of methionine, as described elsewhere (18). For the CTP-Mn and nickel-bound structures, pGIR053 was utilized in E. coli Rosetta 2(DE3) cells. Rosetta 2 cells were grown with agitation at 37 °C in LB containing ampicillin (100 mg·liter−1) until exponential phase (A600 = 0.5–0.6). The culture was transferred to 16 °C, induced with 0.1 mm isopropyl β-d-1-thiogalactopyranoside, and grown for ∼20 h with agitation.
For investigation of LarC1 without LarC2, construct pGIR041 was introduced in Lc. lactis as described above for pGIR031. In addition, the LarC1 construct carrying pGIR043 was introduced in E. coli Rosetta 2 cells as mentioned above.
DNA techniques
We carried out general molecular biology techniques according to standard protocols (19). Transformation of E. coli and Lc. lactis was performed by electrotransformation (20, 21). PCR amplifications used Phusion high-fidelity DNA polymerase (New England Biolabs). The primers used in this study were purchased from Eurogentec and are listed in Table 1. Plasmids bearing variants of larC for expression in Lc. lactis were derived from pGIR031 (3) and provided the 13 variant forms of LarC. For each construction, pGIR031 was first methylated by Dam methylase using S-adenosylmethionine (New England Biolabs), and PCR amplification was performed to obtain a fragment comprising the mutated plasmid. The PCR product was digested with DpnI before transformation in Lc. lactis according to the QuikChange mutagenesis protocol (22). The plasmid sequences were confirmed by sequencing with primer UP_PNZ8048′ and DW_PNZ8048.
For constructing pGIR043 and pGIR053 in E. coli, we amplified, digested, and ligated the larC1 gene, including the N-terminal Strep-II tag into the pET22b vector, similar to the description above. Transformation first in E. coli Dh5α and then E. coli Rosetta 2(DE3) cells followed standard protocols. Constructs were confirmed via sequencing.
Cell lysate preparation
The cells from 1.5 liters of Lc. lactis cultures expressing LarC from pNZ8048 derivatives were collected by centrifugation (5000 × g for 10 min) and washed twice with 50 ml of 100 mm Tris-HCl, pH 7.5, buffer containing 150 mm NaCl (W buffer). Cells were resuspended in 15 ml of W buffer and transferred by 0.5-ml aliquots into 0.5-ml suspensions of glass beads (≤106 μm, Sigma) in W buffer using 2-ml microtubes. Lysis was accomplished by using a FastPrep-24 cell disruptor (MP Biomedicals) twice for 1-min periods, with 5 min cooling on ice between the runs. After lysis, the soluble fractions were collected after centrifugation at 20,000 × g for 15 min at 4 °C. For obtaining denatured cell lysates, the soluble fractions were incubated at 80 °C for 10 min, and following protein precipitation, the supernatants were collected after centrifugation at 20,000 × g for 5 min at room temperature.
Protein purification
LarALp, LarATt, LarC, and LarC variants were purified from 1.5 liters of the appropriate Lc. lactis cultures (containing plasmids that are listed in Table 1). Cell lysates were obtained as described above. Affinity chromatography was performed with gravity flow Strep-Tactin® Superflow® high-capacity columns of 1.5 ml, as described (23), with W buffer. Protein concentrations were measured by the Bradford assay (24) and confirmed by the absorbance at 280 nm for LarC (within 10% range). Only for crystallization purposes, W buffer contained 300 mm NaCl.
LarB and LarE were purified from 1.5 liters of E. coli culture transformed with pGIR026 or pGIR076. The cells were collected, and the pellet was washed twice in W buffer, resuspended in W buffer with 1 g·liter−1 lysozyme, incubated for 30 min at 37 °C, and cooled for 10 min on ice. This treatment was followed by sonication at maximum amplitude with a Vibra Cell 75022 (Bioblock Scientific) using three series of 10 1-s pulses with intervening 2-min pauses on ice. Clear supernatant was obtained after centrifugation at 12,000 × g for 30 min at 4 °C. Affinity chromatography was performed with gravity flow Strep-Tactin® Superflow® high-capacity columns of 1.5 ml, as described (23), with W buffer. 300 mm NaCl was used for LarE purification instead of 150 mm.
LarC and LarC1 were prepared for crystallization purposes from E. coli cultures transformed with pGIR043 and pGIR053. After centrifugation, cells were resuspended in W buffer with 300 mm NaCl and lysed via sonication on ice (40% amplitude, 5-s pulses, 10-s pauses for 12 total min). Centrifugation and purification followed the mentioned LarB and LarE steps with 1.0-ml columns.
In vitro activation of LarA by purified LarB, LarC, and LarE and lysates
We incubated a mixture of LarE (5 μm), LarC (5 μm), LarB (1 μm), NaAD (0.1 mm), ATP (2 mm), MgCl2 (20 mm), and NaHCO3 (50 mm) in Tris-HCl buffer (100 mm, pH 8) with 2-μl lysates from lar-free cells (when indicated) and 1 mm nucleotides (when indicated) in a final volume of 20 μl. After incubation at room temperature for 30 min (or other incubation time, when indicated), 5 μl of the assay mixture was diluted into 45 μl of l-lactate (45 mm) supplemented with LarATt apoprotein (0.8 μm) in HEPES buffer (100 mm, pH 7). The reaction was incubated for 5 min at 50 °C and then stopped by heat treatment at 90 °C. The resulting d-lactate concentration was measured by enzymatic lactate oxidation to pyruvate using a d-lactic acid/l-lactic acid commercial test (Megazyme), as described previously (3). The NADH absorbance was monitored at 340 nm with an Infinite 200 PRO plate reader (Tecan).
Purification of P2TMN and the NPN cofactor
For purification of P2TMN, we incubated 1 ml of a mixture of LarE (200 μm), LarB (10 μm), NaAD (0.2 mm), ATP (2 mm), MgCl2 (20 mm), and NaHCO3 (50 mm) in Tris-HCl buffer (100 mm, pH 7). For purification of the NPN cofactor, we incubated 1 ml of a mixture of LarE (100 μm), LarC (50 μm), LarB (10 μm), NaAD (0.2 mm), CTP (0.2 mm), ATP (2 mm), BME (10 mm), MgCl2 (20 mm), and NaHCO3 (50 mm) in Tris-HCl buffer (100 mm, pH 7). The reactions were incubated for 1 h at room temperature and heat-treated at 80 °C for 10 min, and the supernatant was collected by centrifugation at 20,000 × g for 5 min and loaded onto a 5-ml HiScreen Q HP column (GE Healthcare) using an Akta Purifier (GE Healthcare), washed with Tris-HCl buffer (30 mm, pH 8), and eluted with a gradient of NaCl (0 to 1 m) in Tris-HCl buffer (30 mm, pH 8). For purification of the NPN cofactor, 10 mm BME was added to all buffers. P2TMN and NPN peaks were identified by their absorbance at 330 and 450 nm, respectively.
P2TMN and NPN cofactor analysis
For analysis of P2TMN and the NPN cofactor by MS, 10-μl aliquots were injected into an Accela HPLC system coupled to a Q-Exactive (ThermoFisher Scientific) mass spectrometer. Separations were performed using a Kinetex C18 column (150 × 2.1 mm) in 0.1% formic acid with a gradient of acetonitrile. The run time for each sample was 25 min. For analysis of the UV-visible spectrum of P2TMN, a Cary 50 BIO (SpectroVarian) was used.
For nickel quantification, we used 4-(2-pyridylazo)resorcinol (PAR) (25). The protein sample was denatured for 10 min at 80 °C prior to a 2-min incubation at room temperature with 100 μm PAR in 100 mm Tris-HCl buffer, pH 7.5. The absorbance was read at 496 nm with an Infinite 200 PRO plate reader (Tecan).
In vitro activation of LarA by purified LarC and P2TMN
We incubated a mixture of LarC (2.5 μm), P2TMN (50% v/v), MgCl2 (10 mm) (or MnCl2, when indicated), BME (10 mm), and CTP (0.1 mm) (or other nucleotides, when indicated) in MES buffer (100 mm, pH 6) using a final volume of 10 μl. After incubation at room temperature for 1 min, the reaction was stopped by heat treatment at 80 °C for 10 min, and 5 μl of the assay mixture was diluted into 45 μl of l-lactate (45 mm) supplemented with LarATt apoprotein (0.8 μm) in HEPES buffer (100 mm, pH 7). The reaction was incubated for 5 min at 50 °C and then stopped by heat treatment at 90 °C. The resulting d-lactate concentration was measured as described above.
For optimization of MgCl2, MnCl2, and P2TMN concentrations, the concentration of one component was varied with the other components held static. For the effect of nickel on the LarC reaction, nickel-devoid LarC was purified from cells expressed in the absence of added nickel. For the quantification of P2TMN concentration, the CTP concentration was 1 mm and the incubation time was 5 min.
TLC
We incubated a mixture of LarC (2.5 μm), P2TMN (50% v/v), MgCl2 (10 mm), and CTP (5 μm) containing [α-32P]CTP (1 μCi) in MES buffer (100 mm, pH 6) using a final volume of 10 μl. The reaction was stopped by the addition of 10 μl of 8 m urea, and 5 μl of the sample was spotted on a plate of PEI-cellulose F. The plate was washed twice with deionized water and then eluted with 1 n acetic acid/ethanol (80:20, v/v) containing 0.5 m lithium chloride. The plate was dried, revealed with an Imaging Screen-K (Bio-Rad, Belgium), and read on a Pharos FX Plus (Bio-Rad, Belgium).
Mass spectrometry of proteins
10-μl aliquots were injected into the UPLC system coupled to the QTof MS. Separations were performed on a BetaBasic CN (10 × 1-mm, 5-μm particle size) column with an aqueous phase of 0.1% formic acid and using a gradient of increasing acetonitrile at 30 °C. The run time for each sample was 15 min. The masses were calculated from the ESI-MS spectrum using the advanced maximum entropy (MaxEnt)-based procedure included in the Micromass MassLynx software package.
Protein crystallization
Phases were solved with a selenomethionine-containing crystal obtained from selenomethionine-substituted LarC purified from Lc. lactis. Protein crystals were obtained by mixing 5 μl of ∼10 mg·ml−1 selenomethionine-substituted LarC (50 mm Tris-HCl, pH 7.5, 150 mm NaCl) with 5 μl of reservoir solution. Sitting drop reservoir contained 100 μl of 10% PEG 8000, 100 mm Tris-HCl, pH 7.0, and 200 mm MgCl2. The crystals grew within 2 weeks and were of space group P433 2. In total, over 25 different crystallization conditions, with crystal growth between 1 week and 3 months, were confirmed to be only LarC2 crystals. The crystals varied in space group with P433 2 (one copy in the asymmetric unit), P231 (two copies), and I4 (six copies) observed. The P231 crystals were highly twinned, but using the twinlaw l,-k,h, these crystals ultimately resulted in the highest quality datasets. Each asymmetric unit of the P213 datasets contains two copies of LarC2 chain A and B, which are not 100% identical with an r.m.s.d. of ∼0.7 Å. For the apoprotein structure, 0.13 μl of ∼13 mg·ml−1 LarC (100 mm Tris-HCl, pH 7.5, 300 mm NaCl) was mixed with 0.13 μl of reservoir solution. The sitting drop reservoir contained 100 μl of 0.15 m dl-malic acid and 20% (w/v) PEG 3350. Crystals grew within a week. The crystal was soaked for 1 min in 25% PEG 400, 75% reservoir solution before freezing. For the CTP-Mn structure, 0.34 μl of ∼8 mg·ml−1 LarC (100 mm Tris-HCl, pH 7.5, 300 mm NaCl) were mixed with 0.17 μl of reservoir solution. The sitting drop reservoir contained 100 μl of 0.1 m MES monohydrate, pH 6.5, and 12% (w/v) PEG 20,000. Crystals grew within 2 months. The crystal was soaked in ∼15 mm CTP, ∼54 mm MnCl2, and reservoir solution for 28 min and then for 2 min in 20% PEG 400, 80% reservoir solution. CTP binding was only observed in the presence of magnesium or manganese with no binding or only very weak binding when these salts were substituted for NaCl, NaBr, LiCl, KNO3, or no salt. For the nickel structure, 0.5 μl of ∼5.5 mg·ml−1 LarC (100 mm Tris-HCl, pH 7.5, 300 mm NaCl) were mixed with 0.5 μl of reservoir solution. The hanging drop reservoir contained 100 μl of 0.2 m LiCl, 0.1 m Tris, pH 8.0, 20% (w/v) PEG 6000. Crystals grew within 2 weeks. The crystal was soaked in 16 mm NiCl2 and reservoir solution for 30 min and then for 1 min in 20% PEG 400, 80% reservoir solution.
Diffraction data collection, structure determination, and analysis
X-ray diffraction data were collected at the Advanced Photon Source LS-CAT beamlines (21-ID-D, 21-ID-F, and 21-ID-G) and at beamline 23-ID-B GM/CA. Datasets were processed with xdsapp2.0 (26), and merging and scaling were done using aimless (27). The phase of a selenomethionine-substituted crystal was solved using single wavelength anomalous dispersion in Phenix (28) at 2.5 Å, and molecular replacement was used for all subsequent datasets. Modeling building and refinement were conducted in COOT (29) and Phenix (28). Statistics for the datasets are listed in Table 2. Structure figures were created with UCSF Chimera (30) and LigPlot+ (31).
Table 2.
Crystal statistics for the LarC2 structures
| LarC2 crystals | Apo | MnCTP | Nickel |
|---|---|---|---|
| Data collection | |||
| Beamline | LS-CAT 21-ID-D | GM/CA 23-ID-B | LS-CAT 21-ID-D |
| Wavelength (Å) | 1.127 | 1.033 | 1.033 |
| Detector distance (mm) | 150 | 250 | 170 |
| No. of frames | 56 at 1° | 151 at 0.5° | 166 at 0.5° |
| Space group | P213 | P213 | P213 |
| Unit cell a, b, c (Å) | 97, 97, 97 | 97, 97, 97 | 97, 97, 97 |
| α, β, γ (°) | 90, 90, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolutiona (Å) | 43.33–2.03 (2.08–2.03) | 48.61–1.85 (1.89–1.85) | 48.32–1.81 (1.85–1.81) |
| Unique reflections | 19,861 (1440) | 26,499 (1623) | 27,744 (1665) |
| Redundancya | 6.3 (6.2) | 8.4 (8.5) | 9.4 (9.4) |
| Completenessa (%) | 99.6 (97.8) | 100.0 (99.4) | 99.9 (99.7) |
| I/σIa | 12.0 (2.2) | 11.7 (2.1) | 19.4 (2.2) |
| Rmergea,b | 0.095 (0.833) | 0.105 (0.974) | 0.073 (1.050) |
| Rpima,c | 0.060 (0.540) | 0.057 (0.509) | 0.037 (0.527) |
| CC1/2d | 0.997 (0.553) | 0.997 (0.615) | 0.999 (0.537) |
| Data refinement | |||
| Protein atoms | 2280 | 2260 | 2272 |
| CTP molecules | 2 | ||
| Manganese or nickel atoms | 6 | 4 | |
| Chloride atoms | 3 | ||
| H2O molecules | 98 | 140 | 148 |
| Rwork/Rfreee | 0.140/0.194 | 0.144/0.192 | 0.139/0.192 |
| B-factors (Å2) | 34.9 | 28.1 | 28.7 |
| Protein | 34.8 | 27.8 | 28.3 |
| CTP molecules | 39.0 | ||
| Manganese or nickel atoms | 32.8 | 34.3 | |
| Chloride atoms | 41.4 | ||
| H2O | 38.4 | 34.1 | 34.4 |
| Root mean square deviation in bond lengths (Å) | 0.009 | 0.011 | 0.009 |
| Root mean square deviation in bond angles (°) | 0.911 | 0.986 | 0.937 |
| Ramachandran plot (%) favored | 99.30 | 98.58 | 99.30 |
| Ramachandran plot (%) outliers | 0 | 0 | 0 |
| Rotamer outliers | 0 | 0 | 0 |
| PDB code | 6BWO | 6BWQ | 6BWR |
a Highest resolution shell is shown in parentheses.
b Rmerge = Σhkl Σj |Ij(hkl) − 〈I(hkl)〉|/Σhkl ΣjIj(hkl), where I is the intensity of reflection.
c Rpim = Σhkl (1/(N − 1)]1/2Σj|Ij(hkl) − 〈I(hkl)〉|/Σhkl Σj Ij(hkl), where N is the redundancy of the dataset.
d CC1/2 is the correlation coefficient of the half-datasets.
e Rwork = Σhkl‖Fobs| − |Fcalc‖/Σhkl|Fobs|, where Fobs and Fcalc are the observed and the calculated structure factor, respectively. Rfree is the cross-validation R factor for the test set of reflections (10% of the total) omitted in model refinement.
Author contributions
B. D., M. F., O. R., J. H., P. H., and P. S. conceptualization; B. D., M. F., and J. H. data curation; B. D., M. F., O. R., J. H., R. H., P. H., and P. S. formal analysis; B. D., M. F., J. H., R. H., P. H., and P. S. validation; B. D. and M. F. investigation; B. D., M. F., J. H., and R. H. visualization; B. D. and M. F. methodology; B. D. writing-original draft; B. D., M. F., O. R., J. H., R. H., P. H., and P. S. writing-review and editing; J. H. software; J. H., R. H., P. H., and P. S. supervision; J. H., R. H., and P. H. funding acquisition; R. H. and P. H. project administration.
Acknowledgment
We thank Raoul Rozenberg (Université Catholique de Louvain) for the MS analysis of small molecules.
This work was supported by National Science Foundation Grant CHE-1516126 (to R. P. H. and J. H.) and by Fond National de la Recherche Scientifique and Belgian French Community Actions de Recherche Concertées Programme (to P. S. and P. H.). The authors declare that they have no conflicts of interest with the contents of this article.
The atomic coordinates and structure factors (codes 6BWO, 6BWQ, and 6BWR) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- NPN
- nickel-pincer nucleotide
- P2CMN
- pyridinium-3,5-biscarboxylic acid mononucleotide
- P2TMN
- into pyridinium-3,5-bisthiocarboxylic acid
- NaAD
- nicotinic acid adenine dinucleotide
- PRF
- programmed ribosomal frameshift
- BME
- β-mercaptoethanol
- FeGP
- iron-guanylylpyridinol
- PDB
- Protein Data Bank
- r.m.s.d.
- root mean square deviation
- PAR
- 4-(2-pyridylazo)resorcinol.
References
- 1. Boer J. L., Mulrooney S. B., and Hausinger R. P. (2014) Nickel-dependent metalloenzymes. Arch. Biochem. Biophys. 544, 142–152 10.1016/j.abb.2013.09.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Desguin B., Zhang T., Soumillion P., Hols P., Hu J., and Hausinger R. P. (2015) A tethered niacin-derived pincer complex with a nickel-carbon bond in lactate racemase. Science 349, 66–69 10.1126/science.aab2272 [DOI] [PubMed] [Google Scholar]
- 3. Desguin B., Goffin P., Viaene E., Kleerebezem M., Martin-Diaconescu V., Maroney M. J., Declercq J. P., Soumillion P., and Hols P. (2014) Lactate racemase is a nickel-dependent enzyme activated by a widespread maturation system. Nat. Commun. 5, 3615 10.1038/ncomms4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Desguin B., Soumillion P., Hols P., and Hausinger R. P. (2016) Nickel-pincer cofactor biosynthesis involves LarB-catalyzed pyridinium carboxylation and LarE-dependent sacrificial sulfur insertion. Proc. Natl. Acad. Sci. U.S.A. 113, 5598–5603 10.1073/pnas.1600486113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fellner M., Desguin B., Hausinger R. P., and Hu J. (2017) Structural insights into the catalytic mechanism of a sacrificial sulfur insertase of the N-type ATP pyrophosphatase family, LarE. Proc. Natl. Acad. Sci. U.S.A. 114, 9074–9079 10.1073/pnas.1704967114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Albrecht M. (2010) Cyclometalation using d-block transition metals: fundamental aspects and recent trends. Chem. Rev. 110, 576–623 10.1021/cr900279a [DOI] [PubMed] [Google Scholar]
- 7. Caliskan N., Peske F., and Rodnina M. V. (2015) Changed in translation: mRNA recoding by −1 programmed ribosomal frameshifting. Trends Biochem. Sci. 40, 265–274 10.1016/j.tibs.2015.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Archibald F. (1986) Manganese: its acquisition by and function in the lactic acid bacteria. Crit. Rev. Microbiol. 13, 63–109 10.3109/10408418609108735 [DOI] [PubMed] [Google Scholar]
- 9. Holm L., and Rosenström P. (2010) Dali server: conservation mapping in 3D. Nucleic Acids Res. 38, W545–W549 10.1093/nar/gkq366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fujishiro T., Ermler U., and Shima S. (2014) A possible iron delivery function of the dinuclear iron center of HcgD in [Fe]-hydrogenase cofactor biosynthesis. FEBS Lett. 588, 2789–2793 10.1016/j.febslet.2014.05.059 [DOI] [PubMed] [Google Scholar]
- 11. Fujishiro T., Kahnt J., Ermler U., and Shima S. (2015) Protein-pyridinol thioester precursor for biosynthesis of the organometallic acyl-iron ligand in [Fe]-hydrogenase cofactor. Nat. Commun. 6, 6895 10.1038/ncomms7895 [DOI] [PubMed] [Google Scholar]
- 12. Sigel R. K., and Sigel H. (2007) Nickel and Its Surprising Impact in Nature, pp. 109–180, John Wiley & Sons, Ltd., Chichester, UK [Google Scholar]
- 13. Lecerof D., Fodje M. N., Alvarez León R., Olsson U., Hansson A., Sigfridsson E., Ryde U., Hansson M., and Al-Karadaghi S. (2003) Metal binding to Bacillus subtilis ferrochelatase and interaction between metal sites. J. Biol. Inorg. Chem. 8, 452–458 10.1007/s00775-002-0436-1 [DOI] [PubMed] [Google Scholar]
- 14. Orzel̸ L., Fiedor L., Wolak M., Kania A., van Eldik R., and Stochel G. (2008) Interplay between acetate ions, peripheral groups, and reactivity of the core nitrogens in transmetalation of tetrapyrroles. Chemistry 14, 9419–9430 10.1002/chem.200800991 [DOI] [PubMed] [Google Scholar]
- 15. Zheng Z., Goncearenco A., and Berezovsky I. N. (2016) Nucleotide binding database NBDB–a collection of sequence motifs with specific protein-ligand interactions. Nucleic Acids Res. 44, D301–D307 10.1093/nar/gkv1124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kamberov E. S., Atkinson M. R., and Ninfa A. J. (1995) The Escherichia coli PII signal transduction protein is activated upon binding 2-ketoglutarate and ATP. J. Biol. Chem. 270, 17797–17807 10.1074/jbc.270.30.17797 [DOI] [PubMed] [Google Scholar]
- 17. Palanca C., and Rubio V. (2017) Effects of T-loop modification on the PII-signalling protein: structure of uridylylated Escherichia coli GlnB bound to ATP. Environ. Microbiol. Rep. 9, 290–299 10.1111/1758-2229.12533 [DOI] [PubMed] [Google Scholar]
- 18. Zhang G., Mills D. A., and Block D. E. (2009) Development of chemically defined media supporting high-cell-density growth of lactococci, enterococci, and streptococci. Appl. Environ. Microbiol. 75, 1080–1087 10.1128/AEM.01416-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sambrook J., Fritsch E., and Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 20. Holo H., and Nes I. F. (1989) High-frequency transformation, by electroporation, of Lactococcus lactis subsp. cremoris grown with glycine in osmotically stabilized media. Appl. Environ. Microbiol. 55, 3119–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dower W. J., Miller J. F., and Ragsdale C. W. (1988) High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 16, 6127–6145 10.1093/nar/16.13.6127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng L., Baumann U., and Reymond J. L. (2004) An efficient one-step site-directed and site-saturation mutagenesis protocol. Nucleic Acids Res. 32, e115 10.1093/nar/gnh110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmidt T. G., and Skerra A. (2007) The Strep-tag system for one-step purification and high-affinity detection or capturing of proteins. Nat. Protoc. 2, 1528–1535 10.1038/nprot.2007.209 [DOI] [PubMed] [Google Scholar]
- 24. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 10.1016/0003-2697(76)90527-3 [DOI] [PubMed] [Google Scholar]
- 25. McCall K. A., and Fierke C. A. (2000) Colorimetric and fluorimetric assays to quantitate micromolar concentrations of transition metals. Anal. Biochem. 284, 307–315 10.1006/abio.2000.4706 [DOI] [PubMed] [Google Scholar]
- 26. Sparta K. M., Krug M., Heinemann U., Mueller U., and Weiss M. S. (2016) Xdsapp2.0. J. Appl. Crystallogr. 49, 1085–1092 10.1107/S1600576716004416 [DOI] [Google Scholar]
- 27. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 10.1107/S0907444913000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr D. Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 31. Laskowski R. A., and Swindells M. B. (2011) LigPlot+: multiple ligand–protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 51, 2778–2786 10.1021/ci200227u [DOI] [PubMed] [Google Scholar]
- 32. Thompson J. D., Gibson T. J., and Higgins D. G. (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinformatics 2002, Chapter 2, Unit 2.3 10.1002/0471250953.bi0203s00 [DOI] [PubMed] [Google Scholar]
- 33. de Ruyter P. G., Kuipers O. P., and de Vos W. M. (1996) Controlled gene expression systems for Lactococcus lactis with the food-grade inducer nisin. Appl. Environ. Microbiol. 62, 3662–3667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuipers O. P., de Ruyter P. G., Kleerebezem M., and de Vos W. M. (1998) Quorum sensing-controlled gene expression in lactic acid bacteria. J. Biotechnol. 64, 15–21 10.1016/S0168-1656(98)00100-X [DOI] [Google Scholar]