

Abstract
Mesenchymal stem cells (MSCs) are currently being investigated as candidate cells for regenerative medicine approaches for the repair of damaged articular cartilage. For these cells to be used clinically, it is important to understand how they will react to the complex loading environment of a joint in vivo. In addition to investigating alternative cell sources, it is also important for the structure of tissue‐engineered constructs and the organization of cells within them to be developed and, if possible, improved. A custom built bioreactor was used to expose human MSCs to a combination of shear and compression loading. The MSCs were either evenly distributed throughout fibrin‐poly(ester‐urethane) scaffolds or asymmetrically seeded with a small proportion seeded on the surface of the scaffold. The effect of cell distribution on the production and deposition of cartilage‐like matrix in response to mechanical load mimicking in vivo joint loading was then investigated. The results show that asymmetrically seeding the scaffold led to markedly improved tissue development based on histologically detectable matrix deposition. Consideration of cell location, therefore, is an important aspect in the development of regenerative medicine approaches for cartilage repair. This is particularly relevant when considering the natural biomechanical environment of the joint in vivo and patient rehabilitation protocols. © 2016 The Authors Journal of Tissue Engineering and Regenerative Medicine Published by John Wiley & Sons Ltd.
Keywords: cartilage repair, bioreactor, mesenchymal stem cell, shear, poly‐ester‐urethane, multi‐axial load
1. Introduction
Cell based therapies, based on the use of autologous or donor cells, often but not exclusively in combination with biomaterials are being developed and used clinically (Shin'oka et al., 2001; Cherubino et al., 2003; Atala et al., 2006; Raya‐Rivera et al., 2011). One of the areas in which they have had an impact is the repair of articular cartilage through autologous chondrocyte implantation (ACI) (Brittberg et al., 1994).
However, the use of autologous chondrocytes leads to problems such as donor site morbidity, dedifferentiation during in vitro expansion and the production of fibro‐cartilaginous repair tissue, as well as the need for multiple operations (Holtzer et al., 1960; Benya and Shaffer, 1982; LaPrade et al., 2008; Matricali et al., 2010). This has led to interest in using mesenchymal stem cells (MSCs) as an alternative for chondrocytes in cell based articular cartilage repair (Gardner et al., 2013; Musumeci et al., 2011, 2014). Mesenchymal stem cells have been harvested from a number of tissues, including adipose tissue and bone marrow aspirate, and have been shown to undergo osteogenic, adipogenic and chondrogenic differentiation (Johnstone et al., 1998; Pittenger et al., 1999; Zuk et al., 2002; Musumeci et al., 2011, 2014). The ability to utilize bone marrow MSCs for cartilage repair would allow the collection of cells through an aspiration, rather than the initial harvesting operation required for ACI, as well as decreasing the problems associated with donor site morbidity after cartilage harvesting. If MSCs are to progress towards a clinical application a better understanding of how these cells might behave in vivo is required (Johnstone et al., 2013).
The standard method of inducing chondrogenic differentiation of MSCs in vitro is to culture the cells in a three‐dimensional environment and in the presence of transforming growth factor beta (TGF‐β) (Johnstone et al., 1998) but not in the presence of mechanical stimulation, a key influence within the joint environment. Previous work has shown that MSCs can be directed towards chondrogenic differentiation and matrix deposition in the absence of any growth factors through exposure to a combination of shear and compression load that broadly mimics the loading implants would receive within a joint. Li et al. (2010a) showed that the application of multi‐axial load leads to the endogenous production of TGF‐β1 by MSCs within the loaded scaffolds, which then drives the deposition of cartilage‐like matrix. Subsequent work applied the two components of the load (compression and shear), both individually and, in combination, and showed that the shear component of the load was critical for the induction of chondrogenesis (Schatti et al., 2011). The most common clinical solution for cartilage repair is currently microfracture, where bone marrow is released into the defect by producing holes in the subchondral bone (Steadman et al., 2001). This results in progenitor cells contained within a fibrin clot , a situation broadly mimicked using the culture model system described above. Therefore, being able to study the behaviour of these cells in a mechanical environment that mimics the one they might naturally encounter allows for a greater understanding of how these cells might act within a scaffold or defect in vivo.
The present study further investigates the response of MSCs to physiologically relevant multi‐axial mechanical load that mimics the mechanical environment of an articulating joint. The aim was to investigate whether induction of chondrogenesis and the deposition of cartilage‐like matrix by MSCs in response to multi‐axial load could be improved by asymmetrically seeding scaffolds with a small proportion of the total number of cells seeded onto the scaffold's loaded surface. Rather than take a classical tissue‐engineering approach, whereby stimuli and materials are chosen purely on the basis of maximum matrix production, the present study has taken a more regenerative medicine approach. With this in mind human cells were embedded in a clinically approved material (fibrin) and the only stimulus applied is mechanical, which is possible clinically by way of the applied rehabilitation protocol after surgery. The model used, human progenitor cells embedded in a fibrin gel, would then also offer potential insights into the role of rehabilitation protocols on graft maturation after microfracture for cartilage repair. The MSCs were seeded onto the loaded surface in order to directly expose them to the shear component of the load, which has previously been shown to be important for the induction of MSC chondrogenesis as a result of multi‐axial mechanical loading.
2. Materials and methods
2.1. Preparation of poly(ester‐urethane) scaffolds
The poly(ester‐urethane) was prepared following a one‐step solution poly‐condensation and fabricated into a porous structure via a salt leaching‐phase inverse technique as described previously (Boissard et al., 2009). The poly(ester‐urethane) porous sponge was water‐jet cut (CUTEC AG, Basel, Switzerland) into cylindrical scaffolds 8 mm diameter and 4 mm high, with a porosity above 80% and interconnected macropores ranging from 90 μm to 300 μm. The scaffolds were then sterilized with ethylene oxide in a cold cycle (37°C) and degassed under vacuum for 5 days.
2.2. Isolation of MSCs
Mesenchymal stem cells were isolated from bone marrow aspirates collected during routine operations with full‐ethical approval (KEK‐ZH‐NR: 2010–0444/0). The MSCs were from four different marrow aspirates from vertebral bodies (two female aged 18 years and 49 years, two male aged 22 years and 76 years) and one from tibial plateau (males aged 48 years). Mononuclear cells were isolated from whole bone marrow of each donor using Ficoll density separation (Sigma‐Aldrich, Buchs, Switzerland). Isolated mononuclear cells were then seeded at a density of 50 000 cells/cm2 and left to attach for 96 h in alpha minimum essential medium (αMEM) (Gibco, Carlsbad, CA, USA), 10% MSC tested fetal bovine serum (FBS) (Pan Biotech, Aidenbach, Germany), 5 ng/ml basic fibroblast growth factor (bFGF) (Peprotech, Rocky Hill, CN, USA) and 1% penicillin/streptomycin (Gibco). MSCs isolated from vertebral bodies were used for the main body of experiments, while MSCs isolated from the tibial plateau were used for cell tracking experiments.
2.3. Cell culture, cell membrane labelling and scaffold seeding
Proliferation of MSCs was carried out using αMEM 10% FBS with 5 ng/ml bFGF. Passage 3 cells were seeded into 8 × 4 mm fibrin–poly(ester‐urethane) scaffolds (fibrin from Baxter, Vienna, Austria) (Li et al., 2009). The number and position of cells seeded into each scaffold varied by group (Figure 1). One group of scaffolds contained 4 million cells seeded throughout the scaffold (Uniform). A second group of scaffolds were seeded asymmetrically with 3.6 million cells distributed within the scaffold, and 400 000 cells were allowed to adhere to the loaded surface of the scaffold (Asymmetric). For the final set of scaffolds (Surface‐Only), the scaffolds were filled with fibrin alone, 400 000 cells were then allowed to adhere to the upper face of the scaffolds (Figure 1).
Figure 1.
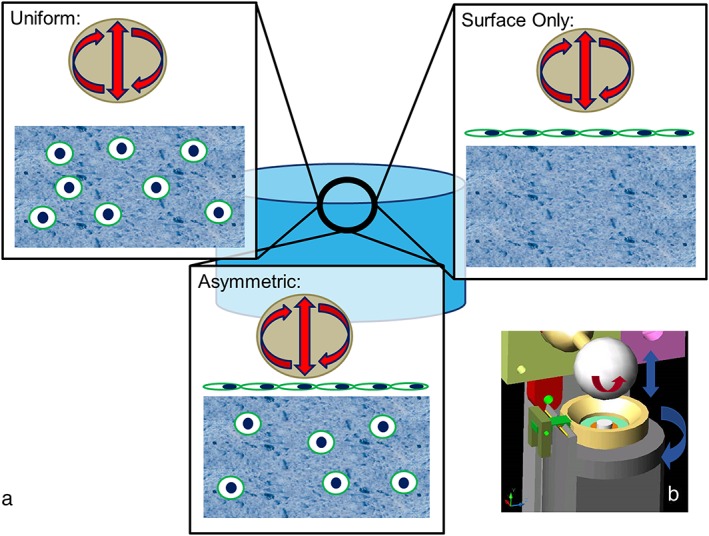
(a) Schematic showing the different seeding patterns used during this work. In Uniform 4 million cells were evenly seeded throughout scaffolds. In Asymmetrically seeded scaffolds, 3.6 million cells were seeded evenly throughout the scaffold and 400 000 cells seeded on the loaded surface of the scaffold. Surface Only scaffolds were not seeded with cells within the scaffold but only with 400 000 cells on the loaded surface. (b) Schematic showing the multi‐axial loading device used in the present study. The hip ball is lowered onto the white disc representing the scaffold. Compression is generated by raising and lowering the ball, and sliding friction is generated by rotating the ball, allowing for a sliding motion over the scaffold. [Colour figure can be viewed at http://wileyonlinelibrary.com]
The scaffold seeding procedure was originally described by Li et al. (2009). In order to seed cells within scaffolds the required number of culture expanded MSCs were first resuspended in 33 mg/ml fibrinogen and transferred to the sterilized cap of an Eppendorf tube. The fibrin cell mixture was then rapidly mixed with an equal volume of 1 unit/ml thrombin. A poly(ester‐urethane) scaffold was then firmly pressed into the Eppendorf lid using a pair of sterile forceps; removing the pressure from the scaffold allowed the poly(ester‐urethane) sponge to regain its original shape and in doing so draw the fibrin cell mixture into the scaffold. The scaffolds were then incubated at 37°C for 1 h to allow polymerization of the fibrin hydrogel. This method has been used previously in a number of studies by this group and results in a homogeneous distribution of cells throughout the scaffold (Li et al., 2009, 2010a; Schatti et al., 2011; Neumann et al., 2013).
To seed MSCs on the scaffold surface 400 000 cells were resuspended in 100 μl of chondropermissive medium. This suspension was then dripped on top of the fully polymerized fibrin poly(ester‐urethane) scaffold and left for 20 min for the cells to adhere. Scaffolds were then transferred to polyether ether ketone holders for the remainder of the culture period.
Six scaffolds were seeded with a mixture of unlabelled cells and cells labelled with the fluorescent cell membrane dye PKH26. The role of the fluorescent label was to establish the morphology and location of MSCs in both free‐swelling and loaded scaffolds after 4 weeks in culture. The MSCs were labelled with PKH26 according to the manufacture's protocol (Sigma‐Aldrich). Two group one scaffolds were seeded with 4 million labelled cells that were evenly distributed throughout the scaffold. Asymmetric scaffolds were seeded with 3.6 million unstained cells inside the scaffold and 400 000 labelled cells on top of the scaffold. Two final scaffolds were seeded as Surface‐Only seeded scaffolds with 400 000 labelled cells on top of otherwise acellular fibrin‐filled scaffolds. Scaffolds were then either mechanically loaded or kept in free‐swelling culture.
Once seeded, the scaffolds were cultured in chondropermissive medium consisting of; Dulbecco's modified Eagle medium 4.5g/l glucose (Gibco), sodium pyruvate 0.11 g/l (Sigma‐Aldrich), l‐ascorbic acid 2‐phosphate sesquimagnesium salt hydrate 50 μg/ml (Sigma‐Aldrich), dexamethasone 1 × 10–7 m (Sigma‐Aldrich), insulin transferrin and selenium 1% (Cyangen, Guangzhou, China), non‐essential amino acids 1% (Gibco), Primocin 0.1% (Invitrogen, San Diego, CA, USA) and 6‐aminocaproic acid 5 μm (Sigma‐Aldrich) to reduce fibrin degradation (Kupcsik et al., 2009). The media were changed three times a week, collected and pooled for analysis.
2.4. Mechanical loading
Control scaffolds were kept in free‐swelling culture for the entire culture period (4 weeks). Designated scaffolds were loaded using a custom‐made multi‐axial load bioreactor based on tribological principals (Wimmer et al., 2004). Loaded scaffolds were exposed to 20 cycles of 10% compression superimposed on top of a 10% pre‐strain and shear loading (± 25°) at 1Hz for 1 h a day, five times a week. The application of multi‐axial mechanical load to fibrin–poly(ester‐urethane) scaffolds in this system has been described previously (Zahedmanesh et al., 2014).
2.5. Sample collection
After 4 weeks of culture, the eight scaffolds from each group were harvested for analyses. Samples were homogenized in TRI reagent (Molecular Research Centre Inc., Cincinnati, OH, USA) for RNA isolation, digested in 0.5 mg/ml proteinase K at 56°C for 16 h (Roche, Basel, Switzerland) for biochemical analysis or fixed in an minimum of 20× the volume of the scaffold in 70% methanol for a minimum of 48 h (Brenntag, Mülheim Germany) for histological processing.
2.6. Glycosaminoglycan and DNA quantification
1,9‐Dimethyl methylene blue (DMMB) Sigma‐Aldrich, Buchs, Switzerland) was used to determine the amount of glycosaminoglycan (GAG) retained within scaffolds from proteinase K‐digested samples, and the amount that was released from the scaffolds into the collected culture media (Farndale et al., 1986). This method uses a standard curve of chondroitin 4‐sulfate sodium salt from bovine trachea (Fluka, St Louis, MO, USA). The standard concentrations used were; 0.01, 0.005, 0.0025, 0.00125, 0.000625 and 0.0003125 mg/ml; 50 μl of each standard was used. The standards were made up in chondropermissive media and a blank of chondropermissive media was used. Into each of the wells containing standard and sample was added 200 μl DMMB (Sigma‐Aldrich). The absorbance of the wells at 535 nm was then read immediately using the Victor 3 Micro Plate Reader (Perkin Elmer, Waltham, MA, USA).
The quantification of GAG in each proteinase K sample was identical to that used for media samples, except that 20 μl of sample and standard were used not 50 μl. The blank used was phosphate‐buffered saline (PBS) and standards (0.0113, 0.00568, 0.00284, 0.00142, 0.00071 and 0.000355 mg/ml) were prepared using PBS.
The DNA content of scaffolds was quantified using Höchst 33258 dye (Polysciences Inc., Warrington, PA, USA) (Labarca and Paigen, 1980). The standard curve was made using calf thymus DNA (Lubio Science, Luzern, Switzerland) in PBS, the concentrations used were: 10, 5, 2.5, 1.25, 0.625 and 0.3125 μg/ml; 40 μl of blank as well as of each standard and sample was pipetted into a white 96‐well plate and 160 μl of 0.01% Höchst dye solution (Polysciences Inc.) added, the plate was then wrapped in aluminium foil and incubated at room temperature for 20 min. Measurements were then made at 360 nm excitation and 465 nm emission using the Victor 3 Micro Plate Reader (Perkin Elmer).
2.7. Histology and immunohistochemistry
Methanol‐fixed samples were frozen in OCT compound (R. Jung GmbH, Nussloch, Germany) before being sectioned (12 μm thick) on a cryotome (Carl Zeiss AG, Oberkochen, Germany). Following sectioning, samples were stained with Safranin O (Chroma, Münster, Germany), counterstained with Weigert's haematoxylin (Merck, Whitehouse Station, NJ, USA) and fast green (Fluka).
The presence of collagen types I, II and X in sections was determined using the following primary antibodies: COL‐1 (C2456; Sigma‐Aldrich), CIICI (Developmental Studies Hybridoma Bank, University of Iowa, Iowa City, IA, USA) and COL‐10 (C7974; Sigma‐Aldrich). For all antibodies, except COL‐10, a biotinylated horse anti‐mouse secondary antibody was then used to detect the primary antibody (Vector Laboratories, Burlingame, CA, USA). For COL‐10, samples were incubated with a biotinylated goat anti‐mouse IgM secondary antibody (Vector Laboratories, Burlingame, USA).
Slides were brought to room temperature and washed in distilled water for 10 min before native peroxidase activity was blocked with 0.3% H2O2 (Fluka) in methanol (Brenntag) for 30 min. Slides were then air dried and washed twice for 5 min in 0.1% Tween‐20 PBS. Slides were incubated in 0.05–0.5 units/ml hyaluronidase (Sigma‐Aldrich) in 0.1% Tween 20 PBS at 37°C for 30 min before being washed three times for 5 min in 0.1% Tween‐20 PBS. Sections were then blocked with an appropriate serum for 1 h at room temperature. For COL‐1 and CIICI this required horse serum diluted 1:20 in 0.1% Tween‐20 PBS; COL‐10 required goat serum diluted 1:20 in 0.1% Tween‐20 PBS (Vector Laboratories). Following the block step, the serum was removed without washing and immediately replaced with the primary antibody. The sections were incubated with the primary antibodies for 30 min at room temperature. The following dilutions with 0.1% Tween‐20 were used for the antibodies: COL‐1 1:2000, CIICI 1:6, 5C6 1:5, C7974 1:2000, MAB240 1:50. Negative controls were incubated with 0.1% Tween‐20 PBS. Slides were then washed three times for 5 min with 0.1% Tween‐20 PBS, before being incubated with the biotinylated anti‐mouse IgG secondary antibody diluted 1:200 in 0.1% Tween‐20 PBS except for C7974, which required an anti‐mouse IgM diluted 1:200. Following this incubation, samples were washed again in 0.1% Tween‐20 and then incubated with ABC solution (Vector Laboratories) for 30 min at room temperature, washed again and then incubated with ImmPACT DAB (Vector Laboratories, Burlingame) for 4 min before being place into distilled water. Samples were the counterstained with Mayer's haematoxylin (Sigma‐Aldrich) for 20 s and blued in tap water for 5 min. Samples were dehydrated in 50%, 70%, 96%, 100%, 100% ethanol before being cleared in xylene and mounted with Eukitt (Sigma‐Aldrich).
2.8. Reverse transcription and real‐time polymerase chain reaction (PCR)
RNA was isolated from samples in TRI reagent (Molecular Research Centre Inc.) as per the manufacturer's instructions.
Reverse transcription was then performed using random hexamer primers and TaqMan reverse transcription reagents (Applied Biosystems, Carlsbad, CA, USA).
Real‐time PCR was performed using the applied biosciences StepOnePlus real‐time PCR system (Applied Biosystems). Primers for collagen type I, collagen type II, collagen type X and aggrecan mRNA were synthesized by Microsynth AG (Balgach, Switzerland), (see the Supplementary material online, Table [Link]). Primers for Sox9 and 18s were purchased from Applied Biosystems. The level of gene expression for each gene was determined relative to day 0 control 18s rRNA via a ΔΔCT comparison.
2.9. Statistical analysis
The data presented represents data from three (real‐time PCR) or four individual experiments (GAG/DNA and TGF‐β1 analysis) each carried out with with MSCs from a different donor. Experiments were performed in quadruplicate (real‐time PCR and TGF‐β1 analysis) or triplicate (GAG/DNA). Statistical analysis was performed GraphPad Prism 6 software (GraphPad Software Inc., La Jolla, CA, USA). Two‐factor analysis of variance (ANOVA) and Bonferroni's test were used on log‐transformed data for comparison between more than two groups. A P‐value of less than 0.05 was considered statistically significant.
3. Results
3.1. Fluorescent membrane labelling
Fluorescent cell labelling was used to detect the location of seeded cells after 4 weeks of culture (Figure 2). In Uniform scaffolds, where all cells seeded within the scaffold were labelled, the labelled MSCs were present around the four edges of the scaffold but not within the centre of the scaffold (Figure 2). Previous work in this model system has shown homogeneous seeding of scaffolds immediately after seeding and at day 7 of culture (Zahedmanesh et al., 2014); therefore, the loss of cells from the centre of the scaffolds appears to occur as a result of longer periods of culture.
Figure 2.
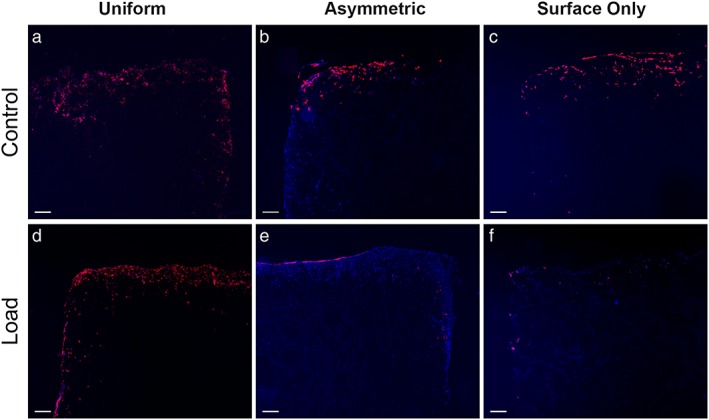
Fluorescent images showing the location of labelled cells after 4 weeks of load. Images (a), (b) and (c) show control scaffolds from Uniform, Asymmetric and Surface Only seeded scaffolds respectively, while images (d), (e) and (f) show loaded scaffolds. Red fluorescence shows the membranes of labelled cells, blue shows nuclei counterstained with 4′,6‐diamidino‐2‐phenylindole (DAPI) (n = 1). Bar, 200 μm. [Colour figure can be viewed at http://wileyonlinelibrary.com]
In Asymmetric and Surface‐Only seeded scaffolds, the cells seeded on to the scaffold surface were labelled whereas cells seeded within the scaffold itself were not. In these two groups, the labelled cells clearly remain on the upper surface of the scaffold after 28 days of culture. In Asymmetric and Surface‐Only control scaffolds, labelled cells were distributed throughout the surface layers of the scaffolds. However, in Asymmetric loaded scaffolds, the distribution of labelled cells was restricted to the uppermost layer of the scaffold surface with few detectable cells visible below this region. This was not replicated in Surface‐Only loaded conditions. The change in cell distribution between Asymmetric control and loaded scaffolds suggests that the application of multi‐axial load modifies the distribution of cells seeded on the scaffold surface in this model.
3.2. Safranin O staining
In order to show the deposition of sulphated GAGs, scaffolds were stained with Safranin O (Figure 3a–f). Positive Safranin O staining was present along the upper surface of Uniform loaded scaffolds and stronger positive staining was present in the same region of Asymmetric loaded scaffolds. No staining was observed in Uniform or Asymmetric control scaffolds or any of the Surface‐Only scaffolds.
Figure 3.
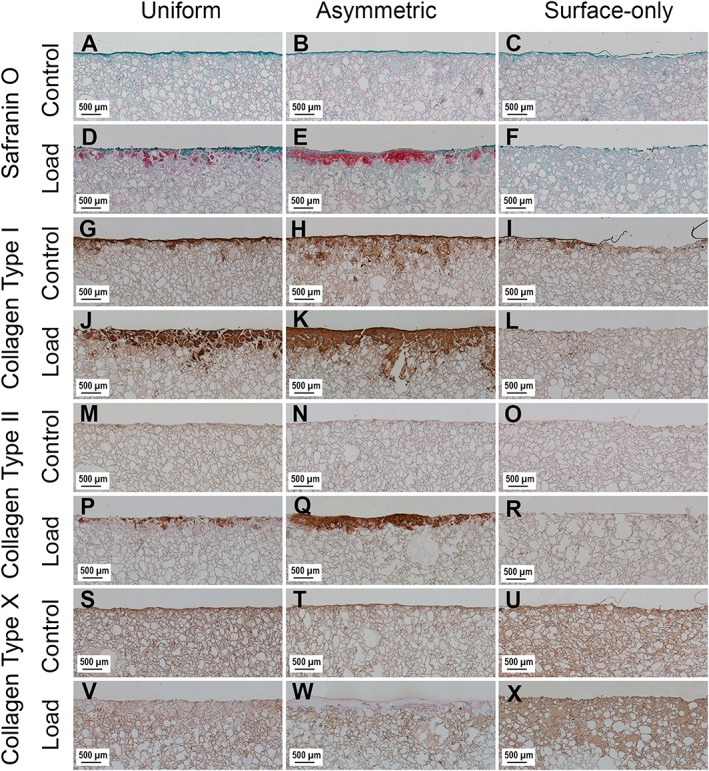
Images showing the surface of scaffolds stained with Safranin O and immunohistochemical labelling for collagen type I, collagen type II and collagen type X after 4 weeks of culture. Images (a), (b) and (c) show Safranin O‐stained control scaffolds from Uniform, Asymmetric and Surface Only seeded scaffolds, respectively, while images (d), (e) and (f) show Safranin O‐stained loaded scaffolds. Images (g), (h) and (i) show control scaffolds labelled for collagen type I, and images (j) (k) and (l) show loaded scaffolds. Images (m), (n) and (o) show control scaffolds labelled for collagen type II and images (p), (q) and (r) show loaded scaffolds. Images (s), (t) and (u) show control scaffolds labelled for collagen type X and images (v), (w) and (x) show loaded scaffolds. [Colour figure can be viewed at http://wileyonlinelibrary.com]
3.3. Immunohistochemistry
Immunohistochemistry showed that collagen type II was in found only in Uniform and Asymmetric loaded scaffolds (Figure 3m–r). The labelling was stronger in Asymmetric loaded scaffolds compared to Uniform loaded scaffolds, matching the pattern of staining seen with Safranin O.
Collagen type I was deposited around the edges of all scaffolds in a pattern that matched the distribution of cells within the scaffolds. In both Uniform and Asymmetric there was an increase in collagen type I deposition in response to mechanical load; however, in Surface‐Only, scaffolds the deposition was greater in control scaffolds (Figure 3g–l).
Non‐specific staining by the antibody used for collagen type X immunohistochemistry of the fibrin component of the scaffolds led to high levels of background labelling, thus interpretation requires caution (Figure 3s–x). However, areas in Uniform and Asymmetric loaded scaffolds that stained positively for sulphated GAG and collagen type II do not stain for collagen type X.
3.4. Glycosaminoglycan and DNA quantification
After 4 weeks in culture the DNA content of all Uniform and Asymmetrical scaffolds remained higher than Surface Only scaffolds (P < 0.05) (Figure 4).
Figure 4.
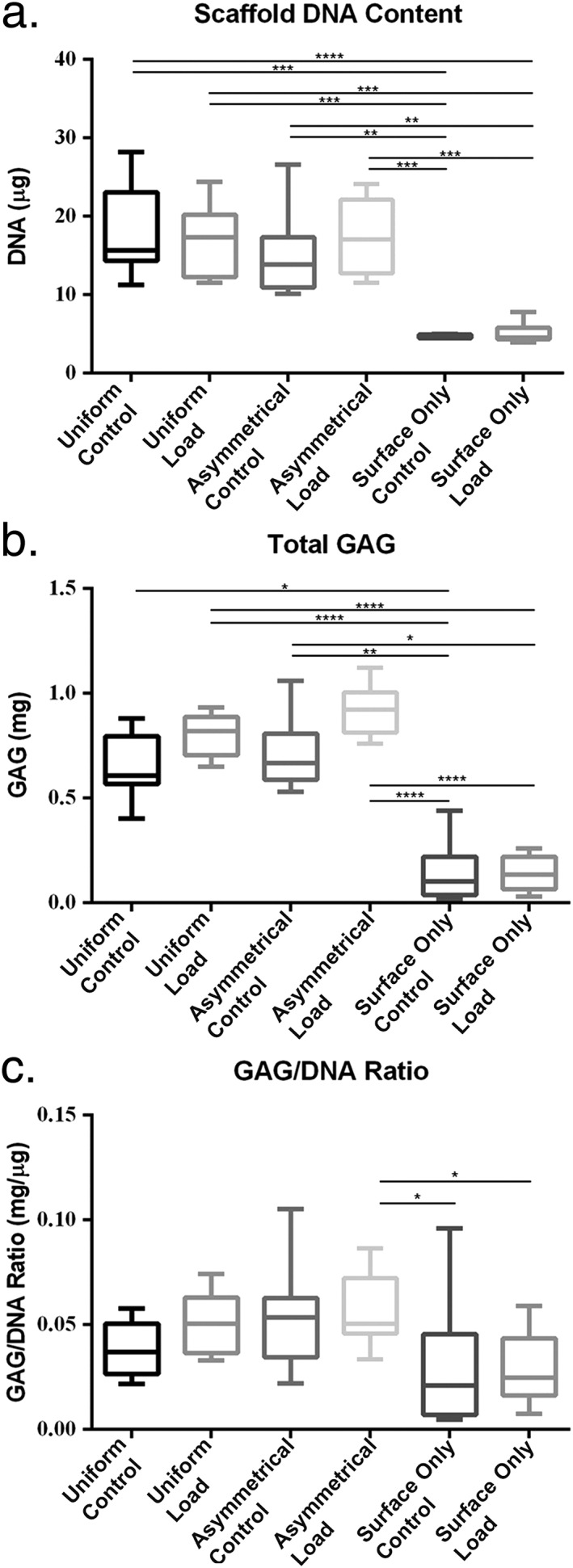
Biochemical analysis of scaffolds after 4 weeks in culture. (a) Höchst 33528 dye was used to quantify the DNA in proteinase K digests of scaffolds. (b) Dimethylmethylene blue (DMMB) was used to determine the total amount of sulphated glycosaminoglycan (GAG) produced by mesenchymal stem cells (MSCs) from both the collected culture media and proteinase K scaffold digests. (c) The GAG/DNA ratio was calculated from total DNA and GAG values to show the production of GAG relative to the MSCs present in each group. Error bars represent standard deviation. Statistical significance was defined as P ≤ 0.05; *P ≤ 0.05, **P ≤ 0.001 and ***P ≤ 0.0001
In Uniform and Asymmetric seeding there was a trend towards increased total GAG in response to mechanical load (Figure 4), although this did not reach significance. There was a significant difference between the total GAG production of control and loaded scaffolds in Uniform and Asymmetric scaffolds compared with Surface‐Only scaffolds (P < 0.05). Calculation of the GAG/DNA ratio in each group trended towards an increased ratio in Uniform and Asymmetric loaded scaffolds but this was not significant (Uniform control mean ± SD, 0.03844 ± 0.01265 mg/μg; group 1 control 0.05084 ± 0.01421 mg/μg; Asymmetric control 0.05216 ± 0.02214 mg/μg; surface‐only control 0.05628 ± 0.01683 mg/μg) (Figure 4).
3.5. Real‐time PCR
Collagen type I expression trended towards an increase with load, however, there were no significant increases within groups when comparing load and control (Figure 5a).
Figure 5.
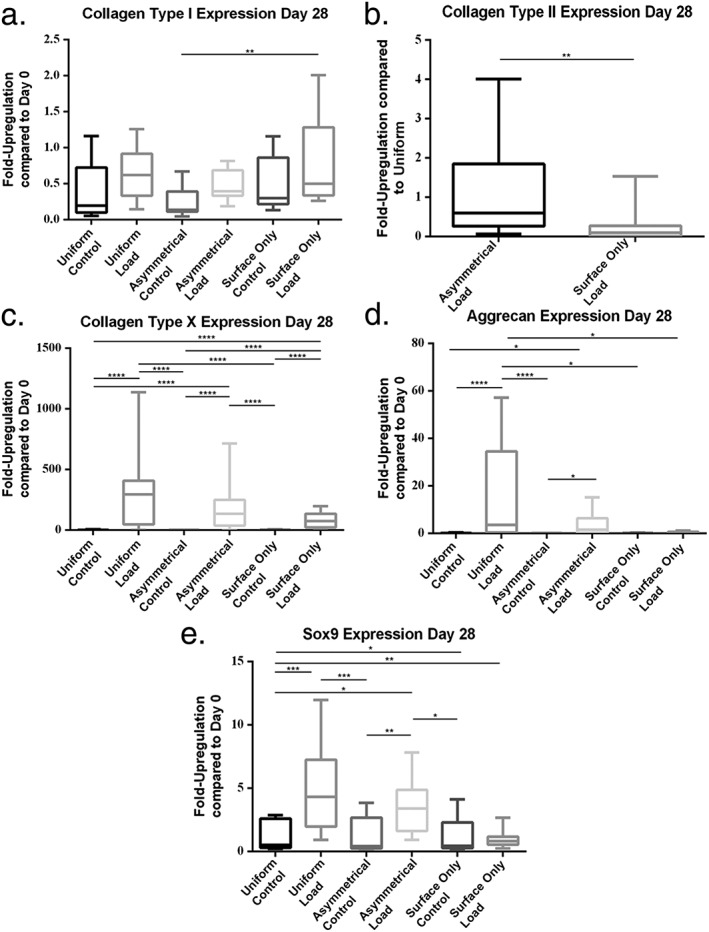
Gene expression measured at day 28 of culture by real‐time polymerase chain reaction (PCR). (a) Collagen type I, (b) collagen type II, (c) collagen type X, (d) aggrecan and (e) Sox9. Error bars represent standard deviation. Statistical significance was defined as P ≤ 0.05; *P ≤ 0.05, **P ≤ P≤0.001 and ***P ≤ 0.0001
Collagen type II expression was only detected in loaded groups. As a result, collagen type II expression was normalized to Uniform loaded scaffolds (Figure 5b). There was significantly greater expression in Asymmetrical than in Surface‐Only groups (P = 0.0041). The expression in group 2 scaffolds was similar to the Uniform group (1.163 ± 1.22) while in the Surface‐Only group it was lower than in the Uniform group (mean ± SD, 0.2460 ± 0.44).
Type X collagen expression significantly increased in all three loaded groups compared with all three control groups (P < 0.001). The expression of collagen type X in control groups was similar to day 0 (mean fold changes ± SD were 1.299 ± 1.90, 0.6267 ± 0.39 and 1.167 ± 0.72 in Uniform, Asymmetrical and Surface‐only groups, respectively), however, expression was much higher in loaded scaffolds (mean ± SD, 299.9 ± 331.0, 216.0 ± 247.4 and 82.00 ± 60.65, respectively) (Figure 5c).
Aggrecan expression was significantly increased in Uniform loaded scaffolds compared with all three control groups (Figure 5d). Aggrecan expression was also significantly increased by load when the cells were seeded asymmetrically. Expression was downregulated when compared with cells at day 0 in all groups except the Uniform and Asymmetrical groups subjected to load (mean ± SD,15.86 ± 22.06 and 3.960 ± 5.18, respectively).
Gene expression analysis of Sox9 shows that there was a significant increase in expression between Uniform and Asymmetrical load and all three control groups (P < 0.05) (Figure 5E). There was no statistically significant increase in response to load in Surface‐Only seeded scaffolds. The level of expression was similar to day 0 in all groups except in loaded Uniform and Asymmetrical scaffolds (mean ± SD, 5.053 ± 3.54 and 3.52 ± 2.06, respectively).
No significant differences were detected between Uniform and Asymmetric loaded scaffolds in any of the genes tested.
4. Discussion
Previous work has shown that when multi‐axial shear and compression loading is applied to MSCs seeded within fibrin–polyurethane scaffolds, the cells undergo chondrogenesis (Kupcsik et al., 2010; Li et al., 2010a, 2010b; Neumann et al., 2013). This is characterized by the upregulation of genes associated with cartilage matrix, such as collagen type II and aggrecan as well as the deposition of cartilage‐like matrix, containing collagen type II and sulphated GAGs, within the scaffolds themselves (Li et al., 2010a). This chondro‐induction was shown to be driven by endogenously produced TGF‐β1 and could be blocked using the TGF‐β receptor 1 (ALK5) inhibitor LY364947 (Li et al., 2010a). Further investigation showed that the shear component of the load was vital in the induction of chondrogenesis. The present work therefore set out to use the shear component of the load to improve the amount of matrix deposition and chondro‐induction in response to mechanical load. In order to do this, scaffolds were seeded asymmetrically with 10% of the total cell number seeded on the loaded surface of the scaffolds in order to directly expose them to the shear component of the load.
Results of histological staining and immunohistochemical labelling showed that asymmetrically seeding scaffolds with a layer of cells on the loaded surface reproducibly increased the amount of cartilage matrix that was deposited in response to load. Safranin O staining showed that there was a clear increase in the deposition of sulphated GAG in Asymmetric loaded scaffolds compared with Uniform loaded scaffolds, this was also true for collagen type II. The increased staining for GAG and collagen type II labelling was consistently seen using multiple donors. This would suggest that scaffolds being designed for articular cartilage repair should take cell distribution into account and incorporate a degree of anisotropy as a potential way to maximise matrix deposition. Collagen type X deposition also appears lower in areas of GAG and collagen type II positive matrix in Asymmetric compared with Uniform scaffolds; however, this interpretation requires caution because of the non‐specific staining of the fibrin component of the scaffold by the primary antibody.
Fluorescence labelling of cells showed that not only do the cells remain in the vicinity of the loaded surface of the scaffolds in Asymmetric and Surface‐Only seeded scaffolds over the time in culture, but also that the application of load directly affects the distribution of the cells seeded on the surface of the scaffold. The MSCs seeded on the surface of non‐loaded control scaffolds were found broadly distributed through the upper layers of the scaffolds. However, cells seeded on the surface of loaded scaffolds, particularly in Asymmetric seeded, remain highly localized to the scaffold surface. This population of cells, which is very close to the surface of the scaffold and to the source of mechanical load, is the key difference between Uniform and Asymmetric scaffolds and may explain the histological differences between these groups. The results collected from Surface‐Only seeded scaffolds show that little cartilage‐like matrix is deposited within the scaffolds or GAG released to the media. The gene expression in this group also shows an increase in the expression of type I and type X collagens without the concomitant rise in chondrogenic markers such as collagen type II and Sox9 seen in Uniform and Asymmetric scaffolds, suggesting a less chondrogenic and more hypertrophic phenotype for the cells in Surface‐Only seeded scaffolds. The results from Surface‐Only scaffolds indicate that a layer of cells seeded on the surface of a scaffold alone cannot lead to matrix deposition of the kind seen in Asymmetrically seeded scaffolds. The increases in matrix deposition in Asymmetric compared with Uniform seeded scaffolds are therefore likely to result from interaction between the population of cells that remain on the surface of the scaffold and the population of cells that were seeded within the scaffold itself.
Analysis of gene expression results showed that there was no significant difference in the expression of any of the genes tested between Uniform and Asymmetrically seeded samples, despite a trend towards greater aggrecan and Sox9 expression in Uniform seeded scaffolds. The results of biochemical analysis of scaffolds and media also showed no significant difference between the production of GAG in response to load between Uniform and Asymmetric scaffolds. Yet, the histological results presented here show that seeding a layer of cells on the surface of a scaffold improves the deposition of matrix in the area directly exposed to mechanical load. This may in part be due to the cells within the scaffolds, which do not respond to the chondrogenic stimulus provided by the mechanical loading (and therefore do not deposit sulphated GAG or type II collagen), diluting out the changes induced in cells the upper layers (which do respond to chondrogenic stimuli) when the scaffolds containing both sets of cells were prepared for real time PCR and biochemical analysis. Alternatively, structural changes in the matrix deposition may also play a role. Changes in crosslinking or matrix maturation caused by the load applied may also explain the differences seen in the histological data. However, further studies would be required to specifically investigate the effect of load on matrix maturation.
The lack of significant differences within GAG/DNA and real‐time PCR data may also result in part from the use of cells from two donors that responded strongly to chondrogenic induction (female 18 years old and male 22 years old) and two donors that responded weakly (female 49 years old, male 48 years old) leading to a reduction in the difference between groups when the results from the four donors were collated and a thus a greater standard deviation. However, more donors would be required to investigate this fully. While this deviation can be reduced by selecting or pooling donors, the frequency with which a result is observed cannot be determined (Stoddart et al., 2012). In addition, in a clinical setting, pooling is not an option.
This work has highlighted the importance of anisotropy within scaffolds for cartilage repair such as that previously described by Sheehy et al. (2013). Seeding a cell layer of MSCs on a three‐dimensional construct containing chondrocytes has also been shown to improve construct properties (Mesallati et al., 2015). This need for anisotropy is even more important in loaded environments; for example, in the loading system used in this work, because the distribution of multicomponent strain – critical in the induction of chondrogenesis – can vary throughout the scaffold (Zahedmanesh et al., 2014).
In summary, the asymmetric seeding of fibrin–polyurethane scaffolds with a small proportion of the total cell number on the surface leads to an increase in matrix deposition in response to multi‐axial mechanical load. This may be important during rehabilitation after microfracture, where progenitors are present within a fibrin clot. Anisotropic scaffold design may further enhance the chondrogenic response of MSCs to complex multi‐axial load in vivo. The limited production of chondrogenic markers in Surface‐Only scaffolds, suggests that the increased deposition of matrix in asymmetrically seeded scaffolds is likely to result from the interaction between the cells seeded on the surface of the scaffolds and those seeded within the scaffold itself. The exact nature of this interaction still needs to be elucidated. These data emphasize the need to establish histological outcome parameters and for chondrogenesis to be investigated using models incorporating a more naturally occurring loading environment. Tribological‐based regenerative medicine and implant testing can reveal differences than cannot be determined in static culture systems.
Conflict of interest
The authors have declared that there is no conflict of interest.
Supporting information
Table S1. Sequences of primers used for real‐time PCR analysis of collected samples.
Acknowledgements
This work was supported by the Acute Cartilage Injury consortium of the AO Foundation and the Swiss National Science Foundation (Grant no. 31003a_146375/1). This study was also supported by a grant from FIR 2014‐2016 (cod. 314509), University of Catania, Italy. We thank M. Glarner (Musculoskeletal Regeneration Programme, AO Research Institute Davos, Davos Platz) for producing the polyurethane scaffolds, Baxter Biosurgery, Vienna, for providing the fibrin components and Dr Jennifer Bara for helping prepare the manuscript. The CIICI antibody was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA, USA.
Gardner, O. F. W. , Musumeci, G. , Neumann, A. J. , Eglin, D. , Archer, C. W. , Alini, M. , and Stoddart, M. J. (2017) Asymmetrical seeding of MSCs into fibrin–poly(ester‐urethane) scaffolds and its effect on mechanically induced chondrogenesis. J Tissue Eng Regen Med, 11: 2912–2921. doi: 10.1002/term.2194.
The copyright line for this article was changed on 09 August 2018 after original online publication
References
- Atala A, Bauer SB, Soker S et al 2006; Tissue‐engineered autologous bladders for patients needing cystoplasty. Lancet 367: 1241–1246. [DOI] [PubMed] [Google Scholar]
- Benya PD, Shaffer JD. 1982; Dedifferentiated chondrocytes reexpress the differentiated collagen phenotype when cultured in agarose gels. Cell 30: 215–224. [DOI] [PubMed] [Google Scholar]
- Boissard I, Bourban PE, Tami AE et al 2009; Nanohydroxyapatite/poly(ester urethane) scaffold for bone tissue engineering. Acta Biomater 5: 3316–3327. [DOI] [PubMed] [Google Scholar]
- Brittberg M, Lindahl A, Nilsson A et al 1994; Treatment of deep cartilage defects in the knee with autologous chondrocyte transplantation. N Engl J Med 331: 889–895. [DOI] [PubMed] [Google Scholar]
- Cherubino P, Grassi FA, Bulgheroni P, et al 2003; Autologous chondrocyte implantation using a bilayer collagen membrane: a preliminary report. J Orthop Surg (Hong Kong) 11: 10–15. [DOI] [PubMed] [Google Scholar]
- Farndale RW, Buttle DJ, Barrett AJ et al 1986; Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta 883: 173–177. [DOI] [PubMed] [Google Scholar]
- Gardner OF, Archer CW, Alini M et al 2013; Chondrogenesis of mesenchymal stem cells for cartilage tissue engineering. Histol Histopathol 28: 23–42. [DOI] [PubMed] [Google Scholar]
- Holtzer H, Abbott J, Lash J, et al 1960; The loss of phenotypic traits by differentiated cells in vitro, I. Dedifferentiation of cartilage cells. Proc Natl Acad Sci U S A 46: 1533–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnstone B, Hering TM, Caplan AI et al 1998; In vitro chondrogenesis of bone marrow‐derived mesenchymal progenitor cells. Exp Cell Res 238: 265–272. [DOI] [PubMed] [Google Scholar]
- Johnstone B, Alini M, Cucchiarini M et al 2013; Tissue engineering for articular cartilage repair – the state of the art. Eur Cell Mater 25: 248–267. [DOI] [PubMed] [Google Scholar]
- Kupcsik L, Alini M, Stoddart MJ. 2009; Epsilon‐aminocaproic acid is a useful fibrin degradation inhibitor for cartilage tissue engineering. Tissue Eng Part A 15: 2309–2313. [DOI] [PubMed] [Google Scholar]
- Kupcsik L, Stoddart MJ, Li Z et al 2010; Improving chondrogenesis: potential and limitations of sox9 gene transfer and mechanical stimulation for cartilage tissue engineering. Tissue Eng Part A 16: 1845–1855. [DOI] [PubMed] [Google Scholar]
- Labarca C, Paigen K. 1980; A simple, rapid, and sensitive DNA assay procedure. Anal Biochem 102: 344–352. [DOI] [PubMed] [Google Scholar]
- Laprade RF, Bursch LS, Olson EJ et al 2008; Histologic and immunohistochemical characteristics of failed articular cartilage resurfacing procedures for osteochondritis of the knee: a case series. Am J Sports Med 36: 360–368. [DOI] [PubMed] [Google Scholar]
- Li Z, Kupcsik L, Yao SJ et al 2009; Chondrogenesis of human bone marrow mesenchymal stem cells in fibrin‐polyurethane composites. Tissue Eng Part A 15: 1729–1737. [DOI] [PubMed] [Google Scholar]
- Li Z, Kupcsik L, Yao SJ et al 2010a; Mechanical load modulates chondrogenesis of human mesenchymal stem cells through the tgf‐beta pathway. J Cell Mol Med 14: 1338–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Yao SJ, Alini M et al 2010b; Chondrogenesis of human bone marrow mesenchymal stem cells in fibrin‐polyurethane composites is modulated by frequency and amplitude of dynamic compression and shear stress. Tissue Eng Part A 16: 575–584. [DOI] [PubMed] [Google Scholar]
- Matricali GA, Dereymaeker GP, Luyten FP. 2010; Donor site morbidity after articular cartilage repair procedures: a review. Acta Orthop Belg 76: 669–674. [PubMed] [Google Scholar]
- Mesallati T, Buckley CT, Kelly DJ. 2015; Engineering cartilaginous grafts using chondrocyte‐laden hydrogels supported by a superficial layer of stem cells. J Tissue Eng Regen Med [DOI] [PubMed] [Google Scholar]
- Musumeci G, Lo Furno D, Loreto C et al 2011; Mesenchymal stem cells from adipose tissue which have been differentiated into chondrocytes in three‐dimensional culture express lubricin. Exp Biol Med (Maywood) 236: 1333–1341. [DOI] [PubMed] [Google Scholar]
- Musumeci G, Mobasheri A, Trovato FM et al 2014; Biosynthesis of collagen I, II, Runx2 and lubricin at different time points of chondrogenic differentiation in a 3D in vitro model of human mesenchymal stem cells derived from adipose tissue. Acta Histochem 116: 1407–1417. [DOI] [PubMed] [Google Scholar]
- Neumann AJ, Alini M, Archer CW et al 2013; Chondrogenesis of human bone marrow‐derived mesenchymal stem cells is modulated by complex mechanical stimulation and adenoviral‐mediated overexpression of bone morphogenetic protein 2. Tissue Eng Part A 19: 1285–1294. [DOI] [PubMed] [Google Scholar]
- Pittenger MF, Mackay AM, Beck SC et al 1999; Multilineage potential of adult human mesenchymal stem cells. Science 284: 143–147. [DOI] [PubMed] [Google Scholar]
- Raya‐Rivera A, Esquiliano DR, Yoo JJ et al 2011; Tissue‐engineered autologous urethras for patients who need reconstruction: an observational study. Lancet 377: 1175–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatti O, Grad S, Goldhahn J et al 2011; A combination of shear and dynamic compression leads to mechanically induced chondrogenesis of human mesenchymal stem cells. Eur Cell Mater 22: 214–225. [DOI] [PubMed] [Google Scholar]
- Sheehy EJ, Vinardell T, Buckley CT et al 2013; Engineering osteochondral constructs through spatial regulation of endochondral ossification. Acta Biomater 9: 5484–5492. [DOI] [PubMed] [Google Scholar]
- Shin'oka T, Imai Y, Ikada Y. 2001; Transplantation of a tissue‐engineered pulmonary artery. N Engl J Med 344: 532–533. [DOI] [PubMed] [Google Scholar]
- Steadman JR, Rodkey WG, Rodrigo JJ. 2001; Microfracture: surgical technique and rehabilitation to treat chondral defects. Clin Orthop Relat Res 301(391 Suppl): S362–S369. [DOI] [PubMed] [Google Scholar]
- Stoddart MJ, Richards RG, Alini M. 2012; In vitro experiments with primary mammalian cells: to pool or not to pool? Eur Cell Mater 24: i–ii. [DOI] [PubMed] [Google Scholar]
- Wimmer MA, Grad S, Kaup T et al 2004; Tribology approach to the engineering and study of articular cartilage. Tissue Eng 10: 1436–1445. [DOI] [PubMed] [Google Scholar]
- Zahedmanesh H, Stoddart M, Lezuo P et al 2014; Deciphering mechanical regulation of chondrogenesis in fibrin‐polyurethane composite scaffolds enriched with human mesenchymal stem cells: a dual computational and experimental approach. Tissue Eng Part A 20: 1197–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuk PA, Zhu M, Ashjian P, De Ugarte DA et al 2002. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell 13: 4279–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequences of primers used for real‐time PCR analysis of collected samples.