

Abstract
GPR3, GPR6 and GPR12 are three orphan receptors that belong to the Class A family of G protein-coupled receptors (GPCRs). These GPCRs share over 60% of sequence similarity among them. Because of their close phylogenetic relationship GPR3, GPR6 and GPR12 share a high percentage of homology with other lipid receptors such as the lysophospholipid and the cannabinoid receptors. On the basis of sequence similarities at key structural motifs, these orphan receptors have been related to the cannabinoid family. However, further experimental data is required to confirm this association.
GPR3, GPR6 and GPR12 are predominantly expressed in mammalian brain. Their high constitutive activation of adenylyl cyclase triggers increases in cAMP levels similar in amplitude to fully activated GPCRs. This feature defines their physiological role under certain pathological conditions.
In this review, we aim to summarize the knowledge attained so far on the understanding of these receptors. Expression patterns, pharmacology, physiopathological relevance, and molecules targeting GPR3, GPR6 and GPR12 will be analyzed herein. Interestingly, certain cannabinoid ligands have been reported to modulate these orphan receptors. The current debate about sphingolipids as putative endogenous ligands will also be addressed.
A special focus will be on their potential role in the brain, particularly under neurological conditions such as Parkinson or Alzheimer’s disease. Reported physiological roles outside the central nervous system will also be covered.
This critical overview may contribute to a further comprehension of the physiopathological role of these orphan GPCRs, hopefully attracting more research towards a future therapeutic exploitation of these promising targets.
Keywords: Orphan receptors, GPR3, GPR6, GPR12, Neurodegenerative diseases, Cannabinoids
Introduction
The molecular cloning of GPR3, GPR6 and GPR12 was reported over two decades ago (Saeki et al. 1993; Eggerickx et al. 1995; Song et al. 1995). The genes encoding these three receptors were located in the human chromosomal regions 1p36.1, 6q21, and 13q12, respectively. Sharing a high percentage of sequence homology among them, GPR3, GPR6 and GPR12 form a subgroup of orphan class A GPCRs (GPR3/6/12) characterized by their high constitutive activity (Eggerickx et al. 1995). Because of their phylogenetic relationship, GPR3/6/12 belongs to the so-called MECA cluster (Fredriksson et al. 2003; Fredriksson and Schio 2005). This group of receptors is formed by the melanocortin receptors (MCRs), the endothelial differentiation G-protein-coupled receptors (EDGR) [currently known as lysophospholipid receptors: sphingosine 1-phosphate (S1PR) and lysophosphatidic acid (LPAR) receptors], the cannabinoid receptors (CBR), the adenosine binding receptors (AR), and the GPR3/6/12 orphan subset (Figure 1). According to the phylogenetic analysis reported by Fredriksson and coworkers, GPR3 and GPR6 share chromosomal positions with the cannabinoid receptors, which suggests that they share a common ancestor (Fredriksson et al. 2003). As detailed in subsequent sections, these receptors share common conserved motifs and specific structural features.
Figure 1.

Phylogenetic tree representation of the MECA cluster. The orphan subset GPR3/6/12 is highlighted using the following color-coding: GPR3 in red; GPR6 in green; GPR12 in blue.
Molecules potently and selectively targeting these receptors remain to be discovered. Very few small molecules have been identified as modulators of these GPCRs. Endocannabinoids such as anandamide or 2-arachidonoylglycerol failed to modulate these orphan receptors (Yin et al. 2009; Laun and Song 2017) whereas cannabidiol (CBD) and other phytocannabinoids displayed activity at GPR3, GPR6 and/or GPR12 (Brown et al. 2017; Laun and Song 2017). Some sphingolipids have been proposed as putative endogenous ligands for GPR3, GPR6 and/or GPR12 (Uhlenbrock et al. 2002; Ignatov et al. 2003a; Ignatov et al. 2003b). However, due to the pharmacological inconsistencies reported among assays, the International Union of Basic and Clinical Pharmacology (IUPHAR) still categorizes these three receptors as orphans (Alexander et al. 2017).
Their close phylogenetic relationship with CB1 and CB2 along with the fact that they may share common ligands, led researchers to postulate a possible association between GPR3, GPR6 and GPR12 and the endocannabinoid system (Pertwee et al. 2010). Nonetheless, extensive studies need to explore this relation, as well as, possible interactions among these GPCRs under specific physiopathological conditions.
Within the past years, numerous studies have linked GPR3, GPR6 and GPR12 to several neurological processes, as well as other functions outside the brain such as oocyte maturation or metabolism. However, the lack of adequate pharmacological tools is delaying the elucidation of their biological relevance and their potential modulation in the treatment of diverse disorders.
The current review intends to provide an organized and critical summary of the literature reported on the characterization of these attractive but complex orphan GPCRs. This global perspective may also help analyzing their role and identifying novel approaches for further investigations.
General features: structure and expression
The orphan receptors GPR3, GPR6 and GPR12 are members of the rhodopsin-like class of GPCRs. These receptors, whose human sequences contain 330, 362, and 334 amino acids respectively, share about 60% of sequence identity among them. GPR3, GPR6 and GPR12 bear over 90% identity with their mouse and rat homologues. Sequence similarities at a transmembrane level with other GPCRs include over 45% homology with the lysophospholipid receptors LPA1, S1P1, and S1P5, and over 40% homology with the cannabinoid receptors CB1 and CB2.1 This high percentage of sequence homology with other lipid-sensing receptors of the MECA cluster leads them to share specific structural features and lack distinct motifs when compared to other receptor subfamilies. For instance, these receptors lack a helix kinking proline residues in the second and fifth transmembrane segments and have only one internal disulfide bridge in the second extracellular loop. GPR3, GPR6 and GPR12 also contain highly conserved residues in the transmembrane helices such as N1.50, D2.50, R3.50, W4.50, P6.50, and P7.50. In addition, they preserve the specific motifs present in TMH3, TMH6 and TMH7 which are DRY, CWXP, and NPXXY motifs, respectively (Morales et al. 2017a).
Despite their structural similarities, sequence differences in specific transmembrane regions, extracellular and intracellular loops have been observed among GPR3, GPR6 and GPR12 (Isawi et al. 2017). In fact, further structural understanding of these differences among receptors may help designing future selective ligands for each of them.
Although no crystal structure has been reported yet for any of these orphan receptors, computational molecular models are being developed based on GPCRs with currently available crystal structures (Morales et al. 2017a). These homology models take into consideration structural differences and similarities with other GPCRs while identifying potential binding pocket residues for GPR3, GPR6 and GPR12. This computational approach may lead to the optimization of known ligands and will aid future drug discovery.
The physiological role of GPR3, GPR6 and GPR12 is clearly defined by their high presence in the central nervous system (CNS) (Saeki et al. 1993; Eggerickx et al. 1995; Heiber et al. 1995). As represented in figure 2, GPR3 is highly expressed in hippocampus, hypothalamus, cerebral cortex and cerebellum. Moreover, GPR6 is mainly located in striatum and hypothalamus, while GPR12 was detected in cerebral cortex, striatum and cerebellum.2
Figure 2.

Schematic representation of GPR3, GPR6 and GPR12 expression. A) Peripheral expression; B) Expression in the CNS. Color coding: GPR3 in red, GPR6 in green, GPR12 in blue; darker colors represent higher expression whereas lighter ones refer to lower expression. White circles refer to very low or no receptor expression.
To a lesser extent, these GPCRs are also found in peripheral tissues. For instance, GPR3 is present in the eye, heart, breast, liver, ovary, testis, adipose tissue and skin. However, in humans, GPR6 and GPR12 have very low expression at a peripheral level. GPR12 can be found in the eye, breast, liver, and skin, while low levels of GPR6 were detected in the stomach and testis.2 It is worth mentioning that GPR3 and GPR12, but not GPR6, were detected in human umbilical vein endothelial cells (Uhlenbrock et al. 2003).
Even though similar expression patterns have been reported in other species, differences in particular tissues and cells have also been observed (Regard et al. 2008). For instance, expression of GPR12 was detected in oocytes of rats and mice but not in humans (Hinckley et al. 2005; DiLuigi et al. 2008).
In summary, since these receptors are highly expressed in human brain regions tightly associated with particular neurological disorders they could represent potential therapeutic targets. Detailed abnormal expression under certain pathological conditions will be described in the biological relevance section.
Pharmacology
A high constitutive activation of adenylyl cyclase has been consistently observed in GPR3, GPR6 and GPR12 (Eggerickx et al. 1995; Uhlenbrock et al. 2002; Martin et al. 2015). In the absence of a ligand, the basal activity of these receptors was found to be similar to the activity of fully activated Gαs-coupled receptors with their corresponding modulators (Eggerickx et al. 1995; Uhlenbrock et al. 2002). Experiments from diverse research groups demonstrated that these three receptors cause a significant increase in intracellular cAMP levels in a wide variety of cell lines (Eggerickx et al. 1995; Uhlenbrock et al. 2002; Ignatov et al. 2003b; Lobo et al. 2007; Tanaka et al. 2007).
Besides their clear ability to couple to Gαs, GPR6 and GPR12 have also been reported to transduce their signal through Gαi/o. This G-protein promiscuity has been observed by independent research groups using different GPR6 or GPR12-transfected cell lines and a variety of functional outcomes (Uhlenbrock et al. 2002; Ignatov et al. 2003a; Ignatov et al. 2003b; Martin et al. 2015). However, this G-protein dual coupling ability was not demonstrated for GPR3.
To the best of our knowledge, there is no further evidence of these orphan receptors coupling with other Gα subunit partners. In fact, co-expression of GPR3, GPR6 or GPR12 with Gαq or Gα16 did not produce substantial intracellular Ca2+ release (Uhlenbrock et al. 2002; Ignatov et al. 2003b).
On the other hand, the G protein independent β-arrestin2 signaling pathway has not been thoroughly investigated at these orphan receptors. GPR3 is the only receptor within this cluster for which this pathway was proved to be of special relevance under certain pathological conditions (Thathiah et al. 2013). This will be further detailed in a following section (see Biological relevance section).
At a molecular level, little is known about the mechanisms that control their signaling activity or potential subcellular localizations. Different reports demonstrated that GPR6 can signal from intracellular compartments. Prasad and collaborators confirmed the intracellular localization of GPR6 using multiple detection methods such as imaging assays, biotinylation or proteolytic enzymes using GPR6-HEK293 cells and striatal neurons (Padmanabhan et al. 2009; Prasad et al. 2010). Further assays demonstrated the Gαs-mediated constitutive activity of GPR6 suggesting its ability to signal from within the cell.
In contrast, Lowther and coworkers suggested that GPR3 does not signal following endocytosis (Lowther et al. 2011; Lowther et al. 2013). They observed that GPR3 signals at the cell surface being susceptible to desensitization by a β-arrestin2 and GRK2 (G protein-coupled receptor kinase)-mediated mechanism. In fact, endocytic inhibition increased cAMP levels and GPR3 accumulation at the cell-surface, whereas overexpression of GRK2 and β-arrestin2 led to lower cAMP levels and a reduced GPR3 cell-surface expression (Lowther et al. 2013).
Further biochemical mechanistic studies are clearly needed for a better pharmacological characterization of GPR3, GPR6 and GPR12. Advances in the development of specific antibodies and potent and selective ligands will certainly guide additional insights into downstream signaling events at these promising targets.
Molecules targeting GPR3/6/12
Thus far, very few ligands have been reported to modulate these orphan receptors. In fact, the lack of potent, efficacious and selective GPR3, GPR6 and GPR12 ligands is delaying their therapeutical exploitation. Therefore, a structural understanding of these molecules and the identification of characteristic features might be crucial for further development of the desirable pharmacological tools. This section offers an overview of reported ligands and their pharmacology.
Putative endogenous ligands
A number of bioactive lipids have been inconclusively tested as potential endogenous molecules at GPR3/6/12 (Yin et al. 2009; Southern et al. 2013). Despite the lack of consensus, several reports point towards the lysophospholipids sphingosine 1-phosphate (S1P), dihydrosphingosine-1-phosphate (DHS1P), and sphingosylphosphorylcholine (SPC) (Figure 3) as putative endogenous ligands for GPR3, GPR6 and/or GPR12. As detailed below for each particular receptor, divergent pharmacological reports are puzzling the deorphanization of these receptors.
Figure 3.

Structure of putative endogenous GPR3/6/12 ligands.
S1P has been proposed as an endogenous GPR3 ligand in different reports. Uhlenbrock and coworkers firstly reported that S1P is able to activate GPR3 in hGPR3-HEK293 using intracellular Ca2+ mobilization and cAMP accumulation assays as readouts (Uhlenbrock et al. 2002). Similar results were obtained in oocytes and mGPR3-COS7 supporting this data (Hinckley et al. 2005; Zhang et al. 2012). However, independent research groups were not able to confirm these results. S1P did not enhance GPR3 activity using cAMP assays in hGPR3-CHO cells (Valverde et al. 2009), nor activate GPR3 in β-arrestin recruitment assays using transfected HEK293, CHO, or HeLa cells (Yin et al. 2009; Southern et al. 2013; Ye et al. 2014). Regarding the sphingolipid SPC, not significant activity was found in the few assays reported so far (Uhlenbrock et al. 2002).
There are contradictory reports around considering S1P as the endogenous ligand for GPR6 as well. It has been demonstrated that in hGPR6-HEK293, S1P and DHS1P elicit intracellular Ca2+ mobilization, whereas no significant changes were observed on cAMP accumulation assays after S1P treatment (Uhlenbrock et al. 2002). Ignatov and coworkers also observed that S1P induces an explicit boost in Ca2+ response in mGPR6-CHO cells, and stimulates GIRK in GPR6 transfected frog oocytes (Ignatov et al. 2003b). According to these reports, S1P displays EC50 values in the nanomolar range at GPR6 (Uhlenbrock et al. 2002; Ignatov et al. 2003b). On the other hand, in the β-arrestin PathHunter™ assay S1P did not show an ability to recruit β-arrestin in GPR6 transfected HEK293 cells or CHO cells (Yin et al. 2009; Southern et al. 2013). Independent assays showed that the bioactive molecule SPC displayed weak (Ignatov et al. 2003b) or no activity (Uhlenbrock et al. 2002) at GPR6 when using Ca2+ mobilization assays.
For GPR12, the endogenous debate is mainly focused on the lysophospholipid SPC. This lipid has been shown to induce significant Ca2+ mobilization in mGPR12-CHO, and to evoke GIRK-mediated current in frog oocytes (Ignatov et al. 2003a). In both assays, SPC displays EC50 values in the nanomolar range. In contrast, other reports found that at GPR12 SPC produced weak or no effects on cAMP (Hinckley et al. 2005), Ca2+ mobilization (Uhlenbrock et al. 2002) or β-arrestin recruitment assays (Yin et al. 2009; Southern et al. 2013), in mGPR12-COS7, hGPR12-HEK293, and hGPR12-CHO cell lines, respectively. On the other hand, S1P exhibited low (Uhlenbrock et al. 2002; Ignatov et al. 2003a) or no efficacy (Yin et al. 2009; Southern et al. 2013) at GPR12 in the reported assays.
Interestingly, the Japanese company Teijin Pharma Limited reported that the neuropeptide Nesfatin-1 (Dore et al. 2017) is also able to activate GPR12 (Mori and Eguchi 2012). This was proved through cAMP accumulation and CREB phosphorylation assays in mGPR12-HeLa and mGPR12-CHO cells respectively.
It is also worth mentioning that endogenous cannabinoid ligands were also tested at GPR3, GPR6 and GPR12. In particular, anandamide, 2-arachidonoylglycerol, virodhamine and noladin ether were assessed using β-arrestin recruitment assays, but no activity was observed at micromolar concentrations (Yin et al. 2009; Laun and Song 2017).
To sum up, the reported results point to the aforementioned sphingolipids as the putative endogenous ligands for these receptors. The controversial pharmacological data may rely on the intrinsic properties of each GPCR, as well as, on the differences among cell-types and functional outcomes. Experimental issues such as the poor solubility of these lipids, or the possible presence of other lipids in the cell media, can also be causing some of these functional discrepancies. Therefore, so far no endogenous ligand has been confirmed for GPR3, GPR6 or GPR12, which are still classified as Class A orphan receptors.
Exogenous ligands
The identification of small molecules targeting GPR3, GPR6 and GPR12 is crucial for the understanding of the physiological role of these receptors. Despite the screening efforts done by academic researchers and pharmaceutical companies, limited hit compounds have been discovered so far. Natural products such as CBD or tyrosol, or chemotypes of synthetic origin such as triazolopyrimidines or pyrazines are among the compounds currently known. Table 1 provides an overview of all the molecules covered in this section.
Table 1.
Summary of GPR3/6/12 ligands and their functionality.
| Ligands | Functionality | Functional Readout | References | ||
|---|---|---|---|---|---|
| GPR3 | GPR6 | GPR12 | |||
| DPI | Agonist* | NA | NA | cAMP accumulation | (Ye et al. 2014) |
| β-arrestin2 recruitment | |||||
| Ca2+ mobilization | |||||
| AF64394 | Inverse agonist** | Inverse agonist* | Inverse agonist* | cAMP accumulation | (Jensen et al. 2014) |
| CBD | Inverse agonist* | Inverse agonist* | NR | β-arrestin2 recruitment | (Laun and Song 2017) |
| NR | NR | Inverse agonist* | cAMP accumulation | (Brown et al. 2017) | |
| 415 | NR | Inverse agonist** | NR | cAMP accumulation | (Hitchcock et al. 2014) |
| 494 | NR | Inverse agonist** | NR | cAMP accumulation | (Hitchcock et al. 2014) |
| 633 | NR | Inverse agonist** | NR | cAMP accumulation | (Hitchcock et al. 2014) |
| 5 | NR | Inverse agonist** | NR | cAMP accumulation | (Hitchcock et al. 2015) |
| 47 | NR | Inverse agonist* | NR | cAMP accumulation | (Brown et al. 2015) |
| 25 | NR | Inverse agonist* | NR | cAMP accumulation | (Adams et al. 2015) |
| ARE111 | NR | Inverse agonist* | NR | cAMP accumulation | (Beeley et al. 2001) |
| GTPγ binding | |||||
| ARE112 | NR | Inverse agonist* | NR | cAMP accumulation | (Beeley et al. 2001) |
| GTPγ binding | |||||
| ARE133 | NR | Inverse agonist** | NR | cAMP accumulation | (Beeley et al. 2001) |
| GTPγ binding | |||||
| ARE136 | NR | Inverse agonist** | NR | cAMP accumulation | (Beeley et al. 2001) |
| GTPγ binding | |||||
| Tyrosol | NR | NR | Agonist** | cAMP accumulation | (Lin et al. 2008) |
NA: No activity found at the concentrations tested; NR: Non-reported;
Potency in the micromolar range;
Potency in the nanomolar range.
Diphenyleneiodoniumchloride
Diphenyleneiodoniumchloride (DPI, figure 4), a known inhibitor of NADPH oxidase (NOX) (Aldieri et al. 2008), was identified as the first small molecule acting as an agonist of GPR3 (Ye et al. 2014). This ligand was discovered from a screening of over 40000 compounds, showing the best agonist dose-dependent activity with EC50 values in the low micromolar range. DPI was shown to promote GPR3 activation through different signaling pathways such as intracellular cAMP accumulation, Ca2+ mobilization, membrane recruitment of β-arrestin2, and receptor desensitization. Interestingly, DPI did not display activity GPR6 and GPR12.
Figure 4.
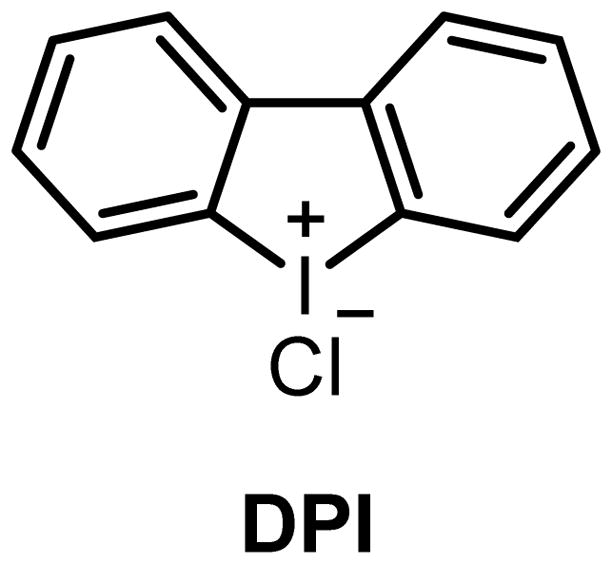
Structure of the GPR3 agonist DPI.
[1,2,4]triazolo[1,5-α]pyrimidine scaffold
The [1,2,4]triazolo[1,5-α]pyrimidine scaffold emerged as a promising GPR3 chemotype from a high throughput screen of the Lundbeck screening collection (Jensen et al. 2014). The identification of compound 1 (Figure 5) as a micromolar inverse agonist of GPR3 inspired the development of a series of triazolopyrimidines in order to fine-tune activity and potency. Numerous derivatives modifying the central bicyclic core, as well as, both phenyl substituents were synthesized and pharmacologically tested. Structure-activity relationships helped to optimize the aromatic substitution pattern at the benzyl group and to understand pharmacophoric requirements such us the presence of a 5-aryl substituent. Among the novel triazolopyrimidine derivatives reported, AF64394 (Figure 5) was the most promising hit identified using homogeneous time-resolved fluorescence (HTRF) cAMP assays in hGPR3-HEK293 cells. This compound was shown to act as a GPR3 inverse agonist with an IC50 value in the mid-nanomolar range. AF64394 showed weak activity at GPR6 or GPR12, but no activity at other class A GPCRs (Jensen et al. 2014).
Figure 5.

Structure of the first triazolopyrimidine identified as a GPR3 inverse agonist, compound 1, and its optimized analog AF64394.
Cannabidiol and derivatives
The well-known phytocannabinoid cannabidiol (CBD, Figure 6) has recently been identified as a modulator of GPR3, GPR6 and GPR12. This non-psychoactive cannabinoid has been shown to act as an inverse agonist at GPR3 and GPR6 using β-arrestin2 recruitment assays (Laun and Song 2017). The same authors later reported that CBD also displays Gαs-mediated inverse agonism at GPR12 using cAMP accumulation assays (Brown et al. 2017). However, CBD does not exhibit high potency at these orphan receptors presenting micromolar IC50s. CBD-related compounds such as cannabidivarin (CBDV, Figure 6) and cannabidiol-2′,6′-dimethyl ether (CBDD, Figure 6) were also tested at GPR12 showing lower activity than CBD (Brown et al. 2017). From the limited GPR12 reported SAR, we can infer that the free hydroxyl groups and the pentyl alkyl chain are crucial for CBD’s activity at this receptor.
Figure 6.

Structure of CBD and its related analogs CBDV and CBDD.
The pharmacology of CBD is complex given its many reported potential therapeutic uses (Morales et al. 2017b; Pisanti et al. 2017; Watt and Karl 2017); its low to moderate activity at GPR3, GPR6 and GPR12 may contribute to CBD’s properties under certain pathological conditions.
It is also important to underscore that related phytocannabinoids, Δ9-tetrahydrocannabinol, cannabinol, and cannabigerol among them, were also evaluated at these receptors, but they did not exhibit activity at the concentrations tested in cAMP accumulation or β-arrestin2 recruitment assays (Brown et al. 2017; Laun and Song 2017). Additionally, synthetic cannabinoid ligands such as HU-210, CP55,940 and WIN55,212-2 were assessed at GPR12 (Brown et al. 2017). These compounds were able to inhibit cAMP accumulation, but only at high micromolar concentrations.
Pyrazine derivatives
Patents from Envoy Therapeutics, Inc. and Takeda Pharmaceutical Company have claimed the use of a wide range of pyrazine analogs as GPR6 modulators (Hitchcock et al. 2014; Adams et al. 2015; Brown et al. 2015; Hitchcock et al. 2015). These inventions claim their use as inverse agonists of GPR6 in the treatment of neurological pathologies such as Parkinson’s disease (PD), Huntington’s disease (HD) and other dyskinesias. The first patent in which this scaffold was identified for the modulation of GPR6 was published in 2014 by Envoy Therapeutics, Inc (Hitchcock et al. 2014). This patent presents over six hundred bicyclic pyrazine derivatives. Among the most potent GPR6 modulators reported were the quinoxaline 415, the pyrido[3,4-b]pyrazine 494, and the pyrazino[2,3-d]pyridazine 633 (Figure 7, the number assigned to each compound relates to the example number reported in the patent). According to cAMP accumulation assays in hGPR6-CHO cells, these three compounds are GPR6 inverse agonists and display IC50 values in the low nanomolar range. Compound 415 was further tested in vivo in a PD rodent model of haloperidol-induced catalepsy. Remarkably, catalepsy was significantly reduced in a dose-dependent manner after intraperitoneal treatment with 415. Structural modifications on the central pyrazine core and its substituents led to the subsequent three patents reported by Takeda Pharmaceutical Company (Adams et al. 2015; Brown et al. 2015; Hitchcock et al. 2015). Novel pyrazino[2,3-d]pyridazines such as 5 showed similar activity to the previous compounds (Hitchcock et al. 2015). However, efforts on tetrahydropyrazines (Brown et al. 2015) or non-bicyclics (Adams et al. 2015) led to a decrease in potency and efficacy at GPR6. For instance, compounds 47 and 25 (Figure 7) showed IC50 values in the micromolar range.
Figure 7.

Structures of the selected pyridazine analogs that modulate GPR6.
All of these patents provide evidence for the potential of the pyrazine scaffold in the development of GPR6 ligands. Nonetheless, selectivity among GPCRs and their activity at other transduction pathways needs to be further investigated.
Imidazolidinethiones and imidazodithiazoles
In 2001 Arena Pharmaceuticals, Inc. patented imidazolidinethione and imidazodithiazole derivatives as GPR6 inverse agonists indicating their potential use in the treatment of obesity (Beeley et al. 2001). In an initial screening using GTPγS and cAMP assays, compounds ARE111 and ARE112 (Figure 8) were identified as hit molecules with IC50 values in the micromolar range. Synthetic fine-tuning of these hits led to the discovery of the GPR6 inverse agonists ARE133 and ARE136 (figure 8), which display nanomolar activity. Compound ARE112 was also tested in vivo demonstrating capacity to reduce body weight and food intake in a rat model (Beeley et al. 2001).
Figure 8.

Structures of imidazolidinethiones ARE111 and ARE133, and imidazodithiazoles ARE112 and ARE136.
Tyrosol
The phenylethanoid antioxidant tyrosol (Figure 9) was identified as a GPR12 ligand by Lin and coworkers (Lin et al. 2008). In this study, fifteen natural products, extracted after isolation of endophytic Streptomyces sp. from a root of Cistanche deserticola, were evaluated using cAMP assays in hGPR12-CHO cells. Tyrosol was the only compound that promoted a GPR12-mediated increase on intracellular cAMP levels at a concentration of 100nM. These findings indicate that tyrosol may be considered a GPR12 ligand and a potential scaffold for the development of more potent GPR12 modulators. (Lin et al. 2008).
Figure 9.
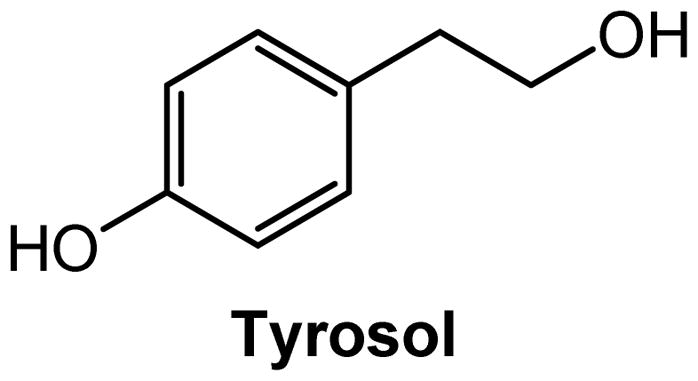
Structure of the GPR12 modulator tyrosol.
The following table summarizes the reported activity of the GPR3, GPR6, and/or GPR12 modulators published thus far.
Biological relevance
The physiological roles of GPR3, GPR6 and GPR12 have not been fully elucidated. However, the increasing research published in the last decade has widely demonstrated the relevance of GPR3, GPR6 and/or GPR12 in a variety of neurological disorders. As previously mentioned, these orphan receptors are highly expressed in particular regions of the brain. This expression pattern is often abnormally regulated under certain physiopathological conditions. Even if their therapeutic potential remains to be fully understood, it is likely that GPR3, GPR6 and GPR12 represent promising targets for the management of the pathological processes described below. Table 2 summarizes the physiopathological implications of each receptor.
Table 2.
Summarized table of the physiopathological role of GPR3, GPR6 and GPR12.
| Target | Physiopathological role | Implication | References |
|---|---|---|---|
| GPR3 | Alzheimer’s Disease | Inhibition of amyloid pathology | (Thathiah et al. 2009) |
| Multiple Sclerosis | Potential biomarker | (Hecker et al. 2011) | |
| Neurite Outgrowth | Promotion of neurite outgrowth and cerebellar development | (Tanaka et al. 2014) | |
| Neuropathic pain | Regulation of neuropathic pain and morphine-induced antinociception | (Ruiz-Medina et al. 2011) | |
| Cell survival | Induction of neuronal survival and antiapoptotic properties | (Tanaka et al. 2007; Tanaka et al. 2014) | |
| Stress | GPR3 deletion triggers anxiety-like and depression-like behavior | (Valverde et al. 2009) | |
| Metabolic Disorders | Impairment of thermogenic response of iBAT and late-onset obesity | (Godlewski et al. 2015) | |
| Oocyte Maturation | Regulation of meiosis | (Mehlmann et al. 2004) | |
| GPR6 | Alzheimer’s Disease | Involvement in the neuroprotective effect of C1q protein | (Benoit et al. 2013) |
| Parkinson’s Disease | GPR6 deletion impacts motor symptoms | (Oeckl et al. 2014) | |
| Huntington’s Disease | GPR6 is downregulated in HD brain | (Hodges et al. 2006) | |
| Schizophrenia | Impairment of striatum P-DARPP-32 | (Oeckl et al. 2014) | |
| Instrumental learning | Regulation of instrumental performance | (Frank and Fossella 2011; Collins and Frank 2012) | |
| Cell survival | Antiapoptotic properties | (Ignatov et al. 2003b) | |
| GPR12 | Neurite Outgrowth | Promotion of neurite outgrowth | (Tanaka et al. 2007; Tanaka et al. 2014) |
| Pain | Sensitivity to heat-related pain | (Carlton et al. 2006) | |
| Cell survival | Antiapoptotic properties | (Adams and Cory 2007; Lu et al. 2012a; Lu et al. 2012b) | |
| Cognitive functions | Regulation of long-term memory | (Peters et al. 2008) | |
| Metabolic Disorders | Appetite control in murine obesity models | (Brennand et al. 2001) | |
| Oocyte Maturation | Regulation of meiosis (only in rodents) | (Hinckley et al. 2005) |
Alzheimer’s Disease
Alzheimer’s disease (AD) is a progressive and chronic brain disorder that accounts for over 60% of the cases of dementia and is the sixth leading cause of death in the United States.3 This neurodegenerative disease is initially characterized by short-term memory loss and progressive impairment of most cognitive functions (Mattson 2004). The etiology of this pathology is associated with the loss of neurons and synapses due to the formation of amyloid plaques and neurofibrillary tangles. These plaques are caused by the accumulation of proteinaceous aggregates composed of amyloid-β (Aβ) peptides (mainly Aβ1–42 but also Aβ1–40 peptides), whereas the neurofibrillary tangles are caused by hyperphosphorylation of the microtubule-associated protein Tau (LaFerla et al. 2007; Karran et al. 2011).
In 2009, Thathiah and coworkers identified a link between GPR3 and AD using a high-throughput functional genomics screen (Thathiah et al. 2009). This receptor was found to modulate one of the aforementioned pathological hallmarks of AD: Aβ peptide generation. In vitro and in vivo studies in AD models demonstrated that GPR3 overexpression triggers an increase in Aβ production, while depletion of this receptor prevents Aβ aggregation. Both Aβ1–40 and Aβ1–42 levels increased in a dose dependent manner with GPR3 expression. Moreover, the expression of GPR3 is elevated in the postmortem brain of sporadic AD patients and this upregulation correlates with the progression of the disease (Thathiah et al. 2009; Huang et al. 2015).
Subsequent findings from the same research group provided further validation of the role of GPR3 in AD (Thathiah et al. 2013; Huang et al. 2015). An array of functional experiments proved that GPR3 regulates Aβ generation via β-arrestin2 via a G protein-independent mechanism (Thathiah et al. 2013). Mutation of the serine amino acids in the C-terminus of GPR3, residues associated with β-arrestin2 recruitment after phosphorylation, resulted in a reduction of Aβ1–40 and Aβ1–42 peptide release, thus confirming that the GPR3 β-arrestin2 pathway is essential for Aβ production. This association was also observed in β-arrestin2 knockout mice since GPR3 only increased Aβ production in the presence of β-arrestin2 in the wild-type (WT) mice (Thathiah et al. 2013).
Pursuing a translational approach, the effect of GPR3 was examined in four different AD transgenic mice models (Huang et al. 2015). An exploration of the role of this receptor in amyloid pathology, as well as, in behavioral studies was undertaken. In the four mice models, GPR3 deletion reduced amyloid plaque burden and deposition as observed in the cortex and hippocampus of these animals. Furthermore, behavioral tests assessing neuromotor coordination, learning and memory capacities, showed that GPR3 genetic ablation alleviates cognitive deficits in AD transgenic mice. Overall, all these results suggest that this orphan receptor is a potential target to be explored in the challenging task of combating AD.
GPR6 was also reported to have a role in the development of AD via the C1q complex (Benoit et al. 2013). The C1q complex is a protein complex that is part of the classical complement pathway (Reid 1983) present in the innate immune system. C1q was found to be associated with AD as it is upregulated in AD models (Alexander et al. 2008; Melchior et al. 2010; Howell et al. 2011). Furthermore, C1q is involved in preventing the neurotoxic effect of fibrillar amyloid-β (fAβ) in vitro (Pisalyaput and Tenner 2007), and it has neuroprotective abilities in early stages of AD models (Benoit and Tenner 2011; Benoit et al. 2013).
The neuroprotective properties of C1q are induced by several mechanisms (Kouser et al. 2015). One of them is modulating GPR6 gene expression. In fAβ-injured neurons, the neuroprotective effect of C1q is accompanied by an increased expression of GPR6. In contrast, the neuroprotective capacity of C1q is diminished when GPR6 is silenced. The same increase in GPR6 expression was exhibited in vivo in the hippocampus of 2–4 months old AD transgenic mice (Benoit et al. 2013). The contribution of GPR6 in this process may arise from the elevated levels of intracellular cAMP production triggered by its increased expression (Heiber et al. 1995; Uhlenbrock et al. 2002). These high levels of cAMP may increase the activation of the central transcription factor pCREB and its subsequent neurotrophic effect (Tong 2004; Kokubo et al. 2009).
Additionally, it is worth mentioning that the biotechnology company Galapagos N. V. patented a method for identifying inhibitors of Aβ production by measuring their potential to modulate GPR3, GPR6 and/or GPR12 (Hoffmann et al. 2006).
Much work remains to be done in the elucidation of the role of these receptors in the pathology of AD. However, we can clearly underscore GPR3 as a potential therapeutic target for the treatment of this neurodegenerative disorder. Pharmacological modulation of this receptor using a G protein independent, β-arrestin2 biased inverse agonist may provide a significant reduction in the amyloid plaques that characterize this disease.
Parkinson’s Disease
Parkinson’s disease (PD) is a neurodegenerative disorder that impacts the motor system causing distinctive symptoms such as tremors, akinesia, bradykinesia, and rigidity. PD is pathologically characterized by degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) and Lewy bodies aggregation (Olanow and Tatton 1999; Jankovic 2008). This cell degeneration impairs the striatonigral and the striatopallidal medium spiny neurons (MSNs), causing dysfunctions in motor coordination (Kreitzer and Malenka 2008).
The role of GPR6 in PD arises from its high expression in the striatum, especially in striatopallidal neurons (Lobo et al. 2007; Heiman et al. 2008; Komatsu et al. 2014). Oeckl and collaborators studied the neurochemical and behavioral effects of GPR6 ablation in mice (Oeckl et al. 2014). GPR6−/− mice showed a decrease of striatal cAMP. In addition, mice lacking GPR6 showed an increase of dopamine concentration in the striatal tissue, while extracellular dopamine levels were not detected to vary. Motor function effects were also observed in these animals. GPR6−/− mice showed higher locomotor activity in the open field with and without haloperidol treatment, a dopamine D2 antagonist that decreases locomotor activity.
Moreover, these authors explored the role of GPR6 in a hemiparkinsonism 6-OHDA (6-hydroxydopamine) mice model. In the absence of GPR6, 6-OHDA mice display a decrease in abnormal involuntary movements suggesting the potential of this receptor in dyskinesia (Oeckl et al. 2014). Dyskinesia is one of the undesirable outcomes of current dopamine PD therapies (Calabresi et al. 2010); therefore, a GPR6 modulator may offer an anti-dyskinesia alternative to dopamine replacement therapy.
In a recent report, the same research group investigated the role of GPR6 in a PD rodent model generated by the dopaminergic neurotoxin MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) (Oeckl and Ferger 2016). GPR6 ablation in these PD mice induced a more prominent reduction in dopamine and 4-hydroxy-3-methoxyphenylacetic acid (HVA). Furthermore, an increase in dopamine turnover was detected in these mice. A significantly higher reduction of the number of tyrosine hydroxylase (TH)-positive neurons in the SNpc was also observed in GPR6−/− mice when compared with their WT counterparts. These findings indicate that GPR6 enhances the dopaminergic neurodegeneration induced by MPTP at a striatal level. Taken together the results reported by Oeckl and coworkers suggest that pharmacological modulation of GPR6 might represent a novel approach for the treatment of PD.
As previously mentioned (exogenous ligands section), patents from Envoy Therapeutics, Inc. and Takeda Pharmaceutical Company (Hitchcock et al. 2014; Adams et al. 2015; Brown et al. 2015; Hitchcock et al. 2015) also claimed the use of GPR6 inverse agonists in the treatment of PD. They showed that pyrazine derivatives are able to reduced haloperidol-induced catalepsy in a rodent PD model.
Even if this data provides solid indications about PD, different models should be used to further demonstrate the relationship between GPR6 and PD and to understand the potential modulation of GPR6 as a molecular target for the treatment of PD.
Huntington’s Disease
Huntington’s disease (HD) is a hereditary disorder that causes a gradual loss of neurons in the basal ganglia (BG), especially in the striatum. Its symptoms are characterized by mood swings, jerky uncontrollable movements, and obsessive-compulsive behavior (Roos 2010).
The neurological dysfunction that HD patients suffer affects the classical pathways of the BG. The striatopallidal pathway MSNs is impaired by this pathology leading to incessantly involuntary motions (Raymond et al. 2011).
Two independent research groups provided evidence of the relation between GPR6 and the HD pathology (Hodges et al. 2006; Le Gras et al. 2017). Analysing 44 human HD brains, Hodges and coworkers observed a significant decrease in GPR6 expression compared to unaffected controls (Hodges et al. 2006). Similarly, in a recent study of striatal transcriptional alterations in HD, the authors detected that the expression of GPR6 eRNA (enhancer RNA) is deregulated in R6/1 HD mouse striatum (Le Gras et al. 2017). They further demonstrated that GPR6 expression is reduced in the striatum of R6/1 and Q140 HD mice models.
Although more research is clearly required, this data suggests that GPR6 may have a role in HD pathology. Therefore, modulation of GPR6 could be a useful therapeutic intervention in the treatment of this disease.
Schizophrenia
Schizophrenia is a long-term psychiatric illness characterized by dysregulation of neurotransmitters such as dopamine, glutamate, serotonin, and GABA in specific brain regions. Its common symptoms include hallucinations, delusions, disorganized thinking and lack of motivation (Patel et al. 2014).
One of the proteins that has been embroiled in schizophrenia pathophysiology is the dopamine and cAMP-regulated phosphoprotein DARPP-32 and its phosphorylated version P-DARPP-32. DARPP-32 and P-DARPP-32 are present in the striatum, especially in MSNs, where they are involved in neo-striatum signaling that affects locomotor behavior (Wang et al. 2017).
Interestingly, in GPR6−/− mice, Oeckl and coworkers observed a two-fold increase in striatum P-DARPP-32 with no alterations in DARPP-32 expression (Oeckl et al. 2014). A decrease in striatal cAMP levels was also detected in these experiments. Therefore, these results suggest that cAMP-independent mechanisms influence DARPP-32 phosphorylation in mice lacking GPR6.
This correlation between GPR6 deletion and P-DARPP-32 only provides the first evidence of the possible implication of this receptor in the treatment of schizophrenia. However, further studies need to identify the effect of GPR6 deficiency on schizophrenia models to provide a clear explanation of its relation with DARPP-32 phosphorylation.
Multiple Sclerosis
Multiple sclerosis (MS) is an inflammatory and degenerative chronic disorder that affects the central nervous system. Even though the cause is not yet clear, its underlying mechanism is characterized by progressive damage to the myelin sheath (demyelination), impairing the conduction of signals between the impacted nerves. This pathology translates into numerous symptoms such as loss of muscle coordination, weakness, numbness, and problems with speech, vision, and bladder control.
Despite the current advances in the field, available drugs are only partially effective, and many patients experience relapses in progressive forms of the disease (Vosoughi and Freedman 2010; Torkildsen et al. 2016). Therefore, there is a clear need for individual molecular biomarkers to better monitor disease evolution and predict therapy response (Graber and Dhib-Jalbut 2011; Coyle 2017). In this context, Hecker and coworkers explored possible disease progression markers in the blood of MS patients (Hecker et al. 2011). They studied an array of 110 genes as potential predictive biomarkers using data sets including 148 subjects. Among these analyses, only GPR3 and IL17RC, a cytokine receptor which binds interleukin 17A (Yao et al. 1997), were validated as potential blood markers of disease progression. Interestingly, the expression of GPR3 was significantly reduced in patients with poor disease progression (more relapses and/or disability increase) in all data sets. Therefore, GPR3 revealed to have a high prognostic value for MS long-term progression (Achiron et al. 2007; Hecker et al. 2011). Similar results were recently observed in another study performed with a group of 87 Egyptian MS patients (Massoud et al. 2017). However, in a study of whole blood samples of 20 relapsing–remitting MS patients, investigators could not confirm a significant reduction in GPR3 expression (Martire et al. 2016).
Therefore, further studies are required to validate the potential of GPR3 as a candidate biomarker for MS progression. Additional functional studies along with an evaluation of how GPR3 depletion affects MS symptoms in animal models may help verify a specific role for GPR3 in this pathology.
Neurite Outgrowth
Neurite outgrowth is one of the essential processes that take place during embryonic development and regeneration of the mammalian nervous system. This process is characterized by the formation of new projections and neurite elongation during neuronal migration and differentiation (Khodosevich and Monyer 2010).
Multiple publications demonstrated that cAMP regulates neurite outgrowth (Cai et al. 1999; Cai et al. 2001; Gao et al. 2003). This finding attracted attention towards the GPR3/6/12 cluster of receptors due to their high constitutive activity and their high abundance in the CNS (Saeki et al. 1993; Eggerickx et al. 1995; Ignatov et al. 2003b). Tanaka and collaborators were the first group that provided evidence of the relation between these receptors and neurite outgrowth (Tanaka et al. 2007). In primary cultures of rat cerebellar granule neurons (CGNs), transfection of GPR3, GPR6 and GPR12 was shown to promote a significant enhancement of neurite outgrowth. GPR12 exhibited the highest increment among the three receptors, exerting its effect by a Gαs and cAMP-dependent protein kinase (PKA) dependent pathway.
Interestingly, GPR3 was shown to be highly expressed in developing rat CGNs, particularly in the internal granule layer. Knocking down endogenous GPR3 in the CGN resulted in a substantial decrease in neurite outgrowth. Cotransfection of either GPR3 or GPR12 was shown to counteract this reduction (Tanaka et al. 2007).
Further reports from this group focused on investigating the role of GPR3 in CGN (Tanaka et al. 2009; Tanaka et al. 2014; Miyagi et al. 2016). These studies showed that this orphan receptor can modulate proliferation and differentiation of cerebellar granule precursors in postnatal development of rodent cerebellum (Tanaka et al. 2009). Further mechanistic insights showed that during development, GPR3 activates the ERK (extracellular signal-regulated protein kinase) and Akt (protein kinase B) signaling pathways. Through these mechanisms, GPR3 is able to protect neurons from apoptosis (Tanaka et al. 2014).
Additional studies demonstrated that in rodent CGN GPR3 can induce upregulation of PKA. Using time-lapse analyses, it was shown that GPR3 is dynamically moved along the neurite tip contributing to local PKA activation in CGN development (Miyagi et al. 2016). Therefore, these results suggest the involvement of GPR3 in crucial neuronal functions, including differentiation and maturation.
A different research group investigated the role of GPR12 overexpression in rat pheochromocytoma PC12 cells (Lu et al. 2012a). They observed that GPR12 promotes neurite outgrowth by inducing differentiation of PC12 into neuron-like cells (increased cell body diameter and neurite length). This effect was accompanied by a significant increment in the expression of multiple neurite outgrowth-related markers (Bcl-xL, Bcl-2, and synaptophysin), and increased ERK1/2 phosphorylation (Lu et al. 2012a).
These reports provide the basis for further investigation of the potential modulation of these orphan receptors in mediating neurite outgrowth and cerebellar development. Targeting these GPCRs may provide a beneficial approach for the treatment of several neurological afflictions or injuries.
Instrumental Conditioning
Human behavior is continuously adjusted by learning and operating processes. The fundamental principles of behaviorism are related to Pavlovian and instrumental conditioning. Pavlovian conditioning is based on pairing a biological stimulus with a previously neutral action. Instrumental conditioning implies that in the process of learning there is an association between behaviors and specific events such as rewards or punishments (Balleine and Dickinson 1998).
As mentioned above, GPR6 is highly expressed in striatopallidal neurons (Lobo et al. 2007; Heiman et al. 2008; Komatsu et al. 2014). Given the importance of striatal involvement in instrumental conditioning (Balleine 2005), Lobo and coworkers decided to explore the role of GPR6 in learning processes (Lobo et al. 2007). In a reward instrumental conditioning assay, WT and GPR6−/− food-deprived mice were trained to bar press for sweet pellets. GPR6−/− mice displayed a lower latency time to press the bar than their WT littermates. Likewise, after more extensive training, GPR6−/− mice reached a higher rate of asymptotic performance. In contrast, using an appetitive Pavlovian conditioning procedure, GPR6 WT and GPR6−/− mice showed comparable results. This data indicates that GPR6 has no effect on Pavlovian conditioning, but it is an important striatopallidal regulator on instrumental conditioning.
Furthermore, by analyzing single nucleotide polymorphisms (SNPs) Frank and collaborators identified GPR6 as a genetic marker of BG (Frank and Fossella 2011; Collins and Frank 2012). In this study, GPR6 was shown to influence reinforcement learning rate on instrumental learning models.
Overall, GPR6 seems to play a role in instrumental performance in mammals. Mechanism, function and possible therapeutic intervention remain to be studied.
Other neurological implications
In addition to the neurodegenerative disorders and neural functions detailed thus far, these orphan receptors have been also implicated in additional brain-related processes such as neuropathic pain, modulation of the early phases of cocaine reinforcement, or cell proliferation.
Pain
Pain mitigation and screening for new molecular targets for antinociception has been a priority among researchers for decades. Both GPR3 and GPR12 have been suggested to play a role in pain modulation.
Ruiz-Medina and coworkers identified a relation between GPR3 and neuropathic pain (Ruiz-Medina et al. 2011). GPR3 was shown to be related to the development and expression of neuropathic pain after sciatic nerve ligature. GPR3−/− mice that suffered a sciatic nerve injury exhibited an increase in sensitivity toward thermal noxious and non-noxious stimuli in both the plantar and cold-plate tests. Additionally, through tail immersion tests in GPR3 WT and GPR3−/− mice, they demonstrated that this orphan receptor is implicated in the analgesia induced by morphine. This suggested that GPR3 is a novel component of a pro-opioid receptor system. These results underscore GPR3 as a potential candidate for the treatment of pain.
GPR12 has been associated with this pathological condition as well. The company Paradigm Therapeutics Limited patented the use of GPR12 modulators for the manipulation of the neuronal and limbic systems and pain treatment (Carlton et al. 2006). In this patent, GPR12−/− mice were found to be more sensitive to heat-induced pain in the tail flick and hot plate tests than WT mice.
These two reports open future avenues for investigating GPR3 and GPR12 in the development of neuropathic pain processes.
Cell proliferation and survival
Diverse studies have demonstrated the participation of GPR3, GPR6 and GPR12 in the control of cell survival and proliferation.
As previously described, Tanaka and collaborators showed that in CGNs these three orphan GPCRs can promote neuronal survival and display antiapoptotic effects (Tanaka et al. 2014). Only GPR3 revealed neuronal antiapoptotic capabilities under a variety of apoptotic stimuli such as hypoxia or reactive oxygen species. As demonstrated in rat CGNs cultures GPR3 mediates this antiapoptotic effect via PKA, ERK, and Akt signaling pathways. These results suggest that the modulation of GPR3 may represent a potential neuroprotective strategy against brain ischemia
Ignatov and coworkers also contributed to this topic studying the implication of GPR6 in apoptotic cell death (Ignatov et al. 2003b). They showed that under serum deprivation or H2O2 apoptosis-inducing stimuli, the presence of S1P causes higher cell viability in GPR6-CHO cells compared to vector-transfected cells. They proposed that GPR6 causes this antiapoptotic effect through the activation of sphingosine kinase and mitogen-activated protein kinase (MAPK).
Regarding GPR12, antiapoptotic properties have also been reported under stress conditions. GPR12 heterologous expression was shown to promote cell proliferation in GPR12-HEK293 cells under serum deprivation (Lu et al. 2012b). Moreover, the antiapoptotic protein B-cell lymphoma/leukemia-2 (Bcl-2) was shown to be upregulated in GPR12-PC12, GPR12-CHO and GPR12-HEK293 cells (Adams and Cory 2007; Lu et al. 2012a; Lu et al. 2012b). Furthermore, endogenously expressed hippocampal GPR12 showed the ability to increase cell proliferation in the presence of SPC (Ignatov et al. 2003a). All these results suggest that GPR12 may participate in the regulation of cell proliferation.
Additional investigations are clearly needed to understand the role of GPR3, GPR6 and GPR12 in cell survival and proliferation in specific cells and tissues. The modulation of these receptors may impact neurological or oncological processes related to abnormal cell regulation.
Additional neural roles of GPR3 and GPR12
Because of their high expression in specific brain regions, GPR3 and GPR12 have attracted the attention of academic researchers and pharmaceutical companies working on related neurological fields.
The role of GPR3 was studied in emotional disorders by Valverde and coworkers (Valverde et al. 2009). This receptor is highly expressed in hippocampus, habenula, cortex, and amygdala, regions tightly involved in behavioral paradigms. They found that GPR3−/− mice exhibit anxiety-like behavior in the open-field and the elevated plus maze. In forced swim and tail suspension tests, GPR3 ablation in mice generated an increase in immobility time indicating depression-related behaviors. Furthermore, increased aggressiveness was detected in GPR3−/− mice in the resident-intruder test. These results indicate that GPR3 ablation affects stress and mood responses. The authors suggest that one reason behind these behavioral results is that variation in monoamine neurotransmitter levels in different brain regions may accompany this deletion (Valverde et al. 2009).
The same research group later reported that GPR3 may regulate behaviors associated with the early phases of cocaine reinforcement (Tourino et al. 2012). GPR3−/− mice showed cocaine-elicited hyperlocomotor responses, enhancement in the rewarding effects in the conditioned place preference (CPP) paradigm and increased reinforcing responses in the self-administration models, compared to WT littermates. This work suggests a possible contribution of GPR3 to drug addiction (Tourino et al. 2012).
The high expression of GPR12 in the limbic system, brain region that controls physiological functions such as memory, emotion or behavior, triggered the exploration of this orphan receptor in such functions. A patent from Helicon Therapeutics claims that GPR12 has a role in regulating long-term memory (Peters et al. 2008). Suppression of hippocampal or amygdala GPR12 in mice caused a significant increase in contextual long-term memory formation, but no differences in short-term memory. Based on these results, they propose that inhibition of GPR12 may be a useful therapeutic approach for the treatment of long-term memory defects.
On the other hand, Frank and coworkers showed no explicit correlation between GPR12 and emotional related behaviors. GPR12−/− and WT mice exhibited comparable results in tests for anxiety-related, depression-like, and locomotor activities (Frank et al. 2012).
The relevance of these orphan receptors in emotional, behavioral or anxiety responses offers novel opportunities for investigating the neurological implication of these receptors. In addition, a role for GPR3 in cocaine addiction has been suggested, opening the door for further investigation in drug addiction.
Functions outside the brain
Besides their clear impact in the CNS, their expression in peripheral tissue along with possible upregulation under certain pathological conditions has led researchers to explore the biological role of GPR3, GPR6 and GPR12 in other diseases such as obesity, dyslipidemia, and ovary aging.
Metabolic disorders
Obesity is one of the major global healthcare burdens worldwide. Consequently, numerous approaches are currently under evaluation in the search of potential treatments. The cannabinoid receptor CB1 appeared to be a potential target for this disease decades ago (Serrano et al. 2012). The phylogenetic relation of GPR3, GPR6 and GPR12 with the cannabinoids attracted researchers to explore the role of these orphan receptors in metabolic disorders. Thus far, only one study has shown a relation between GPR3 and late-onset obesity, whereas diverse reports have examined how GPR12 interplays in these pathologies (Bjursell et al. 2006; Frank et al. 2012; Godlewski et al. 2015). A patent also claimed the use of GPR6 inverse agonists in the treatment of obesity (Beeley et al. 2001).
Kunos and coworkers identified a link between GPR3 weight gain and thermogenesis in old mice (Godlewski et al. 2015). In these studies, mice lacking GPR3 and maintained on standard chow diet showed an obese phenotype after 5 months of life. In fact, 1-year-old GPR3−/− subjects displayed higher adiposity indexes and body weight in addition to reduced energy expenditure and lower body temperature. Further evaluation of these metabolic profiles led them to identify a dysfunction in interscapular brown adipose tissue (iBAT) along with lower expression of UCP1 (uncoupling protein 1), tightly involved in BAT heat generation (Godlewski et al. 2015). These findings suggest that GPR3 deficiency in mice impaired their ability to generate heat by non-shivering thermogenesis. In agreement, previous studies had already associated late-onset obesity with decreased thermogenesis due to a programmed loss of function of iBAT (Waalen and Buxbaum 2011; Rogers et al. 2012). Therefore, these results indicate that GPR3 has a significant function in the thermogenic response of iBAT and may represent a new therapeutic target in age-related metabolic disorders.
Interestingly, and despite the lack of further metabolic studies, a patent from Arena Pharmaceuticals, Inc. claimed the use of GPR6 inverse agonists for the treatment of clinical obesity or overweight disorders in mammals (Beeley et al. 2001). In their studies, the previously mentioned compound ARE112 (figure 8) was able to reduce body weight and food intake after intraperitoneal treatment in rats maintained on standard chow diet.
On the other hand, a patent from AstraZeneca claimed the potential use of GPR12 modulators as appetite control agents in murine obesity models (Brennand et al. 2001). A few years later, researchers from the same company published the first article clearly associating GPR12 with metabolism (Bjursell et al. 2006). They studied food intake, respiratory metabolism, body composition, locomotor activity, and body temperature in mice deficient of GPR12. In these experiments, GPR12−/− mice showed higher body weight, body fat mass, and were dyslipidemic when compared with their WT littermates. The obesity observed in these mice was related to lower energy expenditure since other parameters, such as food intake or physical activity, remained unaltered. These findings suggest that GPR12 may have a role in energy homeostasis, consequently affecting lipid metabolism. It is worth mentioning that these studies were conducted in 3 to 12 weeks-old mice, therefore, these results are not related to aging as in the case of GPR3.
Later studies from the same group of researchers were not able to confirm the relevance of this receptor in the metabolism of GPR12−/− mice (Frank et al. 2012). No significant changes were observed in body weight of GPR12 deficient mice fed with chow or high-fat diets. Little differences were found in other metabolic parameters. Therefore, more research is clearly needed to understand the role of GPR12 in obesity and related processes.
In addition, the Japanese company Teijin Pharma Limited patented a method for screening molecules targeting GPR12 in order to identify Nesfatin-1-like substances for food intake regulation (Mori and Eguchi 2012). Nesfatin-1 is a neuropeptide, mainly produced in the hypothalamus, which is involved in the control of energy homeostasis related with food regulation and water intake (Oh-I et al. 2006; Brailoiu et al. 2007; Dore et al. 2017). The inventors of this patent identified GPR12 as a Nesfatin-1 receptor (Mori and Eguchi 2012), and therefore based their screening method on these findings.
In summary, several studies have suggested a relationship between GPR3/6/12 and metabolic syndromes. However, further exploration of these orphan receptors in the regulation of energy homeostasis are needed to fully understand their roles in obesity, dyslipidemia and related pathologies.
Oocyte maturation
Mammalian oocytes arise and enter meiosis during embryonic development but arrest at prophase I until reproductive maturity (Sánchez and Smitz 2012). During the menstrual cycle, the oocyte completes growth achieving meiotic competence in response to a pre-ovulatory surge of luteinizing hormone (LH) from the pituitary gland that stimulates meiotic resumption to the metaphase II stage (Li and Albertini 2013). It is well recognized that maintenance of meiotic arrest depends on high levels of intracellular cAMP in the oocyte (Mehlmann et al. 2002; Kalinowski et al. 2004), however, the underlying mechanisms are not fully understood.
Mehlmann and coworkers discovered that the constitutively active receptor, GPR3, is directly related to these high levels of cAMP, and therefore it is crucial for the regulation of meiosis (Mehlmann et al. 2004; Freudzon et al. 2005; Mehlmann 2005). In these studies, oocytes from GPR3−/− mice were unable to maintain meiotic arrest. Instead, they underwent spontaneous resumption of meiosis in the absence of an LH surge. Antral follicles from GPR3 WT mice showed prophase-arrested oocytes whereas antral follicles from GPR3−/− ovaries presented oocytes with metaphase II chromosomes and a polar body. In addition, an injection of GPR3 RNA into the oocytes from mice lacking GPR3 was able to prevent resumption of meiosis (Mehlmann et al. 2004).
The expressions of GPR3, GPR6 and GPR12 have been investigated in the ovary, oocytes and follicles of different species. In the mouse oocyte, not only GPR3 (Mehlmann et al. 2004; Hinckley et al. 2005), but also GPR12 (Hinckley et al. 2005) were found, however, only GPR3 proved to be essential to maintain meiosis arrest. On the other hand, in the rat oocyte, only GPR12 expression was detected (Hinckley et al. 2005). This study provides evidence that GPR12 is required to preserve the elevated cAMP levels that prevent meiosis resumption in rat oocytes (Hinckley et al. 2005). Porcine oocytes show analogous results, displaying high GPR3 expression at different stages during porcine oocyte maturation (Yang et al. 2012). Findings from another group, reported the cloning of GPRx, a receptor closely related to GPR12, in frog (Xenopus laevis and Xenopus tropicalisc) oocytes and zebrafish (Danio rerio) oocytes (Rios-Cardona et al. 2008; Nader et al. 2014). Most importantly, studies in human oocytes revealed that RNA encoding GPR3, but not GPR6 or GPR12, is expressed (DiLuigi et al. 2008). In summary, according to expression patterns, GPR3 is the most relevant target for the oocyte maturation process.
Further investigations pointed towards the therapeutic potential of GPR3 in fertility disorders. Ledent and collaborators observed that aging mice lacking GPR3 exhibit a significant fertility decrease (Ledent et al. 2005). GPR3−/− mice showed a progressive decrease in litter size with advancing maternal age. In addition, these female mice displayed lower developmental capacity of embryos, an elevated number of fragmented oocytes, and more signs of reproductive aging. Therefore, GPR3-deficient mice may serve as a premature ovarian failure animal model.
Two other reports examined the role of GPR3 in premature ovarian failure in American (Kovanci et al. 2008) and Chinese women (Zhou et al. 2010), respectively. Kovanci and co-workers examined whether GPR3 mutations were present in a group of 82 American Caucasian women. Similarly, Zhou and collaborators explored the coding region of GPR3 in 100 Chinese patients. In neither study could direct genetic evidence be found to confirm GPR3’s relation to premature ovarian failure (Kovanci et al. 2008; Zhou et al. 2010). Whether GPR3 function and/or expression is upregulated in the ovary of these patients remains to be elucidated.
In summary, over the last decade, diverse investigations have supported the role of GPR3 in the maintenance of meiotic arrest in oocytes. While GPR6 was not detected in oocytes, GPR12 showed meiotic relevance, but only in rats. Therefore, efforts should be focused on additional studies in human ovaries to further validate the therapeutic value of GPR3 in fertility disorders.
Summary and Future Perspectives
Despite their cloning nearly 30 years ago, it has been only in the last decade that research on the therapeutic values of GPR3, GPR6 and GPR12 has begun. Numerous academic research groups and pharmaceutical companies are currently focusing their efforts on a better understanding of the therapeutic role of these receptors.
These three orphan receptors are part of the MECA cluster of Class A GPCRs. Because of this phylogenetic relation, GPR3, GPR6 and GPR12 share a high percentage of homology with the cannabinoid and the lysophospholipid receptors.
GPR3, GPR6 and GPR12 are highly constituvely active GPCRs. In a variety of cell lines, they trigger increases in intracellular cAMP that are comparable in amplitude to activated receptors. In fact, this ability to extensively activate adenylyl cyclase defines their physiological relevance in particular processes.
Despite efforts to identify putative endogenous ligands for GPR3, GPR6 and GPR12, a pharmacological experimental consensus has not yet been reached. This, in turn, is delaying the deorphanization of these receptors (Alexander et al. 2017). Differential GPCR expression among cell lines, G protein-coupling promiscuity, biased agonism or functional outcome heterogeneity might be causing these pharmacological discrepancies.
Novel druggable small molecules are emerging as modulators of these three receptors, nevertheless, potent and selective ligands remained to be discovered. We provide an analysis here of the assorted range of chemotypes reported so far. These insights may help facilitate the elucidation of pharmacophoric structural features and contribute to future rational design of new molecules able to selectively bind GPR3, GPR6 and/or GPR12.
The pathophysiological relevance of these orphan receptors stems mainly from their high expression in the CNS. In certain brain disorders, the expression of these receptors is upregulated and their pharmaceutical intervention may offer a promising therapeutic strategy. As summarized in this review, numerous studies suggest the participation of GPR3, GPR6 and GPR12 in a wide range of neurological conditions. Neurodegenerative diseases such as AD, PD, HD or MS, and neural processes such as neurite outgrowth or instrumental conditioning have been proven to be impacted in different ways by genetic ablation in vitro or in vivo of GPR3, GPR6 and/or GPR12.
It is interesting to highlight that besides their structural similarities, different expression patterns and particular structural differences define their independent genetic implications in each particular disease. Differences among species need also to be considered when evaluating the potential of these GPCRs. For instance, as previously mentioned, differential expression of GPR3 and/or GPR12 has been detected in human, mouse and rat oocytes (Hinckley et al. 2005; DiLuigi et al. 2008).
Because of their expression in peripheral tissues, GPR3 and GPR12 may be potential targets for metabolic and ovary-related pathological conditions. However, much work needs to be conducted to investigate their role under other pathophysiological processes at both central and peripheral levels.
Pharmacological intervention at these receptors represents a promising strategy for the treatment of aforementioned disorders. Even if the lack of accessible pharmacological tools is hampering their appropriate characterization, the knowledge gained so far clearly suggests that modulation of GPR3, GPR6 and/or GPR12 will be beneficial in particular diseases. For example, the outstanding research carried out on the elucidation of the role of GPR3 in the amyloid pathology indicates that selective blockage of the GPR3 G protein independent β-arrestin2 signalling pathway will reduce beta amyloid plaque formation (Thathiah et al. 2013). In contrast, pharmaceutically targeting GPR3 in fertilization processes may require activation of this receptor, ideally through a peripherally restricted mechanism (Mehlmann et al. 2004).
Overall, this work aims to emphasize the therapeutic potential of GPR3, GPR6 and GPR12 through a comprehensive first summary of the state of the art in this field.
Acknowledgments
The authors acknowledge research support from NIH/NIDA grants R01 DA003934 and K05 DA021358 (P.H.R.).
Footnotes
Declaration of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
- Achiron A, Feldman A, Mandel M, Gurevich M. Impaired expression of peripheral blood apoptotic-related gene transcripts in acute multiple sclerosis relapse. Ann N Y Acad Sci. 2007;1107:155–167. doi: 10.1196/annals.1381.017. [DOI] [PubMed] [Google Scholar]
- Adams J, Cory S. Bcl-2-regulated apoptosis: mechanism and therapeutic potential. Curr Opin Immunol. 2007;19:488–496. doi: 10.1016/j.coi.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams ME, Brown JW, Hitchcock S, Kikuchi S, Lam B, Monenschein H, Reichard H, Sun H Takeda Pharmaceutical Company Limited. Japan WO2015123533A1 Pyrazines as modulators of GPR6. 2015 Aug 20;
- Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, Ghigo D. Classical Inhibitors of NOX NAD(P)H Oxidases Are Not Specific. Curr Drug Metab. 2008;9:686–696. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- Alexander JJ, Anderson AJ, Barnum SR, Stevens B, Tenner AJ. The complement cascade: Yin-Yang in neuroinflammation -neuro-protection and -degeneration. J Neurochem. 2008;107:1169–1187. doi: 10.1111/j.1471-4159.2008.05668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Christopoulos A, Davenport AP, Kelly E, Marrion NV, Peters JA, Faccenda E, Harding SD, Pawson AJ, Sharman JL, et al. The concise guide of pharmacology 2017/18: G protein-coupled receptors. Br J Pharmacol. 2017;174:S17–S129. doi: 10.1111/bph.13878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleine B. Neural bases of food-seeking: Affect, arousal and reward in corticostriatolimbic circuits. Physiol Behav. 2005;86:717–730. doi: 10.1016/j.physbeh.2005.08.061. [DOI] [PubMed] [Google Scholar]
- Balleine BW, Dickinson A. Goal-directed instrumental action: contingency and incentive learning and their cortical substrates. Neuropharmacology. 1998;37:407–419. doi: 10.1016/s0028-3908(98)00033-1. [DOI] [PubMed] [Google Scholar]
- Beeley NRA, Behan DP, Chalmers DT, Menzaghi F, Strah-Pleynet S Arena Pharmaceuticals, Inc. USA. WO2001062765A2 Small molecule modulators of G protein-coupled receptor six. 2001 Aug 30;
- Benoit ME, Hernandez MX, Dinh ML, Benavente F, Vasquez O, Tenner AJ. C1q-induced LRP1B and GPR6 Proteins Expressed Early in Alzheimer Disease Mouse Models, Are Essential for the C1q-mediated Protection against Amyloid-β Neurotoxicity. J Biol Chem. 2013;288:654–665. doi: 10.1074/jbc.M112.400168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit ME, Tenner AJ. Complement Protein C1q-Mediated Neuroprotection Is Correlated with Regulation of Neuronal Gene and MicroRNA Expression. J Neurosci. 2011;31:3459–3469. doi: 10.1523/JNEUROSCI.3932-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjursell M, Gerdin AK, Jönsson M, Surve VV, Svensson L, Huang XF, Törnell J, Bohlooly-Y M. G protein-coupled receptor 12 deficiency results in dyslipidemia and obesity in mice. Biochem Biophys Res Commun. 2006;348:359–366. doi: 10.1016/j.bbrc.2006.07.090. [DOI] [PubMed] [Google Scholar]
- Brailoiu GC, Dun SL, Brailoiu E, Inan S, Yang J, Jaw KC, Dun NJ. Nesfatin-1: Distribution and interaction with a G protein-coupled receptor in the rat brain. Endocrinology. 2007;148:5088–5094. doi: 10.1210/en.2007-0701. [DOI] [PubMed] [Google Scholar]
- Brennand JC, Charles AD, Hart KA AstraZeneca. Germany. WO2001048483A2 Method for screening of appetite control agents. 2001 Jul 5;
- Brown J, Hitchcock S, Hopkins M, Kikuchi S, Monenschein H, Reichard H, Schleicher K, Sun H, Macklin T Takeda Pharmaceutical Company Limited. Japan. WO2015095728A1 Tetrahydropyridopyrazines as modulators of GPR6. 2015 Jun 25;
- Brown KJ, Laun AS, Song Z-h. Cannabidiol, a novel inverse agonist for GPR12. Biochem Biophys Res Commun. 2017;493:451–454. doi: 10.1016/j.bbrc.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci. 2001;21:4731–4739. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai D, Yingjing S, De Bellard ME, Tang S, Filbin MT. Prior exposure to neurotrophins blocks inhibition of axonal regeneration by MAG and myelin via a cAMP-dependent mechanism. Neuron. 1999;22:89–101. doi: 10.1016/s0896-6273(00)80681-9. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Filippo MD, Ghiglieri V, Tambasco N, Picconi B. Levodopa-induced dyskinesias in patients with Parkinson’s disease: filling the bench-to-bedside gap. The Lancet Neurology. 2010;9:1106–1117. doi: 10.1016/S1474-4422(10)70218-0. [DOI] [PubMed] [Google Scholar]
- Carlton M, Dixon J, Hendrick A, Messager S, Zahn D, Horwood JM Paradigm Therapeutics Limited. UK. WO 2005027628 Sphingosylphosphorylcholine receptor GPR12, GPR12 knockout animals for the neuronal and limbic system drug screening, and therapeutic and diagnostic uses. 2006 Mar 31;
- Collins AGE, Frank MJ. How much of reinforcement learning is working memory, not reinforcement learning? A behavioral, computational, and neurogenetic analysis. Eur J Neurosci. 2012;35:1024–1035. doi: 10.1111/j.1460-9568.2011.07980.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle PK. Pharmacogenetic Biomarkers to Predict Treatment Response in Multiple Sclerosis: Current and Future Perspectives. Multiple Sclerosis International. 2017;2017:1–33. doi: 10.1155/2017/6198530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiLuigi A, Weitzman VN, Pace MC, Siano LJ, Maier D, Mehlmann LM. Meiotic arrest in human oocytes is maintained by a Gs signaling pathway. Biol Reprod. 2008;78:667–672. doi: 10.1095/biolreprod.107.066019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dore R, Levata L, Lehnert H, Schulz C. Nesfatin-1: Functions and physiology of a novel regulatory peptide. J Endocrinol. 2017;232:R45–R65. doi: 10.1530/JOE-16-0361. [DOI] [PubMed] [Google Scholar]
- Eggerickx D, Denef JF, Labbe O, Hayashi Y, Refetoff S, Vassart G, Parmentier M, Libert F. Molecular cloning of an orphan G-protein-coupled receptor that constitutively activates adenylate cyclase. The Biochemical Journal. 1995;309:837–843. doi: 10.1042/bj3090837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank E, Wu Y, Piyaratna N, Body WJ, Snikeris P, South T, Gerdin AK, Bjursell M, Bohlooly-Y M, Storlien L, et al. Metabolic parameters and emotionality are little affected in G-protein coupled receptor 12 (Gpr12) mutant mice. PLoS ONE. 2012;7:1–8. doi: 10.1371/journal.pone.0042395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank MJ, Fossella JA. Neurogenetics and pharmacology of learning, motivation, and cognition. Neuropsychopharmacology: official publication of the American College of Neuropsychopharmacology. 2011;36:133–152. doi: 10.1038/npp.2010.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredriksson R, Lagerström MC, Lundin L-G, Schiöth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Schio HB. The Repertoire of G-Protein – Coupled Receptors in Fully. Mol Pharmacol. 2005;67:1414–1425. doi: 10.1124/mol.104.009001. [DOI] [PubMed] [Google Scholar]
- Freudzon L, Norris RP, Hand AR, Tanaka S, Saeki Y, Jones TLZ, Rasenick MM, Berlot CH, Mehlmann LM, Jaffe LA. Regulation of meiotic prophase arrest in mouse oocytes by GPR3, a constitutive activator of the Gs G protein. J Cell Biol. 2005;171:255–265. doi: 10.1083/jcb.200506194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Nikulina E, Mellado W, Filbin MT. Neurotrophins elevate cAMP to reach a threshold required to overcome inhibition by MAG through extracellular signal-regulated kinase-dependent inhibition of phosphodiesterase. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:11770–11777. doi: 10.1523/JNEUROSCI.23-37-11770.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewski G, Jourdan T, Szanda G, Tam J, Resat C, Harvey-White J, Liu J, Mukhopadhyay B, Pacher P, Ming Mo F, et al. Mice lacking GPR3 receptors display late-onset obese phenotype due to impaired thermogenic function in brown adipose tissue. Scientific Reports. 2015;5:14953. doi: 10.1038/srep14953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graber JJ, Dhib-Jalbut S. Biomarkers of disease activity in multiple sclerosis. J Neurol Sci. 2011;305:1–10. doi: 10.1016/j.jns.2011.03.026. [DOI] [PubMed] [Google Scholar]
- Hecker M, Paap BK, Goertsches RH, Kandulski O, Fatum C, Koczan D, Hartung HP, Thiesen HJ, Zettl UK. Reassessment of blood gene expression markers for the prognosis of relapsing-remitting multiple sclerosis. PLoS ONE. 2011;6:e29648. doi: 10.1371/journal.pone.0029648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heiber M, Docherty JM, Shah G, Nguyen T, Cheng R, Heng HHQ, Márchese A, Tsui L-C, Shi X, George SR, et al. Isolation of Three Novel Human Genes Encoding G Protein-Coupled Receptors. DNA Cell Biol. 1995;14:25–35. doi: 10.1089/dna.1995.14.25. [DOI] [PubMed] [Google Scholar]
- Heiman M, Schaefer A, Gong S, Peterson JD, Day M, Ramsey KE, Suárez-Fariñas M, Schwarz C, Stephan DA, Surmeier DJ, et al. A Translational Profiling Approach for the Molecular Characterization of CNS Cell Types. Cell. 2008;135:738–748. doi: 10.1016/j.cell.2008.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinckley M, Vaccari S, Horner K, Chen R, Conti M. The G-protein-coupled receptors GPR3 and GPR12 are involved in cAMP signaling and maintenance of meiotic arrest in rodent oocytes. Dev Biol. 2005;287:249–261. doi: 10.1016/j.ydbio.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Hitchcock S, Hopkins M, Kikuchi S, Monenschein H, Reichard H, Sun H, Macklin T Takeda Pharmaceutical Company Limited. Japan. WO2015123505A1 Pyridopyrazines modulators of GPR6. 2015 Aug 20;
- Hitchcock S, Monenschein H, Reichard H, Sun H, Kikuchi S, Macklin T, Hopkins M Envoy Therapeutics, Inc. USA. WO2014028479A1 Quinoxaline derivatives as GPR6 modulators. 2014 Feb 20;
- Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, et al. Regional and cellular gene expression changes in human Huntington’s disease brain. Hum Mol Genet. 2006;15:965–977. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- Hoffmann M, Merchiers PG, Spittaels KFF, Galapagos NV. Belgium. WO2005103713A3 Methods, compositions and compound assays for inhibiting amyloid-beta protein production. 2006 Nov 3;
- Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, Barbay JM, King BL, Marchant JK, Hibbs M, et al. Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J Clin Invest. 2011;121:1429–1444. doi: 10.1172/JCI44646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Skwarek-Maruszewska A, Horre K, Vandewyer E, Wolfs L, Snellinx A, Saito T, Radaelli E, Corthout N, Colombelli J, et al. Loss of GPR3 reduces the amyloid plaque burden and improves memory in Alzheimer’s disease mouse models. Science Translational Medicine. 2015;7:309ra164. doi: 10.1126/scitranslmed.aab3492. [DOI] [PubMed] [Google Scholar]
- Ignatov A, Lintzel J, Hermans-Borgmeyer I, Kreienkamp H-J, Joost P, Thomsen S, Methner A, Schaller HC. Role of the G-protein-coupled receptor GPR12 as high-affinity receptor for sphingosylphosphorylcholine and its expression and function in brain development. The Journal of neuroscience. 2003a;23:907–914. doi: 10.1523/JNEUROSCI.23-03-00907.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatov A, Lintzel J, Kreienkamp H-J, Chica Schaller H. Sphingosine-1-phosphate is a high-affinity ligand for the G protein-coupled receptor GPR6 from mouse and induces intracellular Ca2+ release by activating the sphingosine-kinase pathway. Biochem Biophys Res Commun. 2003b;311:329–336. doi: 10.1016/j.bbrc.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Isawi I, Morales P, Laun A, et al. Towards a structural understanding of the orphan receptor GPR6: Relationship with the Cannabinoids. Southeastern Regional Meeting of the American Chemical Society; Nov 7–11; Charlotte, NC. 2017. [Google Scholar]
- Jankovic J. Parkinson’s disease: clinical features and diagnosis. Journal of Neurology, Neurosurgery & Psychiatry. 2008;79:368–376. doi: 10.1136/jnnp.2007.131045. [DOI] [PubMed] [Google Scholar]
- Jensen T, Elster L, Nielsen SM, Poda SB, Loechel F, Volbracht C, Klewe IV, David L, Watson SP. The identification of GPR3 inverse agonist AF64394; The first small molecule inhibitor of GPR3 receptor function. Bioorg Med Chem Lett. 2014;24:5195–5198. doi: 10.1016/j.bmcl.2014.09.077. [DOI] [PubMed] [Google Scholar]
- Kalinowski RR, Berlot CH, Jones TLZ, Ross LF, Jaffe LA, Mehlmann LM. Maintenance of meiotic prophase arrest in vertebrate oocytes by a G s protein-mediated pathway. Dev Biol. 2004;267:1–13. doi: 10.1016/j.ydbio.2003.11.011. [DOI] [PubMed] [Google Scholar]
- Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nature reviews Drug discovery. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- Khodosevich K, Monyer H. Signaling involved in neurite outgrowth of postnatally born subventricular zone neurons in vitro. BMC Neurosci. 2010;11:1–11. doi: 10.1186/1471-2202-11-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokubo M, Nishio M, Ribar TJ, Anderson KA, West AE, Means AR. BDNF-Mediated Cerebellar Granule Cell Development Is Impaired in Mice Null for CaMKK2 or CaMKIV. J Neurosci. 2009;29:8901–8913. doi: 10.1523/JNEUROSCI.0040-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsu H, Maruyama M, Yao S, Shinohara T, Sakuma K, Imaichi S, Chikatsu T, Kuniyeda K, Siu FK, Peng LS, et al. Anatomical transcriptome of G protein-coupled receptors leads to the identification of a novel therapeutic candidate gpr52 for psychiatric disorders. PLoS ONE. 2014;9:e90134. doi: 10.1371/journal.pone.0090134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouser L, Madhukaran SP, Shastri A, Saraon A, Ferluga J, Al-Mozaini M, Kishore U. Emerging and Novel Functions of Complement Protein C1q. Frontiers in Immunology. 2015;6:317. doi: 10.3389/fimmu.2015.00317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovanci E, Simpson JL, Amato P, Rohozinski J, Heard MJ, Bishop CE, Carson SA. Oocyte-specific G-protein-coupled receptor 3 (GPR3): no perturbations found in 82 women with premature ovarian failure (first report) Fertility and Sterility. 2008;90:1269–1271. doi: 10.1016/j.fertnstert.2007.07.1373. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Malenka RC. Striatal Plasticity and Basal Ganglia Circuit Function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFerla FM, Green KN, Oddo S. Intracellular amyloid-β in Alzheimer’s disease. Nature Reviews Neuroscience. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- Laun AS, Song Z-H. GPR3 and GPR6, novel molecular targets for cannabidiol. Biochem Biophys Res Commun. 2017;490:17–21. doi: 10.1016/j.bbrc.2017.05.165. [DOI] [PubMed] [Google Scholar]
- Le Gras S, Keime C, Anthony A, Lotz C, De Longprez L, Brouillet E, Cassel J-C, Boutillier A-L, Merienne K. Altered enhancer transcription underlies Huntington’s disease striatal transcriptional signature. Scientific Reports. 2017;7:42875. doi: 10.1038/srep42875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledent C, Demeestere I, Blum D, Petermans J, Hämäläinen T, Smits G, Vassart G. Premature ovarian aging in mice deficient for GPR3. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:8922–8926. doi: 10.1073/pnas.0503840102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Albertini DF. The road to maturation: somatic cell interaction and self-organization of the mammalian oocyte. Nature Reviews Molecular Cell Biology. 2013;14:141–152. doi: 10.1038/nrm3531. [DOI] [PubMed] [Google Scholar]
- Lin Z-J, Lu X-M, Zhu T-J, Fang Y-C, Gu Q-Q, Zhu W. GPR12 Selections of the metabolites from an endophytic Streptomyces sp. Asociated with Cistanches deserticola. Archives of Pharmacal Research. 2008;31:1108–1114. doi: 10.1007/s12272-001-1276-4. [DOI] [PubMed] [Google Scholar]
- Lobo MK, Cui Y, Ostlund SB, Balleine BW, Yang XW. Genetic control of instrumental conditioning by striatopallidal neuron-specific S1P receptor Gpr6. Nat Neurosci. 2007;10:1395–1397. doi: 10.1038/nn1987. [DOI] [PubMed] [Google Scholar]
- Lowther KM, Nikolaev VO, Mehlmann LM. Endocytosis in the mouse oocyte and its contribution to cAMP signaling during meiotic arrest. Reproduction. 2011;141:737–747. doi: 10.1530/REP-10-0461. [DOI] [PubMed] [Google Scholar]
- Lowther KM, Uliasz TF, Götz KR, Nikolaev VO, Mehlmann LM. Regulation of Constitutive GPR3 Signaling and Surface Localization by GRK2 and β-arrestin-2 Overexpression in HEK293 Cells. PLoS ONE. 2013;8:e65365. doi: 10.1371/journal.pone.0065365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Zhang N, Dong S, Hu Y. Involvement of GPR12 in the induction of neurite outgrowth in PC12 cells. Brain Res Bull. 2012a;87:30–36. doi: 10.1016/j.brainresbull.2011.09.020. [DOI] [PubMed] [Google Scholar]
- Lu X, Zhang N, Meng B, Dong S, Hu Y. Involvement of GPR12 in the regulation of cell proliferation and survival. Mol Cell Biochem. 2012b;366:101–110. doi: 10.1007/s11010-012-1287-x. [DOI] [PubMed] [Google Scholar]
- Martin AL, Steurer MA, Aronstam RS. Constitutive Activity among Orphan Class-A G Protein Coupled Receptors. PLoS ONE. 2015;10:e0138463. doi: 10.1371/journal.pone.0138463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire S, Navone ND, Montarolo F, Perga S, Bertolotto A. A gene expression study denies the ability of 25 candidate biomarkers to predict the interferon-beta treatment response in multiple sclerosis patients. J Neuroimmunol. 2016;292:34–39. doi: 10.1016/j.jneuroim.2016.01.010. [DOI] [PubMed] [Google Scholar]
- Massoud H, Medhat D, Eldeen ES, Khayat ZE, Osman MA. G protein coupled membrane receptor expression and IL - 17 A in multiple sclerosis. BIoscience Research. 2017;14:282–290. [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer’s disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlmann LM. Oocyte-specific expression of Gpr3 is required for the maintenance of meiotic arrest in mouse oocytes. Dev Biol. 2005;288:397–404. doi: 10.1016/j.ydbio.2005.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehlmann LM, Jones TLZ, Jaffe LA. Meiotic Arrest in the Mouse Follicle Maintained by a Gs Protein in the Oocyte. Science. 2002;297:1343–1345. doi: 10.1126/science.1073978. [DOI] [PubMed] [Google Scholar]
- Mehlmann LM, Saeki Y, Tanaka S, Brennan TJ, Evsikov AV, Pendola FL, Knowles BB, Eppig JJ, Jaffe LA. The Gs-linked receptor GPR3 maintains meiotic arrest in mammalian oocytes. Science. 2004;306:1947–1950. doi: 10.1126/science.1103974. [DOI] [PubMed] [Google Scholar]
- Melchior B, Garcia AE, Hsiung B-K, Lo KM, Doose JM, Thrash JC, Stalder AK, Staufenbiel M, Neumann H, Carson MJ. Dual Induction of TREM2 and Tolerance-Related Transcript, Tmem176b, in Amyloid Transgenic Mice: Implications for Vaccine-Based Therapies for Alzheimer’s Disease. ASN Neuro. 2010;2:157–170. doi: 10.1042/AN20100010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyagi T, Tanaka S, Hide I, Shirafuji T, Sakai N. The Subcellular Dynamics of the Gs-Linked Receptor GPR3 Contribute to the Local Activation of PKA in Cerebellar Granular Neurons. PLoS ONE. 2016;11:e0147466. doi: 10.1371/journal.pone.0147466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales P, Hurst DP, Reggio PH. Methods for the Development of In Silico GPCR Models. In: Reggio PH, editor. Cannabinoids and their receptors. Methods in Enzymology ed. London, UK: Elsevier; 2017a. pp. 405–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales P, Hurst DP, Reggio PH. In: Molecular Targets of the Phytocannabinoids: A Complex Picture. Kinghorn AD, Gibbons S, editors. Switzerland: Springer International Publishing; 2017b. pp. 103–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Eguchi H Teijin Pharma Limited. Japan. US8221990B2 Screening GPR12 receptor for substances having Nesfatin-1-like action, or which regulate Nesfatin-1 action. 2012 Jul 17;
- Nader N, Dib M, Daalis A, Kulkarni RP, Machaca K. Role for endocytosis of a constitutively active GPCR (GPR185) in releasing vertebrate oocyte meiotic arrest. Dev Biol. 2014;395:355–366. doi: 10.1016/j.ydbio.2014.08.036. [DOI] [PubMed] [Google Scholar]
- Oeckl P, Ferger B. Increased susceptibility of G-protein coupled receptor 6 deficient mice to MPTP neurotoxicity. Neuroscience. 2016;337:218–223. doi: 10.1016/j.neuroscience.2016.09.021. [DOI] [PubMed] [Google Scholar]
- Oeckl P, Hengerer B, Ferger B. G-protein coupled receptor 6 deficiency alters striatal dopamine and cAMP concentrations and reduces dyskinesia in a mouse model of Parkinson’s disease. Exp Neurol. 2014;257:1–9. doi: 10.1016/j.expneurol.2014.04.010. [DOI] [PubMed] [Google Scholar]
- Oh-I S, Shimizu H, Satoh T, Okada S, Adachi S, Inoue K, Eguchi H, Yamamoto M, Imaki T, Hashimoto K, et al. Identification of nesfatin-1 as a satiety molecule in the hypothalamus. Nature. 2006;443:709–712. doi: 10.1038/nature05162. [DOI] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG. Etiology and pathogenesis of parkinson’s disease. Annu Rev Neurosci. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- Padmanabhan S, Myers AG, Prasad BM. Constitutively active GPR6 is located in the intracellular compartments. FEBS Lett. 2009;583:107–112. doi: 10.1016/j.febslet.2008.11.033. [DOI] [PubMed] [Google Scholar]
- Patel KR, Cherian J, Gohil K, Atkinson D. Schizophrenia: Overview and Treatment Options. Pharmacy and Therapeutics. 2014;39:638–645. [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG, Howlett AC, Abood ME, Alexander SPH, Marzo VD, Elphick MR, Greasley PJ, Hansen HS, Kunos G. International Union of Basic and Clinical Pharmacology. LXXIX. Cannabinoid Receptors and Their Ligands: Beyond CB1 and CB2. Pharmacol Rev. 2010;62:588–631. doi: 10.1124/pr.110.003004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters M, Scott REM, Tully TP Helicon Therapeutics, Inc. USA. WO2008144453A1 Methods of treating cognitive disorders by inhibition of Gpr12, and screening method for Gpr12 modulators. 2008 Nov 27;
- Pisalyaput K, Tenner AJ. Complement component C1q inhibits β-amyloid- and serum amyloid P-induced neurotoxicity via caspase- and calpain-independent mechanisms. J Neurochem. 2007;104:696–707. doi: 10.1111/j.1471-4159.2007.05012.x. [DOI] [PubMed] [Google Scholar]
- Pisanti S, Malfitano AM, Ciaglia E, Lamberti A, Ranieri R, Cuomo G, Abate M, Faggiana G, Proto MC, Fiore D, et al. Cannabidiol: State of the art and new challenges for therapeutic applications. Pharmacol Ther. 2017;175:133–150. doi: 10.1016/j.pharmthera.2017.02.041. [DOI] [PubMed] [Google Scholar]
- Prasad BM, Hollins B, Lambert NA. Methods in Enzymology. London, UK: Elsevier; 2010. Chapter 10, Methods to Detect Cell Surface Expression and Constitutive Activity of GPR6. Constitutive Activity in Receptors and Other Proteins, Part A; pp. 179–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond LA, André VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS. Pathophysiology of Huntington’s disease: time-dependent alterations in synaptic and receptor function. Neuroscience. 2011;198:252–273. doi: 10.1016/j.neuroscience.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regard JB, Sato IT, Coughlin SR. Anatomical Profiling of G Protein-Coupled Receptor Expression. Cell. 2008;135:561–571. doi: 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid KBM. Proteins involved in the activation and control of the two pathways of human complement. Biochem Soc Trans. 1983;11:1–12. doi: 10.1042/bst0110001. [DOI] [PubMed] [Google Scholar]
- Rios-Cardona D, Ricardo-Gonzalez RR, Chawla A, Ferrell JE. A role for GPRx, a novel GPR3/6/12-related G-protein coupled receptor, in the maintenance of meiotic arrest in Xenopus laevis oocytes. Dev Biol. 2008;317:380–388. doi: 10.1016/j.ydbio.2008.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers NH, Landa A, Park S, Smith RG. Aging leads to a programmed loss of brown adipocytes in murine subcutaneous white adipose tissue. Aging Cell. 2012;11:1074–1083. doi: 10.1111/acel.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos RA. Huntington’s disease: a clinical review. Orphanet Journal of Rare Diseases. 2010;5:40–40. doi: 10.1186/1750-1172-5-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Medina J, Ledent C, Valverde O. GPR3 orphan receptor is involved in neuropathic pain after peripheral nerve injury and regulates morphine-induced antinociception. Neuropharmacology. 2011;61:43–50. doi: 10.1016/j.neuropharm.2011.02.014. [DOI] [PubMed] [Google Scholar]
- Saeki Y, Ueno S, Mizuno R, Nishimura T, Fujimura H, Nagai Y, Yanagihara T. Molecular cloning of a novel putative G protein-coupled receptor (GPCR21) which is expressed predominantly in mouse central nervous system. FEBS Lett. 1993;336:317–322. doi: 10.1016/0014-5793(93)80828-i. [DOI] [PubMed] [Google Scholar]
- Sánchez F, Smitz J. Molecular control of oogenesis. Biochimica et Biophysica Acta - Molecular Basis of Disease. 2012;1822:1896–1912. doi: 10.1016/j.bbadis.2012.05.013. [DOI] [PubMed] [Google Scholar]
- Serrano A, Pavon FJ, Suarez J, Romero-Cuevas M, Baixeras E, Goya P, de Fonseca FR. Obesity and the Endocannabinoid System: Is There Still a Future for CB1 Antagonists in Obesity? Current Obesity Reports. 2012;1:216–228. [Google Scholar]
- Song Z-H, Modi W, Bonner TI. Molecular Cloning and Chromosomal Localization of Human Genes Encoding Three Closely Related G Protein-Coupled Receptors. Genomics. 1995;28:347–349. doi: 10.1006/geno.1995.1154. [DOI] [PubMed] [Google Scholar]
- Southern C, Cook JM, Neetoo-Isseljee Z, Taylor DL, Kettleborough CA, Merritt A, Bassoni DL, Raab WJ, Quinn E, Wehrman TS, et al. Screening β-Arrestin Recruitment for the Identification of Natural Ligands for Orphan G-Protein–Coupled Receptors. J Biomol Screen. 2013;18:599–609. doi: 10.1177/1087057113475480. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Ishii K, Kasai K, Sung OY, Saeki Y. Neural expression of G protein-coupled receptors GPR3, GPR6, and GPR12 up-regulates cyclic AMP levels and promotes neurite outgrowth. J Biol Chem. 2007;282:10506–10515. doi: 10.1074/jbc.M700911200. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Miyagi T, Dohi E, Seki T, Hide I, Sotomaru Y, Saeki Y, Antonio Chiocca E, Matsumoto M, Sakai N. Developmental expression of GPR3 in rodent cerebellar granule neurons is associated with cell survival and protects neurons from various apoptotic stimuli. Neurobiology of Disease. 2014;68:215–227. doi: 10.1016/j.nbd.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Tanaka S, Shaikh IM, Chiocca EA, Saeki Y. The Gs-Linked Receptor GPR3 Inhibits the Proliferation of Cerebellar Granule Cells during Postnatal Development. PLoS ONE. 2009;4:e5922. doi: 10.1371/journal.pone.0005922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thathiah A, Horré K, Snellinx A, Vandewyer E, Huang Y, Ciesielska M, De Kloe G, Munck S, De Strooper B. β-arrestin 2 regulates Aβ generation and γ-secretase activity in Alzheimer’s disease. Nat Med. 2013;19:43–49. doi: 10.1038/nm.3023. [DOI] [PubMed] [Google Scholar]
- Thathiah A, Spittaels K, Hoffmann M, Staes M, Cohen A, Horré K, Vanbrabant M, Coun F, Baekelandt V, Delacourte A, et al. The orphan G protein-coupled receptor 3 modulates amyloid-beta peptide generation in neurons. Science (New York, NY) 2009;323:946–951. doi: 10.1126/science.1160649. [DOI] [PubMed] [Google Scholar]
- Tong L. B-Amyloid Peptide at Sublethal Concentrations Downregulates Brain-Derived Neurotrophic Factor Functions in Cultured Cortical Neurons. J Neurosci. 2004;24:6799–6809. doi: 10.1523/JNEUROSCI.5463-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torkildsen O, Myhr KM, Bø L. Disease-modifying treatments for multiple sclerosis - a review of approved medications. European Journal of Neurology. 2016;23:18–27. doi: 10.1111/ene.12883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourino C, Valjent E, Ruiz-Medina J, Herve D, Ledent C, Valverde O. The orphan receptor GPR3 modulates the early phases of cocaine reinforcement. Br J Pharmacol. 2012;167:892–904. doi: 10.1111/j.1476-5381.2012.02043.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlenbrock K, Gassenhuber H, Kostenis E. Sphingosine 1-phosphate is a ligand of the human gpr3, gpr6 and gpr12 family of constitutively active G protein-coupled receptors. Cell Signal. 2002;14:941–953. doi: 10.1016/s0898-6568(02)00041-4. [DOI] [PubMed] [Google Scholar]
- Uhlenbrock K, Huber J, Ardati A, Busch AE, Kostenis E. Fluid shear stress differentially regulates gpr3, gpr6, and gpr12 expression in human umbilical vein endothelial cells. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2003;13:75–84. doi: 10.1159/000070251. [DOI] [PubMed] [Google Scholar]
- Valverde O, Célérier E, Baranyi M, Vanderhaeghen P, Maldonado R, Sperlagh B, Vassart G, Ledent C. GPR3 Receptor, a Novel Actor in the Emotional-Like Responses. PLoS ONE. 2009;4:e4704. doi: 10.1371/journal.pone.0004704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosoughi R, Freedman MS. Therapy of MS. Clin Neurol Neurosurg. 2010;112:365–385. doi: 10.1016/j.clineuro.2010.03.010. [DOI] [PubMed] [Google Scholar]
- Waalen J, Buxbaum JN. Is older colder or colder older? The association of age with body temperature in 18,630 individuals. Journals of Gerontology - Series A Biological Sciences and Medical Sciences. 2011;66A:487–492. doi: 10.1093/gerona/glr001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Farhan M, Xu J, Lazarovici P, Zheng W. The involvement of DARPP-32 in the pathophysiology of schizophrenia. Oncotarget. 2017;8:53791–53803. doi: 10.18632/oncotarget.17339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt G, Karl T. In vivo Evidence for Therapeutic Properties of Cannabidiol (CBD) for Alzheimer’s Disease. Frontiers in Pharmacology. 2017;8:1–7. doi: 10.3389/fphar.2017.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CR, Wei Y, Qi ST, Chen L, Zhang QH, Ma JY, Luo YB, Wang YP, Hou Y, Schatten H, et al. The G protein coupled receptor 3 is involved in cAMP and cGMP signaling and maintenance of meiotic arrest in porcine oocytes. PLoS ONE. 2012;7:e38807. doi: 10.1371/journal.pone.0038807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Spriggs MK, Derry JM, Strockbine L, Park LS, VandenBos T, Zappone JD, Painter SL, Armitage RJ. Molecular characterization of the human interleukin (IL)-17 receptor. Cytokine. 1997;9:794–800. doi: 10.1006/cyto.1997.0240. [DOI] [PubMed] [Google Scholar]
- Ye C, Zhang Z, Wang Z, Hua Q, Zhang R, Xie X. Identification of a novel small-molecule agonist for human G protein-coupled receptor 3. The Journal of pharmacology and experimental therapeutics. 2014;349:437–443. doi: 10.1124/jpet.114.213082. [DOI] [PubMed] [Google Scholar]
- Yin H, Chu A, Li W, Wang B, Shelton F, Otero F, Nguyen DG, Caldwell JS, Chen YA. Lipid G Protein-coupled Receptor Ligand Identification Using β-Arrestin PathHunter™ Assay. J Biol Chem. 2009;284:12328–12338. doi: 10.1074/jbc.M806516200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B-l, Li Y, Ding J-h, Dong F-l, Hou Y-j, Jiang B-c, Shi F-x, Xu Y-x. Sphingosine 1-phosphate acts as an activator for the porcine Gpr3 of constitutively active G protein-coupled receptors. Journal of Zhejiang University Science B. 2012;13:555–566. doi: 10.1631/jzus.B1100353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou S, Wang B, Ni F, Wang J, Cao Y, Xu M. GPR3 may not be a potential candidate gene for premature ovarian failure. Reproductive BioMedicine Online. 2010;20:53–55. doi: 10.1016/j.rbmo.2009.10.013. [DOI] [PubMed] [Google Scholar]