

SUMMARY
The PD-1 pathway, consisting of the co-inhibitory receptor PD-1 on T cells and its ligand (PD-L1) on antigen-presenting cells (APCs), is a major mechanism of tumor immune evasion. PD-1 and PD-L1 blockade antibodies have produced remarkable clinical activities against a subset of cancers. Binding between T cell-intrinsic PD-1 and APC-intrinsic PD-L1 triggers inhibitory signaling to attenuate the T cell response. Here, we report that PD-1 is co-expressed with PD-L1 on tumor cells and tumor-infiltrating APCs. Using reconstitution and cell culture assays, we demonstrate that the co-expressed PD-1 binds to PD-L1 in cis. Such interaction inhibits the ability of PD-L1 to bind T cell-intrinsic PD-1 in trans and, in turn, represses canonical PD-L1/PD-1 inhibitory signaling. Selective blockade of tumor-intrinsic PD-1 frees up tumor-intrinsic PD-L1 to inhibit T cell signaling and cytotoxicity. Our study uncovers another dimension of PD-1 regulation, with important therapeutic implications.
In Brief
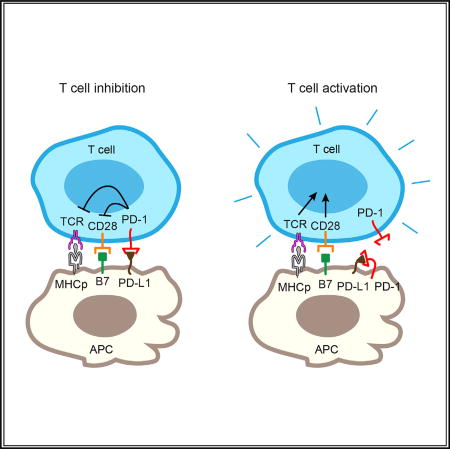
Zhao et al. show that the T cell inhibitory receptor PD-1 expressed on tumor cells and tumor-infiltrating APCs neutralizes its ligand, PD-L1, in cis to inhibit canonical PD-1 signaling. Selective blockade of tumor-intrinsic PD-1 frees up tumor PD-L1 for T cell suppression.
INTRODUCTION
Recent years have seen the exciting progress in harnessing the immune system to combat human cancer. A highly successful modality is to reactivate the immune system that is aberrantly repressed by cancers. A key cancer immunotherapy target is programmed cell death protein-1 (PD-1), best known as a T cell co-inhibitory receptor. The interaction between PD-1 on T cells and its ligand PD-L1, which is highly expressed on several types of human tumor cells and tumor infiltrating immune cells, restrains the activity of effector T cells against human cancers and chronic virus infections (Baitsch et al., 2011; Chen and Mellman, 2013; Pardoll, 2012; Pauken and Wherry, 2015; Sharma and Allison, 2015; Zou et al., 2016). Antibodies that block PD-L1/ PD-1 interactions have produced durable clinical benefit in several cancer indications in a small subset of patients.
To date, mechanistic studies of PD-1 have been largely focused on its role on T cells. Absent on naive T cells, PD-1 is inducibly expressed on T cells by T cell antigen receptor (TCR) signal and then acts as a molecular brake to prevent uncontrolled T cell activity. Upon binding to its ligand PD-L1 on the antigen-presenting cell (APC), a pair of tyrosines within the cytoplasmic tail of PD-1 becomes phosphorylated and recruits the protein tyrosine phosphatases SHP2 and SHP1, which dephosphorylate both the TCR and co-stimulatory signaling components (Hui et al., 2017; Parry et al., 2005; Sheppard et al., 2004; Yokosuka et al., 2012). These biochemical events ultimately lead to the attenuation of T cell proliferation, cytokine production, and cytolytic activities (Keir et al., 2008).
Despite the widely accepted notion that PD-1 primarily functions as a T cell inhibitory receptor, PD-1 has also been found to be expressed on other types of immune and non-immune cells, including B cells, macrophages, dendritic cells (DCs), and even some tumor cells (Keir et al., 2008; Kleffel et al., 2015). Mounting recent evidence indicates important roles of PD-1 on non-T cells in regulating the survival of DCs, the phagocytosis of macrophages, and the glycolysis of tumor cells (Gordon et al., 2017; Kleffel et al., 2015; Park et al., 2014). Similarly, PD-L1, the PD-1 ligand well known to be expressed on tumor cells and professional APCs (e.g., B cells, macrophages, and DCs), is also expressed on activated T cells at low levels (Keir et al., 2008). Hence, PD-L1 and PD-1 might be co-expressed on multiple cell types, raising the questions of whether they can interact with each other in cis and how this putative cis interaction might regulate immune responses.
In stark contrast to the intensively studied PD-L1/PD-1 trans interaction, the existence and functional consequence of the cis interaction are unknown. Challenges for this effort include the co-expression of PD-L1 and PD-1 on both APCs and T cells, signaling in both directions, and the potential crosstalk with other signaling axes.
In this work, we investigated whether PD-1 and PD-L1 interact in cis and how the potential cis interaction regulates classical PD-1 signaling outputs using well-defined in vitro reconstitution, cellular reconstitution, and cell culture assays. In both HEK293T cells and liposomes reconstituted with both PD-1 and PD-L1, we determined their molecular proximity using Förster resonance energy transfer (FRET). We next asked whether the presence of cis-PD-1 impacts the ability of PD-L1 to engage PD-1 in trans, using a liposome–bilayer conjugation assay, a cell-bilayer assay, and APC-T cells assays with multiple signaling readouts. Finally, we determined how antibody blockade of cis-PD-1 affects T cell signaling and cytotoxicity.
RESULTS
PD-1 and PD-L1 Are Co-expressed on a Subset of Tumor Cells and Tumor-Infiltrating APCs
We first analyzed PD-1 and PD-L1 co-expression on multiple cell types in the context of tumors using flow cytometry. In one non-small cell lung cancer (NSCLC) human patient, PD-1 and PD-L1 are co-expressed on a subset of tumor cells and tumor infiltrating professional APCs, including DCs, macrophages, and myeloid derived suppressor cells (MDSCs) (Figure 1). Interestingly, in another NSCLC patient, PD-1 and PD-L1 co-expression was detected on tumor-infiltrating professional APCs, but not on tumor cells (Figure S1). Little to no PD-1/PD-L1 double-positive cells were found on peripheral blood mononuclear cells (PBMCs) from either patients or healthy individual controls. We also found that PD-1 and PD-L1 are co-expressed on 99.8% of EL4 mouse lymphoma cells (Figure S2). Using fluorescent beads as standards, we then quantitated the expression levels of both PD-1 and PD-L1 (STAR Methods) and found that PD-1 is generally expressed at a higher level than PD-L1 on PD-1/PD-L1 double-positive cells isolated from NSCLC tumor sites (Table S1).
Figure 1. Co-expression of PD-1 and PD-L1 in a Lung Cancer Patient.

(A) Gating strategy for analyzing PD-1 and PD-L1 expression in human patient samples where CD45+/CD3+ cells were classified as T cells, CD45+/CD3− cells as B cells, CD45+CD11c+ cells as DCs, CD45+CD11b+ cells as macrophages (Macs) and MDSCs, and CD45− cells within the tumor site as tumor cells.
(B) Expression of PD-1 and PD-L1 on the indicated cell types derived from PBMCs from a healthy human individual, a lung cancer patient, and the tumor site of the same patient.
See also Figures S1 and S2 and Table S1.
PD-1 and PD-L1 Bind to Each Other in cis
Co-expression of PD-1 and PD-L1 raises the possibility that they might bind in cis on cell membranes. We tested this idea using FRET analysis with confocal microscopy. To this end, we co-transfected CLIP-tagged PD-L1 and SNAP-tagged PD-1 into HEK293T cells and labeled them orthogonally with an energy donor (Dy547) and acceptor (Alexa Fluor 647 [AF647]), respectively. Using flow cytometry and fluorescent beads, we found that PD-1 and PD-L1 are expressed at 72 and 91 molecules/µm2 respectively, which is comparable to their levels in NSCLC tumor sites (Table S1). Using confocal microscopy, we found that photobleaching of PD-1-conjugated AF647 substantially increases the fluorescence of PD-L1 conjugated Dy547 (Figure 2A). The recovery of donor fluorescence after acceptor photobleaching suggests molecular proximity of PD-1 and PD-L1. Similar levels of FRET signal were also detected between PD-1 and PD-L2, a second ligand of PD-1 (Figure 2B). Replacement of PD-1 with a mutant version (K78A) with defective PD-L1 binding (Lázár-Molnár et al., 2008) or with B7.2, a structurally related APC surface protein with no reported PD-L1 binding activity, significantly decreased the FRET efficiency (p < 0.001). These data suggest that PD-1 interacts in cis with both PD-L1 and PD-L2 on cell membranes.
Figure 2. PD-1 Associates with PD-L1 and PD-L2 in cis on Cell Membranes.

(A) An acceptor-photobleaching FRET assay showing the molecular nearness of PD-1 and PD-L1 on the same cell membrane. Shown are confocal microscopy images of a HEK293T cell co-transduced with PD-L1 (labeled with Dy547, the energy donor, indicated by green) and either PD-1, PD-1(K78A) or B7.2 (labeled with AF647, the energy acceptor, indicated by red). Shown from left to right are excited donor (Dy547*PD-L1) images before bleaching, excited acceptor (AF647*PD-1, AF647*PD-1 (K78A) or AF647*B7.2) images before bleaching, excited donor images after bleaching, excited acceptor images after bleaching, calculated FRET efficiency images (pseudo-color, with red to blue color spectrum represents strong to weak FRET efficiency), and differential interference contrast (DIC) images (STAR Methods). A bar graph on the right summarizes the FRET efficiencies as mean ± SEM from at least 25 cells from three independent experiments.
(B) An acceptor-photobleaching FRET assay showing the molecular nearness of PD-1 and PD-L2 on the same cell membrane. Experiments were performed in the same manner as in (A), except PD-L1 was replaced with PD-L2.
Scale bars, 10 µm.
Due to the complex environment of a cell membrane, it remains possible that the molecular proximity between PD-1 and its ligands is mediated by other proteins or is driven by lipid microdomains such as rafts. To rule out these possibilities, we next assessed the molecular nearness of PD-L1 and PD-1 in a cell-free membrane reconstitution system using purified recombinant proteins (Figure 3A). We pre-attached nickel chelating lipid (DGS-NTA-Ni) containing large unilamellar vesicles (LUVs) with a purified His10-tagged extracellular domain of PD-L1 labeled with an energy donor (SNAP-Cell-505) (SC505*PD-L1EX-His). Subsequent addition of PD-1EX-His, labeled with an energy acceptor (SNAP-Cell-TMR [tetramethylrhodamine]) (TMR*PD-1EX-His) triggered a rapid and robust quenching of the PD-L1 fluorescence (Figure 3B, black trace). By contrast, the addition of TMR-labeled PD-1(K78A)EX mutant or B7.2EX elicited a much weaker quenching of the PD-L1 fluorescence (Figure 3B, orange and gray traces). As expected, similar levels of PD-1-mediated quenching were observed when the LUVs were pre-attached with the donor-labeled, His10-tagged extracellular domain of PD-L2 (SC505*PD-L2EX-His) (Figure 3C).
Figure 3. PD-1 Directly Interacts with PD-L1 and PD-L2 in cis on Reconstituted LUV Membranes.

(A) Cartoon depicting the experiment scheme of a kinetic FRET assay. DGS-NTA-Ni containing LUVs were pre-attached with SC505 (energy donor)-labeled PD-L1EX-His (SC505*PD-L1EX-His). Subsequently, TMR (energy acceptor)-labeled PD-1EX-His (TMR*PD-1EX-His) was added and bound to the LUVs, causing SC505*PD-L1EX and TMR*PD-1EX to co-exist on the same LUVs. The donor (SC505) fluorescence was monitored throughout the process (STAR Methods).
(B) Representative time course of SC505*PD-L1 EX-His fluorescence intensity (black trace) showing a robust quenching induced by TMR*PD-1EX-His addition. Orange trace is the same as the black trace condition, except TMR*PD-1EX-His was replaced with TMR*PD-1(K78A)EX -His. Gray trace is the same as the black trace condition, except TMR*PD-1EX-His was replaced with TMR*B7.2EX-His. Shown is one representative result from three independent replicates. Data were normalized as described in STAR Methods.
(C) Same as (B), except SC505*PD-L1EX-His-attached LUVs were replaced with SC505*PD-L2EX-His-attached LUVs.
See also Figures S3 and S4.
It is possible that PD-1EX-His competes with PD-L1EX-His for binding sites on the LUVs, causing a fluorescence change. However, we ruled out this possibility by using unlabeled PD-1EX-His, which failed to alter PD-L1 fluorescence (Figure S3, black). Indeed, the DGS-NTA-Ni concentration (16.6 µM) was 500-fold higher than the total protein concentration (33.3 nM), which is more than enough to bind both proteins without competition. The energy transfer depends on the membrane localization of PD-1, because removing the His tag from PD-1 abolished the quenching (Figure S3, gray). This result indicates that binding between LUV-attached PD-L1 and solution PD-1 was minimal. Indeed, the bulk concentration of PD-1 (25 nM) was two orders of magnitude lower than the reported dissociation constant (770–7,800 nM; Butte et al., 2007; Cheng et al., 2013; Lázár-Molnár et al., 2017; Maute et al., 2015) of the PD-L1/PD-1 interaction. Finally, PD-L1 and PD-1 binding in trans via LUV conjugation led to a much slower and smaller decrease of PD-L1 fluorescence (Figure S3, orange), indicating that the rapid, robust PD-L1 quenching observed with TMR*PD-1EX addition was due to direct cis interaction with PD-1 from the same membrane.
Next, we determined the affinity of the PD-L1/PD-1 cis interaction. To this end, we fixed the concentration of LUV-bound SC505*PD-L1EX-His at 8.3 nM and measured its percent quenching as a function of the level of TMR*PD-1EX-His (Figure S4). Equal concentrations of TMR*B7.2EX-His were run in parallel to reflect quenching due to crowding effect (Figure S4). Plotting the percent quenching (calculated by subtracting the PD-1 signals from the corresponding B7.2 signals, see STAR Methods) against PD-1 concentration yielded a dissociation constant (KD) of 18 ± 1 nM, which is much lower than the solution KD reported previously. These data suggest that confinement of PD-1 and PD-L1 on the same membrane can facilitate their interaction in cis. The curve fitting also revealed a Hill coefficient of 2.4 ± 0.2 (Figure S4), indicating a weak cooperativity of the PD-1/PD-L1 cis interaction.
PD-L1/PD-1 cis Interaction Inhibits the Ability of PD-L1 to Bind PD-1 in trans
Having demonstrated that PD-L1 directly binds to PD-1 in cis, we next determined whether the cis interaction affects the ability of PD-L1 to engage PD-1 from a different membrane (i.e., trans interaction). Accordingly, we developed a microscopy assay to measure membrane apposition driven by the PD-1/PD-L1 trans interaction. In this assay, two forms of lipid bilayers, LUVs (containing Bodipy-PE as the probe) and a supported lipid bilayer (SLB), were reconstituted with PD-L1EX-His and AF647-labeled PD-1EX-His, respectively (STAR Methods). Notably, after a 5-min incubation with the PD-L1EX-coupled LUVs and extensive washes, the SLB captured a number of PD-L1EX-coupled LUVs, each registered as a bright green dot in the total internal reflection fluorescence (TIRF) field, owing to the Bodipy fluorescence (Figure 4A, Bodipy channel). In addition, AF647-labeled PD-1EX-His was clearly enriched under the PD-L1EX-coupled LUVs (Figure 4A, AF647 channel and merged), suggesting that the SLB-LUV association was mediated by the PD-1/PD-L1 trans interaction. We confirmed this notion by using B7.2EX -coupled LUVs, which barely bound to PD-1EX functionalized SLB (Figures 4B and 4F). Importantly, an equivalent concentration of LUVs attached with both PD-L1EX and PD-1EX exhibited a substantially weaker SLB binding (Figures 4C and 4F), indicating that the PD-1/PD-L1 cis interaction inhibits the ability of PD-L1 to bind PD-1 in trans. Moreover, replacement of wild-type (WT) PD-1 with an equal concentration of either PD-1(K78A)EX or B7.2EX on the LUVs restored LUV-bilayer conjugation (Figures 4D–4F). By titrating the level of cis-PD-1, we found that an ~3-fold excess of PD-1 is sufficient to block the PD-1/PD-L1 trans interaction (Figure S5).
Figure 4. PD-L1/PD-1 cis Interaction Prevents PD-L1 from Binding to PD-1 in trans.

(A–E) Left: cartoons depicting a SC647*PD-1EX-coupled SLB overlaid with Bodipy/DGS-NTA-Ni LUVs pre-attached with PD-L1EX (A), B7.2EX (B), PD-L1EX/PD-1EX combined (1:3 molar ratio) (C), PD-L1EX/PD-1(K78A)EX combined (1:3 molar ratio) (D), or PD-L1EX/B7.2EX combined (1:3 molar ratio) (E). Right: representative TIRF images of the SC647*PD-1EX-coupled SLB after incubation with the designed LUVs and a wash (STAR Methods) in the Bodipy channel (showing LUVs bound to the SLB), the AF647 channel (showing SC647*PD-1EX on the SLB), and the two channels combined.
(F) A bar graph summarizing the fluorescence intensity (F.I.) of the Bodipy channel under each condition (A–E), normalized to the intensity in (A). Data are shown as mean ± SEM from 10 independent TIRF fields.
Scale bars, 5 µm. See also Figure S5.
Cis-PD-1 Neutralizes the Ability of PD-L1 to Trigger PD-1 Microclusters in T Cells
We next determined how the PD-L1/PD-1 cis interaction affects the ability of PD-L1 to trigger PD-1 signaling in living T cells. A hallmark event of PD-1 activation is the formation of PD-1 microclusters, as revealed by microscopy studies using SLB as an artificial APC (Groves and Dustin, 2003; Hui et al., 2017; Yokosuka et al., 2012). Here, we used this T cell/SLB system to determine how the PD-L1/PD-1 cis interaction affects PD-1 microcluster formation in OT-I CD8+ cytotoxic T cells. We functionalized DGS-NTA-Ni containing SLB with peptide (SIINFEKL) linked major histocompatibility complex class I (MHC-I) H2Kb (abbreviated as pMHC, for TCR activation), PD-L1EX-His (for PD-1 activation), and B7.2EX-His as the filler molecule. Using TIRF microscopy, we found that the pMHC/PD-L1EX/B7.2EX SLB elicited plasma membrane microclusters of both TCR and PD-1 (Figures 5A and 5D). The TCR and PD-1 microclusters appeared to be partially colocalized, in agreement with our recent report (Hui et al., 2017). Strikingly, replacement of B7.2EX-His by equal concentrations of PD-1EX-His (three-fold the concentration of PD-L1EX) on SLB largely abolished PD-1 clusters, while leaving TCR clusters intact (Figures 5B and 5D). This result indicates that PD-L1/PD-1 cis interaction prevents PD-L1 from engaging PD-1 on T cells. In support of this model, treatment of SLB PD-1 with a PD-1 blockade antibody J43 partially recovered the PD-1 microclusters (Figures 5C and 5D).
Figure 5. Cis-PD-1 Inhibits the Ability of PD-L1 to Trigger PD-1 Microclusters in CD8+ T Cells.

(A) A cell-bilayer assay showing that ligand-functionalized SLB triggers TCR and PD-1 clusters in OT-I CD8+ cells. PD-1–mCherry transduced OT-I cells, labeled with TCR-β antibody (H57-597*AF647), were plated on an SLB attached with peptide (SIINFEKL)-linked MHC-I H2Kb (pMHC), B7.2EX, and PD-L1EX (see cartoon on the left and STAR Methods for details). Shown on the right are representative TIRF images of PD-1–mCherry (rendered in green) and H57-597*AF647 (rendered in purple, indicating TCR distribution) 30 s after the cell-bilayer contact.
(B) Same as (A), except B7.2EX was replaced with equivalent concentrations of PD-1EX on the SLB. The pMHC/PD-L1EX/PD-1EX reconstituted SLB was plated with PD-1–mCherry transduced OT-I (see cartoons on the left). Representative TIRF images of PD-1–mCherry and H57-597*AF647 are shown on the right.
(C) Same as (B), except PD-1EX was preincubated with blockade antibody J43 (see cartoons on the left). Representative TIRF images of PD-1–mCherry and H57-597*AF647 are shown on the right.
(D) Bar graphs showing the clustering index of PD-1 and TCR in each condition (A–C). Data are the means ± SEM of at least 20 cells from three independent replicates.
Scale bars, 5 µm.
Co-expression of PD-1 with PD-L1 on APCs Blunts PD-1 Signaling in T Cells
We next turned to an APC-T cell co-culture system to further investigate the roles of PD-1 on APCs using superantigen-loaded Raji B cells as the APCs for Jurkat T cells (Tian et al., 2015). Because neither Raji nor Jurkat cells express PD-1 or PD-L1, the Raji-Jurkat system offers a clean platform for dissecting the roles of cis and trans PD-L1/PD-1 interactions. Recently, we showed that trans interaction between virally transduced PD-L1 on Raji cells and virally transduced PD-1 on Jurkat cells causes the suppression of TCR/CD28 signaling and interleukin-2 (IL-2) production (Hui et al., 2017). Here, we examined how co-expression of PD-L1 with PD-1 (cis) on Raji cells affects the ability of PD-L1 to activate PD-1 (trans) on Jurkat cells. Specifically, we created two types of Raji cells via lentiviral transduction and fluorescence activated cell sorting (FACS): PD-L1-positive PD-1-negative Raji cells (PD-L1+) and PD-L1/PD-1 double-positive Raji cells (PD-L1+/PD-1+) (Figure 6A). We then stimulated PD-1-transduced Jurkat cells with either type of Raji cells preloaded with superantigen and measured classical PD-1 signaling readouts in Jurkat cells. Antigen-loaded parental Raji cells, which lack both PD-L1 and PD-1, were used as controls. First, using confocal microscopy, we found that PD-1–mGFP transduced Jurkat cells form a conjugate with parental Raji cells, with no enrichment of PD-1–mGFP to the Jurkat-Raji interface (Figures 6B and 6E). As expected, PD-1–mGFP became strongly enriched to the conjugate interface when PD-L1–mCherry was expressed on Raji cells (Figures 6C and 6E), owing to PD-L1/PD-1 trans interaction. Remarkably, when PD-1 was co-expressed with PD-L1–mCherry on Raji cells, the interface enrichment of PD-1–mGFP significantly decreased (Figures 6D and 6E), consistent with the results in the T cell-SLB assay (Figure 5). On the Raji APC side, PD-1 appeared to co-cluster with PD-L1 on the cell membrane, with no interface enrichment (Figure S6). The basis for PD-1/PD-L1 co-clustering is unclear, but it might be due to intracellular signaling events initiated by the cis interaction. We also found that the PD-1/PD-L1 cis interaction also appears to occur on T cells, because co-expressing PD-L1 on PD-1+ Jurkat cells inhibited the synaptic enrichment of PD-1 (Figure S7).
Figure 6. Co-expression of PD-1 with PD-L1 on APCs Inhibits PD-1 Signaling in T Cells.

(A) Left: cartoon showing a cell culture assay in which PD-1 transduced Jurkat cells were stimulated with three types of antigen-loaded Raji B cells: (1) parental Raji cells that express neither PD-L1 nor PD-1, (2) Raji cells transduced with only PD-L1 (PD-L1+), and (3) Raji transduced with both PD-L1 and PD-1 (PD-L1+/PD-1+). On the right are FACS histograms showing PD-1 and PD-L1 surface expression of parental Jurkat cells, PD-1–mGFP transduced Jurkat (PD-1+) cells, parental Raji cells, PD-L1–mCherry transduced Raji (PD-L1+) cells, and Raji cells co-transduced with both PD-1–mGFP and PD-L1–mCherry (PD-L1+/PD-1+). a.u., arbitrary units.
(B–D) Jurkat cells expressing PD-1–mGFP (PD-1+) were conjugated with Raji B cells (parental) (B), Raji cells transduced with only PD-L1–mCherry (PD-L1+) (C), or Raji cells co-transduced with PD-L1–mCherry and unlabeled PD-1 (PD-L1+/PD-1+) (D), as illustrated in the cartoon on the left (CD28 and B7 are omitted in this cartoon for simplicity). Shown on the right are confocal images of the cell conjugate acquired 2 min after cell-cell contact. mGFP and mCherry signals are shown as green and magenta, respectively. Scale bars, 10 µm.
(E) Bar graph summarizing the interface enrichment indices (calculated as described in STAR Methods) of the three conditions shown in (B). Data are shown as mean ± SEM; n = 35 cells from three independent experiments. See also Figures S6 and S7.
(F) A representative western blot showing the levels ZAP70-Y493 phosphorylation and p85 (PI3K regulatory subunit) co-immunoprecipitated (IP) with CD28 from the lysates of the indicated Jurkat-Raji co-culture. Jurkat cells expressing PD-1–mGFP (PD-1+) were stimulated with Raji cells (parental), Raji transduced with PD-L1–mCherry (PD-L1+), or Raji cells co-transduced with PD-L1–mCherry and PD-1–mGFP (PD-L1+/PD-1+); the times at which the co-culture was lysed are indicated (STAR Methods). WCL, whole cell lysate.
(G and H) Bar graphs summarizing immunoblots in (F), including pY493-ZAP70 immunoblot (G) and CD28 coIP p85 immunoblot (H). The optical density corresponding to each band was quantified by ImageJ and normalized to the conditions in which parental Raji cells were used as the APCs. Data are presented as mean ± SEM from three independent replicates.
(I) Bar graph summarizing IL-2 levels in the medium of the indicated Jurkat-Raji co-culture, as described in (B)–(D), 24 hr after cell-cell contact. Data are presented as mean ± SEM from three independent measurements, with each run performed in triplicate.
Next, we assayed for membrane-proximal intracellular signaling events associated with PD-1 activation, including the dephosphorylation of ZAP70 and CD28, key targets of PD-1 bound phosphatases. In agreement with our recent report (Hui et al., 2017), ZAP70 phosphorylation (measured by an anti-pY493 antibody) and CD28 phosphorylation (measured by co-immunoprecipitated p85) were both induced by Raji-Jurkat contact, becoming detectable at 2 min and increased at 5 min (Figure 6F–6H, Raji parental cells). Replacement of the parental Raji cells with Raji (PD-L1+) cells substantially decreased the phosphorylation of ZAP70 and CD28 at 2 min [Figures 6F–6H, Raji (PD-L1+) cells]. Notably, when equal numbers of Raji (PD-L1+/ PD-1+) cells were used as the APCs, phosphorylation of ZAP70 and CD28 were largely recovered [Figures 6F–6H, Raji (PD-L1+/PD-1+)]. Consistent with these membrane-proximal signaling outputs, IL-2) secretion, a distal output of TCR/CD28 signaling, also recovered significantly when PD-1 was co-expressed with PD-L1 on Raji APCs (Figure 6I). Collectively, these data demonstrate that APC-intrinsic PD-1 inhibits the ability of PD-L1 to trigger PD-1 signaling in T cells.
Selective Blockade of APC-Intrinsic PD-1 Inhibits T Cell Signaling
Conceivably, PD-1 blockade antibodies might act on both T cell-intrinsic PD-1 and APC-intrinsic PD-1 in vivo. We next sought to decouple these two blockade actions and to determine their respective effects on the T cell response. First, we asked in the Raji-Jurkat conjugation assay how blockade of Raji PD-1 or Jurkat PD-1 affects the synaptic enrichment of Jurkat PD-1. To this end, we began with the condition used in Figure 6D, in which co-expression of PD-1 with PD-L1 on Raji cells inhibits the synaptic enrichment of PD-1 from Jurkat cells (Figures 7A and 7E). Remarkably, preincubation of the PD-1 blockade antibody pembrolizumab with PD-1+/PD-L1+ Raji significantly enhanced the synaptic enrichment of Jurkat PD-1 and Raji PD-L1 (Figures 7B and 7E), suggesting that blockade of APC-intrinsic PD-1 frees up PD-L1 for engaging T cell intrinsic PD-1. By contrast, preincubation of pembrolizumab with only Jurkat cells, or with both Raji and Jurkat cells, eliminated the interface enrichment of Jurkat PD-1 and Raji PD-L1 (Figures 7C–7E), confirming that the synaptic enrichment is a consequence of PD-L1/PD-1 trans interaction.
Figure 7. Blockade of APC-Intrinsic PD-1 Enhances the Synaptic Enrichment of T Cell-Intrinsic PD-1 and Inhibits T Cell-Mediated Cytotoxicity.

(A) Left: cartoon showing a Jurkat cell expressing PD-1–mGFP (shown as PD-1 in green) conjugated with a Raji cell co-transduced with PD-L1–mCherry and unlabeled PD-1. Right: representative confocal images of the conjugate at the indicated channel acquired 2 min after cell-cell contact. Scale bars, 10 µm.
(B–D) Same as (A), except that Raji (PD-L1+/PD-1+) cells (B), Jurkat (PD-1–mGFP) cells (C), or both (D) were preincubated with pembrolizumab and washed extensively prior to conjugation. Scale bars, 10 µm.
(E) Bar graph comparing the interface enrichment indices (calculated as described in STAR Methods) of the four conditions shown in (A)–(D). Data are expressed as mean ± SEM; n = 40 cells from three independent experiments.
(F) Cartoon illustrating the cytotoxicity assay, in which EL4 cells were used as the target for OT-I cytotoxic T cells.
(G) Bar graph summarizing the OT-I cytotoxicity under indicated conditions, with PD-1 blockade antibody J43 or isotype antibody (Iso) preincubated with neither cell, only EL4, only OT-I, or both cell types. See STAR Methods for details. Cytotoxicity was normalized to the no blockade antibody condition (i.e., EL4 and OT-I preincubated with the isotype control). n.s., not significantly different. Data are presented as mean ± SEM from three independent replicates.
(H) Left: FACS histograms showing PD-1 and PD-L1 surface expression in parental EL4 (EL4 WT), PD-1 knockout EL4 (EL4 PD-1 KO). Right: bar graph summarizing the OT-I cytotoxicity under indicated conditions. OT-I cells preincubated with either PD-1 blockade antibody J43 or its isotype was co-cultured with parental EL4 (EL4 WT) and PD-1 knockout EL4 (EL4 KO), as described in STAR Methods. Cytotoxicity was normalized to the condition containing isotype-treated OT-I and WT EL4 (i.e., OT-I [Iso] + EL4 [WT]. n.s., not significant. Data are presented as mean ± SEM from three independent replicates.
Finally, we determined the functional significance of APC-intrinsic PD-1 and antibody blockade effects in a T cell-mediated tumor lysis assay. For this purpose, we analyzed the cytotoxicity of murine OT-I CD8+ T cells using EL4 lymphoma cells, a well-established OT-I target (Figure 7F). Unlike Jurkat or Raij cells, WT EL4 cells co-express PD-1 and PD-L1 (Figure S2). In the absence of PD-1 blockade antibodies, co-culturing OT-I with peptide-loaded EL4 led to lysis of EL4 (Figure 7G, black; STAR Methods). Strikingly, preincubation of EL4 with a murine PD-1 blockade antibody (J43) significantly decreased the cytotoxicity of OT-I (Figure 7G, white), suggesting EL4-intrinsic PD-1 promotes T cell cytotoxicity. This positive role of PD-1 contrasts the conventional view of PD-1 as a negative regulator for T cell signaling. Moreover, the effect of EL4-specific PD-1 blockade depends on the presence of T cell-intrinsic PD-1, because preincubation of J43 with both OT-I and EL4 eliminated the effect of EL4 PD-1 blockade (Figure 7G, dark gray). Of note, preincubation of J43 with OT-I alone also failed to affect OT-I cytotoxicity compared to the no-treatment condition (Figure 7G, light gray versus black), suggesting that PD-L1 on EL4 was completely quenched by PD-1 in cis. Indeed, using fluorescent beads as standards, we found that PD-1 (2,975 molecules/µm2) is much more highly expressed than PD-L1 (137 molecules/µm2) on EL4 cells (Table S1). Finally, we confirmed the antibody blockade effects using PD-1 knockout (KO) EL4 cells (Figures 7H and S2). Collectively, these data demonstrate that tumor-intrinsic PD-1 quenches PD-L1 in cis to promote T cell cytotoxicity.
DISCUSSION
The data presented here demonstrate that PD-1 and PD-L1 interact directly in cis with high affinity on cell membranes and that this cis interaction competes with their trans interaction to inhibit canonical PD-1 signaling. Our study suggests that the availability of APC-intrinsic PD-L1 for triggering the PD-1 pathway in T cells is negatively regulated by PD-1 on APCs. Through quantitative measurements, we show that a 3-fold molar excess of cis PD-1 is sufficient to neutralize PD-L1, suggesting that the cis interaction is a strong regulatory mechanism for the PD-L1/PD-1 pathway.
Previous work has established the bidirectional signaling of the PD-L1/PD-1 axis across the immunological synapse. On one hand, PD-L1 on APCs activates PD-1 on T cells to negatively regulate the T cell response (Dong et al., 1999; Freeman et al., 2000; Mazanet and Hughes, 2002). This also represents the best-understood mechanism of the signaling axis. On the other hand, the PD-L1 on T cells might activate PD-1 on APCs to impact the function of APCs, including inhibiting the survival of DCs (Park et al., 2014), suppressing the phagocytic activity of macrophages (Gordon et al., 2017), and enhancing the metabolism of tumor cells (Kleffel et al., 2015).
Our work has uncovered a positive role of APC-intrinsic PD-1 and another dimension of regulation of the PD-1 pathway. Our data with lentivirally transduced Jurkat cells also show that the PD-L1/PD-1 cis interaction can occur on T cells (Figure S7). However, due to the low expression of PD-L1 on naturally occurring T cells (Keir et al., 2008) (Figures 1 and S1), we speculate that the PD-L1/PD-1 cis interaction on T cells has little impact on the effective level of PD-1. However, we cannot rule out the possibility that PD-L1 is expressed on T cells at a higher level under certain circumstances. It is also possible that the PD-L1/PD-1 cis interaction can trigger productive signaling (Figure S6) in the absence of trans ligands or receptors, and this topic warrants further investigation. The net outcome of the PD-L1/PD-1 pathway likely depends on their expression levels on both APCs and T cells.
PD-L1 expression on APCs (tumors and immune cells) has been used as a predictive and prognostic marker (Herbst et al., 2014; Patel and Kurzrock, 2015; Ribas and Hu-Lieskovan, 2016), but little to no correlation between PD-L1 expression and therapeutic response was observed under several scenarios (Kim et al., 2016; Maleki Vareki et al., 2017; Mu et al., 2011; Song et al., 2013). Of note, high expression of PD-1 and PD-L1 in tumor tissues was found to be associated with better prognosis in breast cancer, colorectal cancer, human papillomavirus (HPV)-associated head and neck cancer, and follicular lymphoma patients (Badoual et al., 2013; Carreras et al., 2009; Li et al., 2016; Sabatier et al., 2015). In particular, high levels of both PD-1 and PD-L1 on ovarian cancer cells correlate with favorable prognosis (Darb-Esfahani et al., 2016). In line with these findings, our work shows that PD-1 expressed on tumor cells or professional APCs would effectively quench PD-L1 to disrupt PD-L1/PD-1 signaling to T cells. This finding suggests that the PD-L1 level alone is insufficient to predict whether the PD-1 pathway contributes to tumor immune evasion. Co-expressed PD-1 and other potential cis regulators need to be co-measured.
Our study has shown that both cis and trans PD-1/PD-L1 interactions are susceptible to antibody blockade. Selective blockade of the APC intrinsic PD-1 and T cell intrinsic PD-1 produced the opposite effects on the interface recruitment of PD-1 (Figure 7E). The net effect of PD-1 blockade antibody in vivo would thus depend on the relative expression levels of cis and trans PD-1. In the future, it might be possible to develop agents to selectively block the PD-L1/PD-1 trans interaction for a better therapeutic response.
Finally, we also speculate that PD-L1 might be regulated by other membrane proteins in cis. These cis interactions might apply to other ligand-receptor pairs that are co-expressed on immune cells and could represent a general mechanism to regulate immune response. Indeed, cis interactions have been described (or suggested) with several other signaling receptors and their ligands, including Notch/Delta in cell fate decisions (Sprinzak et al., 2010), Eph/Ephrin and Plexin-A4/Sema6A in neuron guidance (Haklai-Topper et al., 2010; Kao and Kania, 2011), and MHC-I/Ly49, SLAMF6/SLAMF6, and HVEM/BTLA in threshold modulation of immune cells activation (Cheung et al., 2009; Held and Mariuzza, 2008; Wu et al., 2016). In these cases, the magnitude of trans-activated receptor signaling is downregulated by cis interactions to determine the cellular responses to environmental cues. This in turn helps cell pattern formation during development, the navigation of neurons to their targets, and homeostasis of immune cells. These established examples, together with our findings here, suggest that multidimension regulation is a common mechanism by which various signaling systems fine-tune cellular responses.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
CONTACT FOR REAGENT AND RESOURCE SHARING
- EXPERIMENTAL MODEL AND SUBJECT DETAILS
-
○Cell Cultures
-
○Human Lung Cancer Samples
-
○
- METHOD DETAILS
-
○Flow Cytometry Based Profiling and Quantification
-
○FRET Assay with Confocal Microscopy
-
○Recombinant Proteins
-
○LUVs Reconstitution and FRET Assays
-
○LUVs–SLB Conjugation Assays
-
○Virus Production and Transduction
-
○OT-I–SLB TIRF microscopy assay
-
○Jurkat–Raji Conjugation Assay
-
○Phosphorylation Assay and IL-2 Secretion Assay
-
○OT-I Cytotoxicity Assay
-
○Knockout PD-1 from EL4 cells
-
○
QUANTIFICATION AND STATISTICAL ANALYSIS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD28 antibody | Thermo Fisher Scientific | Cat # 16-0289-85; RRID: AB_468927 |
| PI3 Kinase p85 antibody | Cell Signaling Technology | Cat # 4292; RRID: AB_329869 |
| Alexa Fluor 647 CD3ε antibody | BioLegend | Cat # 317312; RRID: AB_571883 |
| Pacific Blue PD-1 antibody | BioLegend | Cat # 329915; RRID: AB_1877194 |
| PE/Cy7 PD-L1 antibody | BioLegend | Cat # 329717; RRID: AB_2561686 |
| ZAP70 pY493 antibody | Cell Signaling Technology | Cat # 2704S; RRID: AB_2217457 |
| PE mouse PD-1 antibody | BioLegend | Cat # 109103; RRID: AB_313420 |
| APC mouse PD-1 antibody | BioLegend | Cat # 109111; RRID: AB_10613470 |
| PE mouse PD-L1 antibody | BioLegend | Cat # 124307; RRID: AB_2073557 |
| PE mouse isotype antibody | BioLegend | Cat # 400607; RRID: N/A |
| APC mouse isotype antibody | BioLegend | Cat # 400611; RRID: N/A |
| Pacific Blue PD-1 antibody | BioLegend | Cat # 329915; RRID: AB_1877194 |
| PE-Cy7 PD-L1 antibody | BioLegend | Cat # 329717; RRID: AB_2561686 |
| Pacific Blue isotype antibody | BioLegend | Cat # 400151; RRID: N/A |
| PE-Cy7 isotype antibody | BioLegend | Cat # 400302; RRID: N/A |
| TruStain FcX | BioLegend | Cat # 422301; RRID: N/A |
| PE PD-1 antibody | Thermo Fisher Scientific | Cat # 12-9969-41; RRID: AB_10733013 |
| PE PD-L1 antibody | Thermo Fisher Scientific | Cat # 12-5983-41; RRID: AB_11042721 |
| BV421 CD45 antibody | BioLegend | Cat # 368521; RRID: AB_2687374 |
| Alexa700 CD11b antibody | BioLegend | Cat # 101222; RRID: AB_493705 |
| PE/Cy5 CD11c antibody | BioLegend | Cat # 301609; RRID: AB_493579 |
| Alexa488 CD3 antibody | BioLegend | Cat # 300319; RRID: AB_493690 |
| PE PD-L1 antibody | BioLegend | Cat # 329705; RRID: AB_940366 |
| APC PD-1 antibody | BioLegend | Cat # 329907; RRID: AB_940473 |
| PE isotype antibody | BioLegend | Cat # 400313; RRID: N/A |
| APC isotype antibody | BioLegend | Cat # 400119; RRID: N/A |
| AF647 mouse TCRβ antibody | BioLegend | Cat # 109217; RRID: AB_493347 |
| Mouse PD-1 blockade antibody (J43) | Thermo Fisher Scientific | Cat # 16-9985-82; RRID: AB_469307 |
| Isotype antibody for J43 | Thermo Fisher Scientific | Cat # 16-4888-81; RRID: AB_470171 |
| Human PD-1 blockade antibody (Pembrolizumab) | BioVison Inc | Cat # A1306; RRID: N/A |
| Isotype antibody for Pembrolizumab | BioLegend | Cat # 403701; RRID: N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) | Avanti Polar Lipids | Cat # 850457C |
| 1,2-dioleoyl-sn-glycero-3-[(N-(5-amino-1-carboxypentyl) iminodiacetic acid) succinyl] (nickel salt, DGS-NTA-Ni) | Avanti Polar Lipids | Cat # 790404C |
| 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N- (lissamine Rhodamine B sulfonyl) (ammonium salt, Rhodamine-PE) | Avanti Polar Lipids | Cat # 810158C |
| N-(4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s-Indacene-3-Propionyl)-1,2-Dihexadecanoyl-sn-Glycero-3-Phosphoethanolamine (Triethylammonium Salt, BODIPY-PE) | Thermo Fisher Scientific | Cat # D3800 |
| Poly-D-Lysine | Sigma-Aldrich | Cat # P6407 |
| CLIP-Surface 547 | New England Biolabs | Cat # S9233S |
| SNAP-Surface Alexa Fluor 647 | New England Biolabs | Cat # S9136S |
| SNAP-Cell 505-Star | New England Biolabs | Cat # S9103S |
| SNAP-Cell TMR-Star | New England Biolabs | Cat # S9105S |
| SNAP-Cell 647-SiR | New England Biolabs | Cat # S9102S |
| SEE super antigen | Toxin Technology | Cat # ET404 |
| Strep-SNAP-PD-1EX-His10 | This study | N/A |
| Strep-SNAP-PD-1EX | This study | N/A |
| Strep-SNAP-PD-1(K78A)EX -His10 | This study | N/A |
| Strep-SNAP-PD-L1EX-His10 | This study | N/A |
| Strep-SNAP-PD-L2EX-His10 | This study | N/A |
| Strep-SNAP-B7.2EX-His10 | This study | N/A |
| Mouse MHC-I H2Kb | Enfu Hui | N/A |
| Mouse PD-1EX-His | Sino Biological | Cat # 50124-m08h |
| Mouse PD-L1EX-His | Sino Biological | Cat # 50010-m08h |
| Mouse B7.2EX-His | Sino Biological | Cat # 50068-m08h |
| Mouse ICAMEX-His | Sino Biological | Cat # 50440-m08h |
| SIINFEKL peptide | Anaspec | Cat # AS-60193-1 |
| Live/Dead Aqua | Thermo Fisher Scientific | Cat # L34966 |
| Critical Commercial Assays | ||
| Human IL-2 ELISA MAX Deluxe | BioLegend | Cat # 431804 |
| CytoTox 96 Non-Radioactive Cytotoxicity Assay kit | Promega | Cat # G1780 |
| Cell Line Nucleofector Kit L | LONZA | Cat # VACA-1005 |
| Quantum R-PE MESF | Bangs Laboratories Inc | Cat # 827 |
| Quantum APC MESF | Bangs Laboratories Inc | Cat # 823 |
| Experimental Models: Cell Lines | ||
| HEK293T | Ronald Vale | N/A |
| Jurkat E6.1 T cells | Arthur Weiss | N/A |
| Raji B cells | Ronald Vale | N/A |
| HEK293F | Andrew Ward | N/A |
| Jurkat T cells with PD-1-mGFP | Enfu Hui | N/A |
| Raji B cells with PD-L1-mCherry | Enfu Hui | N/A |
| OT-I | Ananda Goldrath | N/A |
| EL4 | Ira Mellman | N/A |
| Oligonucleotides | ||
| See Table S2 for the list of Oligos | N/A | N/A |
| Recombinant DNA | ||
| See Table S3 for the list of recombinant DNA | N/A | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Micro-Manager | Open Imaging | https://micro-manager.org/ |
| AccPbFRET | (Roszik et al., 2008) | http://biophys.med.unideb.hu/accpbfret/ |
| GraphPad Prism 5 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
| FlowJo | FlowJo, LLC | https://www.flowjo.com/ |
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Enfu Hui (enfuhui@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Cultures
HEK293T cells and Raji B cells were obtained from Dr. Ronald Vale (University of California San Francisco), and Jurkat T cells from Dr. Arthur Weiss (University of California San Francisco). HEK293F cells were a generous gift from Dr. Andrew Ward (Scripps Research Institute). HEK293T cells were maintained in DMEM medium (DMEM supplemented with 10% fetal bovine serum, 100 U/mL of Penicillin, and 100 µg/mL of Streptomycin) at 37°C / 5% CO2. Jurkat T cells and Raji B cells were maintained in RPMI medium (RPMI 1640 supplemented with 10% fetal bovine serum, 100 U/mL of Penicillin, and 100 µg/mL of Streptomycin) at 37°C / 5% CO2. HEK293F cells were maintained in FreeStyle 293 Expression Medium at 37°C / 8% CO2. OT-I splenocytes were harvested from C57BL/6-Tg (TcraTcrb) 1100Mjb/J (OT-I) mice (Jackson Laboratory) and maintained in OT-I culture medium (RPMI 1640 supplemented with 10% fetal bovine serum, 1 mM Sodium Pyruvate, 50 µM β-mecaptoethanol, 100 U/mL of Penicillin, and 100 µg/mL of Streptomycin) at 37°C / 5% CO2. EL4 cells were maintained in EL4 culture medium (RPMI 1640 supplemented with 10% fetal bovine serum, 50 µM β-mecaptoethanol, 100 U/mL of Penicillin, and 100 µg/mL of Streptomycin) at 37°C / 5% CO2.
Human Lung Cancer Samples
Peripheral blood and tumor tissues were obtained from de-identified human NSCLC patients. Study with these samples was reviewed and approved by the Institutional Review Board (IRB) of the University of Chicago.
METHOD DETAILS
Flow Cytometry Based Profiling and Quantification
For data shown in Figure 1 and Figure S1, flow cytometry was used to determine the expressions of PD-1 and PD-L1 of healthy and cancer samples. To this end, PBMCs from de-identified NSCLC patients or healthy donors were isolated with Ficoll (Sigma-Aldrich) gradient separation. Tumor tissues were cut into small pieces and grinded into single-cell suspension. PBMCs and tumor cell suspension were stored in liquid nitrogen tank until use. Prior to flow cytometry, frozen PBMCs and tumor tissue cells were thawed in 37°C water bath, washed, and re-suspended in FACS buffer (PBS with 1% BSA and 0.1% NaN3), and pre-incubated with Human TruStain FcX (BioLegend, Cat # 422301) to prevent non-specific labeling by Fc receptor-antibody binding. Cells were then incubated with an antibody mixture containing PE-labeled anti-PD-L1 (or isotype) (BioLegend, Cat # 329705; Cat # 400313), APC-labeled anti-PD-1 (or isotype) (BioLegend, Cat # 329907; Cat # 400119), BV421-labeled anti-CD45 (BioLegend, Cat # 368521), Alexa700-labeled anti-CD11b (BioLegend, Cat # 101222), PE/Cy5-labeled anti-CD11c (BioLegend, Cat # 301609), Alexa488-labeled anti-CD3 (BioLegend, Cat # 300319), and aqua live/dead cell stain (Thermo Fisher Scientific, Cat # L34966). Stained cells were analyzed on an LSRFortessa cell analyzer (BD Biosciences) and gated as shown in Figure 1A. Cell surface expression of PD-1 and PD-L1 on double positive cells were quantified using the Quantum APC MESF kit and Quantum R-PE MESF kit respectively (Bangs Laboratories Inc), following manufacturer’s instructions. Briefly, fluorescence beads standards and antibody stained cells were run in parallel using identical setting and the data were analyzed by FlowJo software (BD Biosciences). To quantify the surface expression levels of PD-1 and PD-L1 on EL4 cells and transfected HEK293T as shown in Table S1, cells were stained with either PE-labeled anti-PD-1 (BioLegend, Cat # 109103 and Thermo Fisher Scientific, Cat # 12-9969-41), or PE-labeled anti-PD-L1 (BioLegend, Cat # 124307 and Thermo Fisher Scientific, Cat # 12-5983-41), and the expression levels were quantified using the Quantum R-PE MESF kit. Molecular densities were calculated assuming the following diameters: 13 µm for HEK293T (Zimmermann et al., 2006), 8 µm for EL4 (Nath et al., 2016), 10.5 µm for lung cancer cells (Sikdar et al., 2014; Yan et al., 2013), 7 µm for human T and B cells (Dimeloe et al., 2016), 12.5 µm for DCs (Dumortier et al., 2005), and 14.5 µm for Macs & MDSCs (Fernandez et al., 2016). For data shown in Figure 6, parental and lentivirally transduced Jurkat T and Raji B cell lines were stained with Pacific Blue labeled anti-PD-1 (or isotype) (BioLegend, Cat # 329915; Cat # 400151) and PE-Cy7 labeled anti-PD-L1 (or isotype) (BioLegend, Cat # 329717; Cat # 400302) according to manufacturer instructions. For data shown in Figure 7, parental and PD-1 KO EL4 cells were stained with APC labeled anti-PD-1 (or isotype) (BioLegend, Cat # 109111; Cat # 400611) and PE labeled anti-PD-L1 (or isotype) (BioLegend, Cat # 124307; Cat # 400607) according to manufacturer’s instructions. Flow cytometry data were acquired with an LSRFortessa cell analyzer and analyzed with FlowJo software (BD Biosciences).
FRET Assay with Confocal Microscopy
For data shown in Figure 2, pHR plasmid encoding CLIP tagged full-length PD-L1 (CLIP–PD-L1) or PD-L2 (CLIP–PD-L2) was co-transfected with pHR encoding either SNAP-tagged full-length PD-1 (SNAP–PD-1), PD-1(K78A) [SNAP–PD-1(K78A)], or B7.2 (SNAP–B7.2) into HEK293T cells using polyethylenimine, following protocols as described (Tanenbaum et al., 2014). Plasmids and related primers are listed in Tables S2 and S3. 72-hour after transfection, cells were trypsinized and seeded on Poly-D-lysine (Sigma) treated 96-wells plate with glass bottom (Dot Scientific, Cat # MGB096-1-2-LG-L). 24 hr later, cells were labeled with CLIP-Surface 547 (NEB) and SNAP-Surface Alexa Fluor 647 (NEB) at 37°C / 5% CO2 for 30 min, and washed 3 times with 1 × phosphate buffered saline (PBS, pH 7.4). Labeled cells were then fixed with 4% paraformaldehyde (PFA) and used for the FRET assay. Images were acquired with an FV1000 confocal microscope (Olympus) by exciting CLIP-Surface 547 (energy donor) at 543 nm and SNAP-Surface Alexa Fluor 647 (energy acceptor) at 635 nm. Donor images before and after acceptor bleaching were acquired for FRET analysis using ImageJ (Fiji) with the AccPbFRET plugin, as previously described (Roszik et al., 2008).
Recombinant Proteins
pPPI4 plasmid encoding the extracellular portion of either human PD-1 (aa 21–170, designated as PD-1EX), human PD-1 (K78A) (aa 21–170, K78A, designated as PD-1 (K78A)EX), human PD-L1 (aa 19–239, designated as PD-L1EX), human PD-L2 (aa 20–220, designated as PD-L2EX), or human B7.2 (aa 24–247, designated as B7.2EX) was transfected to HEK293F cell using polyethylenimine, as described previously (Murin et al., 2014). Plasmids and related primers are listed in Tables S2 and S3. The N terminus of each extracellular segment was fused with the signal peptide of HIV envelope glycoprotein gp120 followed by a twinstrep tag (amino acids sequence: WSHPQFEKGGGSGGGSGGSAWSHPQFEK) and a SNAP-tag. The C terminus of each extracellular segment was fused with a decahistidine (His10) tag. Under some conditions when His-tag free PD-1 was desired, the His-tag coding sequence was removed from the expression construct. Six days after transfection, the His10-tagged protein was purified from the cell culture medium using HisTrap Excel column (GE Healthcare) and eluted with 0.5 M imidazole. His10-tag free PD-1 extracellular domain was purified with a StrepTrap HP column (GE Healthcare) in 100 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, pH 8.0 and eluted with the same buffer containing 2.5 mM desthiobiotin. The extracellular domain of mouse MHC-I molecule H2Kb was produced as a disulfide stabilized single chain trimer with a covalently linked ovalbumin (OVA) peptide SIINFEKL (Mitaksov et al., 2007), and a C-terminal His10 tag, using the Bac-to-Bac baculovirus expression system, as previously described (Hui et al., 2017). For OT-I/SLB microscopy described in Figure 5, His10-tagged extracellular domains of recombinant murine PD-1, PD-L1, B7.2, and ICAM were purchased from Sino Biologicals. All affinity-purified proteins were size-exclusion-purified using a Superdex 200 Increase 10/300 GL column (GE Healthcare) in HEPES buffered saline (50mMHEPES-NaOH, pH 7.5, 150mMNaCl, 10% glycerol). Gel filtered proteins were labeled with either SNAP-Cell 505 (NEB), SNAP-Cell TMR (NEB) or SNAP-Cell 647 (NEB) following manufacturer’s instructions. Free dyes were then removed using a PD-10 desalting column (GE Healthcare). All proteins were quantified by SDS-PAGE and Coomassie blue staining, using bovine serum albumin (BSA) as a standard.
LUVs Reconstitution and FRET Assays
To prepare LUVs for experiments in Figure 3, phospholipids (80% POPC + 20% DGS-NTA-Ni) were mixed in chloroform, dried under a stream of nitrogen, desiccated for 1 hr in a vacuum container and then resuspended in PBS. LUVs were generated by extrusion 20 times through a pair of polycarbonate filters containing pores of 200 nm diameter, as described previously (Hui and Vale, 2014). 8.3 nM SNAP-Cell-505-labeled PD-L1EX-His (SC505*PD-L1EX-His) or SNAP-Cell-505-labeled PD-L2EX-His (SC505*PD-L2EX-His) was mixed with 0.23 nM LUVs harboring DGS-NTA-Ni in PBS containing 1.5 mg/mL BSA and 1mM TCEP, and incubated at room temperature in a 96-well solid white microplate (Greiner Bio-One Catalog # 655075), during which the SNAP-Cell-505 fluorescence was monitored in real time using a plate reader (Tecan Spark20) with 504-nm excitation and 540-nm emission. Following 90 min incubation, the fluorescence reading was paused and the second protein component (25 nM unlabeled PD-1EX-His, SNAP-Cell-TMR-labeled PD-1EX-His (TMR*PD-1EX-His), SNAP-Cell-TMR-labeled PD-1(K78A)EX (TMR*PD-1(K78A)EX-His), SNAP-Cell-TMR-labeled B7.2EX (TMR*B7.2EX-His), or SNAP-Cell-TMR-labeled PD-1EX without His tag) was injected and the fluorescence was further monitored for another 80 min. For the trans-interaction control, the second component was equal amounts of SNAP-Cell-TMR-labeled PD-1EX pre-bound to LUVs (via 90-minute incubation), so that PD-L1EX could only interact with PD-1EX in trans. For PD-1 titration FRET assay, SNAP-Cell-TMR PD-1EX with a range of concentrations (5–100 nM) was mixed with LUVs pre-bound with 8.3 nM SNAP-Cell-505 PD-L1EX, and SNAP-Cell-505 fluorescence was monitored as above. SNAP-Cell-TMR B7.2EX with the same concentration of PD-1EX was used as control to correct the fluorescence quenching due to a molecular crowding effect on LUVs. TMR*B7.2EX-His mediated quenching of SC505*PD-L1EX-His was subtracted from the TMR*PD-1EX-His signal to get the corrected quenching signal. The titration curve was calculated with the average of last ten fluorescence intensity values of each concentration and fitted with GraphPad Prism 5.0 using the “Specific binding with Hill Slope,” yielding the dissociation constant (Kd) and Hill coefficient (nH) of the PD-1/PD-L1 cis-interaction.
LUVs–SLB Conjugation Assays
SLBs were formed in Hellmanex and hydroxide washed 96-well glass-bottomed plates as described previously (Taylor et al., 2017) with modifications. Briefly, the plate was incubated with 5% Hellmanex III (Hëlma Analytics) overnight on a 50°C heatpad, thoroughly rinsed with ddH2O and sealed with Nunc sealing tape (Thermo Fisher Scientific, Cat # 232698). The desired wells were washed twice with 5 M NaOH (each 30 min), and three times with 500 µL ddH2O followed by equilibration with PBS. Freshly prepared small unilamellar vesicles (SUVs; lipid composition: 97.5% POPC, 2% DGS-NTA-Ni and 0.5% PEG5000 PE) were added to the cleaned wells containing 200 µL 1× PBS, and incubated for 90 min at 50°C to induce SLB formation. The SLBs were then rinsed thoroughly with PBS to remove excess SUVs, and blocked with 1 mg/mL BSA in PBS for 30 min at 37°C. 200 µL 1.5 nM SNAP-Cell-647-labeled PD-1EX-His (SC647*PD-1EX) was overlaid onto SLBs. After 1-hour incubation at 37°C, the unbound proteins were washed away with excess PBS containing 1 mg/mL BSA. The plate was incubated at 37°C for another 30 min and washed again with PBS containing 1 mg/mL BSA to remove dissociated SC647*PD-1EX, leaving the bilayer with stably bound SC647*PD-1EX (Nye and Groves, 2008).
LUVs containing both DGS-NTA-Ni and Bodipy-PE (89.7% POPC + 10% DGS-NTA-Ni + 0.3% Bodipy-PE) were prepared by the aforementioned extrusion method. 0.23 nM LUVs with Bodipy-PE were incubated with PD-L1EX-His alone (8.3 nM), B7.2EX-His alone (8.3 nM), PD-L1EX-His / PD-1EX-His mixture (8.3 nM and 25 nM, respectively), PD-L1EX-His /PD-1(K78A)EX -His mixture (8.3 nM and 25 nM, respectively), or PD-L1EX-His/B7.2EX-His mixture (8.3 nM and 25 nM, respectively), for 90-minute in the presence of 1 mg/mL BSA and 1mMTCEP at room temperature. For titration assay shown in Figure S5, 0.23 nM LUVs with Bodipy-PE were incubated with PD-L1EX-His (4.2 nM or 8.3 nM) as well as increasing concentration of PD-1EX-His. B7.2EX-His was used as a filler to keep the total His-tag protein concentration constant, thereby avoiding artifact due to potential competition of binding sites. The high DGS-NTA-Ni content (10%) in conjunction with the 90-minute incubation ensured that all His10-tagged molecules bind to the LUVs in a kinetically stable manner. The protein-bound LUVs were then added onto SC647*PD-1EX-His functionalized SLBs. After 5-minute incubation, unbound LUVs were washed away with excess PBS and the SLB-captured LUVs visualized and recorded by a Nikon Eclipse Ti TIRF microscope equipped with a 100× Apo TIRF 1.49 NA objective, controlled by the Micro-Manager software (Edelstein et al., 2014). The molecular densities of PD-1 and PD-L1 were computed as described previously (Hui and Vale, 2014). The fluorescence intensity of LUVs from the Bodipy (488 nm) channel in the TIRF field was calculated using the ImageJ software.
Virus Production and Transduction
Lentiviral transduction was used to introduce PD-L1–mCherry, PD-1–mGFP, or PD-1–SNAP into Raji B cells, and PD-L1–SNAP or PD-1–mGFP into Jurkat T cells, essentially as described (Hui et al., 2017). Plasmids and related primers are listed in Tables S2 and S3. To produce lentiviruses, cDNA encoding the gene of interest was cloned into the pHR vector, and co-transfected with the envelope plasmid pMD2.G and the packaging plasmid psPAX2 into HEK293T cells using polyethylenimine in DMEM medium. 18-hour after transfection, the medium was replaced with complete RPMI medium and the virus supernatants harvested after another 54 hr. To transduce Jurkat T cells, 0.5 million cells were pelleted at 600 × g for four minutes and resuspended in 1 mL of fresh virus supernatant, and incubated overnight at 37°C / 5% CO2 before adding another 9 mL of complete RPMI medium. To transduce Raji B cells, 0.5 million cells were pelleted at 600 × g for four minutes and resuspended in 1 mL of fresh virus supernatant containing 8 µg/mL Lipofectamine in a 24-well plate. The virus–cell mixture was centrifuged at 35°C, 1000 × g for 60 min, and incubated at 37°C / 5% CO2 overnight before transferred into a T25 flask containing 9 mL fresh complete RPMI medium. The transduced cells were sorted out via FACS at least one week after the lentiviral transduction.
For OT-I/SLB imaging assays shown in Figure 4, full-length murine PD-1–mCherry was introduced into OT-I cells via retrovirus transduction. The retrovirus was produced as described previously (Hui et al., 2017). Freshly purified OT-I splenocytes were stimulated with 10 nM SIINFEKL peptide in OT-I culture medium supplemented with 100 U/ml mouse recombinant IL-2 at 37°C / 5% CO2 incubator. 36 hr later, cells were resuspended in retrovirus supernatants containing 8 µg/ml Lipofectamine and 100 U/ml mouse recombinant IL-2, spin-infected at 35°C, 1000 × g for 120 min, and incubated at 37°C / 5% CO2 overnight. The virus supernatant was replaced with fresh OT-I culture medium supplemented with 10 nM SIINFEKL peptide and 100 U/ml mouse recombinant IL-2 the second day and cells incubated for another 48–96 hr before microscopy.
OT-I–SLB TIRF microscopy assay
SLBs were prepared as described above and incubated for 1 hr at 37°C with a mixture of 5 nM pMHC-I-His, 2 nM mouse ICAMEX-His, 3 nM mouse PD-L1EX-His and 9 nM mouse PD-1EX-His, or with a mixture of 5 nM pMHC-I-His, 2 nM mouse ICAMEX-His, 3 nM mouse PD-L1EX-His, and 9 nM mouse B7.2EX-His. For blockade antibody treatment, 170 ng mouse PD-1EX was pre-incubated with 6 µg J43 at room temperature for 30 min. Then 9 nM of J43-treated PD-1EX was mixed with 5 nM mouse pMHC-I-His, 2 nM ICAMEX-His, and 3 nM PD-L1EX-His for bilayer functionalization. After 1-hour incubation at 37°C, excess unbound proteins were removed by extensive washes with PBS. PD-1–mCherry transduced OT-I cells were harvested via centrifugation at 200 × g for 4 min, incubated with 10 µg/ml AF647 labeled mouse TCRβ antibody (H57-597) for 30 min on in ice, washed three times with imaging buffer (Hui et al., 2017), and then plated onto functionalized SLBs. TIRF microscopy images were acquired at 37°C on a Nikon Eclipse Ti microscope equipped with a 100× Apo TIRF 1.49 NA objective, controlled by the Micro-Manager software and analyzed with ImageJ. Clustering index was calculated by dividing the fluorescence intensity of PD-1 or TCR microclusters with the total fluorescence intensity of PD-1 or TCR of the entire cell.
Jurkat–Raji Conjugation Assay
For cell conjugation assay shown in Figure 6B, Jurkat cells expressing PD-1–mGFP were mixed with Raji parental cells, Raji cells expressing either PD-L1–mCherry alone or Raji cells expressing both PD-L1–mCherry and PD-1–SNAP (unlabeled). The latter Raji cell line was generated by lentivirally transducing PD-1–SNAP into PD-L1–mCherry expressing Raji B cells. For cell conjugation assay shown in Figure S6, PD-1–SNAP expressing Jurkat cells were lentivirally transduced with PD-1–SNAP (unlabeled) to generate PD-1+ Jurkat cells, and PD-1+/PD-L1+ RajiB cells were generated by by lentivirally transducing PD-1–mGFP into PD-L1–mCherry expressing Raji B cells. For cell conjugation assay shown in Figure S7, PD-1–mGFP expressing Jurkat cells were lentivirally transduced with PD-L1–SNAP (unlabeled) to generate PD-1+/PD-L1+ Jurkat cells, which were then mixed with PD-L1–mCherry expressing Raji cells. Jurkat cells expressing only PD-1–mGFP were used as a control. Prior to the conjugation assay, Raji B cells were pre-incubated with 30 ng/mL staphylococcal enterotoxin E (SEE) superantigen (Toxin Technology) in RPMI medium for 30 min at 37°C. For blockade treatment in Figure 7, both SEE-loaded Raji and Jurkat cells were treated with 2 µg of either Pembrolizumab or isotype antibody per million cells on ice for 45 min before mixing them together. 0.55 million antigen-loaded Raji B cells and 0.75 million Jurkat T cells were precooled on ice and mixed in a 96-well plate. The plate was centrifuged at 290× g for one minute at 4°C to initiate cell–cell contact, and immediately transferred to a 37°C water bath. Two minutes later, cells were resuspended and fixed with 1% PFA and loaded into a 96-well glass-bottom plates for confocal microscopy assays. Images were acquired with FV1000 confocal microscope and processed, and quantified using ImageJ. Interface enrichment index of PD-1 on Jurkat cells were computed by dividing the fluorescence density at the interface divided with fluorescence density of the cell membrane excluding the interface. Fluorescence density was calculated as fluorescence intensity divided by area. The interface was defined as the conjugated area between Jurkat and Raji cells based on the DIC images.
Phosphorylation Assay and IL-2 Secretion Assay
For assaying the phosphorylation states of ZAP70 and CD28, PD-1–mGFP expressing Jurkat T cells were stimulated with either SEE-loaded Raji parental cells, Raji cells expressing PD-L1–mCherry, or Raji cells expressing both PD-1–mGFP and PD-L1–mCherry, following procedures described previously (Hui et al., 2017). Briefly, Raji B cells were pre-incubated with 30 ng/mL SEE in serum free RPMI medium for 30 min at 37°C. Jurkat cells were starved in serum free RPMI medium at 37°C for 3 hr to reduce the phosphorylation background. 5.5 million SEE-loaded Raji B cells and 7.5 million serum starved Jurkat T cells were precooled on ice and then mixed in a 96-well plate, followed by centrifugation at 290× g for one minute at 4°C to initiate the cell–cell contact. Immediately after centrifugation, the cell mixture plate was transferred to a 37°C water bath. The reactions were terminated with NP40 lysis buffer (125 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% NP40, 1 mM EDTA, 5% glycerol, 1 mM PMSF, supplemented with Roche PhosSTOP phosphatase inhibitor cocktail) at indicated time points and the lysates were subjected to co-immunoprecipitation assay with anti-CD28 antibody (Thermo Fisher Scientific, Cat # 16-0289-85). Equal fractions of the immunoprecipitates were subjected to SDS-PAGE and blotted with PI3 Kinase p85 antibody (Cell Signaling Technology, Cat # 4292). The whole cell lysates were blotted with anti-ZAP70-pY493 antibody (Cell Signaling Technology, Cat # 2704S). The optical density corresponding to p85α and ZAP70-pY493 bands were quantified by ImageJ, and normalized to the conditions in which parental Raji cells were used as the APCs.
For IL-2 secretion assays, Raji B cells were pre-loaded with 30 ng/mL SEE for 30 min at 37°C. 0.2 million serum starved Jurkat T cells were co-cultured with 0.05 million antigen-loaded Raji B cells in a 96-well plate in triplicate wells and the supernatants were collected after 24 hr. IL-2 was quantified by ELISA using Human IL-2 ELISA MAX Deluxe kit (BioLegend).
OT-I Cytotoxicity Assay
OT-I splenocytes were harvested from C57BL/6-Tg (TcraTcrb) 1100Mjb/J (OT-I) mice (Jackson Laboratory) and stimulated with 10 nM SIINFEKL peptide in the presence of 100 U/ml IL-2. 60 to 72 hr later, 1 million OT-I cells were incubated with either 1 µg of J43 antibody or isotype at 37°C / 5% CO2 for 1 hr, and washed twice with 10 mL culture medium. Similarly, 1 million EL4 cells were incubated with 1 nM SIINFEKL peptide at 37°C / 5% CO2 for 1 hr together with 2 µg of either J43 antibody or isotype, and washed twice with 10 mL culture medium. Immediately after the wash, OT-I cells were mixed with EL4 cells at ratio of 12.5:1 and incubate at 37°C / 5% CO2 for 4 hr. Cytotoxicity was measured using the CytoTox 96 Non-Radioactive Cytotoxicity Assay kit (Promega, Cat # G1780), following manufacturer’s instructions.
Knockout PD-1 from EL4 cells
To generate PD-1 knockout EL4, two reported mouse PD-1 single guide (sgRNA) (Seki and Rutz, 2018) were each inserted into a modified PX330 plasmid containing a GFP coding sequence, and electroporated into EL4 cells using the Cell Line Nucleofector Kit L (LONZA, Cat # VACA-1005). The electroporated cells were recovered in culture medium at 37°C / 5% CO2 for two days, after which GFP-positive cells were enriched by FACS and maintained in culture medium at 37°C / 5% CO2. One week later, PD-1 knockout cells were sorted by staining cells with mouse PD-1 PE (BioLegend, Cat # 109103).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were shown as mean ± SEM, and number of replicates were indicated in figure legends. Curve fitting and normalization were performed in GraphPad Prism 5. Statistical significance was evaluated by two-tailed Student’s t test (*, p < 0.05; **, p < 0.01; ***, p < 0.001) in GraphPad Prism 5. Data with p < 0.05 are considered statistically significant.
Supplementary Material
Highlights.
A subset of tumor cells and infiltrating APCs in NSCLC co-express PD-1 and PD-L1
PD-1 and PD-L1 on the same cell bind in cis with high affinity
PD-1/PD-L1 cis interaction on APCs prevents PD-L1 from triggering T cell PD-1
Selective blockade of tumor intrinsic PD-1 frees up PD-L1 for T cell inhibition
Acknowledgments
We thank Dr. Ananda Goldrath and Toan Nguyen for providing OT-I cells and cell sorting assistance, Dr. Andrew Ward and Dr. Daniel Murin for providing the HEK293F expression system, Dr. Haopeng Wang and Fei Wang for providing CRISPR-Cas9 knockout plasmids, Dr. Li-Fan Lu and Sunglim Cho for flow cytometry assistance, Zijun Wu for providing an ImageJ script to analyze LUVs–SLB conjugation assays, Sandra Rabat and Nathan Jayne for critically reading the manuscript, and Jennifer Santini and Miranda Bohm for help with confocal microscopy at the UCSD School of Medicine Microscopy Core, which is supported by a NINDS P30 grant (NS047101). This work was supported by UCSD startup fund (to E.H.), NIH grants R00AI106941 and R21AI120010 (to J.H.), and NSF CAREER award 1653782 (to J.H.). Y.Z. is supported by a CRI Irvington Postdoctoral Fellowship. D.L.H. is supported by NIH grant T32-EB009412. E.H. is a Searle Scholar and a Pew Biomedical Scholar.
Footnotes
Supplemental Information includes seven figures and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.06.054.
AUTHOR CONTRIBUTIONS
Y.Z. and E.H. designed the study, conducted experiments (except for those shown in Figure 1), and wrote the paper. D.L.H. and J.H. measured PD-1 and PD-L1 expression in NSCLC samples provided by J.L. Y.S. prepared DNA constructs for the experiments shown in Figure 2 and assisted with protein purifications for other experiments.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Badoual C, Hans S, Merillon N, Van Ryswick C, Ravel P, Benhamouda N, Levionnois E, Nizard M, Si-Mohamed A, Besnier N, et al. PD-1-expressing tumor-infiltrating T cells are a favorable prognostic biomarker in HPV-associated head and neck cancer. Cancer Res. 2013;73:128–138. doi: 10.1158/0008-5472.CAN-12-2606. [DOI] [PubMed] [Google Scholar]
- Baitsch L, Baumgaertner P, Devêvre E, Raghav SK, Legat A, Barba L, Wieckowski S, Bouzourene H, Deplancke B, Romero P, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J. Clin. Invest. 2011;121:2350–2360. doi: 10.1172/JCI46102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butte MJ, Keir ME, Phamduy TB, Freeman GJ, Sharpe AH. PD-L1 interacts specifically with B7-1 to inhibit T cell proliferation. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carreras J, Lopez-Guillermo A, Roncador G, Villamor N, Colomo L, Martinez A, Hamoudi R, Howat WJ, Montserrat E, Campo E. High numbers of tumor-infiltrating programmed cell death 1-positive regulatory lymphocytes are associated with improved overall survival in follicular lymphoma. J. Clin. Oncol. 2009;27:1470–1476. doi: 10.1200/JCO.2008.18.0513. [DOI] [PubMed] [Google Scholar]
- Chen DS, Mellman I. Oncology meets immunology: the cancer-immunity cycle. Immunity. 2013;39:1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- Cheng X, Veverka V, Radhakrishnan A, Waters LC, Muskett FW, Morgan SH, Huo J, Yu C, Evans EJ, Leslie AJ, et al. Structure and interactions of the human programmed cell death 1 receptor. J. Biol. Chem. 2013;288:11771–11785. doi: 10.1074/jbc.M112.448126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung TC, Oborne LM, Steinberg MW, Macauley MG, Fukuyama S, Sanjo H, D’Souza C, Norris PS, Pfeffer K, Murphy KM, et al. T cell intrinsic heterodimeric complexes between HVEM and BTLA determine receptivity to the surrounding microenvironment. J. Immunol. 2009;183:7286–7296. doi: 10.4049/jimmunol.0902490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darb-Esfahani S, Kunze CA, Kulbe H, Sehouli J, Wienert S, Lindner J, Budczies J, Bockmayr M, Dietel M, Denkert C, et al. Prognostic impact of programmed cell death-1 (PD-1) and PD-ligand 1 (PD-L1) expression in cancer cells and tumor-infiltrating lymphocytes in ovarian high grade serous carcinoma. Oncotarget. 2016;7:1486–1499. doi: 10.18632/oncotarget.6429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimeloe S, Mehling M, Frick C, Loeliger J, Bantug GR, Sauder U, Fischer M, Belle R, Develioglu L, Tay S, et al. The immune-metabolic basis of effector memory CD4+ T cell function under hypoxic conditions. J. Immunol. 2016;196:106–114. doi: 10.4049/jimmunol.1501766. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med. 1999;5:1365–1369. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- Dumortier H, van Mierlo GJD, Egan D, van Ewijk W, Toes REM, Offringa R, Melief CJM. Antigen presentation by an immature myeloid dendritic cell line does not cause CTL deletion in vivo, but generates CD8+ central memory-like T cells that can be rescued for full effector function. J. Immunol. 2005;175:855–863. doi: 10.4049/jimmunol.175.2.855. [DOI] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, Stuurman N. Advanced methods of microscope control using µManager software. J. Biol. Methods. 2014;1:e10. doi: 10.14440/jbm.2014.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez IE, Greiffo FR, Frankenberger M, Bandres J, Heinzelmann K, Neurohr C, Hatz R, Hartl D, Behr J, Eickelberg O. Peripheral blood myeloid-derived suppressor cells reflect disease status in idiopathic pulmonary fibrosis. Eur. Respir. J. 2016;48:1171–1183. doi: 10.1183/13993003.01826-2015. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, Gupta R, Tsai JM, Sinha R, Corey D, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495–499. doi: 10.1038/nature22396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves JT, Dustin ML. Supported planar bilayers in studies on immune cell adhesion and communication. J. Immunol. Methods. 2003;278:19–32. doi: 10.1016/s0022-1759(03)00193-5. [DOI] [PubMed] [Google Scholar]
- Haklai-Topper L, Mlechkovich G, Savariego D, Gokhman I, Yaron A. Cis interaction between Semaphorin6A and Plexin-A4 modulates the repulsive response to Sema6A. EMBO J. 2010;29:2635–2645. doi: 10.1038/emboj.2010.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Held W, Mariuzza RA. Cis interactions of immunoreceptors with MHC and non-MHC ligands. Nat. Rev. Immunol. 2008;8:269–278. doi: 10.1038/nri2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Vale RD. In vitro membrane reconstitution of the T-cell receptor proximal signaling network. Nat. Struct. Mol. Biol. 2014;21:133–142. doi: 10.1038/nsmb.2762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, Sasmal DK, Huang J, Kim JM, Mellman I, Vale RD. T cell costimulatory receptor CD28 is a primary target for PD-1-mediated inhibition. Science. 2017;355:1428–1433. doi: 10.1126/science.aaf1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao T-J, Kania A. Ephrin-mediated cis-attenuation of Eph receptor signaling is essential for spinal motor axon guidance. Neuron. 2011;71:76–91. doi: 10.1016/j.neuron.2011.05.031. [DOI] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu. Rev. Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HR, Ha S-J, Hong MH, Heo SJ, Koh YW, Choi EC, Kim EK, Pyo KH, Jung I, Seo D, et al. PD-L1 expression on immune cells, but not on tumor cells, is a favorable prognostic factor for head and neck cancer patients. Sci. Rep. 2016;6:36956. doi: 10.1038/srep36956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleffel S, Posch C, Barthel SR, Mueller H, Schlapbach C, Guenova E, Elco CP, Lee N, Juneja VR, Zhan Q, et al. Melanoma cell-intrinsic PD-1 receptor functions promote tumor growth. Cell. 2015;162:1242–1256. doi: 10.1016/j.cell.2015.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázaár-Molnár E, Yan Q, Cao E, Ramagopal U, Nathenson SG, Almo SC. Crystal structure of the complex between programmed death-1 (PD-1) and its ligand PD-L2. Proc. Natl. Acad. Sci. USA. 2008;105:10483–10488. doi: 10.1073/pnas.0804453105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázár-Molnár E, Scandiuzzi L, Basu I, Quinn T, Sylvestre E, Palmieri E, Ramagopal UA, Nathenson SG, Guha C, Almo SC. Structure-guided development of a high-affinity human Programmed Cell Death-1: Implications for tumor immunotherapy. E Bio Medicine. 2017;17:30–44. doi: 10.1016/j.ebiom.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Liang L, Dai W, Cai G, Xu Y, Li X, Li Q, Cai S. Prognostic impact of programed cell death-1 (PD-1) and PD-ligand 1 (PD-L1) expression in cancer cells and tumor infiltrating lymphocytes in colorectal cancer. Mol. Cancer. 2016;15:55. doi: 10.1186/s12943-016-0539-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maleki Vareki S, Garrigós C, Duran I. Biomarkers of response to PD-1/PD-L1 inhibition. Crit. Rev. Oncol. Hematol. 2017;116:116–124. doi: 10.1016/j.critrevonc.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Maute RL, Gordon SR, Mayer AT, McCracken MN, Natarajan A, Ring NG, Kimura R, Tsai JM, Manglik A, Kruse AC, et al. Engineering high-affinity PD-1 variants for optimized immunotherapy and immuno-PET imaging. Proc. Natl. Acad. Sci. USA. 2015;112:E6506–E6514. doi: 10.1073/pnas.1519623112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazanet MM, Hughes CCW. B7-H1 is expressed by human endothelial cells and suppresses T cell cytokine synthesis. J. Immunol. 2002;169:3581–3588. doi: 10.4049/jimmunol.169.7.3581. [DOI] [PubMed] [Google Scholar]
- Mitaksov V, Truscott SM, Lybarger L, Connolly JM, Hansen TH, Fremont DH. Structural engineering of pMHC reagents for T cell vaccines and diagnostics. Chem. Biol. 2007;14:909–922. doi: 10.1016/j.chembiol.2007.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu C-Y, Huang J-A, Chen Y, Chen C, Zhang X-G. High expression of PD-L1 in lung cancer may contribute to poor prognosis and tumor cells immune escape through suppressing tumor infiltrating dendritic cells maturation. Med. Oncol. 2011;28:682–688. doi: 10.1007/s12032-010-9515-2. [DOI] [PubMed] [Google Scholar]
- Murin CD, Julien J-P, Sok D, Stanfield RL, Khayat R, Cupo A, Moore JP, Burton DR, Wilson IA, Ward AB. Structure of 2G12 Fab2 in complex with soluble and fully glycosylated HIV-1 Env by negative-stain single-particle electron microscopy. J. Virol. 2014;88:10177–10188. doi: 10.1128/JVI.01229-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath S, Christian L, Tan SY, Ki S, Ehrlich LIR, Poenie M. Dynein separately partners with NDE1 and dynactin to orchestrate T cell focused secretion. J. Immunol. 2016;197:2090–2101. doi: 10.4049/jimmunol.1600180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nye JA, Groves JT. Kinetic control of histidine-tagged protein surface density on supported lipid bilayers. Langmuir. 2008;24:4145–4149. doi: 10.1021/la703788h. [DOI] [PubMed] [Google Scholar]
- Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer. 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SJ, Namkoong H, Doh J, Choi JC, Yang BG, Park Y, Chul Sung Y. Negative role of inducible PD-1 on survival of activated dendritic cells. J. Leukoc. Biol. 2014;95:621–629. doi: 10.1189/jlb.0813443. [DOI] [PubMed] [Google Scholar]
- Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, Linsley PS, Thompson CB, Riley JL. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol. Cell. Biol. 2005;25:9543–9553. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel SP, Kurzrock R. PD-L1 expression as a predictive biomarker in cancer immunotherapy. Mol. Cancer Ther. 2015;14:847–856. doi: 10.1158/1535-7163.MCT-14-0983. [DOI] [PubMed] [Google Scholar]
- Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265–276. doi: 10.1016/j.it.2015.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Hu-Lieskovan S. What does PD-L1 positive or negative mean? J. Exp. Med. 2016;213:2835–2840. doi: 10.1084/jem.20161462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roszik J, Szöllosi J, Vereb G. AccPbFRET: an ImageJ plugin for semi-automatic, fully corrected analysis of acceptor photobleaching FRET images. BMC Bioinformatics. 2008;9:346. doi: 10.1186/1471-2105-9-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatier R, Finetti P, Mamessier E, Adelaide J, Chaffanet M, Ali HR, Viens P, Caldas C, Birnbaum D, Bertucci F. Prognostic and predictive value of PDL1 expression in breast cancer. Oncotarget. 2015;6:5449–5464. doi: 10.18632/oncotarget.3216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki A, Rutz S. Optimized RNP transfection for highly efficient CRISPR/Cas9-mediated gene knockout in primary T cells. J. Exp. Med. 2018;215:985–997. doi: 10.1084/jem.20171626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, Qiu Y, Jussif JM, Carter LL, Wood CR, Chaudhary D. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574:37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- Sikdar S, Kumar Saha S, Rahman Khuda-Bukhsh A. Relative apoptosis-inducing potential of homeopathic Condurango 6C and 30C in H460 lung cancer cells in vitro: apoptosis induction by homeopathic Condurango in H460 cells. J. Pharmacopuncture. 2014;17:59–69. doi: 10.3831/KPI.2014.17.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Chen D, Lu B, Wang C, Zhang J, Huang L, Wang X, Timmons CL, Hu J, Liu B, et al. PTEN loss increases PD-L1 protein expression and affects the correlation between PD-L1 expression and clinical parameters in colorectal cancer. PLoS ONE. 2013;8:e65821. doi: 10.1371/journal.pone.0065821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprinzak D, Lakhanpal A, Lebon L, Santat LA, Fontes ME, Anderson GA, Garcia-Ojalvo J, Elowitz MB. Cis-interactions between Notch and Delta generate mutually exclusive signalling states. Nature. 2010;465:86–90. doi: 10.1038/nature08959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanenbaum ME, Gilbert LA, Qi LS, Weissman JS, Vale RD. A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell. 2014;159:635–646. doi: 10.1016/j.cell.2014.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MJ, Husain K, Gartner ZJ, Mayor S, Vale RD. A DNA-based T cell receptor reveals a role for receptor clustering in ligand discrimination. Cell. 2017;169:108–119.e20. doi: 10.1016/j.cell.2017.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian R, Wang H, Gish GD, Petsalaki E, Pasculescu A, Shi Y, Mollenauer M, Bagshaw RD, Yosef N, Hunter T, et al. Combinatorial proteomic analysis of intercellular signaling applied to the CD28 T-cell costimulatory receptor. Proc. Natl. Acad. Sci. USA. 2015;112:E1594–E1603. doi: 10.1073/pnas.1503286112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu N, Zhong M-C, Roncagalli R, Pérez-Quintero L-A, Guo H, Zhang Z, Lenoir C, Dong Z, Latour S, Veillette A. A hematopoietic cell-driven mechanism involving SLAMF6 receptor, SAP adaptors and SHP-1 phosphatase regulates NK cell education. Nat. Immunol. 2016;17:387–396. doi: 10.1038/ni.3369. [DOI] [PubMed] [Google Scholar]
- Yan X, Luo H, Zhou X, Zhu B, Wang Y, Bian X. Identification of CD90 as a marker for lung cancer stem cells in A549 and H446 cell lines. Oncol. Rep. 2013;30:2733–2740. doi: 10.3892/or.2013.2784. [DOI] [PubMed] [Google Scholar]
- Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase SHP2. J. Exp. Med. 2012;209:1201–1217. doi: 10.1084/jem.20112741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann D, Terpitz U, Zhou A, Reuss R, Müller K, Sukhorukov VL, Gessner P, Nagel G, Zimmermann U, Bamberg E. Biophysical characterisation of electrofused giant HEK293-cells as a novel electrophysiological expression system. Biochem. Biophys. Res. Commun. 2006;348:673–681. doi: 10.1016/j.bbrc.2006.07.112. [DOI] [PubMed] [Google Scholar]
- Zou W, Wolchok JD, Chen L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016;8:328rv4. doi: 10.1126/scitranslmed.aad7118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.