

Abstract
Interspecies hybridization is an important evolutionary mechanism in yeasts. The genus Zygosaccharomyces in particular contains numerous hybrid strains and/or species. Here, we investigated the genome of Zygosaccharomyces strain MT15, an isolate from Maotai-flavor Chinese liquor fermentation. We found that it is an interspecies hybrid and identified it as Zygosaccharomyces pseudobailii. The Z. bailii species complex consists of three species: Z. bailii, which is not a hybrid and whose 10 Mb genome is designated ‘A’, and two hybrid species Z. parabailii (‘AB’ genome, 20 Mb) and Z. pseudobailii (‘AC’ genome, 20 Mb). The A, B and C subgenomes are all approximately 7%–10% different from one another in nucleotide sequence, and are derived from three different parental species. Despite being hybrids, Z. pseudobailii and Z. parabailii are capable of mating and sporulating. We previously showed that Z. parabailii regained fertility when one copy of its MAT locus became broken into two parts, causing the allodiploid hybrid to behave as a haploid gamete. In Z. pseudobailii, we find that a very similar process occurred after hybridization, when a deletion of 1.5 kb inactivated one of the two copies of its MAT locus. The half-sibling species Z. parabailii and Z. pseudobailii therefore went through remarkably parallel but independent steps to regain fertility after they were formed by separate interspecies hybridizations.
Keywords: evolution, hybridization, MAT locus, HO endonuclease, Zygosaccharomyces
Several natural interspecies hybrids in Zygosaccharomyces overcame sterility by inactivating a MAT locus and undergoing whole-genome duplication.
INTRODUCTION
Advances in genome sequencing have led to a growing recognition that interspecies hybridization has occurred many times during budding yeast evolution (Morales and Dujon 2012; Campbell et al.2016; Mixão and Gabaldón 2018). Many such hybrids have been discovered—for example, considering only the subphylum Saccharomycotina, natural interspecies hybrids (allodiploids) have been identified in the genera Saccharomyces (Hittinger 2013; Lopandic 2018; Peris et al.2018), Zygosaccharomyces (Mira et al.2014; Ortiz-Merino et al.2017; Watanabe et al.2017), Millerozyma (Leh Louis et al.2012), Saccharomycopsis (Choo et al.2016), Pichia (Smukowski Heil et al.2018), the Candida orthopsilosis/metapsilosis clade (Pryszcz et al.2015; Schroder et al.2016) and probably also in Metschnikowia (Piombo et al.2018; Venkatesh et al.2018). These interspecies hybrids were formed by mating, but in most cases they are sterile (unable to produce viable spores) or infertile (unable to sporulate) due to post-zygotic mechanisms such as chromosome mis-segregation during meiosis (Watanabe et al.2017; Boynton, Janzen and Greig 2018). They are therefore stuck in an asexual state and can only reproduce mitotically (Dujon and Louis 2017; Marsit et al.2017). The genomes of these allodiploid hybrids generally correspond to the sum of the genomes of their two parental species with little gene loss, although sequence homogenization (loss of heterozygosity) can occur. Because they were formed by mating, they typically have a MATa/α genotype at the mating-type locus, where the two MAT alleles come from the two parental species.
Recent studies on Zygosaccharomyces (Ortiz-Merino et al.2017; Watanabe et al.2017) have identified an evolutionary route by which interspecies hybrids can regain a complete sexual cycle. In this route, one allele at the MAT locus of an interspecies allodiploid hybrid becomes damaged. For example, a spontaneous deletion in a MATα allele can convert a MATa/α allodiploid genotype into a MATa/- hemizygous genotype. A hybrid cell with this damage would behave as a MATa haploid, seeking a MATα partner to mate with. If the hybrid's genome also contains silent mating-type-like loci (HML/HMR) and an HO endonuclease gene, it can generate a partner by mating-type switching (Haber 2012; Hanson and Wolfe 2017). Mating then produces a cell that has two identical copies of every chromosome from both of the parental species, and a MATa/α genotype, so it is functionally diploid. Meiosis can occur by pairing the two copies of each chromosome, leading to viable spores that are functionally haploid (MATa or MATα), bearing one copy of all chromosomes from both parents. This evolutionary mechanism of restoring fertility is referred to as whole-genome duplication after interspecies hybridization (Ortiz-Merino et al.2017; Mixão and Gabaldón 2018) and recapitulates a process first proposed by Øjvind Winge (Winge 1917; Clausen and Goodspeed 1925).
Although it may seem convoluted, this route of interspecies hybridization followed by MAT locus damage and restoration of fertility has occurred at least twice, in natural hybrids discovered in two separate groups of Zygosaccharomyces species (the Z. rouxii species complex (Watanabe et al.2017), and the Z. bailii species complex (Ortiz-Merino et al.2017)). In both cases, a new species was formed that has a haploid/diploid life cycle and a doubled genome size. The same mechanism has been proposed to have caused the ancient whole-genome duplication that occurred in an ancestor of Saccharomycescerevisiae (Scannell et al.2006; Marcet-Houben and Gabaldón 2015; Wolfe 2015). A similar process has been detected in interspecies hybrids (allotetraploids) constructed between Saccharomyces species in the laboratory by Sipiczki and colleagues. In these hybrids, fertility was restored by spontaneous loss of one MAT-containing chromosome (rather than by damage to one MAT locus), followed by mating-type switching at the remaining MAT locus, and mother–daughter mating (Pfliegler, Antunovics and Sipiczki 2012; Karanyicz et al.2017).
One of the species that was formed by this evolutionary route in the Zygosaccharomyces genus is Z. parabailii, which is of economic importance as a food spoilage agent (Mira et al.2014; Ortiz-Merino et al.2017). Zygosaccharomycesparabailii was formed by interspecies hybridization between Z. bailii (genome A, 10 Mb) and an unidentified Zygosaccharomyces species (genome B, 10 Mb). Zygosaccharomycesparabailii has a hybrid genome (AB, 20 Mb), but the MAT locus of the B-subgenome became damaged, leaving the MAT locus of the A-subgenome as the only functional MAT locus in the hybrid, which led to mating-type switching and mother–daughter mating. Zygosaccharomycesparabailii now has a life cycle in which haploids have a 20 Mb genome (AB) and diploids have a 40 Mb genome (AABB).
The Z. bailii species complex consists of three known species: Z. bailii and Z. parabailii, whose genomes have been sequenced, and Z. pseudobailii whose genome is uncharacterized (Galeote et al.2013; Suh et al.2013; Mira et al.2014; Ortiz-Merino et al.2017; Palma et al.2017). All three species have complete sexual cycles that include mating-type switching, mating and sporulation (Suh et al.2013; Ortiz-Merino et al.2017). In recent work, Xu et al. (2017) sequenced the genome of yeast strain MT15, which was initially identified as Z. bailii. MT15 was isolated from grain fermentation used to distil Maotai, a Chinese strong liquor with characteristic soy-sauce aroma (Wu, Chen and Xu 2013; Jin, Zhu and Xu 2017). Here, we show that MT15 is a strain of Z. pseudobailii, and that this species has a hybrid genome (AC) where one parent is Z. bailii and the other is an unidentified Zygosaccharomyces species that is different from the B-parent of Z. parabailii. We show that the Z. pseudobailii MAT locus obtained from the A-parent is intact and functional, but the MAT locus obtained from the C-parent has been damaged by a small deletion. We confirmed that this deletion is also present in the type strain of Z. pseudobailii. Thus, Z. pseudobailii and Z. parabailii are independent interspecies hybrids that regained fertility by separate but similar routes involving damage to one of their MAT alleles.
MATERIALS AND METHODS
Illumina sequencing of the MT15 genome and assembly using Newbler and SSPACE was previously reported by Xu et al. (2017). For this study, we also reassembled the Illumina raw data using SPAdes v3.11.1 (Bankevich et al.2012) and compared the assemblies. We used the SPAdes data to make minor manual edits to the MAT and HML/HMR regions of the Newbler/SSPACE scaffolds, and to provide coverage information. The edited Newbler/SSPACE scaffolds have been submitted to the International Nucleotide Sequence Database Collaboration databases with accession numbers OVGK01000001–OVGK01000095.
Strain CBS2856T was purchased from the Westerdijk Institute, Netherlands. PCR primer sequences used for amplification of its MAT loci were SLA2A (ACAGGTAGCGTTATGGC), SLA2C (TAACAGGTGGTATTGTAGGA), MATa2A (AAATCCTTGTGGTTTTCTGG), MATalpha1 (ATTCATTTCCTCAGTGTACG) and DIC1 (AGGCAAGCTACGATACC). PCR was carried out using Q5 high-fidelity 2x master mix (New England Biolabs) with annealing temperature 55°C. Sequences of the three CBS2856TMAT locus PCR products have been submitted to the databases with accession numbers MH330392–MH330394.
Phylogenetic trees were constructed by maximum likelihood using PhyML as implemented in the Seaview package (Gouy, Guindon and Gascuel 2010), with default parameters. Support levels shown for internal branches are approximate likelihood ratio test support in Fig. 1, and bootstrap support in Fig. 2. The trees in Figs 1 and 2 were constructed from nucleotide and amino acid sequences, respectively.
Figure 1.

MT15 is a strain of Z. pseudobailii. (A) Unrooted phylogenetic tree of the ITS region of ribosomal DNA. The sequences analyzed were ITSs of multiple strains of Z. pseudobailii (Zpse), Z. parabailii (Zpar) and Z. bailii (Zbai), including the type strains of each species, from Suh et al. (2013), as well as ITS sequences identified in the genome assemblies of MT15 (one locus) and Z. parabailii ATCC60483 (two loci, on chromosomes 4 and 11; Ortiz-Merino et al.2017). Nodes represent individual ITS sequence variants; many strains have identical sequences. The scale bar indicates numbers of nucleotide substitutions per site. (B) Phylogenetic trees for RPB1, RPB2 and TUB2 genes. The A, B and C subgenomes are color coded as in panel A. Asterisks beside NCBI sequence accession numbers indicate type strain sequences that originally contained ambiguous sites (Suh et al.2013), for which we inferred the B- or C-copy sequence by subtracting the A-copy sequence. The scale bars indicate numbers of nucleotide substitutions per site. (C) Triangular relationship among the genomes and species in the Z. bailii species complex.
Figure 2.
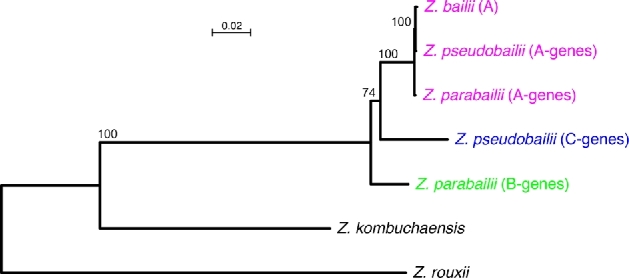
Phylogenetic relationship among the A-, B- and C-subgenomes. The tree was constructed from concatenated amino acid sequence alignments of the six largest proteins (total 21 933 residues) using PhyML as implemented in Seaview (Gouy, Guindon and Gascuel 2010), with default parameters. Support levels from 100 bootstrap replicates are shown for internal branches. The scale bar indicates numbers of amino acid substitutions per site.
RESULTS
MT15 is a strain of Z. pseudobailii and is an interspecies hybrid
The genome sequence and phenotypic properties of Zygosaccharomyces strain MT15 were reported by Xu et al. (2017). The genome was sequenced by paired-end Illumina sequencing of four genomic libraries with insert sizes of up to 8 kb, and assembled using Newbler and SSPACE. The size of the genome (20 Mb) and the presence of many duplicated genes indicated that it could be an interspecies hybrid (Xu et al.2017). To investigate the MT15 genome more extensively, we annotated its scaffolds using YGAP (Proux-Wéra et al.2012), and analyzed its phylogenetic relationship to other species in the Z. bailii species complex. YGAP predicted 9957 genes in the MT15 genome, including orthologs of 4502 ‘Ancestral’ genes from the pre-Whole Genome Duplication ancestor (Gordon, Byrne and Wolfe 2009). Most of these ancestral genes (93%) were present in two copies in the MT15 genome, consistent with a hybrid origin. Typically, one of the two copies is almost identical to the orthologous gene in Z. bailii CLIB213T, and the other is about 7%–10% different in nucleotide sequence. This situation resembles the genome of Z. parabailii ATCC60483 (Ortiz-Merino et al.2017). However, the sequences of the extra gene copies in MT15 were dissimilar to the extra (‘B’) gene copies in Z. parabailii (Xu et al.2017), which indicates that MT15 is a different type of interspecies hybrid.
We hypothesized that MT15 could be a strain of Z. pseudobailii, the only known species in the Z. bailii complex whose genome has not yet been reported. The only genes that have previously been sequenced from Z. pseudobailii are some phylogenetic marker genes sequenced by Suh et al. (2013) in their initial description of this species, so we compared these markers to MT15.
The sequence of the ITS region of ribosomal DNA from the MT15 genome is identical to ITS sequences from Z. pseudobailii strains, including the type strain ATCC56074T (Suh et al.2013), and is different from the ITS sequences of Z. bailii and Z. parabailii (Fig. 1A).
The sequences of the RPB1, RPB2 and TUB2 genes previously reported from strains of Z. pseudobailii, including the type strain, all contain multiple ambiguous nucleotides. Since these sequences were obtained by direct sequencing of PCR products (Suh et al.2013), the ambiguous sites indicate a high level of heterozygosity and a possible hybrid nature of the species. The MT15 genome sequence contains two copies of each of these genes. At each ambiguous site in the sequences from the type strain of Z. pseudobailii, one of the possible nucleotides matches the A-subgenome sequence from Z. bailii, so we inferred the sequence of the second allele in the type strain by subtracting the nucleotides contributed by the A-subgenome. We then constructed a phylogenetic tree for each gene (Fig. 1B).
The trees for RPB1, RPB2 and TUB2 show that MT15 is a strain of Z. pseudobailii, and that Z. pseudobailii is an interspecies hybrid. MT15 and the Z. pseudobailii type strain both have an A-subgenome derived from Z. bailii, and a second subgenome that comes from a different source, which we designated the C-subgenome. Zygosaccharomycesbailii contains only the A genome, Z. parabailii is an AB hybrid and Z. pseudobailii is an AC hybrid. The relationship among the genomes and species in the Z. bailii species complex can be summarized in a triangular diagram (Fig. 1C) similar to the famous ‘Triangle of U’ for plant species in the genus Brassica (U 1935).
Phylogenetic relationship among the A-, B- and C-subgenomes of the Z. bailii species complex
To investigate the relationship among the A-, B-, and C-subgenomes, we constructed a phylogenetic tree from concatenated alignments of the six largest Zygosaccharomyces proteins—Mdn1, Dyn1, Tra1, Tom1, Vps13 and Ira1 (Fig. 2). The tree was inferred by maximum likelihood using amino acid sequences. For each protein, the data included one sequence from Z. bailii, two from Z. parabailii, two from Z. pseudobailii, and one each from Z. kombuchaensis and Z. rouxii as outgroups (Souciet et al.2009; Goncalves et al.2018). As expected, the A-subgenome sequences from the three species are almost identical (Fig. 2). The B-subgenome sequences (from Z. parabailii) lie slightly outside the A- and C- subgenome sequences, although bootstrap support for the internal branch is relatively low (74%). In trees constructed from each of the six proteins individually, five of the six (all except Dyn1) showed the same topology as the concatenated tree. The same topology was also seen in the RPB1, RPB2 and TUB2 trees (Fig. 1B). We conclude that the parental species from which the A- and C-subgenomes originated are slightly more closely related to each other than to the parent of the B-subgenome.
We also examined levels of nucleotide substitution in genes. We aligned 3916 trios of gene sequences, where each trio is a set of homologs from the A-, B- and C-subgenomes, using the CLIB213T, ATCC60483 and MT15 genome sequences as the respective sources. For each trio, we calculated the non-synonymous (KA) and synonymous (KS) sequence divergence per site, using the yn00 method in PAML (Yang 2007). The median KA and KS values are lower for the A-B comparison than for the A-C and B-C comparisons (Table 1). This result highlights the asymmetry of the branch lengths in the trees (Fig. 2 and Fig. 1B). Because the C-subgenome is at the end of a relatively long branch, the A-C distance exceeds the A-B distance, even though the A-C split is younger than the A-B split. Thus, the rate of molecular evolution in the C-lineage has been faster than in the A- and B-lineages.
Table 1.
Median levels of non-synonymous and synonymous sequence divergence among the three subgenomes, in 3916 trios of genes, and their standard deviations.
| Non-synonymous divergence (KA) | Synonymous divergence (KS) |
|||
|---|---|---|---|---|
| Subgenome pair | Median | s. d. | Median | s. d. |
| A vs B | 0.017 | 0.015 | 0.155 | 0.035 |
| A vs C | 0.021 | 0.021 | 0.215 | 0.048 |
| B vs C | 0.023 | 0.023 | 0.244 | 0.056 |
Structure of MAT and HMR/HML loci in Z. pseudobailii MT15
We investigated the structure of the MAT locus in Z. pseudobailii MT15, using BLAST searches with Z. bailii and Z. parabailii genes as queries (Fig. 3). We identified scaffolds in the MT15 genome assembly that contain the MAT loci derived from the A- and C-parental species that combined to form the hybrid. We also identified silent HMR and HML loci. As in many other budding yeasts (Gordon et al.2011; Watanabe, Uehara and Mogi 2013), the MAT locus is located between the SLA2 and DIC1 genes, whereas HMR and HML are beside telomere-associated genes (CHA1, VAC17, ATG2). The MATa1 gene is not present anywhere in the MT15 genome, consistent with our previous inference that it was lost in the most recent common ancestor of the Z. bailii species complex (Ortiz-Merino et al.2017).
Figure 3.

Gene organization at MAT-related loci in Z. pseudobailii (not to scale). (A) Structure of the damaged C-subgenome MAT locus. (B) Inferred structure of the MATa allele of the A-subgenome. (C) Inferred structure of the MATα allele of the A-subgenome. (D, E)HMRa loci. (F)HMLα locus. Scaffold numbers refer to the genome assembly of strain MT15. Panels A–C include PCR data from CBS2856T, the type strain of Z. pseudobailii. The pink and blue arrows indicate genes assigned to the A- and C-subgenomes, respectively, based on their divergence from Z. bailii CLIB213T. The red and green arrows indicate full-length α-genes, and full-length a-genes, respectively. The brown arrows represent sections of genes whose association with the A- or C-subgenome cannot be determined. Prime symbols in gene names (e.g. SLA2’) indicate incomplete fragments of genes.
Protein-coding genes near the MAT and HMR/HML loci of MT15 were assigned to either the A- or the C-subgenome, depending on their level of divergence from Z. bailii CLIB213T. Genes were assigned to the A-subgenome (pink genes in Fig. 3) if they had ≥98% DNA sequence identity to genes in Z. bailii, or assigned to the C-subgenome (blue) if they were more divergent. We also identified the Z and X repeat regions that, by analogy to other species, are expected to guide the DNA exchanges that occur during mating-type switching. Similar to Z. parabailii (Ortiz-Merino et al.2017), the Z region contains the 3΄ ends of the SLA2 and MATα1 genes, while the X region contains a central portion of the MATa2 gene (Fig. 3). The sequences of the five Z regions, and the six X regions, in MT15 are virtually identical, so it is not possible to assign them to the A- or C-subgenome. The same situation was found in Z. parabailii and is presumably caused by homogenization of these repeat regions during mating-type switching. The X and Z sequences are also virtually identical among the three species in the complex.
The two copies of the Z. parabailii MAT locus, derived from the two parental species, are contained in scaffolds 26 and 27 of the MT15 genome assembly (Fig. 3A and B). However, scaffold 26 contains a MAT allele from which the Z and Y regions have been completely deleted. It contains an unusual junction between a point in SLA2 and a point in MATa2, with the result that the 3΄ end of SLA2 (codons 373–454) and the 5΄ end of MATa2 (codons 1–135) are missing. Approximately 1.5 kb has been deleted, extending from 62 bp before the Z region to 96 bp after the Y region (Fig. S1, Supporting Information). It is impossible to tell whether the scaffold 26 sequence was originally a MATa or a MATα allele, because the deletion removed the entire Y region. Furthermore, the deletion in scaffold 26 coincides with a transition between DNA derived from the C-subgenome (to the left of the MAT locus) to DNA derived from the A-subgenome (to the right of the MAT locus). The DIC1-GNEAS1 region to the right of MAT appears to have undergone gene conversion, because there is only one copy (an A-copy) of these genes in the genome assembly, and the sequence coverage of this region was twice the average for the genome. Further to the right, there are separate A- and C- copies of the next gene, PEX2, in scaffolds 37 and 38 (Fig. 3A and B).
The HMRa loci derived from the two parents are located in scaffolds 29 and 30 (Fig. 3D and E). The gene CHA1, located to the right of HMRa, has undergone gene conversion so that there are two identical A-copies of CHA1 and no C-copy. Scaffold 53 contains an HMLα sequence from the A-subgenome (Fig. 3F), but there is no equivalent sequence from the C-subgenome. The C-copy of HMLα seems to have been deleted from the genome, so the only copies of the α1 and α2 genes in the MT15 genome are the ones from the A-subgenome.
The type strain of Z. pseudobailii has the same deletion of the C-subgenome MAT locus.
To verify that the MAT locus structures we inferred for MT15 are shared with other Z. pseudobailii strains, we used PCR to amplify and sequence the MAT loci of the type strain of Z. pseudobailii, CBS2856T (synonymous with ATCC56074T).
PCR amplification confirmed that CBS2856T has the same 1.5-kb deletion as MT15, and the same junction between C-subgenome and A-subgenome DNA (Fig. 3A). Thus, the MAT locus of the C-subgenome of Z. pseudobailii must be non-functional, because the only remaining sequence is a partial MATa2 gene that lacks a promoter and 5΄ end. Additionally, the absence of a Z region means that mating-type switching cannot occur at this locus.
In contrast, PCR showed that the MAT locus of the A-subgenome of CBS2856T is intact, and also that mating-type switching occurs at this locus. Using PCR primers specific for the A-subgenome copies of SLA2, MATa2 and MATα1, we were able to obtain both a SLA2-MATa2 PCR product and a SLA2-MATα1 PCR product (Fig. 3B and C). Sequencing these products confirmed that they come from the A-subgenome. This result indicates that the culture of CBS2856T from which we extracted DNA included cells that had switched mating type. Therefore, this locus, derived from the A-subgenome, is the active MAT locus of Z. pseudobailii.
One copy of the HO endonuclease gene is a pseudogene in both Z. pseudobailii and Z. parabailii
The MT15 genome contains two intact copies of most genes, which suggests that the interspecies hybridization that formed it occurred relatively recently. However, it has only one intact copy of the HO gene, which codes for the endonuclease that initiates mating-type switching by cleaving the MAT locus at the junction between the Y and Z regions (Haber 2012). The A-subgenome copy of HO contains a premature stop codon and is therefore a pseudogene, whereas the C-copy of HO is intact. This mutation is interesting because a similar inactivation of the A-copy of HO occurred in Z. parabailii (Ortiz-Merino et al.2017). The two mutations are different so they must have occurred independently after the hybridizations. Zygosaccharomycesparabailii has a frameshift at amino acid 157, and Z. pseudobailii has a premature stop codon at amino acid 444, of the 586-residue HO protein.
DISCUSSION
We have shown that MT15 is a strain of Z. pseudobailii, so genome sequences are now available for all three species known to exist in the Z. bailii species complex. Zygosaccharomycespseudobailii and Z. parabailii are hybrid species that are half-siblings, i.e. they share one of their two parents (the ‘A’ parent, Z. bailii). As well as the three known species, it is likely that the Z. bailii species complex also contains undetected species corresponding to pure genomes B and C, and possibly a BC hybrid (Fig. 1C). Similar half-sibling relationships have been found among naturally occurring hybrids in the genus Saccharomyces (Hittinger 2013). Most of the Saccharomyces natural hybrids are allodiploids or allotriploids and are unable to produce spores with high viability (Boynton, Janzen and Greig 2018; Peris et al.2018). One S. cerevisiae × S. kudriavzevii natural hybrid, PB7, has high spore viability and is an almost perfect allotetraploid with two copies of each chromosome from each parental species (Peris et al.2012). PB7 may have gone through a process similar to what we describe for Zygosaccharomyces.
Both of the Zygosaccharomyces hybridizations appear to have occurred recently. We previously estimated that Z. parabailii is <1000 years old (Ortiz-Merino et al.2017). The low level of gene inactivation in Z. pseudobailii after hybridization indicates a similarly recent origin for that species.
There are extensive similarities between how Z. pseudobailii and Z. parabailii evolved after the separate hybridizations that formed them (Fig. 4). The hybrids were probably initially sterile because chromosomal rearrangements between the A, B and C genomes made the formation of viable meiotic spores impossible (Ortiz-Merino et al.2017). However, both of the hybrids regained fertility by a process of MAT locus damage that converted the initial allodiploid MATa/α genotypes into hemizygous genotypes (MATa/– or MATα/–), thereby converting the cells from zygotes (allodiploid with 2 × 10 Mb DNA content) into gametes (haploid with 1 × 20 Mb DNA content). The gametes were able to switch mating type at their surviving MAT locus, after which mother–daughter mating occurred, producing diploid zygote cells (autodiploid with 2 × 20 Mb DNA content, a MATa/α genotype at the functional MAT locus and a ‘–/–’ genotype at the damaged locus which cannot be described as a MAT locus any more). Since every chromosome in these cells is present in two identical pairs, productive meiosis and sporulation are possible and the two hybrid species are fully fertile, leading to the current life cycles of Z. pseudobailii and Z. parabailii (Fig. 4). Mating and spore formation have been observed by microscopy in both Z. pseudobailii (Suh et al.2013) and Z. parabailii (Suh et al.2013; Ortiz-Merino et al.2017).
Figure 4.

Model for evolution of genomes and species in the Z. bailii species complex. See text for details. The black dots on chromosomes represent intact MAT loci. The numbers in parentheses indicate approximate DNA content per cell.
Another intriguing similarity between the two hybrids is the fact that one copy of the HO endonuclease gene has been inactivated in both species. We do not understand the significance of this observation, but it should be emphasized that very few genes have acquired inactivating mutations in either Z. pseudobailii or Z. parabailii since hybridization.
The only significant difference in the evolutionary history of the two hybrids, apart from the involvement of the B versus C parent, is in the details of the DNA damage at the MAT locus. In Z. parabailii, the damage was very clearly caused by cleavage of the B-subgenome MAT locus by HO endonuclease (Ortiz-Merino et al.2017). Instead of a normal mating-type switching event, the cleaved locus interacted with another site in the genome and caused a translocation, leaving the B-subgenome MAT locus broken into two halves on different chromosomes. In Z. pseudobailii, the C-subgenome MAT locus was almost completely deleted, but the chromosome was repaired without causing a translocation. The deleted region includes the recognition site for HO, but it is impossible to know if the deletion was caused by HO activity or was spontaneous. However, the location of the deletion is consistent with cleavage by HO at the Y/Z junction, followed by resection of the chromosome in both directions (Haber 2012). The endpoints of the deletion occur at a 4 bp repeated sequence (Fig. S1, Supporting Information), consistent with repair by microhomology-mediated end joining (Yu and Gabriel 2003; McVey and Lee 2008). In both Z. pseudobailii and Z. parabailii, the damaged MAT locus includes a junction between the two different subgenomes (A-C or A-B), which shows that the damage cannot have occurred in a parent before the hybrids were formed.
The mechanism of fertility restoration seen in Z. pseudobailii and Z. parabailii is also very similar to the mechanism reported in interspecies hybrids in the Z. rouxii species complex (Watanabe et al.2017). These hybrids, represented by strains NBRC1876 and NBRC110957, contain two subgenomes (T and P) corresponding to haploid parental species Z. rouxii and ‘Z. pseudorouxii’. The T and P subgenomes are about 15% different in nucleotide sequence (Gordon and Wolfe 2008; Watanabe et al.2017). In these hybrids, restoration of fertility was caused by ectopic mitotic recombination between one of the two MAT loci and either an HML locus (in NBRC1876) or an HMR locus (in NBRC110957), resulting in a translocation between two chromosomes. Each strain contains two chimeric MAT-HM loci that were created by this exchange, but they are transcriptionally silent, with the result that the unrearranged MAT locus is now the only functional MAT locus in the hybrid strains (Watanabe et al.2017). A ‘TP’ hybrid zygote was therefore converted into a gamete by damage to one of its homologous MAT loci, similar to what occurred in Z. pseudobailii and Z. parabailii. Interestingly, another ‘TP’ hybrid may have successfully formed a viable meiotic spore without genome doubling (Watanabe, Uehara and Tsukioka 2018), which suggests that 15% sequence divergence may constitute an approximate upper limit for the ability of homologous chromosomes to pair in meiosis (allosyndetic chromosome pairing; Karanyicz et al.2017). In hexaploid wheat plants, meiotic pairing or non-pairing of homologous chromosomes is a variable trait that is controlled by a single genetic locus, Ph1 (Griffiths et al.2006). It would be of interest to know if a similar control mechanism exists in yeasts.
SUPPLEMENTARY DATA
Supplementary data are available at FEMSYR online.
{kind=link}
FUNDING
This work was supported by Science Foundation Ireland [13/IA/1910], the European Union FP7 Marie Curie Initial Training Network YEASTCELL [606795], and CONACyT, Mexico [440667].
Conflict of interest. None declared.
REFERENCES
- Bankevich A, Nurk S, Antipov D et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 2012;19:455–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boynton PJ, Janzen T, Greig D. Modeling the contributions of chromosome segregation errors and aneuploidy to Saccharomyces hybrid sterility. Yeast 2018;35:85–98. [DOI] [PubMed] [Google Scholar]
- Campbell MA, Ganley AR, Gabaldon T et al. The case of the missing ancient fungal polyploids. Am Nat 2016;188:602–14. [DOI] [PubMed] [Google Scholar]
- Choo JH, Hong CP, Lim JY et al. Whole-genome de novo sequencing, combined with RNA-Seq analysis, reveals unique genome and physiological features of the amylolytic yeast Saccharomycopsis fibuligera and its interspecies hybrid. Biotechnol Biofuels 2016;9:246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen RE, Goodspeed TH. Interspecific hybridization in Nicotiana. II. A tetraploid glutinosa-tabacum hybrid, an experimental verification of Winge's hypothesis. Genetics 1925;10:278–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon BA, Louis EJ. Genome diversity and evolution in the budding yeasts (Saccharomycotina). Genetics 2017;206:717–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galeote V, Bigey F, Devillers H et al. Genome sequence of the food spoilage yeast Zygosaccharomyces bailii CLIB 213T. Genome Announcements 2013;1:e00606–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves C, Wisecaver JH, Kominek J et al. Evidence for loss and reacquisition of alcoholic fermentation in a fructophilic yeast lineage. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL, Armisen D, Proux-Wera E et al. Evolutionary erosion of yeast sex chromosomes by mating-type switching accidents. Proc Natl Acad Sci USA 2011;108:20024–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL, Byrne KP, Wolfe KH. Additions, losses, and rearrangements on the evolutionary route from a reconstructed ancestor to the modern Saccharomyces cerevisiae Genome. PLoS Genet 2009;5:e1000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL, Wolfe KH. Recent allopolyploid origin of Zygosaccharomyces rouxii strain ATCC 42981. Yeast 2008;25:449–56. [DOI] [PubMed] [Google Scholar]
- Gouy M, Guindon S, Gascuel O. SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 2010;27:221–4. [DOI] [PubMed] [Google Scholar]
- Griffiths S, Sharp R, Foote TN et al. Molecular characterization of Ph1 as a major chromosome pairing locus in polyploid wheat. Nature 2006;439:749–52. [DOI] [PubMed] [Google Scholar]
- Haber JE. Mating-type genes and MAT switching in Saccharomyces cerevisiae. Genetics 2012;191:33–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SJ, Wolfe KH. An evolutionary perspective on yeast mating-type switching. Genetics 2017;206:9–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hittinger CT. Saccharomyces diversity and evolution: a budding model genus. Trends Genet 2013;29:309–17. [DOI] [PubMed] [Google Scholar]
- Jin G, Zhu Y, Xu Y. Mystery behind Chinese liquor fermentation. Trends Food Sci Technol 2017;63:18–28. [Google Scholar]
- Karanyicz E, Antunovics Z, Kallai Z et al. Non-introgressive genome chimerisation by malsegregation in autodiploidised allotetraploids during meiosis of Saccharomyces kudriavzevii x Saccharomyces uvarum hybrids. Appl Microbiol Biot 2017;101:4617–33. [DOI] [PubMed] [Google Scholar]
- Leh Louis V, Despons L, Friedrich A. Pichia sorbitophila , an interspecies yeast hybrid, reveals early steps of genome resolution after polyploidization. G3 2012;2:299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopandic K. Saccharomyces interspecies hybrids as model organisms for studying yeast adaptation to stressful environments. Yeast 2018;35:21–38. [DOI] [PubMed] [Google Scholar]
- McVey M, Lee SE. MMEJ repair of double-strand breaks (director's cut): deleted sequences and alternative endings. Trends Genet 2008;24:529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcet-Houben M, Gabaldón T. Beyond the whole-genome duplication: phylogenetic evidence for an ancient interspecies hybridization in the baker's yeast lineage. PLoS Biol 2015;13:e1002220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsit S, Leducq JB, Durand E et al. Evolutionary biology through the lens of budding yeast comparative genomics. Nat Rev Genet 2017;18:581–98. [DOI] [PubMed] [Google Scholar]
- Mira NP, Munsterkotter M, Dias-Valada F et al. The genome sequence of the highly acetic acid-tolerant Zygosaccharomyces bailii-derived interspecies hybrid strain ISA1307, isolated from a sparkling wine plant. DNA Res 2014;21:299–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mixão V, Gabaldón T. Hybridization and emergence of virulence in opportunistic human yeast pathogens. Yeast 2018;35:5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales L, Dujon B. Evolutionary role of interspecies hybridization and genetic exchanges in yeasts. Microbiol Mol Biol R 2012;76:721–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Merino RA, Kuanyshev N, Braun-Galleani S et al. Evolutionary restoration of fertility in an interspecies hybrid yeast, by whole-genome duplication after a failed mating-type switch. PLoS Biol 2017;15:e2002128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma M, Munsterkotter M, Peca J et al. Genome sequence of the highly weak-acid-tolerant Zygosaccharomyces bailii IST302, amenable to genetic manipulations and physiological studies. FEMS Yeast Res 2017;17:fox025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris D, Lopes CA, Belloch C et al. Comparative genomics among Saccharomyces cerevisiae × Saccharomyces kudriavzevii natural hybrid strains isolated from wine and beer reveals different origins. BMC Genomics 2012;13:407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peris D, Perez-Torrado R, Hittinger CT et al. On the origins and industrial applications of Saccharomyces cerevisiae × Saccharomyces kudriavzevii hybrids. Yeast 2018;35:51–69. [DOI] [PubMed] [Google Scholar]
- Pfliegler WP, Antunovics Z, Sipiczki M. Double sterility barrier between Saccharomyces species and its breakdown in allopolyploid hybrids by chromosome loss. FEMS Yeast Res 2012;12:703–18. [DOI] [PubMed] [Google Scholar]
- Piombo E, Sela N, Wisniewski M et al. Genome sequence, assembly and characterization of two Metschnikowia fructicola strains used as biocontrol agents of postharvest diseases. Front Microbiol 2018;9:593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proux-Wéra E, Armisén D, Byrne KP et al. A pipeline for automated annotation of yeast genome sequences by a conserved-synteny approach. BMC Bioinformatics 2012;13:237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryszcz LP, Nemeth T, Saus E et al. The genomic aftermath of hybridization in the opportunistic pathogen Candida metapsilosis. PLoS Genet 2015;11:e1005626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scannell DR, Byrne KP, Gordon JL et al. Multiple rounds of speciation associated with reciprocal gene loss in polyploid yeasts. Nature 2006;440:341–5. [DOI] [PubMed] [Google Scholar]
- Schroder MS, Martinez de San Vicente K, Prandini TH et al. Multiple origins of the pathogenic yeast Candida orthopsilosis by separate hybridizations between two parental species. PLoS Genet 2016;12:e1006404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smukowski Heil C, Burton JN, Liachko I et al. Identification of a novel interspecific hybrid yeast from a metagenomic spontaneously inoculated beer sample using Hi-C. Yeast 2018;35:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souciet JL, Dujon B, Gaillardin C et al. Comparative genomics of protoploid Saccharomycetaceae. Genome Res 2009;19:1696–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SO, Gujjari P, Beres C et al. Proposal of Zygosaccharomyces parabailii sp. nov. and Zygosaccharomyces pseudobailii sp. nov., novel species closely related to Zygosaccharomyces bailii. Int J Syst Evol Microbiol 2013;63:1922–9. [DOI] [PubMed] [Google Scholar]
- U N. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Japan J Bot 1935;7:389–452. [Google Scholar]
- Venkatesh A, Murray AL, Boyle AB et al. Draft genome sequence of a highly heterozygous yeast strain from the Metschnikowia pulcherrima subclade, UCD127. Genome Announc 2018;6:e00550–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe J, Uehara K, Mogi Y. Diversity of mating-type chromosome structures in the yeast Zygosaccharomyces rouxii caused by ectopic exchanges between MAT-like loci. PLoS One 2013;8:e62121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe J, Uehara K, Mogi Y et al. Mechanism for restoration of fertility in hybrid Zygosaccharomyces rouxii generated by interspecies hybridization. Appl Environ Microb 2017;83:e01187–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe J, Uehara K, Tsukioka Y. Can interspecies hybrid Zygosaccharomyces rouxii produce an allohaploid gamete? Appl Environ Microb 2018;84:e01845–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winge O. The chromosomes. Their numbers and general significance. CR Lab Carls 1917;13:131–275. [Google Scholar]
- Wolfe KH. Origin of the yeast whole-genome duplication. PLoS Biol 2015;13:e1002221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Chen L, Xu Y. Yeast community associated with the solid state fermentation of traditional Chinese Maotai-flavor liquor. Int J Food Microbiol 2013;166:323–30. [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhi Y, Wu Q et al. Zygosaccharomyces bailii is a potential producer of various flavor compounds in Chinese Maotai-flavor liquor fermentation. Front Microbiol 2017;8:2609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol 2007;24:1586–91. [DOI] [PubMed] [Google Scholar]
- Yu X, Gabriel A. Ku-dependent and Ku-independent end-joining pathways lead to chromosomal rearrangements during double-strand break repair in Saccharomyces cerevisiae. Genetics 2003;163:843–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.