

Abstract
HIV-1 causes premature aging in chronically infected patients. Despite effective anti-retroviral therapy, around 50% of patients suffer HIV-associated neurocognitive disorders (HAND), which likely potentiate aging-associated neurocognitive decline. Microglia support productive HIV-1 infection in the brain. Elevated markers of cellular senescence, including p53 and p21, have been detected in brain tissues from patients with HAND, but the potential for microglia senescence during HIV-1 infection has not been investigated. We hypothesized that HIV-1 can induce senescence in microglia. Primary human fetal microglia were exposed to single-round infectious HIV-1 pseudotypes or controls, and examined for markers of senescence. Post-infection, microglia had significantly elevated: senescence-associated β-galactosidase activity, p21 levels, and production of cytokines such as IL-6 and IL-8, potentially indicative of a senescence-associated secretory phenotype. We also found increased detection of p53-binding protein foci in microglia nuclei post-infection. Additionally, we examined mitochondrial reactive oxygen species (ROS) and respiration, and found significantly increased mitochondrial ROS levels and decreased ATP-linked respiration during HIV-1 infection. Supernatant transfer from infected cultures to naïve microglia resulted in elevated p21 and caveolin-1 levels, and IL-8 production. Finally, nucleoside treatment reduced senescence markers induction in microglia. Overall, HIV-1 induces a senescence-like phenotype in human microglia, which could play a role in HAND.
Keywords: Microglia senescence, HIV-1-associated neurocognitive disorders, Mitochondrial ROS, Mitochondrial respiration, Nucleoside treatment
Although dementia associated with chronic human immunodeficiency virus type 1 (HIV-1) infection has gradually become a rare clinical occurrence during the combination antiretroviral therapy (cART) era, approximately half of all infected patients continue to develop central nervous system (CNS) manifestations of varying severity, commonly known as HIV-associated neurocognitive disorders (HAND) (1). Remarkably, the severity of HAND is expected to rise due to the aging of the HIV-1-infected population, and it has been shown that even the mildest form of HAND, asymptomatic neurocognitive impairment, can predict development of more serious neurocognitive deficits as the patients age, and likely potentiate age-associated cognitive impairment (2). Currently, there is no therapy designed specifically for the treatment of HAND. Therefore, more studies are needed to reveal mechanistic insights that could lead to the design and development of targeted, protective, and adjunctive therapies, which could more effectively ameliorate the neurocognitive impairments during chronic HIV-1 infection.
Microglia, the brain-resident macrophages, constitutes a major target of productive HIV-1 infection in the CNS and could potentially serve as a cellular reservoir during chronic HIV-1 infection refractory to cART (3). Microglia activation is an essential component of their cellular function to maintain CNS homeostasis, and a plethora of studies have demonstrated that microglia become activated during HIV-1 infection, as evidenced by upregulation of activation markers and release of pro-inflammatory cytokines (4). However, persistent and unregulated microglia activation can be deleterious for the CNS and, during life-long HIV-1 infection, is likely to contribute to the development of HAND. It is also known that microglia undergo aging-associated functional changes, and persistent, low-level microglia activation could be an important component of the microglia aging program in both healthy subjects and patients with aging-associated neurocognitive impairments (5,6). Indeed, dysfunctional, aged microglia with characteristics resembling those found in cells undergoing cellular senescence, have been reported (7,8). Cellular senescence is known to play important roles in regulating human organ and tissue aging and aging-related pathology (9,10). Senescent phenotypes have been observed in various cell types such as bronchial cells, adipocytes, pancreatic β cells, and astrocytes, and development of senescence in those cells has been shown to contribute to and/or to be associated with the onset of pulmonary fibrosis, obesity, type 2 diabetes, and Alzheimer’s disease, respectively (11–16). Although cellular senescence has been typically associated with cells or cell types undergoing active proliferation, there is also now evidence for non-replicative senescence or “senescence-after-differentiation”, for example, in neurons (17). In addition, recent studies have shown that astrocytes may undergo senescence in the context of HIV-1 and with antiretroviral drugs (18,19). Previous studies have also suggested that microglia could develop a senescence-like phenotype during HAND, since elevated p53-p21 pathway, a characteristic biomarker of cellular senescence, was found in brain tissues of HIV-1-infected patients who developed HAND, as compared to the tissues from those that remained unimpaired (20,21). However, microglia senescence and the potential role that HIV-1 infection may play on it, remain obscure.
To explore whether microglia could undergo cellular senescence during HIV-1 infection, we examined a panel of biomarkers that have been associated with the development of a senescence program in other cell types (9,10). We found that HIV-1 infection of microglia cultures induces elevated senescence-associated β-galactosidase (SA-β-gal) activity and p21 expression, formation of DNA damage-associated p53-binding protein 1 (53BP1) foci, and the release of cytokines/chemokines that could be part of a senescence-associated secretory phenotype (SASP). Since mitochondrial dysfunction is a crucial component of organismal aging and cellular senescence (22,23), we examined the production of mitochondrial reactive oxygen species (ROS) and mitochondrial electron transport chain (ETC) respiration, and found both elevated mitochondrial ROS production and reduced basal, maximal, and ATP-linked respiration in microglia cultures during HIV-1 infection. Interestingly, we observed that naïve microglia exposed to supernatants from HIV-1-infected microglia cultures also developed senescence biomarkers, potentially suggesting a role for SASP components in microglia senescence. Overall, our results indicate that microglia develop a senescence-like phenotype in the context of HIV-1 infection, which could contribute to the development of HAND, potentially opening new opportunities to target the neurological complications of HIV-1 infection.
Materials and Methods
Cell Culture
Human fetal microglia, thereafter referred to as microglia, derived from multiple fetal brain tissues (gestational age, 16–18 weeks), were obtained from the Temple University Comprehensive NeuroAIDS Center Basic Science Core-1, which operates in full compliance with National Institutes of Health and Temple University ethical guidelines. Drexel University Institutional Review Board determined that these studies do not constitute human subjects research. Microglia were cultured at 37°C and 5% CO2 in microglia growth medium, consistent of DMEM:F12 (1:1), supplemented with 15% heat-inactivated fetal bovine serum (BenchMark FBS, Gemini Bio-Products; certified to have extremely low levels of hemoglobin and endotoxin to prevent cellular activation), 2 mM L-glutamine, 50 µg/mL Gentamicin, 5 ug/mL Fungizone, 10 µg/mL Insulin, 10 µg/L D-biotin, and 10 mL/L N1 supplement (Sigma). Microglia had been purified by shaking prior to aliquoting, and after thawing, the cultures contained >96% ionized calcium-binding adaptor molecule 1 (Iba1, a microglia- and tissue macrophage-specific calcium-binding protein) positive cells, as determined by both immunofluorescence and flow cytometry using an anti-Iba1 antibody (Ab) (Abcam), while all cells were negative for CD14, a marker of monocytes and macrophages that is absent or has an extremely low expression in microglia, except under certain fully activating conditions (24).
Viral Production and Infection
To produce Env-pseudotyped, luciferase-reporter viruses, 293T cells were co-transfected using calcium phosphate precipitation (ProFection Mammalian Transfection System; Promega) with a vesicular stomatitis virus glycoprotein (VSV-G)-expression vector and with the HIV-1 envelope glycoprotein (Env)-deficient, pNL4-3-luc+env− (pNL43) provirus that contains a luciferase reporter gene inserted in the Nef open reading frame. pNL43 was developed by N. Landau by introducing a frameshift mutation in the gene encoding the HIV-1 Env of pNL4-3-luc+ vector. VSV-G expression vector is utilized to facilitate cellular entry. For controls, 293T cells were transfected with either VSV-G or pNL43 expression vectors alone. Culture supernatants containing the single-round infectious pseudotyped particles or controls were collected 48–72 h after transfection, clarified by centrifugation, aliquoted, and stored at −80°C until use. Microglia were exposed to a 1:2 dilution of these supernatants in fresh medium at 37°C and 5% CO2. To assess infectivity, at 6 days post-infection, cells were washed with phosphate buffered saline (PBS) and lysed, and infectivity was measured by detecting luciferase activity (Luciferase Assay System, Promega) in a microplate luminometer (GloMax, Promega). Multiplicity of infection (MOI) of 0.005 was used. MOI was assessed based on TCID50 values. TCID50 assays were conducted in microglia plated at 2 × 104 cells per well in a 96-well plate. 12 1:2 serial dilutions of viral stocks were performed in quadruplicate. Luciferase activities were used to determine TCID50/mL using the Spearman and Kärber algorithm (25). MOI was then calculated using the following equation:
SA-β-gal Activity
The SA-β-gal activity was evaluated as previously described (26). Briefly, following exposure to HIV-1 pseudotypes or controls, microglia were fixed in 2% formaldehyde/0.2% glutaraldehyde for 5 min and stained overnight using X-gal as substrate. Images taken were used to select the positive (blue) and negative cells using ImageJ, and then exported to CellProfiler for analysis. At least 200 cells were counted per condition, in triplicate. Neocarzinostatin (NCS), a DNA damage-inducing agent (27), was used at 0.5–1 µg/mL as a positive control for induction of senescence. The percentages of positive cells were graphed.
Immunoblotting
Microglia were incubated with HIV-1 or controls in microglia media. After 4 days, media was changed and cells washed with PBS. Cell lysates were collected 8 days post-treatment in RIPA buffer containing protease and phosphatase inhibitors. Western blot analysis was performed under standard conditions using 20 µg of total protein and stained for specific proteins using anti-p16INK4a (1:200, BP Pharmingen), anti-p21 (1:200, H-164, Santa Cruz Biotechnology), anti-p53 (1:500, Ab-6, Calbiochem) Abs, as well as anti-β-actin (1:500, Sigma), or anti-GAPDH (1:500, Calbiochem) Abs as loading controls.
Immunofluorescence
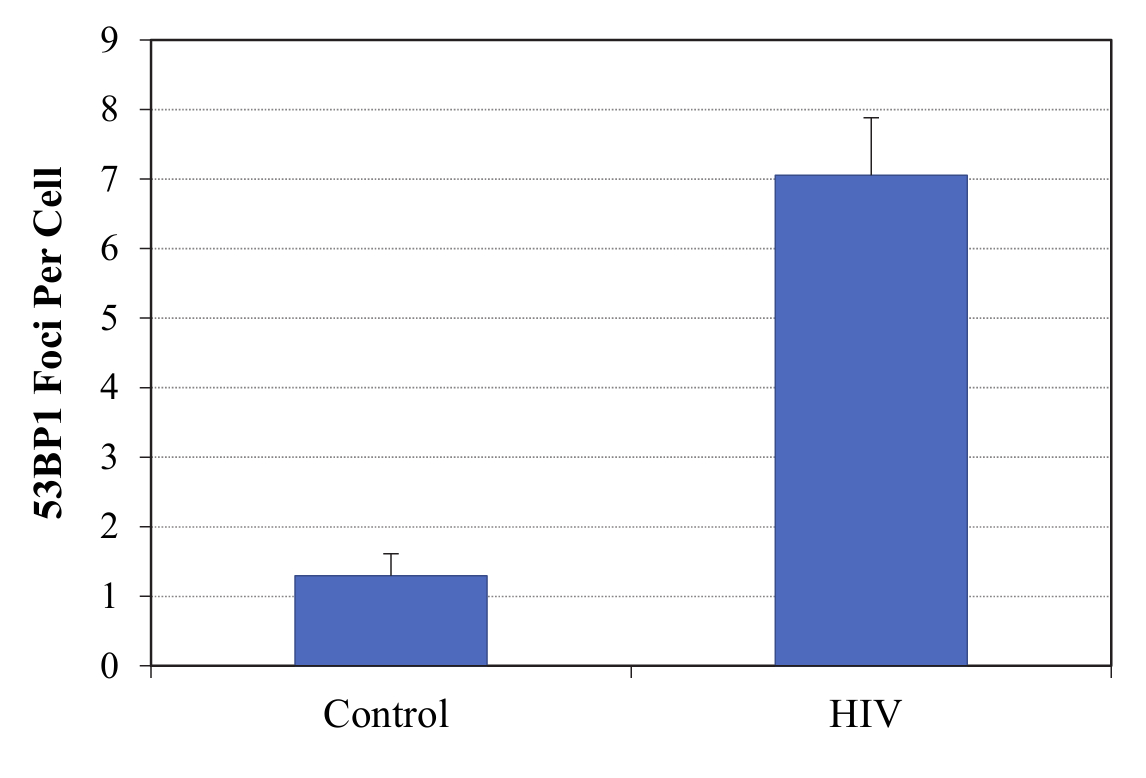
Cells were plated in culture slides (Corning) at 104 cells per chamber post-treatment. Cells were first fixed for 10 min with 4% paraformaldehyde at room temperature (RT) and then permeabilized for 15 min with 0.2% TritonX-100. Slides were blocked in 5% normal goat serum, 0.1% PBS–bovine serum albumin (BSA) for 2 h, then incubated overnight in a humidified chamber at 4°C with primary rabbit antibody anti-53BP1 (Novus Biologicals) diluted at 1:500 in PBS 0.1% BSA. Slides were washed three times for 5 min in 0.1% BSA and 0.1% Triton X-100, then incubated for 1 h in the dark at RT with the secondary antibody goat anti-rabbit Alexa Fluor 555 (Invitrogen). After 3 × 5-min washes in 1× PBS with 0.1% BSA and 0.1% Triton X-100, slides were incubated with DAPI, and mounted with Vectashield Mounting Medium (Vector Laboratories, Inc.). Cells were visualized using an Olympus BX61 fluorescence microscope coupled with a Hamamatsu ORCA-ER camera and using Slide Book 4 software version 4.0.1.44 (Intelligent Innovations, Inc.). We performed a quantitative analysis of 53BP1 foci formation by obtaining an average of foci per cell and comparing the average between untreated and HIV-1-exposed cells.
Mitochondrial ROS Analysis
For mitochondrial ROS studies, cells were incubated with 5 μM MitoSox superoxide anion indicator (Molecular Probes), a dye specific for mitochondrial ROS (28), in microglia medium at 37°C in 5% CO2 for 30min. Microglia were then harvested in 2.5% trypsin–EDTA, re-suspended in 200 μL of complete growth medium, and analyzed immediately with a Guava Easy-Cyte Mini using the Guava Express Plus program (Millipore). Cells were first gated based on size and cellularity and then analyzed using InCyteTM (Millipore) and mean fluorescence intensities were obtained for comparative analysis.
Mitochondrial ETC Function
For real-time analysis of oxygen consumption rate (OCR), microglia were analyzed with an XF-96 Extracellular Flux Analyzer (Seahorse Bioscience), as previously described (29). Briefly, either triplicate or quadruplicate wells were seeded for each condition depending on the experiment, and three consecutive measurements were obtained under basal conditions and after the sequential addition of 5 µM oligomycin, to inhibit mitochondrial ATP synthase; 4 µM FCCP (fluoro-carbonyl cyanide phenylhydrazone), a protonophore that uncouples ATP synthesis from proton gradient generated by the electron-transport chain through respiration; and 1.8 µM rotenone plus antimycin A, which inhibit complexes I and III, respectively, of the ETC, completely suppressing mitochondrial respiration (Supplementary Figure 1). In this assay, basal oxygen consumption can be established as the measured OCR in the absence of drugs by subtracting non-mitochondrial respiration. Reduced OCR after the addition of oligomycin is used to derive ATP-linked respiration (by subtracting the oligomycin rate from baseline cellular OCR). The oligomycin-insensitive portion of baseline OCR represents proton leak. Maximal OCR occurs after the addition of FCCP due to the attempt of the cells to maintain a proton gradient across the inner mitochondrial membrane by increasing the consumption of oxygen. This also defines the reserve or spare respiratory capacity (by subtracting basal from maximal OCR), which is the amount of extra ATP that can be produced by oxidative phosphorylation in case of a sudden increase in energy demand. Finally, addition of rotenone/antimycin A reveals the non-mitochondrial respiration. The OCR is represented as absolute values (pMoles/min) normalized by the number of cells.
Analysis of Cytokine/Chemokine Secretion
Control and infected microglia were washed twice with PBS and supernatants were replaced with serum-free MCDB105 media (Sigma-Aldrich) 4 days post-treatment. After 24-h incubation, conditioned media were collected and cells were trypsinized and counted to determine cell number for normalization. Human cytokine antibody array (RayBiotech) was used to detect cytokines present in the collected media according to the product manual. The intensity of the signal on the array membranes was quantified by densitometry using ImageJ (NIH) and then normalized by cell number. Supernatants from three cases were analyzed to confirm the secretory profile. An enzyme-linked immunosorbent assay (ELISA) for Interleukin (IL)-8 (eBioscience) was also performed. Results were normalized to the number of cells and represented in ng/mL per 106 cells.
Supernatant Transfer Assay
To examine whether components of the microglia supernatant post-infection affect innocent bystander cells, supernatant transfer assays were performed. Specifically, supernatants from uninfected, infected, and control cells were collected 4 days post-infection. Treatment-naïve microglia were incubated with supernatants diluted with microglia media at 1:3. One day post-treatment, cell lysates were collected and examined for p21 and caveolin (Cav)1 protein expression (rabbit anti-Cav1, 1:300, Cell Signaling).
Nucleoside Treatment
Treatment with nucleosides has been shown to prevent or reverse oncogene-induced cellular senescence (30). Thus, we tested the potential beneficial effects of nucleoside treatment in the senescence-like phenotype in microglia. Microglia were treated with 50 or 250 nM of nucleoside cocktail (gift from Dr. Rugang Zhang, Wistar Institute, Philadelphia, PA) at the same time of infection, and replenished every 4 days.
Viability Staining
Microglia were trypsinized and 4 × 104 cells were stained with ViaCount Reagent (Millipore) and analyzed with Guava ViaCount software (Millipore). The percentages of nucleated and viable cells were graphed. Heat-killed cells were used as positive control.
Statistical Analysis
Non-parametric tests, either Mann–Whitney U unpaired (two-tailed) or paired Wilcoxon’s signed rank test (two-tailed) where appropriate, were performed with SPSS (IBM). p values less than or equal to .05 were considered significant and are annotated on the graphs.
Results
Development of Senescence-like Phenotype in Microglia During HIV-1 Infection
To determine whether microglia develop a senescence or senescence-like phenotype during HIV-1 infection, we examined various senescence markers including SA-β-gal activity, expression of cell cycle regulators including p21, p53, and p16, as well as 53BP1 foci formation. Microglia were incubated for 8 days with VSVg-pseudotyped HIV-1, fresh media and transfection controls containing VSVg only, or the pNL43 pro-viral backbone only, and then stained for SA-β-gal activity. Cells were also treated with NCS at 0.5–1 µg/mL, a DNA damage-inducing agent (27), as a positive control for induction of senescence. Images were analyzed and the percentages of cells positive for SA-β-gal activity, as indicated by blue color development, were calculated. As shown in Figure 1A, microglia exposed to HIV-1 pseudotypes developed increased SA-β-gal activity, similar to that induced by treatment with NCS, while this was not observed in untreated cells or in those exposed to VSV-G or pNL43 controls. Using cells from multiple cases, we found that microglia infected with VSVg-pseudotyped HIV-1 showed significantly higher percentage of positive cells for SA-β-gal activity (median proportion of positive cells: 25.1%) compared to various controls (untreated: 10.3%; pNL43: 14.6%; VSVg: 10.7%), and similar to that induced by NCS (32.3%) (Figure 1B). We did not observe any gross morphological changes, other than some enlargement of the cells in the cultures becoming senescent, nor the finer cytoplasmic or dendritic changes typical of dystrophic microglia seen in aging brains (7). However, the increase in SA-β-gal activity post-HIV-1 infection suggests that either infection itself, and/or the exposure to soluble factors associated with the viral infection, could potentially result in the development of changes consistent with cellular senescence in microglia.
Figure 1.

Senescence biomarkers in microglia post-HIV-1 infection. SA-β-gal activity was detected in human fetal microglia (HFM) exposed to HIV-1 pseudotypes or controls. Representative pictures of microglia cultures (A) and summarized data from multiple cases (B), demonstrate increased SA-β-gal activity in microglia post-HIV-1 infection. Total of eight cases were used but not all experimental conditions could be done with all cases [group size: untreated, n = 8; pNL43, n = 4; VSVg, n = 5; VSVg pNL43, n = 7; Neocarzinostatin (NCS), n = 3]. Black bars represent the median for each experimental group. P values were calculated using the non-parametric Wilcoxon’s signed rank test (two-tailed). Representative Western blot for detection of p53, p21, and p16 levels, and GAPDH as loading control, in microglia (C). Densitometry values were normalized to the loading control and then expressed as fold-change with respect to untreated cells. Summarized Western blot data for p21 expression from multiple cases (D). Total of seven cases were used, but not all experimental conditions could be done with all cases (group size: untreated, n = 7; VSVg, n = 6; pNL43, n = 3; VSVg pNL43, n = 7; NCS, n = 4). Black bars represent the median for each experimental group. 53BP1 foci formation was evaluated post-HIV-1 infection (E). Immunofluorescence staining for DNA damage foci was performed, and 53BP1 (red) and nuclei marker DAPI (blue) are shown. Viability data of multiple microglia cases for each experimental group (F). The percentages of nucleated and viable cells were measured in singlicate (single measurement per condition per case) and are shown as mean ± SD. Heat-killed cells were used as positive control. Nucleosides (NS) were used at 250 nM. NCS concentrations were 1.0 µg/mL (HFM22-24) or 0.5 µg/mL (HFM25, 26).
To perform a more comprehensive analysis of the potential senescence phenotype in microglia, protein levels of the cell cycle regulators p16, p53, and p21 were analyzed upon incubation with VSVg-pseudotyped HIV-1 or controls. Increased levels of p16, p53, and/or p21 have been observed during the development of cellular senescence in multiple cell types. Similarly, we found that, in the context of HIV-1 infection, microglia usually had higher levels of p21 and p53, but not p16, than the untreated cells or those exposed to controls (representative Western blot, Figure 1C). With cells from multiple cases, we found that microglia exhibited significantly elevated p21 levels as compared to untreated (median 3.14-fold increase) and to VSVg (median 1.14-fold increased over untreated) controls (Figure 1D). Although with HIV-1 infection microglia also express higher levels of p21 compared to the pNL43 control (median 0.95-fold change with respect to untreated), the difference did not reach statistical significance. No statistically significant differences were found in p53 and p16 levels (data not shown). These results seem to suggest that HIV-1 infection might induce microglia senescence through the p21 pathway and not the p16-mediated signaling pathway. This is further supported by the observation of elevated 53BP1 foci formation in microglia in the context of HIV-1 infection (Figure 1E; Supplementary Figure 2), which often indicates the activation of DNA damage response upstream of the p21 pathway (31). 53BP1 foci formation was not observed in cells exposed to VSV-G or pNL43 controls (data not shown). It is known that HIV-1 infection induces a DNA damage response that is needed to complete its viral life cycle, and 53BP1, a DNA damage checkpoint factor required for DNA damage repair, is often observed in senescent cells (32,33). Despite the markers of DNA damage, which could result in cell death, microglia retained viability comparable to untreated controls in all the experimental treatments (Figure 1F). Overall, elevated SA-β-gal activity, increased p21 expression and formation of 53BP1 foci all support the development of a senescence phenotype in microglia during HIV-1 infection.
Elevated Mitochondrial ROS Production is Associated with Microglia Senescence
It is well established that there is an association between mitochondrial dysfunction and the onset and maintenance of cellular senescence in multiple cell types (22,23). To explore whether mitochondrial function in microglia is altered in the context of cellular senescence associated with HIV-1 infection, we measured the mitochondrial ROS production post-HIV-1 infection. Microglia were infected with HIV-1 or exposed to controls and then incubated with MitoSOX at 10 days post-treatment, and analyzed by flow cytometry. Microglia were gated based on size and cellularity (Figure 2A) and showed that HIV-1 infected cells had higher mean fluorescence intensity (MFI) than untreated, VSVg and pNL43 controls (Figure 2B shows a representative case). With cells from multiple cases, we found that in the context of HIV-1 infection, microglia had significantly elevated mitochondrial ROS (median MFI: 53) compared to untreated cells (median MFI: 26) (Figure 2C). Although infected cells from several independent cases also had higher MFI than the corresponding VSVg (median: 38) and pNL43 (median: 47.8) controls, the differences were not statistically significant, likely due to the smaller sample size in the VSVg and pNL43 controls, which were not significantly different from the untreated cells.
Figure 2.

Elevated mitochondrial ROS production post-HIV-1 infection. Flow cytometry was used to analyze MitoSOX staining intensity. Microglia was gated based on size and cellularity (A). Representative histogram of fluorescence intensity for MitoSOX staining (B) shows unstained controls (dotted lines) and stained samples (solid lines) of various treatment groups. Summarized data from multiple cases (C) show increased mean fluorescence intensity in the context of infection compared to controls. Black bars represent the median for each treatment group. P values were calculated using the non-parametric Wilcoxon’s signed rank test (two-tailed).
HIV-1 Infection Alters Mitochondrial Respiration
Since the majority of mitochondrial ROS is generated by the ETC, it is possible that microglia in the context of HIV-1 infection might present with an altered ETC homeostasis. Thus, we decided to assess mitochondrial ETC function. The OCR were measured in real time under basal condition and when challenged with inhibitors of various ETC complexes including oligomycin, FCCP, and antimycine A/rotenone, and were compared between HIV-1 infection and control cells. Basal respiration, ATP-linked respiration, proton leak, maximal respiration, and spare respiration were calculated based on OCR, as previously described (29) (Supplementary Figure 1). The non-mitochondrial respiration was reduced to similar levels for infected as well as VSVg and pNL43 control-treated cells, suggesting that the supernatants from transfected 293T cells can reduce cellular respiration that is not dependent on mitochondria (Figure 3A). Basal respiration in VSVg-pNL43 infected cells was significantly lower than in VSVg control, and also lower than in pNL43 and untreated controls, but without statistical significance (Figure 3B). ATP-linked respiration in VSVg-pNL43 infected cells was significantly lower than in untreated and VSVg controls, and also lower than in the pNL43 control, but without statistical significance (Figure 3C). Maximal respiration was only significantly reduced in VSVg-pNL43 infected cells when compared to the VSVg control, and did not differ from untreated or pNL43 controls (Figure 3D). No significant differences were found in proton leak between the controls and infected microglia (Figure 3E). Finally, spare respiration (or reserve capacity) seemed to be reduced in pNL43 control and infected cells, compared to untreated and VSVg control, but the differences were not statistically significant (Figure 3F). Reduced basal, ATP-linked, and maximal respiration in the context of HIV-1 infection of microglia, compared to at least some of the controls, and combined with the observed increase in mitochondrial ROS, seem to suggest an impaired mitochondrial function, as well as mitochondrial-linked cellular metabolism, in microglia in the context of HIV-1 infection.
Figure 3.

Altered mitochondrial respiration post-HIV-1 infection. Mitochondrial ETC was measured using a Seahorse Bioanalyzer and oxygen consumption rate (OCR) normalized by cell number (pmoles/min per 105 cells) was plotted as a function of time (minutes) (A). Black arrows from left to right indicate addition of 5 µM oligomycin, 4 µM FCCP, and 1.8 µM rotenone/antimycin A. Each data point represents the average of multiple cases (N = 5–7). Basal respiration (B), ATP-linked respiration (C), maximal respiration (D), proton leak (E), and spare respiration (F) were calculated (as described in the text and also shown in Supplementary Figure 1) and plotted for each experimental group. Black bars represent the median for each treatment group. p values were calculated using the non-parametric Wilcoxon’s signed rank test (two-tailed).
Secretory Phenotype of Microglia in the Context of HIV-1 Infection
Senescent cells usually develop a unique, cell type-specific secretory phenotype, known as SASP, which both serves as a marker of cellular senescence and also plays an important role in the propagation of functional changes associated with senescence (34,35). To examine whether the secretory profile by microglia during HIV-1 infection could be related with cellular senescence, supernatants from three cases of HIV-1-infected senescent microglia, and their respective non-senescent untreated controls, were analyzed using a membrane-based antibody array. Thirteen soluble factors were found to be increased by at least twofold in senescent vs. non-senescent cells, in all three cases studied (Figure 4A). These factors included cytokines IL-6, IL-7, and macrophage migration inhibitory factor (MIF), chemokines IL-8 (CXCL8), and growth-regulated oncogene (GRO)-α,β,γ (CXCL1-3), and other growth factors and regulators such as vascular endothelial growth factor A (VEGF-A), granulocyte-macrophage colony-stimulating factor (GM-CSF), and tissue inhibitor of metalloproteinases 2 (TIMP2), which have been classically associated with the development of cellular senescence (34). IL-6 and IL-8 were present in non-senescent supernatants but increased to higher levels post-infection, while others were absent in untreated cells but secreted at detectable albeit low levels post-infection. To quantitatively confirm the levels of secreted cytokines, we measured protein concentrations of secreted IL-8 using ELISA. Using supernatants from seven cases, IL-8 levels were found to be significantly higher in microglia post-infection than in untreated, VSVg and pNL43 controls (Figure 4B). Overall, it seems that the secretory profile of microglia post-HIV-1 infection could potentially reflect the induction of senescence and be consistent with a classical SASP.
Figure 4.

SASP and effects of nucleosides (NS) treatment. The secretory profile post-HIV-1 infection was investigated with an antibody array (A), and the plot shows the fold changes in densitometry values between HIV-1 infection and untreated control for 13 soluble products that increased (>twofold) in all three cases studied. IL-8 levels were also tested by ELISA in all experimental conditions for seven cases (B). Black bars represent the median for each treatment group. p values were calculated using the non-parametric Wilcoxon’s signed rank test (two-tailed). Representative western blots for detection of p21 and Caveolin-1 levels in naïve microglia exposed to supernatants from infected or control cells (C), and p21 in microglia infected or exposed to controls (D), with GAPDH as loading control, show that 250 nM NS treatment reduces p21 and Caveolin-1 levels post-exposure to supernatants from infected cells, and p21 levels post-infection. NS treatment also result in a reduced IL-8 production in microglia post-infection (E), as measured by ELISA and shown as mean ± SD. Luciferase activity in microglia infected or treated with controls (mean ± SD), shows that there is no difference in infectivity with and without NS treatment (F).
SASP-Induced Senescence-like Phenotype Is Attenuated with Nucleoside Treatment
It is known that components of SASP can both reinforce cellular senescence in cells that are already senescent as well as initiate or promote cellular senescence in nonsenescent cells (34,35). We decided to examine whether supernatants from senescent microglia could induce senescence in uninfected, naïve microglia. In addition to p21, we also examined Caveolin (Cav)-1 levels, which has been shown to be elevated in various diseases associated with organismal aging and to regulate cellular senescence through activation of the p53 pathway (36), in naïve microglia following treatment with conditioned media from HIV-1 infected, senescent microglia, and from untreated, non-senescent control. Both p21 and Cav-1 levels were higher in microglia exposed to supernatants from HIV-1 infected, senescent microglia, compared to supernatants from untreated, non-senescent cells (Figure 4C). This result suggests that molecules secreted by senescent microglia post-HIV-1 infection could lead or contribute to the induction of cellular senescence in neighboring cells.
It was previously reported that a decrease in deoxyribonucleotide triphosphate (dNTP) levels underlies oncogene-induced stable senescence-associated cell growth arrest (30), and that it was caused by oncogene-induced repression of ribonucleotide reductase subunit M2 (RRM2), a rate-limiting protein in dNTP synthesis. Furthermore, RRM2 repression and knockdown were reported to correlate with, and to drive, respectively, senescence of melanoma cells. Importantly, treatment with nucleosides was shown to prevent and/or reverse the oncogene-induced cellular senescence (30). In addition, p21 seems to block dNTP biosynthesis (37). Thus, we tested the potential beneficial effects of nucleoside treatment in the senescence-like phenotype in microglia. Interestingly, the increases in p21 and Cav-1 levels observed in naïve microglia exposed to supernatants from HIV-1 infected, senescent microglia, were attenuated when the naïve cells were also treated with nucleosides post-exposure to the conditioned medium (Figure 4C). We also tested whether nucleoside treatment could affect the induction of microglia senescence during infection, by exposing cells simultaneously to VSVg-pseudotyped HIV-1 (or controls) and nucleosides. Although nucleoside treatment by itself resulted in slightly elevated p21 levels in microglia, the combination of nucleoside treatment with HIV-1 infection resulted in a large decrease in the levels of p21, as compared to those present in the context of infection alone (Figure 4D). Furthermore, 250 nM nucleoside treatment also reduced IL-8 secretion by microglia during HIV-1 infection (Figure 4E). The partial reversal of microglia senescence-associated phenotypes with nucleoside treatment was not due to reduced pseudotype infectivity, since luciferase activity indicative of infection was not altered (Figure 4F). Overall, these results suggest that nucleoside treatment could partially reverse selected phenotypes of HIV-1 infection-induced cellular senescence in microglia and potentially attenuate bystander effects of factors secreted by senescent microglia.
Discussion
Chronic HIV-1-infected individuals display an accelerated aging compared to their uninfected counterparts (38), and this has been linked to changes in age-associated parameters including altered DNA methylation in blood and brain tissue (39,40), reduced telomere stability (41,42) and elevated immune activation (43). Accumulation of senescent cells is known to contribute to tissue/organ dysfunction and organismal aging. Thus, it is possible that development of cellular senescence in multiple cell types may be another factor underlying this accelerated biological aging in HIV-1 infected patients. However, a direct contribution of infection to this phenotype has not been established. In human fetal microglia, we have found that markers of cellular senescence, including an increase in SA-β-gal activity, higher levels of the cell cycle regulatory protein p21, and formation of DNA damage-associated foci of 53BP1 protein, develop after infection with single-round HIV-1 pseudotypes, suggesting the onset of cellular senescence. An increase in p21 protein levels during HIV-1 infection has been previously observed in primary human CD4+ T cells (44). Although it might have been expected that p16 would be also increased in the context of HIV-1 infection, it is known that p21 is critical to the establishment of senescence (45), and it is possible that human fetal microglia, in the context of HIV-1 infection, respond preferentially through this pathway. p21 expression is known to be regulated by the transcription factor p53, but we did not find a consistent induction of p53 in microglia post-HIV-1 infection at the time of our analyses (days 8–10 post-infection). Garden et al. (21) demonstrated increased immunoreactivity of p53 in the nucleus of neurons, microglia, and astrocytes, in human brain tissues from patients with HIV-associated dementia, the most severe form of HAND, suggesting an involvement of increased activation of the p53 signaling pathway. They also showed that p53 activation in murine microglia is required for gp120-induced neurotoxicity. It is possible that p53 might have been transiently increased earlier after infection, and had returned to basal levels at the time of our studies, especially considering that its levels need to be tightly regulated and it has been shown that it is quickly degraded due to its naturally unstable structure (46). Given the important role of p53 in regulating HIV-1 Tat activities, and subsequently HIV-1 viral replication (47), it would be of interest to examine the kinetics of p53 in microglia upon HIV-1 infection, and its potential roles in modulation of viral replication, p21 induction, and cellular senescence. It is possible that activation of cellular senescence subsequent to HIV-1 infection creates a cellular environment unfavorable for active viral replication through, at least in part, inactivation of Tat, perhaps leading to viral latency. This would be of great interest especially considering the potential role of microglia as a cellular reservoir for HIV-1 in the post-cART era. If true, HIV-1-induced cellular senescence could be a potential target for suppressing latent reservoirs and ultimately eradicating HIV-1.
Although these experiments were performed with fetal microglia due to the limited availability of adult microglia, a recent transcriptome profiling study showed a striking similarity between primary human fetal and adult microglia, and induced pluripotent stem cell-derived microglia-like cells (48). This could suggest that similar effects might have been observed with human adult microglia, albeit this remains to be investigated. In addition, our studies were performed with VSV-g pseudotyped, single-round infectious virions, instead of fully infectious viruses. This allowed us to facilitate microglia infection by-passing the limitation of, and potential differences in, receptor availability in primary cells, and to avoid the continuous production of new virions resulting in new infections throughout the culture period. It is possible that the effects we describe upon single-round infection in microglia might be compounded in the context of fully infectious HIV-1, which could result in an increase in the number of infected cells, over time, in the in vitro culture. The pseudotypes we used also lack the viral protein Nef, which is dispensable for in vitro infection, but could play a role in the microglia response to infection. These are important experiments that need to be performed to further characterize the changes induced in microglia during HIV-1 infection.
Mitochondrial dysfunction is closely associated with the development of cellular senescence (22,23,35), and interventions that impact key aspects of metabolism have the potential to delay cellular senescence and to extend cellular lifespan (49). Altered mitochondrial-mediated energy metabolism including reduced ATP synthesis could contribute to the progression of cellular senescence. We observed elevated mitochondrial ROS production, as well as reduced mitochondrial respiration linked to ATP production, in microglia during HIV-1 infection, both of which are aspects of mitochondrial dysfunction known to contribute to the development of cellular senescence. These changes in mitochondrial function are also consistent with results obtained with samples from HIV-1-infected patients. Compromised mitochondrial ETC function has been observed in peripheral blood mononuclear cells from both treated and treatment-naïve patients (50,51), and in the brains of patients with HAND, abnormal mitochondrial morphology, and altered mitochondrial dynamics have been reported (52). Thus, it is plausible that mitochondrial dysfunction in senescent microglia during HIV-1 infection may play a role in the development and/or progression of HAND.
SASP development is a central aspect of cellular senescence. The most classic cytokines of SASP include IL-6, IL-8, and IL-1α. In particular, IL-1α has been shown to be a key component of SASP regulating IL-6/IL-8 secretion during cellular senescence (53,54). However, despite finding increased levels of IL-6 and IL-8, among other cytokines, chemokines and soluble factors produced by senescent microglia, we did not detect IL-1α production into the microglia supernatants post-HIV-1 infection. Recent evidence demonstrates, however, that mitochondrial dysfunction seems to induce an IL-1α-independent SASP profile that is distinct from that induced by DNA damage (55). Since mitochondrial dysfunction was observed in microglia during HIV-1 infection, although it is unclear whether it is a cause or effect, it is possible that the SASP profile of microglia senescence subsequent to HIV-1 infection may not involve IL-1α production. The exact of role of mitochondrial dysfunction in the development of the microglia senescence phenotype during HIV-1 infection needs to be clarified in future studies. Furthermore, it would of interest to distinguish between the effects of DNA damage and mitochondrial dysfunction, both of which are observed in microglia during HIV-1 infection, with regard to mechanistic pathways underlying SASP development.
The observation that naïve microglia, when exposed to supernatants from HIV-1-infected, senescent microglia, present markers of senescence, seems to suggest that soluble factors released by senescent microglia into the extracellular environment can promote cellular senescence in other cells. In this in vitro study this is observed in the context of single-round infections with HIV-1 pseudotypes, thus in the absence of new infections. However, infected cells may release not only SASP components, but also viral proteins that might potentially contribute to this phenotype. In this sense, microglia exposed to the pNL43 control (supernatants from cells transfected with the pNL43 backbone construct alone) showed small changes in various experiments, such as in SA-β-gal activity and in mitochondrial ROS production and respiration. However, because this construct expresses all viral proteins except Env and Nef, viral-like particles may be produced and some viral proteins may be released as well into the supernatants, and could potentially have an effect on microglia. Further experiments would be required to determine if endocytosis of viral-like particles, or specific viral proteins, could mediate some of the observed effects in microglia. In addition, this may have physiological significance, since despite a potentially small proportion of microglia being likely infected in vivo in HIV-1-infected patients, the subsequent effects of such a limited infection could still be broad and long-lasting. Additionally, the elevated Cav-1 levels in cells exposed to SASP could have significant ramifications with regards to HIV-1 infection and replication, since it is known that HIV-1 infection affects Cav-1 processing and conversely Cav-1 restricts HIV-1 infection (56). Finally, Cav-1 is being increasingly shown to modulate multiple aspects of microglial function including amyloid beta phagocytosis, activation, and mitochondrial respiration (57,58). Thus, the effects of Cav-1 induction in naïve microglia by supernatants from senescent microglia need to be further investigated to define their own potential senescence phenotype and functional alterations, which might potentially contribute to HAND development.
It is also interesting the finding that addition of nucleosides to microglia has a beneficial effect in terms of p21 induction and IL-8 secretion post-infection, and on p21 and Cav-1 induction in naïve microglia exposed to culture medium from senescent microglia. These results seem to suggest that nucleoside treatment partially counteracts the signals that lead to the development of senescence. Future studies are required to elucidate the mechanism(s) of action of the addition of nucleosides, which can be taken up across the cell membrane through nucleoside transporter proteins (59). Considering the impairment of ATP-linked mitochondrial respiration in microglia during HIV-1 infection, it is possible that impaired ATP production may contribute to the development of microglia senescence. As a result, addition of the nucleoside adenosine, which is an ATP precursor, might result in a beneficial effect by preserving ATP levels and preventing or ameliorating the senescence phenotype. Future studies should therefore examine whether adenosine independently can result in prevention or improvement of the senescence phenotype in microglia. Furthermore, since adenosine is also an important neurotransmitter regulating various microglial functions (eg, phagocytosis and migration) through activation of purinergic receptors expressed on the surface of human microglia (60), it will be important to clarify whether adenosine may modulate microglial phenotypes through direct activation of purinergic receptors instead of, or in addition to, contributing to the synthesis of intracellular ATP.
Overall, our studies have shown functional alterations in microglia during HIV-1 infection that suggests the development of a senescence-like phenotype (Supplementary Figure 3). Caldeira et al. (61) found an age-like phenotype in neonatal microglia after 16 days in vitro. Our studies were performed at 8–10 days post-infection, and the differences observed between HIV-1 infection and the controls seem to suggest that, even though a contribution of the time in culture might be possible, the reported phenotypes are related to the presence of HIV-1 infection. How other microglia functions such as autophagy, cell migration, or phagocytosis may be affected, remains to be investigated. These alterations could be an additional mechanism contributing to the accelerated aging phenotype of HIV-1 infected patients, and could potentially play a role in the development of HAND during chronic HIV-1 infection.
Supplementary Material
Supplementary data is available at The Journals of Gerontology, Series A: Biological Sciences and Medical Sciences online.
Funding
This work was supported in part by the National Institutes of Health grants NS065727, AI098549, and AG051296 (to J.M.G.), and NS078283, NS078283-S1, and AG046943 (to C.T.); and by internal Drexel funds. N.C.C. has been partially supported by the National Institutes of Health grant T32 MH079785, Interdisciplinary and Translational Research Training in NeuroAIDS. A.T.P. has been partially supported by a Drexel University Aging Initiative Fellowship.
Conflict of Interest
The authors declare that no competing interest exists.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
We are grateful to Dr. Rugang Zhang (The Wistar Institute, Philadelphia, PA) for providing the nucleosides cocktail used to treat microglia.
References
- 1. Chan P, Brew BJ. HIV associated neurocognitive disorders in the modern antiviral treatment era: Prevalence, characteristics, biomarkers, and effects of treatment. Curr HIV/AIDS Rep. 2014;11:317–324. doi:10.1007/s11904-014-0221-0 [DOI] [PubMed] [Google Scholar]
- 2. Grant I, Franklin DR Jr, Deutsch R et al. ; CHARTER Group Asymptomatic HIV-associated neurocognitive impairment increases risk for symptomatic decline. Neurology. 2014;82:2055–2062. doi:10.1212/WNL.0000000000000492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. González-Scarano F, Martín-García J. The neuropathogenesis of AIDS. Nat Rev Immunol. 2005;5:69–81. doi:10.1038/nri1527 [DOI] [PubMed] [Google Scholar]
- 4. Chen NC, Partridge AT, Sell C, Torres C, Martín-García J. Fate of microglia during HIV-1 infection: From activation to senescence?Glia. 2017;65:431–446. doi:10.1002/glia.23081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schuitemaker A, van der Doef TF, Boellaard R et al. Microglial activation in healthy aging. Neurobiol Aging. 2012;33:1067–1072. doi:10.1016/j.neurobiolaging.2010.09.016 [DOI] [PubMed] [Google Scholar]
- 6. Wong WT. Microglial aging in the healthy CNS: Phenotypes, drivers, and rejuvenation. Front Cell Neurosci. 2013;7:22. doi:10.3389/fncel.2013.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Streit WJ, Sammons NW, Kuhns AJ, Sparks DL. Dystrophic microglia in the aging human brain. Glia. 2004;45:208–212. doi:10.1002/glia.10319 [DOI] [PubMed] [Google Scholar]
- 8. Bisht K, Sharma KP, Lecours C et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia. 2016;64:826–839. doi:10.1002/glia.22966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Muñoz-Espín D, Serrano M. Cellular senescence: From physiology to pathology. Nat Rev Mol Cell Biol. 2014;15:482–496. doi:10.1038/nrm3823 [DOI] [PubMed] [Google Scholar]
- 10. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi:10.1083/jcb.201009094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhat R, Crowe EP, Bitto A et al. Astrocyte senescence as a component of Alzheimer’s disease. PLoS One. 2012;7:e45069. doi:10.1371/journal.pone.0045069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chesnokova V, Wong C, Zonis S et al. Diminished pancreatic beta-cell mass in securin-null mice is caused by beta-cell apoptosis and senescence. Endocrinology. 2009;150:2603–2610. doi:10.1210/en.2008-0972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chinta SJ, Lieu CA, Demaria M, Laberge RM, Campisi J, Andersen JK. Environmental stress, ageing and glial cell senescence: A novel mechanistic link to Parkinson’s disease?J Intern Med. 2013;273:429–436. doi:10.1111/joim.12029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Minagawa S, Araya J, Numata T et al. Accelerated epithelial cell senescence in IPF and the inhibitory role of SIRT6 in TGF-β-induced senescence of human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2011;300:L391–L401. doi:10.1152/ajplung.00097.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Minamino T, Orimo M, Shimizu I et al. A crucial role for adipose tissue p53 in the regulation of insulin resistance. Nat Med. 2009;15:1082–1087. doi:10.1038/nm.2014 [DOI] [PubMed] [Google Scholar]
- 16. Sone H, Kagawa Y. Pancreatic beta cell senescence contributes to the pathogenesis of type 2 diabetes in high-fat diet-induced diabetic mice. Diabetologia. 2005;48:58–67. doi:10.1007/s00125-004-1605-2 [DOI] [PubMed] [Google Scholar]
- 17. Jurk D, Wang C, Miwa S et al. Postmitotic neurons develop a p21-dependent senescence-like phenotype driven by a DNA damage response. Aging Cell. 2012;11:996–1004. doi:10.1111/j.1474-9726.2012.00870.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cohen J, D’Agostino L, Wilson J, Tuzer F, Torres C. Astrocyte senescence and metabolic changes in response to HIV antiretroviral therapy drugs. Front Aging Neurosci. 2017;9:281. doi:10.3389/fnagi.2017.00281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Yu C, Narasipura SD, Richards MH, Hu XT, Yamamoto B, Al-Harthi L. HIV and drug abuse mediate astrocyte senescence in a β-catenin-dependent manner leading to neuronal toxicity. Aging Cell. 2017;16:956–965. doi:10.1111/acel.12593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jayadev S, Yun B, Nguyen H, Yokoo H, Morrison RS, Garden GA. The glial response to CNS HIV infection includes p53 activation and increased expression of p53 target genes. J Neuroimmune Pharmacol. 2007;2:359–370. doi:10.1007/s11481-007-9095-x [DOI] [PubMed] [Google Scholar]
- 21. Garden GA, Guo W, Jayadev S et al. HIV associated neurodegeneration requires p53 in neurons and microglia. FASEB J. 2004;18:1141–1143. doi:10.1096/fj.04-1676fje [DOI] [PubMed] [Google Scholar]
- 22. Correia-Melo C, Passos JF. Mitochondria: Are they causal players in cellular senescence?Biochim Biophys Acta. 2015;1847:1373–1379. doi:10.1016/j.bbabio.2015.05.017 [DOI] [PubMed] [Google Scholar]
- 23. Ziegler DV, Wiley CD, Velarde MC. Mitochondrial effectors of cellular senescence: Beyond the free radical theory of aging. Aging Cell. 2015;14:1–7. doi:10.1111/acel.12287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Melief J, Koning N, Schuurman KG et al. Phenotyping primary human microglia: Tight regulation of LPS responsiveness. Glia. 2012;60:1506–1517. doi:10.1002/glia.22370 [DOI] [PubMed] [Google Scholar]
- 25. Ramakrishnan MA. Determination of 50% endpoint titer using a simple formula. World J Virol. 2016;5:85–86. doi:10.5501/wjv.v5.i2.85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bitto A, Sell C, Crowe E et al. Stress-induced senescence in human and rodent astrocytes. Exp Cell Res. 2010;316:2961–2968. doi:10.1016/j.yexcr.2010.06.021 [DOI] [PubMed] [Google Scholar]
- 27. Hewitt G, Jurk D, Marques FD et al. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012;3:708. doi:10.1038/ncomms1708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Mukhopadhyay P, Rajesh M, Haskó G, Hawkins BJ, Madesh M, Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nat Protoc. 2007;2:2295–2301. doi:10.1038/nprot.2007.327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rose S, Frye RE, Slattery J et al. Oxidative stress induces mitochondrial dysfunction in a subset of autism lymphoblastoid cell lines in a well-matched case control cohort. PLoS One. 2014;9:e85436. doi:10.1371/journal.pone.0085436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Aird KM, Zhang G, Li H et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep. 2013;3:1252–1265. doi:10.1016/j.celrep.2013.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lossaint G, Besnard E, Fisher D, Piette J, Dulić V. Chk1 is dispensable for G2 arrest in response to sustained DNA damage when the ATM/p53/p21 pathway is functional. Oncogene. 2011;30:4261–4274. doi:10.1038/onc.2011.135 [DOI] [PubMed] [Google Scholar]
- 32. Roshal M, Kim B, Zhu Y, Nghiem P, Planelles V. Activation of the ATR-mediated DNA damage response by the HIV-1 viral protein R. J Biol Chem. 2003;278:25879–25886. doi:10.1074/jbc.M303948200 [DOI] [PubMed] [Google Scholar]
- 33. d’Adda di Fagagna F, Reaper PM, Clay-Farrace L et al. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi:10.1038/nature02118 [DOI] [PubMed] [Google Scholar]
- 34. Coppé JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: The dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi:10.1146/annurev-pathol-121808-102144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Salama R, Sadaie M, Hoare M, Narita M. Cellular senescence and its effector programs. Genes Dev. 2014;28:99–114. doi:10.1101/gad.235184.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zou H, Stoppani E, Volonte D, Galbiati F. Caveolin-1, cellular senescence and age-related diseases. Mech Ageing Dev. 2011;132:533–542. doi:10.1016/j.mad.2011.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Allouch A, David A, Amie SM et al. p21-mediated RNR2 repression restricts HIV-1 replication in macrophages by inhibiting dNTP biosynthesis pathway. Proc Natl Acad Sci USA. 2013;110:E3997–E4006. doi:10.1073/pnas.1306719110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Desquilbet L, Jacobson LP, Fried LP et al. HIV-1 infection is associated with an earlier occurrence of a phenotype related to frailty. J Gerontol A Biol Sci Med Sci. 2007;62:1279–1286. [DOI] [PubMed] [Google Scholar]
- 39. Rickabaugh TM, Baxter RM, Sehl M et al. Acceleration of age-associated methylation patterns in HIV-1-infected adults. PLoS One. 2015;10:e0119201. doi:10.1371/journal.pone.0119201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Horvath S, Levine AJ. HIV-1 infection accelerates age according to the epigenetic clock. J Infect Dis. 2015;212:1563–1573. doi:10.1093/infdis/jiv277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu JC, Leung JM, Ngan DA et al. Absolute leukocyte telomere length in HIV-infected and uninfected individuals: Evidence of accelerated cell senescence in HIV-associated chronic obstructive pulmonary disease. PLoS One. 2015;10:e0124426. doi:10.1371/journal.pone.0124426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chou JP, Ramirez CM, Wu JE, Effros RB. Accelerated aging in HIV/AIDS: Novel biomarkers of senescent human CD8+ T cells. PLoS One. 2013;8:e64702. doi:10.1371/journal.pone.0064702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Martin GE, Gouillou M, Hearps AC et al. Age-associated changes in monocyte and innate immune activation markers occur more rapidly in HIV infected women. PLoS One. 2013;8:e55279. doi:10.1371/journal.pone.0055279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Guha D, Mancini A, Sparks J, Ayyavoo V. HIV-1 infection dysregulates cell cycle regulatory protein p21 in CD4+ T cells through miR-20a and miR-106b regulation. J Cell Biochem. 2016;117:1902–1912. doi:10.1002/jcb.25489 [DOI] [PubMed] [Google Scholar]
- 45. Stein GH, Drullinger LF, Soulard A, Dulić V. Differential roles for cyclin-dependent kinase inhibitors p21 and p16 in the mechanisms of senescence and differentiation in human fibroblasts. Mol Cell Biol. 1999;19:2109–2117. doi:10.1128/MCB.19.3.2109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tsvetkov P, Reuven N, Shaul Y. Ubiquitin-independent p53 proteasomal degradation. Cell Death Differ. 2010;17:103–108. doi:10.1038/cdd.2009.67 [DOI] [PubMed] [Google Scholar]
- 47. Yoon CH, Kim SY, Byeon SE et al. p53-derived host restriction of HIV-1 replication by protein kinase R-mediated Tat phosphorylation and inactivation. J Virol. 2015;89:4262–4280. doi:10.1128/JVI.03087-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Abud EM, Ramirez RN, Martinez ES et al. iPSC-derived human microglia-like cells to study neurological diseases. Neuron. 2017;94:278–293.e9. doi:10.1016/j.neuron.2017.03.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Nacarelli T, Sell C. Targeting metabolism in cellular senescence, a role for intervention. Mol Cell Endocrinol. 2017;455:83–92. doi:10.1016/j.mce.2016.08.049 [DOI] [PubMed] [Google Scholar]
- 50. Miró O, López S, Pedrol E et al. Mitochondrial DNA depletion and respiratory chain enzyme deficiencies are present in peripheral blood mononuclear cells of HIV-infected patients with HAART-related lipodystrophy. Antivir Ther. 2003;8:333–338. [PubMed] [Google Scholar]
- 51. Miró O, López S, Martínez E et al. Mitochondrial effects of HIV infection on the peripheral blood mononuclear cells of HIV-infected patients who were never treated with antiretrovirals. Clin Infect Dis. 2004;39:710–716. doi:10.1086/423176 [DOI] [PubMed] [Google Scholar]
- 52. Fields JA, Serger E, Campos S et al. HIV alters neuronal mitochondrial fission/fusion in the brain during HIV-associated neurocognitive disorders. Neurobiol Dis. 2016;86:154–169. doi:10.1016/j.nbd.2015.11.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci USA. 2009;106:17031–17036. doi:10.1073/pnas.0905299106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McCarthy DA, Clark RR, Bartling TR, Trebak M, Melendez JA. Redox control of the senescence regulator interleukin-1α and the secretory phenotype. J Biol Chem. 2013;288:32149–32159. doi:10.1074/jbc.M113.493841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wiley CD, Velarde MC, Lecot P et al. Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell Metab. 2016;23:303–314. doi:10.1016/j.cmet.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mergia A. The role of caveolin 1 in HIV infection and pathogenesis. Viruses. 2017;9:129. doi:10.3390/v9060129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Niesman IR, Zemke N, Fridolfsson HN et al. Caveolin isoform switching as a molecular, structural, and metabolic regulator of microglia. Mol Cell Neurosci. 2013;56:283–297. doi:10.1016/j.mcn.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jang SK, Yu JM, Kim ST et al. An Aβ42 uptake and degradation via Rg3 requires an activation of caveolin, clathrin and Aβ-degrading enzymes in microglia. Eur J Pharmacol. 2015;758:1–10. doi:10.1016/j.ejphar.2015.03.071. [DOI] [PubMed] [Google Scholar]
- 59. King AE, Ackley MA, Cass CE, Young JD, Baldwin SA. Nucleoside transporters: from scavengers to novel therapeutic targets. Trends Pharmacol Sci. 2006;27:416–425. doi:10.1016/j.tips.2006.06.004 [DOI] [PubMed] [Google Scholar]
- 60. Koizumi S, Ohsawa K, Inoue K, Kohsaka S. Purinergic receptors in microglia: functional modal shifts of microglia mediated by P2 and P1 receptors. Glia. 2013;61:47–54. doi:10.1002/glia.22358 [DOI] [PubMed] [Google Scholar]
- 61. Caldeira C, Oliveira AF, Cunha C et al. Microglia change from a reactive to an age-like phenotype with the time in culture. Front Cell Neurosci. 2014;8:152. doi:10.3389/fncel.2014.00152 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.